

ABSTRACT
Ribosome biogenesis is an energy consuming process which takes place mainly in the nucleolus. By producing ribosomes to fuel protein synthesis, it is tightly connected with cell growth and cell cycle control. Perturbation of ribosome biogenesis leads to the activation of p53 tumor suppressor protein promoting processes like cell cycle arrest, apoptosis or senescence. This ribosome biogenesis stress pathway activates p53 through sequestration of MDM2 by a subset of ribosomal proteins (RPs), thereby stabilizing p53. Here, we identify human HEATR1, as a nucleolar protein which positively regulates ribosomal RNA (rRNA) synthesis. Downregulation of HEATR1 resulted in cell cycle arrest in a manner dependent on p53. Moreover, depletion of HEATR1 also caused disruption of nucleolar structure and activated the ribosomal biogenesis stress pathway – RPL5 / RPL11 dependent stabilization and activation of p53. These findings reveal an important role for HEATR1 in ribosome biogenesis and further support the concept that perturbation of ribosome biosynthesis results in p53-dependent cell cycle checkpoint activation, with implications for human pathologies including cancer.
KEYWORDS: ribosome biogenesis, HEATR1, p53, ribosome biogenesis stress, cancer
Introduction
Protein synthesis is a fundamental feature of life that relies on specialized organelles, the ribosomes. The ribosomes are large protein-RNA complexes, formed by about 80 ribosomal proteins (RPs) and four types of ribosomal RNAs (rRNAs) to compose a platform for protein translation. In eukaryotes, each ribosome consists of a 40 S small and a 60 S large subunit [1–3]. While the matured ribosomes are localized in the cytoplasm, biosynthesis and assembly of the ribosomal subunits takes place mainly in the nucleolus. This membrane-less organelle is located within the nucleus and organized around ribosomal DNA (rDNA) genes. Ribosome biogenesis is initiated by the transcription of rDNA, performed by RNA Polymerase I (RNA Pol I). The resulting 47S pre-rRNA transcript is further processed and modified to give rise to several species of rRNAs: 18S rRNA – component of the small ribosomal subunit – 5.8S and 28S rRNAs – components of the large subunit. As an exception, transcription of another rRNA species – 5S rRNA, part of the large ribosomal subunit – occurs in the nucleus and is carried out by RNA Polymerase III. RP genes are transcribed by RNA Polymerase II in the nucleus and translated in the cytoplasm. Both the RPs and the 5S rRNA are first imported into the nucleolus where assembly of the ribosomal subunits begins. Precise maturation of the rRNAs and appropriate assembly of the ribosomal subunits requires high energy investment and depends on the action of multiple nucleolar accessory and assembly factors – primarily non-ribosomal proteins and small nucleolar RNAs (snoRNAs) [4–6].
Due to the tremendous energy demand for ribosome biogenesis and the continuous need for protein synthesis, production of the ribosomes must be tightly regulated and synchronized with physiological conditions of the cells. Numerous studies showed that perturbation of various steps in the complex proteosynthetic process, a scenario that is commonly referred to as nucleolar or ribosome biogenesis stress [7–11] leads to cell cycle arrest and eventually to senescence or apoptosis through activation of the p53 tumor suppressor protein. Under normal growth conditions p53 is engaged in a complex with, and ubiquitilated by, the MDM2 E3 ubiquitin ligase, leading to p53's continuous proteasomal degradation [12–14]. However, impaired ribosome biogenesis triggers the release of several RPs – most notably RPL5 and RPL11 – to the nucleoplasm, where these proteins bind and sequester MDM2, resulting in p53 stabilization and activation [9,11,15].
In this study we investigated human HEAT repeat containing 1 (HEATR1) protein, its role in ribosome biogenesis and impact of its downregulation on cells. The human HEATR1 gene is located at chromosome 1q43 and encodes a large (236 kDa) protein consisting of 2144 amino acids. The HEATR1 protein contains one HEAT repeat on its C-terminal end (http://www.uniprot.org/uniprot/Q9H583). HEAT repeats are found in other proteins – e.g. huntingtin, elongation factor 3 or protein phosphatase 2A – and considered to be involved in protein-protein interactions [16]. UTP10, the yeast homolog of HEATR1 also contains a HEAT repeat at its C-terminus, and was shown to be involved in rDNA transcription and small ribosomal subunit maturation [17–20]. Unlike UTP10 or human HEATR1 protein, the Zebrafish homolog Bap28 contains 8 HEAT repeats [21]. Despite the structural differences, similarly to the yeast or human protein [17,19,20,22], Bap28 was also suggested to be involved in the regulation of rRNA synthesis and maturation. Furthermore, expression of a deletion mutant of Bap28 in Zebrafish embryos severely impaired organismal development, possibly through the activation of p53-dependent apoptosis [21]. In addition, human HEATR1 has been implicated in cancer, through stimulation of cytotoxic T lymphocyte responses in gliomas [23] and promotion of AKT dephosphorylation in the mTOR signaling pathway in pancreatic ductal adenocarcinomas (PDAC) [24].
Here, we report that human HEATR1 is a nucleolar protein with a prominent role in ribosome biogenesis. Downregulation of HEATR1 leads to impaired ribosome biogenesis, thereby inducing disruption of the nucleolar structure and triggering cellular ribosome biogenesis stress response.
Results
Knockdown of HEATR1 activates and stabilizes p53
To identify human proteins involved in ribosome biogenesis, we performed a high-content RNA interference (RNAi) screen and found HEATR1 among prominent hits, whose depletion led to increased p53 level. To validate this observation, we used three independent siRNAs against HEATR1, all of which reduced the endogenous level of the protein and resulted in the elevation of p53 and its target, p21 to the same extent in human osteosarcoma (U2OS) cell line (Figure 1A). In order to reduce potential off-target effects [25], we also used a pool of the three siRNAs here (Figure 1A) and in all further experiments. Pooled siHEATR1 efficiently reduced the endogenous level of HEATR1, not only in U2OS but in normal diploid human fibroblast (BJ) cells, as well (Figure 1A and Figure S1A, B). Furthermore, knockdown of HEATR1 did not lead to DNA damage induction or activation of DNA damage signaling, as we did not observe any increased γH2AX (an established biomarker of DNA damage) in these HEATR1-depleted samples (Figure S1C).
Figure 1.
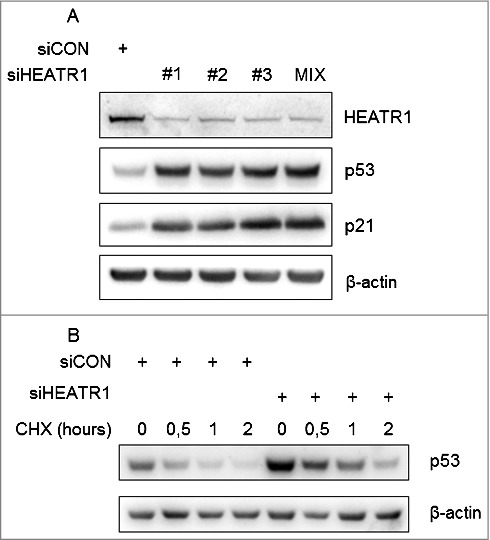
Depletion of HEATR1 stabilizes p53 A. U2OS cells were transfected with control or HEATR1 siRNAs, cell lysates were prepared 72 h after transfection and immunoblotted with indicated antibodies. B. U2OS cells were transfected with control or HEATR1 siRNAs, treated with 50 μg/ml cycloheximide (CHX) 72 h later and harvested at indicated time points. Cell lysates were immunoblotted with indicated antibodies.
Next we wondered whether the observed increased abundance of the p53 protein reflects its enhanced stability. To address this issue, we applied cycloheximide – an inhibitor of protein synthesis – to examine turnover of p53 in control versus HEATR1-depleted cells. Indeed, absence of HEATR1 led to slower degradation of p53 protein (Figure 1B), as its half-life increased from 56 minutes to 106 minutes, suggesting that p53 upregulation upon HEATR1 knockdown might be a consequence of its elevated stability. These results demonstrated that ablation of HEATR1 leads to activation and stabilization of p53.
Depletion of HEATR1 leads to impaired proliferation and induces p53-dependent cell cycle arrest
To assess any impact of HEATR1 status on cell cycle progression, we first examined proliferation rate of control and HEATR1-depleted U2OS cells. Cells deficient in HEATR1 showed impaired growth rate compared to control, as determined by total cell counts at 2, 4 and 6 days after siRNA transfection (Figure 2A). This impairment of the overall cell proliferation upon HEATR1 depletion was not cell-type restricted, as ablation of HEATR1 led to growth arrest also in normal diploid BJ cells (Figure S2A). Further analyses showed that HEATR1 knockdown led to altered cell cycle progression, documented by a dramatic decrease of cells in S phase and enhanced subpopulation of cells in G1 (Figure 2B). Notably, co-depletion of HEATR1 and p53 restored normal cell cycle profile (Figure 2B), suggesting that p53 is causally linked to the observed G1-phase accumulation of HEATR1-depleted cells. In an independent parallel set of experiments, we confirmed the reduced fraction of replicating cells upon HEATR1 knockdown by monitoring 5-ethynyl-2′-deoxyuridine (EdU) incorporation (Figure 2C). Importantly, depletion of p53 efficiently reduced the level of p53 without affecting abundance of HEATR1 protein (Figure S2B). In contrast to U2OS cells, downregulation of HEATR1 in human cervical carcinoma (HeLa) cell line did not induce cell cycle arrest, as similar fractions (29% and 31%, respectively) of the control mock-treated and HEATR1-depleted cells were present in S phase and the overall cell cycle profiles were very similar (Figure S2C). From these experiments, we concluded that the apparent lack of the p53-dependent G1 accumulation in HEATR1-depleted HeLa cells likely reflects the absence of functional p53 in HeLa cells, caused by the endogenous expression of the human papilloma virus E6 oncoprotein [26,27]. Overall, these data indicated that HEATR1 knockdown leads to accumulation and activation of p53 that induces cell cycle arrest and impairs cell growth in p53-proficient human normal and tumor cells.
Figure 2.
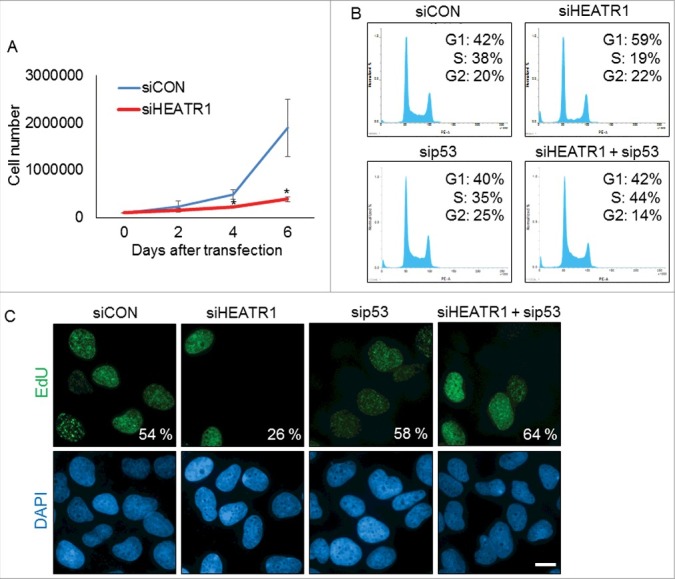
Knockdown of HEATR1 leads to impaired proliferation and induces p53-dependent cell cycle arrest A. U2OS cells were transfected with control or HEATR1 siRNAs and 100000 cells were seeded. Cell counts were determined at the indicated time points after transfection. Error bars represent SDs, n = 3. Significance determined by two-tailed student's t-test: * P<0,05. B. U2OS cells were transfected with the indicated siRNAs and cell cycle profiles were assessed by flow cytometry 72 h after transfection. Results are representative of three independent experiments. C. U2OS cells were transfected with the indicated siRNAs and labeled with 10 μM 5-ethynyl-2′-deoxyuridine (EdU) for 30 min. The cells were fixed and incorporated EdU was visualized by click chemistry. The nuclei were stained by DAPI. Results are representative of three independent experiments. Bar, 10 µm.
HEATR1 is a nucleolar protein
Next, we investigated the localization of HEATR1 in cultured human cells. Immunostaining of the endogenous HEATR1 protein in exponentially growing U2OS cells revealed that HEATR1 is localized in the nuclei, with a pronounced accumulation in the nucleoli, the latter validated by co-staining for nucleophosmin (NPM), a nuclear protein with preferential accumulation in nucleoli (Figure 3A). The nucleolar localization of HEATR1 was specific, as depletion of HEATR1 by siRNA led to the disappearance of the staining signal from the nucleoli, while the weak nucleoplasmic background staining signal remained unchanged (Figure 3B). The observed nucleolar localization of HEATR1 was also shared by two distinct and widely used strains of human diploid fibroblasts: BJ and MRC-5 (Figure S3A and B). Transfection of siRNA targeting HEATR1 into such normal cells abolished again only the nucleolar staining signal, thereby validating the results obtained with the U2OS cells (Figure S1B and data not shown). During these experiments, we noted that the protein expression level of HEATR1 is lower in normal cells, compared to either U2OS or HeLa cells. Indeed, a comparative Western blotting analysis of total cell lysates from exponentially growing cultures confirmed higher levels of the HEATR1 protein in U2OS and HeLa cells, in contrast to very low, barely detectable levels seen in either BJ or MRC-5 cell extracts examined in parallel (Figure S3C). On the other hand, these differences may reflect distinct overall size/contribution of nucleolar compartments in cancer versus normal cells, as we found a similar trend towards enhanced relative abundance of other nucleolar proteins in the cancer cell lines (Figure S3C).
Figure 3.
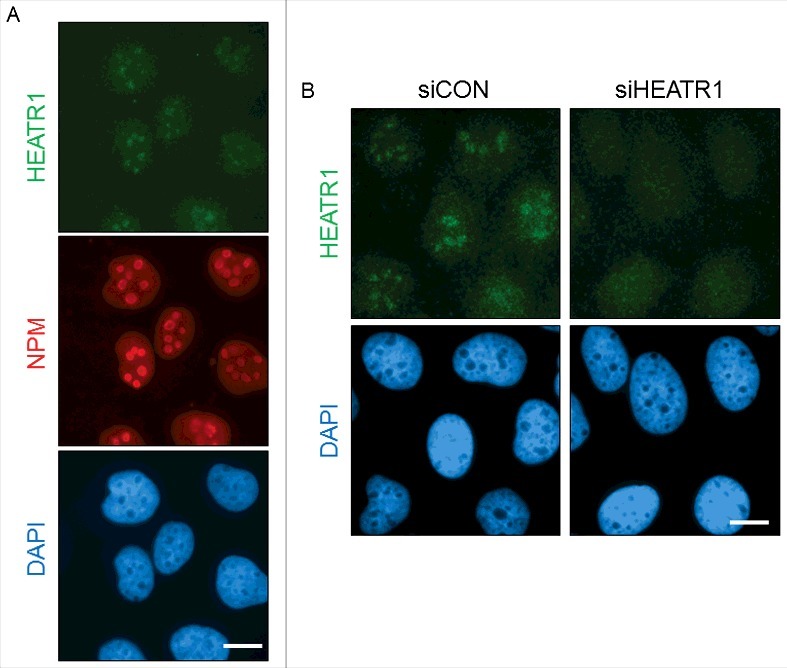
HEATR1 is localized to the nucleoli A. U2OS cells were fixed and immunostained with HEATR1 and nucleophosmin (NPM) antibodies. Nuclei were visualized by DAPI staining. Bar, 10 µm. B. U2OS cells were transfected with indicated siRNAs and fixed and immunostained with HEATR1 antibody 72 after transfection. Nuclei were visualized by DAPI staining. Bar, 10 µm.
We conclude from these analyses that human HEATR1 is a largely nucleolar protein that is overexpressed in cancerous cell lines compared to normal cells, consistent with cancer-associated over-abundance of other nucleolar proteins.
HEATR1 knockdown leads to disruption of nucleolar structure and induces ribosome biogenesis stress
Nucleolar localization of HEATR1, along with induced p53 and cell-cycle arrest upon HEATR1 knockdown suggested that HEATR1 might be involved in ribosomal biogenesis. To test whether accumulation and activation of p53 upon HEATR1 knockdown is a consequence of ribosome biogenesis stress, we first analyzed the integrity of the nucleolus by immunofluorescence in control and HEATR1-depleted cells. Although disruption of nucleolar structure is not required for p53 activation upon ribosome biogenesis stress [28], perturbation of ribosome biosynthesis can result in dramatic alterations of nucleolar morphology [9,29–31]. To characterize the impact of HEATR1 depletion on nucleolar structure, we followed established markers of nucleolar disintegration. Whereas fibrillarin (FBL), NPM and nucleolin (NCL) all showed predominantly nucleolar localization in control siRNA-treated U2OS and HeLa cells (Figure 4A, B and Figure S4A, B), parallel cultures treated with of HEATR1-targeting siRNA caused redistribution of these proteins in both cell lines. Specifically, NPM and NCL became released from nucleoli into the nucleoplasm, while the FBL staining signal relocated to nucleolar caps, structures characteristic for cells with inhibited RNA Pol I-mediated transcription [32–34] (Figure 4A, B and Figure S4 A, B). In addition, we also confirmed induction of p53 in HEATR1 deficient U2OS cells by immunofluorescence staining (Figure 4B).
Figure 4.
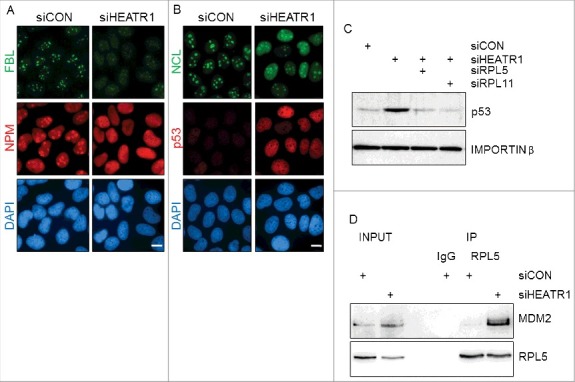
HEATR1 knockdown induces ribosomal stress A, B. U2OS cells were transfected with indicated siRNAs, fixed and immunostained with fibrillarin (FBL), nucleophosmin (NPM), nucleolin (NCL) and p53 antibodies 72 h after transfection. Nuclei were visualized with DAPI staining. Bar, 10 µm. C. U2OS cells were transfected with indicated siRNA, lysed and immunoblotted with indicated antibodies 72 h after transfection. D. U2OS cells were transfected with control or HEATR1 siRNAs. Cell lysates were immunoprecipitated 72 h after transfection with control (IgG) or RPL5 antibodies and immunoblotted with indicated antibodies.
To test whether HEATR1 depletion triggers the canonical RPL5/RPL11-MDM2-dependent response to ribosome biogenesis stress, we performed simultaneous depletion of HEATR1 and either RPL5 or RPL11 and examined the p53 response. Indeed, knockdown of either RPL5 or RPL11 prevented the p53 accumulation seen in HEATR1-depleted U2OS cells, without affecting HEATR1 protein itself (Figure 4C and Figure S4C). Furthermore, co-immunoprecipitation experiments detected a complex of RPL5 with MDM2 in HEATR1-deficient, but not in control cells (Figure 4D). These observations supported our working hypothesis that depletion of HEATR1 leads to ribosome biogenesis stress accompanied with the disruption of the nucleolar structure. This, in turn, leads to release of some nucleolar and ribosomal proteins into the nucleoplasm, while a subset of nucleolar proteins is redistributed, forming nucleolar caps. Cells react to this stressful scenario by triggering the RPL5/RPL11-MDM2-mediated stabilization and activation of p53, leading to G1 cell cycle arrest.
The role of HEATR1 in ribosome biogenesis
To provide further insights into the potential role(s) of HEATR1 in the nucleolus, we analyzed nascent RNA synthesis in control versus HEATR1-depleted cells. The incorporation of the nucleotide analogue, 5-ethynyl uridine (EU) into RNA was most prominent in nucleolar regions in control cells, as ribosomal RNAs are the most abundant RNA species in the cell [35] (Figure 5A and Figure S5). In contrast, siRNA-mediated depletion of HEATR1 impaired the nascent transcription in the nucleoli, evidenced by the severe depletion of the nucleolar EU signal (Figure 5A and Figure S5). The strong suppression of rRNA transcription caused by ablation of HEATR1 was comparable to effects of treatment with low concentrations of actinomycin D (ActD, Figure 5A), which is known to specifically inhibit the activity of RNA Pol I [36,37].
Figure 5.
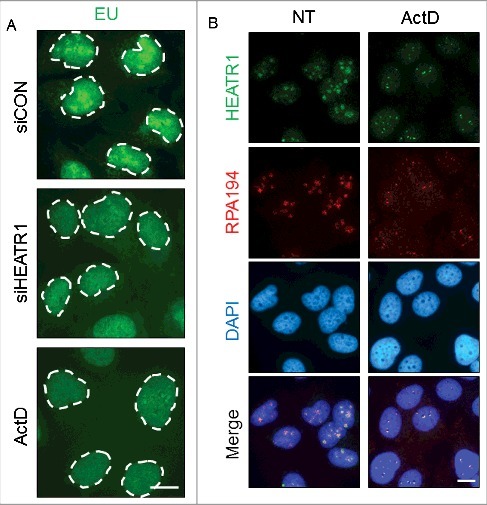
HEATR1 is involved in rRNA synthesis. A. U2OS cells were transfected with indicated siRNAs or treated with 5 nM ActD overnight and labeled with 1 mM 5-ethynyl uridine (EU) for 30 min. EU detection was performed by click chemistry. Bar, 10 µm. B. U2OS cells were mock- or actinomycin D- (ActD, 5nM) treated overnight, then fixed and immunostained with indicated antibodies. Nuclei were visualized by DAPI staining. Bar, 10 µm.
Proteins associated with the Pol I transcription machinery co-localize in the fibrillar center of the nucleolus and are redistributed to small, so-called fibrillar caps upon transcription inhibition [34]. Since HEATR1 seemed to be required for proper rRNA synthesis, we asked whether: i) HEATR1 co-localizes with Pol I under unperturbed growth; and ii) if yes, how is such localization pattern affected by the low-ActD treatment. To address these questions, we visualized RNA Pol I by immunostaining with antibodies against the RPA194 subunit of Pol I (Figure 5B). Under normal growth conditions, HEATR1 and RPA194 co-localized in the nucleoli, presumably in the fibrillar centers (Figure 5B). When low concentration of ActD was applied to inhibit rDNA transcription, both HEATR1 and RPA194 were redistributed to the periphery of nucleoli, forming small, overlaying nucleolar caps (Figure 5B). Considering the emerging potential of small molecule inhibitors of ribosome biogenesis in anti-cancer treatment, we evaluated the effect of one such promising compound, BMH-21 [38,39] on HEATR1 localization. BMH-21 induces ribosome biogenesis stress by intercalating into GC rich region of the DNA, thereby inhibiting rRNA synthesis and promoting degradation of RPA194 [38,39]. Similarly to ActD treatment, exposure of human cells to BMH-21 induced redistribution of HEATR1 into nucleolar caps, while RPA194 was degraded (Figure S5B). These data collectively indicated that HEATR1 plays a prominent role in rRNA transcription, as knockdown of the protein led to impaired rRNA synthesis. Moreover, our results showed that HEATR1 co-localizes with Pol I under unperturbed conditions, and undergoes the same pattern of topological redistribution as Pol I upon ActD treatment, thereby further supporting HEATR1's involvement in rRNA transcription.
Discussion
In this study, we investigated the role of human HEATR1, so-far a very poorly characterized protein, which we identified as one of the prominent hits in a high-throughput siRNA screen for human proteins whose depletion leads to p53 stabilization (the complete dataset from the screen will be reported separately). Here, we showed that depletion of HEATR1 leads to activation and stabilization of the tumor suppressor protein p53. Thus, RNAi-mediated knockdown of HEATR1 caused a nearly two-fold increase of p53 protein half-life, accompanied by induced expression of p53's transcriptional target, the CDK inhibitor p21. At the cellular level, this sequence of molecular events resulted in decreased cell proliferation and cell cycle arrest in G1. Importantly, such activation of the cell cycle checkpoint was not due to the canonical DNA damage-induced phosphorylation of p53, since HEATR1 depletion did not lead to DNA damage. These results are complementary to, and consistent with, our previous report that the U2OS cells cannot induce the p53-dependent G1 checkpoint after DNA damage, despite they express wild-type p53. Such malfunction of the DNA-damage dependent p53 activation is due to the overexpression in U2OS cells of a truncated, mutant form of Wip1, a phosphatase that potently counteracts the otherwise functional phosphorylation of p53 under DNA damage conditions [40]. On the other hand, in response to HEATR1 depletion, p53 becomes stabilized via a phosphorylation-independent pathway (see below) and therefore the overexpressed mutant Wip1 does not prevent activation of the p53-p21 checkpoint under such ribosomal stress conditions. Furthermore, the absence of DNA damage signaling documented by the lack of γH2AX in HEATR1-depleted cells also indicates that the DNA damage response (DDR) is not activated. ATM kinase is a key component of the DDR network and negatively regulates abundance of the alternative reading frame (ARF) tumor suppressor protein [41]. ARF is another nucleolar protein which – similarly to some RPs – is able to activate p53-dependent pathways upon stress stimuli, by disrupting MDM2's interaction with p53 [42]. ARF also inhibits ribosome biogenesis independently of p53, by preventing nucleolar import of RNA polymerase I transcription termination factor (TTF-I) [43]. Since HEATR1 ablated cells did not show DDR activation, one possibility was that ARF is induced and elicits its dual effect on p53 activation and repression of ribosome biogenesis. However, U2OS cells used in our experiments do not express ARF due to promoter hypermethylation [44]. Thus, both the observed repression of ribosome biogenesis and activation of p53 seem to be independent of ARF, further signifying the importance of the p53 activation pathway via the RPL5/RPL11-MDM2 axis upon perturbation of ribosome biogenesis (see below). Additionally, we show here that the observed cell-cycle arrest induced by HEATR1 silencing could be rescued by co-depletion of p53. All these data indicated that p53 is activated through a DDR- and ARF-independent mechanism. Moreover, in the human papilloma virus (HPV) positive HeLa cells knockdown of HEATR1 did not result in cell-cycle arrest, an observation that reflects the chronically impaired function of p53 in this cell line by the HPV-encoded E6 oncoprotein [26,27]. Most relevant for our present data related to ribosomal stress responses, recent reports suggested existence of both p53-dependent, but also alternative, p53-independent checkpoint activation pathways upon impaired ribosome biogenesis (reviewed in ref. [5,45]). Perturbed ribosome biogenesis results in the release of several nucleolar RPs into the nucleoplasm where they bind MDM2. Binding of MDM2 by the RPL11 initiates not only the stabilization of p53 (through the canonical RPL5/RPL11 dimer-mediated sequestration of MDM2), but reportedly also degradation of the E2F-1 transcription factor, leading to cell-cycle arrest even in p53-deficient cells [46]. Moreover, another RP, RPL3 was reported to activate p21 independently of p53, inducing cell cycle arrest and apoptosis upon ribosome biogenesis stress [47,48]. While we cannot exclude the possibility of such alternative checkpoint activation at later time points in HeLa cells, depletion of HEATR1 in our experiments did not result in any detectable cell cycle arrest in Hela cells, suggesting that the predominant mode of the p53 response to ribosomal stress upon HEATR1 knockdown reflects the RPL5/RPL11-MDM2 pathway.
A recent study suggested that HEATR1 is a regulator of the AKT pathway, whereby HEATR1 facilitates the interaction between AKT and its inactivating phosphatase, PP2A by acting as a scaffold. Unlike our findings presented here, this study suggested that HEATR1 is localized exclusively in the cytoplasm, a prerequisite for its co-localization and interaction with AKT [24]. In contrast, we found that HEATR1 is localized predominantly in the nucleoli of all cell types examined, both normal and cancerous, a result that is consistent with the report by Prieto and McStay [22]. Although our immunostaining of HEATR1 revealed some weaker signal also in the nucleoplasm and occasionally in the cytoplasm, our siRNA targeting HEATR1 abolished only the nucleolar staining signal, suggesting that HEATR1 mainly resides in the nucleolus. However, we cannot exclude the possibility that siRNAs used here preferentially target the ‘nucleolar form’ of HEATR1, or that HEATR1 may be more abundant in the cytosol of pancreatic cancer cells, the only cell type examined in the study reporting the HEATR1-AKT cytosolic interplay [24].
Perturbation of ribosome biogenesis triggers p53 activation through the RPL5/RPL11-MDM2 axis [9,11,15], often accompanied by disintegration of the nucleolus [9,29–31,34]. In our present experiments, depletion of HEATR1 disrupted the structure of the nucleolus and, consequently, the nucleolar proteins NPM and NCL were released into the nucleoplasm, while FBL redistributed to nucleolar caps. Although it is not a prerequisite for p53 activation, nucleolar disintegration is a robust surrogate marker of ribosome biogenesis stress [9,28–31,34]. Consistently, the increased level of p53 caused by RNAi-mediated depletion of HEATR1 could be reversed by co-depletion of either RPL5 or RPL11. Moreover, ablation of HEATR1 also led to the increased interaction between RPL5 and MDM2, providing further evidence that deficiency of HEATR1 results in ribosome biogenesis stress.
HEAT repeats are expressed by a wide variety of eukaryotic proteins involved in diverse cellular processes, however protein-protein interaction is considered to represent the main function for these structures [16]. Broadly consistent with our present findings, UTP10, a yeast HEAT repeat protein was reported to localize in the nucleolus [19]. UTP10 – similarly to other UTPs (U3 small nucleolar RNA-associated proteins) – binds to U3 snoRNA and thereby contributes to maturation of 18S rRNA and assembly of the small ribosomal subunit [19,20]. Furthermore, UTP10 was also shown to regulate rDNA transcription [17]. Zebrafish Bap28 which shows 34% sequence similarity to yeast UTP10 was also characterized as a protein required for rRNA synthesis and 18S rRNA maturation [21]. Here, we showed that human HEATR1 is a positive regulator of transcription by RNA Pol I. Indeed, depletion of HEATR1 caused inhibition of rRNA synthesis to the same extent as low concentration of ActD, a widely used and selective treatment to block Pol I activity. Moreover, we found that HEATR1 was co-localized with Pol I both under normal conditions and upon inhibition of transcription by ActD. In the latter case, HEATR1 and Pol I became redistributed to the periphery of the nucleoli, into cap-like structures. Our data are also consistent with the work of Prieto and McStay who reported nucleolar co-localization of HEATR1 with the Pol I transcription machinery, in a manner independent of active transcription yet dependent on the integrity of the upstream binding factor (UBF) [22]. Last but not least, the model we propose was further supported by the responses we observed upon treatment with the ribosome biogenesis inhibitor BMH-21. While treatment with BMH-21 led to rapid degradation of RPA194, HEATR1 became redistributed to form cap-like structures, similarly to what we observed in response to ActD.
Aberrantly enlarged nucleoli and upregulation of ribosome biogenesis are common features of various types of cancer. Due to its intimate link with cell growth and cell cycle regulation, enhanced ribosomal biogenesis can impair differentiation and promote tumorigenesis [6,49–52]. Our observation that HEATR1 is elevated in cancer cell lines, compared to normal non-cancerous cells, is also consistent with the concept of the global deregulation of ribosomal biogenesis in cancer. While in the two cancer cell lines we examined, elevation of HEATR1 was accompanied by enhanced levels of other nucleolar proteins, it is possible that HEATR1 is specifically overexpressed in certain types of cancer. For instance, a recent report demonstrated that HEATR1 is overexpressed in glioblastoma cells, providing an excellent target for T-cell mediated immunotherapy [23]. In contrast, aberrantly low levels of ribosome biogenesis represent the hallmark of the so-called ribosomopathies, a group of human diseases characterized by hyperactivation of p53, reduced cell growth and protein synthesis [53–55]. Abnormal downregulation of HEATR1 seems to correlate with poor prognosis in patients with pancreatic ductal carcinoma, however, this phenotype was attributed to the consequence of HEATR1's involvement in AKT phosphorylation (see above for the discussion of cytosolic HEATR1 in pancreatic cell lines reported in this study) and may be unrelated to its function in ribosome biogenesis [24]. Nonetheless, further studies are clearly warranted to elucidate any potential contribution of HEATR1 alteration(s) to pathogenesis of various cancer types.
Taken together, we report here that human HEATR1 is a nucleolar protein that plays a prominent positive role in regulation of rRNA synthesis. Deficiency of HEATR1 leads to perturbation of ribosome biogenesis, resulting in DNA damage-independent activation of the p53-p21 checkpoint response through the RPL5/RPL11-MDM2 axis. Our study provides fresh insights into the complex nucleolar events and the ways malfunction of ribosomal biogenesis can lead to growth and cell-cycle arrest, with implications for pathologies including cancer.
Material and methods
Cell culture
Cell lines used in this work were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (Life Technologies) and penicillin/streptomycin (Sigma-Aldrich) in humidified atmosphere of 5% CO2 at 37°C. For BJ and MRC-5 culturing, DMEM was supplemented with 1% MEM Non-essential amino acids (Life Technologies), as well. All cell lines were purchased from ATCC.
Chemicals and antibodies
Actinomycin D (A1410) and cycloheximide (C4859) were purchased from Sigma-Aldrich. BMH-21 (S7718) was purchased from Selleckchem.
Antibodies used in this study included rabbit antibodies: HEATR1 (HPA046917, Sigma-Aldrich), p21 (2947, Cell Signaling), nucleolin (ab70493, Abcam), nucleostemin (ab70346, Abcam), fibrillarin (ab5821, Abcam), RPL5 (ab157099, Abcam) and mouse antibodies: β-actin (sc-47778, Santa Cruz), Importin-β (ab2811, Abcam), p53 (sc-126, Santa Cruz), γH2AX (05–636, Millipore), nucleophosmin (ab10530, Abcam), and MDM2 (ab16895, Abcam).
RNA interference
All siRNA transfections were performed using Lipofectamine RNAiMAX (Invitrogen) according to the manufacturer's instructions. All siRNA duplexes were purchased from Ambion: siCON (negative control #1, AM4635, 5′-AGUACUGCUUACGAUACGGTT-3′), siHEATR1#1 (s30230, 5′-CCACUUUCCAUUUGCGAUATT-3′), siHEATR1#2 (s30231, 5′-GAUGUUGUUUUGUCGGCUATT-3′), siHEATR1#3 (s30232, 5′-CACUUUCCAUUUGCGAUAATT-3′), sip53 (s605, 5′-GUAAUCUACUGGGACGGAATT -3′), siRPL5 (s56733, 5′-CAGUUCUCUCAAUACAUAATT-3′), and siRPL11 (s12169, 5′-CAACUUCUCAGAUACUGGATT -3′).
Cell cycle analysis
Cells were fixed in 70% ethanol and stained with propidium iodide for flow cytometric analysis. Fixed cells were analyzed on a FACS Verse instrument (BD Biosciences) and cell cycle distribution was assigned using the FACSuite software (BD Biosciences).
Immunoblotting
Immunoblotting was performed as previously described [56]. WCLs were prepared in Laemmli sample buffer (LSB; 50 mM Tris, pH 6.8, 100 mM DTT, 2% SDS, 0.1% bromophenol blue, and 10% glycerol), separated by SDS-PAGE, and transferred to nitrocellulose membranes (GE Healthcare). The membranes were blocked with 5% (wt/vol) dry milk in 0.1% (vol/vol) Tween-20 in PBS and probed with the primary antibodies, followed by HRP-labeled secondary antibodies (GE Healthcare), and visualized using ECL detection reagents (GE Healthcare).
Immunofluorescence
Immunofluorescence analysis was performed as previously described with minor changes [57]. Cells grown on 12-mm coverslips were fixed with 4% paraformaldehyde in PBS for 15 min and then permeabilized with PBS containing 0.2% (v/v) Triton X-100 for 5 min. Fixed cells were blocked with 5% (v/v) fetal bovine serum in PBS for 30 minutes and incubated overnight at 4 °C with primary antibodies (diluted in 5% (w/v) bovine serum albumin in PBS). Coverslips were washed 3 times in PBS supplemented with 0,1% (v/v) Tween 20, once with PBS and then incubated with an appropriate secondary goat anti-rabbit or goat anti-mouse Alexa Fluor 488 or Alexa Fluor 568 conjugated (Invitrogen) secondary antibody (diluted in in 5% (w/v) bovine serum albumin in PBS) for 60 min at RT. Slips were then washed as above and mounted onto slides using the DAPI containing Vectashield mounting reagent (Vector Laboratories). Slides were visualized by the Axio Observer.Z1/Cell Observer Spinning Disc microscopic system (Yokogawa and Zeiss) equipped with an Evolve 512 Photometrix) EMCCD camera. Zeiss Plan Apochromat 100x/1.40 NA objective was used.
EdU and EU staining
Cells were labelled either with 10 μM EdU or with 1 mM EU (Sigma-Aldrich) for 30 min, then fixed with 4% paraformaldehyde in PBS for 15 min and permeabilized with 0.2% Triton-X100 for 5 min, followed by incubation with staining solution (100 mM Tris-HCl, pH = 8.5, 1 mM CuSO4, 1 μM azide conjugated fluorochrome, 100 mM ascorbic acid) for 30 min. Nuclei were stained with DAPI. Image acquisition and analysis was performed using ScanR system (Olympus).
Immunoprecipitation
Immunoprecipitation was performed as previously described with minor changes [58]. Briefly, cells were washed three times in PBS and lysed in TNE buffer (150 mM NaCl, 50 mM Tris-HCl, pH 8.0, 1 mM EDTA, 0.5% NP-40) supplemented with cOmplete and PhosSTOP tablets (Roche). After 30 min incubation on ice, lysates were cleared by centrifugation. Where appropriate, antibodies were added to lysate and incubated for 16 h at 4°C. Lysates were then incubated with 25 µl of Dynabeads M-280 Sheep anti Rabbit IgG (Novex) for 1 h at 4°C. Ig–antigen complexes were washed extensively before elution in 2 × LSB before SDS-PAGE.
Supplementary Material
Funding Statement
This work was supported by the Grant Agency of the Czech Republic under grants 17–14743S and 17–25976S, the MEYS CR (CZ.02.1.01/0.0/0.0/16_013/0001775 Czech-BioImaging), Czech National Program of Sustainability (LO1304), UP IGA_LF_2017_002, the Danish Cancer Society, the Danish Council for Independent Research, the Swedish Research Council, CancerFonden, the Lundbeck Foundation, and the Danish National Research Foundation (Center of Excellence, project CARD).Czech Science Foundation (GACR) [grant number 17-14743S]
Disclosure of potential conflict of interest
No potential conflicts of interest were disclosed.
References
- [1].Budkevich TV, El'skaya AV, Nierhaus KH. Features of 80S mammalian ribosome and its subunits. Nucleic Acids Res. 2008;36:4736-4744. doi: 10.1093/nar/gkn424. PMID:18632761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Klinge S, Voigts-Hoffmann F, Leibundgut M, et al.. Atomic structures of the eukaryotic ribosome. Trends Biochem Sci. 2012;37:189–198. doi: 10.1016/j.tibs.2012.02.007. PMID:22436288 [DOI] [PubMed] [Google Scholar]
- [3].Wilson DN, Doudna Cate JH. The structure and function of the eukaryotic ribosome. Cold Spring Harb Perspect Biol. 2012;4:1–17. doi: 10.1101/cshperspect.a011536. PMID:22550233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Kressler D, Hurt E, Bassler J. A puzzle of life: crafting ribosomal subunits. Trends Biochem Sci. 2017;42:640–654. doi: 10.1016/j.tibs.2017.05.005. PMID:28579196 [DOI] [PubMed] [Google Scholar]
- [5].James A, Wang Y, Raje H, et al.. Nucleolar stress with and without p53. Nucleus. 2014;5:402–426. doi: 10.4161/nucl.32235. PMID:25482194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Derenzini M, Montanaro L, Trere D. Ribosome biogenesis and cancer. Acta Histochem. 2017;119:190–197. doi: 10.1016/j.acthis.2017.01.009. PMID:28168996 [DOI] [PubMed] [Google Scholar]
- [7].Yang L, Song T, Chen L, et al.. Nucleolar repression facilitates initiation and maintenance of senescence. Cell Cycle. 2015;14:3613–3623. doi: 10.1080/15384101.2015.1100777. PMID:26505814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Morgado-Palacin L, Llanos S, Urbano-Cuadrado M, et al.. Non-genotoxic activation of p53 through the RPL11-dependent ribosomal stress pathway. Carcinogenesis 2014;35:2822–2830. doi: 10.1093/carcin/bgu220. PMID:25344835 [DOI] [PubMed] [Google Scholar]
- [9].Bursac S, Donati G, Brdovcak MC, et al.. Activation of the tumor suppressor p53 upon impairment of ribosome biogenesis. Biochim Biophys Acta. 2013;1842:817–830. PMID:24514102 [DOI] [PubMed] [Google Scholar]
- [10].Dong Z, Zhu C, Zhan Q, et al.. The roles of RRP15 in nucleolar formation, ribosome biogenesis and checkpoint control in human cells. Oncotarget. 2017;8:13240–13252. PMID:28099941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Fumagalli S, Ivanenkov VV, Teng T, et al.. Suprainduction of p53 by disruption of 40S and 60S ribosome biogenesis leads to the activation of a novel G2/M checkpoint. Genes Dev. 2012;26:1028–1240. doi: 10.1101/gad.189951.112. PMID:22588717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Honda R, Tanaka H, Yasuda H. Oncoprotein MDM2 is a ubiquitin ligase E3 for tumor suppressor p53. FEBS Lett. 1997;420:25–27. doi: 10.1016/S0014-5793(97)01480-4. PMID:9450543 [DOI] [PubMed] [Google Scholar]
- [13].Michael D, Oren M. The p53-Mdm2 module and the ubiquitin system. Semin Cancer Biol. 2003;13:49–58. doi: 10.1016/S1044-579X(02)00099-8. PMID:12507556 [DOI] [PubMed] [Google Scholar]
- [14].Haupt Y, Maya R, Kazaz A, et al.. Mdm2 promotes the rapid degradation of p53. Nature. 1997;387:296–299. doi: 10.1038/387296a0. PMID:9153395 [DOI] [PubMed] [Google Scholar]
- [15].Golomb L, Volarevic S, Oren M. p53 and ribosome biogenesis stress: the essentials. FEBS Lett. 2014;588:2571–2579. doi: 10.1016/j.febslet.2014.04.014. PMID:24747423 [DOI] [PubMed] [Google Scholar]
- [16].Yoshimura SH, Hirano T. HEAT repeats – versatile arrays of amphiphilic helices working in crowded environments? J Cell Sci. 2016;129:3963–3970. PMID:27802131 [DOI] [PubMed] [Google Scholar]
- [17].Gallagher JE, Dunbar DA, Granneman S, et al.. RNA polymerase I transcription and pre-rRNA processing are linked by specific SSU processome components. Genes Dev 2004;18:2506–2517. doi: 10.1101/gad.1226604. PMID:15489292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Oeffinger M, Dlakic M, Tollervey D. A pre-ribosome-associated HEAT-repeat protein is required for export of both ribosomal subunits. Genes Dev 2004;18:196–209. doi: 10.1101/gad.285604. PMID:14729571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Dez C, Dlakic M, Tollervey D. Roles of the HEAT repeat proteins Utp10 and Utp20 in 40S ribosome maturation. Rna 2007;13:1516–1527. doi: 10.1261/rna.609807. PMID:17652137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Krogan NJ, Peng WT, Cagney G, et al.. High-definition macromolecular composition of yeast RNA-processing complexes. Mol Cell. 2004;13:225–239. doi: 10.1016/S1097-2765(04)00003-6. PMID:14759368 [DOI] [PubMed] [Google Scholar]
- [21].Azuma M, Toyama R, Laver E, et al.. Perturbation of rRNA synthesis in the bap28 mutation leads to apoptosis mediated by p53 in the zebrafish central nervous system. J Biol Chem. 2006;281:13309–13316. doi: 10.1074/jbc.M601892200. PMID:16531401 [DOI] [PubMed] [Google Scholar]
- [22].Prieto JL, McStay B. Recruitment of factors linking transcription and processing of pre-rRNA to NOR chromatin is UBF-dependent and occurs independent of transcription in human cells. Genes Dev. 2007;21:2041–2054. doi: 10.1101/gad.436707. PMID:17699751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Wu ZB, Qiu C, Zhang AL, et al.. Glioma-associated antigen HEATR1 induces functional cytotoxic T lymphocytes in patients with glioma. J Immunol Res. 2014;2014:131494. doi: 10.1155/2014/131494. PMID:25126583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Liu T, Fang Y, Zhang H, et al.. HEATR1 negatively regulates Akt to help sensitize pancreatic cancer cells to chemotherapy. Cancer Res. 2016;76:572–581. doi: 10.1158/0008-5472.CAN-15-0671. PMID:26676747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Jackson AL, Bartz SR, Schelter J, et al.. Expression profiling reveals off-target gene regulation by RNAi. Nat Biotechnol. 2003;21:635–637. doi: 10.1038/nbt831. PMID:12754523 [DOI] [PubMed] [Google Scholar]
- [26].Scheffner M, Werness BA, Huibregtse JM, et al.. The E6 oncoprotein encoded by human papillomavirus Type-16 and Type-18 promotes the degradation of P53. Cell. 1990;63:1129–1136. doi: 10.1016/0092-8674(90)90409-8. PMID:2175676 [DOI] [PubMed] [Google Scholar]
- [27].Werness BA, Levine AJ, Howley PM. Association of human papillomavirus types 16 and 18 E6 proteins with p53. Science 1990;248:76–79. doi: 10.1126/science.2157286. PMID:2157286 [DOI] [PubMed] [Google Scholar]
- [28].Fumagalli S, Di Cara A, Neb-Gulati A, et al.. Absence of nucleolar disruption after impairment of 40S ribosome biogenesis reveals an rpL11-translation-dependent mechanism of p53 induction. Nat Cell Biol. 2009;11:501–508. doi: 10.1038/ncb1858. PMID:19287375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Yuan X, Zhou Y, Casanova E, et al.. Genetic inactivation of the transcription factor TIF-IA leads to nucleolar disruption, cell cycle arrest, and p53-mediated apoptosis. Mol Cell. 2005;19:77–87. doi: 10.1016/j.molcel.2005.05.023. PMID:15989966 [DOI] [PubMed] [Google Scholar]
- [30].Boulon S, Westman BJ, Hutten S, et al.. The nucleolus under stress. Mol Cell. 2010;40:216–227. doi: 10.1016/j.molcel.2010.09.024. PMID:20965417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Rubbi CP, Milner J. Disruption of the nucleolus mediates stabilization of p53 in response to DNA damage and other stresses. EMBO J. 2003;22:6068–6077. doi: 10.1093/emboj/cdg579. PMID:14609953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Tajrishi MM, Tuteja R, Tuteja N. Nucleolin: the most abundant multifunctional phosphoprotein of nucleolus. Commun Integr Biol. 2011;4:267–275. doi: 10.4161/cib.4.3.14884. PMID:21980556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Colombo E, Alcalay M, Pelicci PG. Nucleophosmin and its complex network: a possible therapeutic target in hematological diseases. Oncogene. 2011;30:2595–2609. doi: 10.1038/onc.2010.646. PMID:21278791 [DOI] [PubMed] [Google Scholar]
- [34].Shav-Tal Y, Blechman J, Darzacq X, et al.. Dynamic sorting of nuclear components into distinct nucleolar caps during transcriptional inhibition. Mol Biol Cell. 2005;16:2395–2413. doi: 10.1091/mbc.E04-11-0992. PMID:15758027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Jao CY, Salic A. Exploring RNA transcription and turnover in vivo by using click chemistry. P Natl Acad Sci USA. 2008;105:15779–15784. doi: 10.1073/pnas.0808480105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Burger K, Muhl B, Harasim R T, et al.. Chemotherapeutic drugs inhibit ribosome biogenesis at various levels. J Biol Chem. 2010;285:12416–12425. doi: 10.1074/jbc.M109.074211. PMID:20159984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Hernandez-Verdun D, Roussel P, Thiry M, et al.. The nucleolus: structure/function relationship in RNA metabolism. Wiley Interdiscip Rev RNA. 2010;1:415–431. doi: 10.1002/wrna.39. PMID:21956940 [DOI] [PubMed] [Google Scholar]
- [38].Peltonen K, Colis L, Liu H, et al.. A targeting modality for destruction of rna polymerase i that possesses anticancer activity. Cancer Cell. 2014;25:77–90. doi: 10.1016/j.ccr.2013.12.009. PMID:24434211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Peltonen K, Colis L, Liu H, et al.. Identification of novel p53 pathway activating small-molecule compounds reveals unexpected similarities with known therapeutic agents. Plos One. 2010;5:1–11. doi: 10.1371/journal.pone.0012996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Kleiblova P, Shaltiel IA, Benada J, et al.. Gain-of-function mutations of PPM1D/Wip1 impair the p53-dependent G1 checkpoint. J Cell Biol. 2013;201:511–521. doi: 10.1083/jcb.201210031. PMID:23649806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Velimezi G, Liontos M, Vougas K, et al.. Functional interplay between the DNA-damage-response kinase ATM and ARF tumour suppressor protein in human cancer. Nat Cell Biol. 2013;15:967–977. doi: 10.1038/ncb2795. PMID:23851489 [DOI] [PubMed] [Google Scholar]
- [42].Sherr CJ. Divorcing ARF and p53: an unsettled case. Nat Rev Cancer. 2006;6:663–673. doi: 10.1038/nrc1954. PMID:16915296 [DOI] [PubMed] [Google Scholar]
- [43].Lessard F, Morin F, Ivanchuk S, et al.. The ARF tumor suppressor controls ribosome biogenesis by regulating the RNA polymerase I transcription factor TTF-I. Mol Cell. 2010;38:539–550. doi: 10.1016/j.molcel.2010.03.015. PMID:20513429 [DOI] [PubMed] [Google Scholar]
- [44].Badal V, Menendez S, Coomber D, et al.. Regulation of the p14ARF promoter by DNA methylation. Cell Cycle. 2008;7:112–119. doi: 10.4161/cc.7.1.5137. PMID:18196972 [DOI] [PubMed] [Google Scholar]
- [45].Russo A, Russo G. Ribosomal proteins control or bypass p53 during nucleolar stress. Int J Mol Sci. 2017;18:1–16. doi: 10.3390/ijms18010140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Donati G, Brighenti E, Vici M, et al.. Selective inhibition of rRNA transcription downregulates E2F-1: a new p53-independent mechanism linking cell growth to cell proliferation. J Cell Sci. 2011;124:3017–3028. doi: 10.1242/jcs.086074. PMID:21878508 [DOI] [PubMed] [Google Scholar]
- [47].Russo A, Pagliara V, Albano F, et al.. Regulatory role of rpL3 in cell response to nucleolar stress induced by Act D in tumor cells lacking functional p53. Cell Cycle. 2016;15:41–51. doi: 10.1080/15384101.2015.1120926. PMID:26636733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Russo A, Esposito D, Catillo M, et al.. Human rpL3 induces G(1)/S arrest or apoptosis by modulating p21 (waf1/cip1) levels in a p53-independent manner. Cell Cycle. 2013;12:76–87. doi: 10.4161/cc.22963. PMID:23255119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Rossetti S, Wierzbicki AJ, Sacchi N. Mammary epithelial morphogenesis and early breast cancer. Evidence of involvement of basal components of the RNA Polymerase I transcription machinery. Cell Cycle. 2016;15:2515–2526. doi: 10.1080/15384101.2016.1215385. PMID:27485818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Boudra R, Lagrafeuille R, Lours-Calet C, et al.. mTOR transcriptionally and post-transcriptionally regulates Npm1 gene expression to contribute to enhanced proliferation in cells with Pten inactivation. Cell Cycle. 2016;15:1352–1362. doi: 10.1080/15384101.2016.1166319. PMID:27050906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Orsolic I, Jurada D, Pullen N, et al.. The relationship between the nucleolus and cancer: Current evidence and emerging paradigms. Semin Cancer Biol. 2016;37–38:36–50. doi: 10.1016/j.semcancer.2015.12.004. PMID:26721423 [DOI] [PubMed] [Google Scholar]
- [52].Stepinski D. Nucleolus-derived mediators in oncogenic stress response and activation of p53-dependent pathways. Histochem Cell Biol. 2016;146:119–139. doi: 10.1007/s00418-016-1443-6. PMID:27142852 [DOI] [PubMed] [Google Scholar]
- [53].Nakhoul H, Ke J, Zhou X, et al.. Ribosomopathies: mechanisms of disease. Clin Med Insights Blood Disord. 2014;7:7–16. doi: 10.4137/CMBD.S16952. PMID:25512719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Danilova N, Gazda HT. Ribosomopathies: how a common root can cause a tree of pathologies. Dis Model Mech. 2015;8:1013–1026. doi: 10.1242/dmm.020529. PMID:26398160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Koch S, Garcia Gonzalez O, Assfalg R, et al.. Cockayne syndrome protein A is a transcription factor of RNA polymerase I and stimulates ribosomal biogenesis and growth. Cell Cycle. 2014;13:2029–2037. doi: 10.4161/cc.29018. PMID:24781187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Oplustilova L, Wolanin K, Mistrik M, et al.. Evaluation of candidate biomarkers to predict cancer cell sensitivity or resistance to PARP-1 inhibitor treatment. Cell Cycle. 2012;11:3837–3850. doi: 10.4161/cc.22026. PMID:22983061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Moudry P, Lukas C, Macurek L, et al.. Ubiquitin activating enzyme UBA1 is required for cellular response to DNA damage. Cell Cycle. 2012;11:1573–1582. doi: 10.4161/cc.19978. PMID:22456334 [DOI] [PubMed] [Google Scholar]
- [58].Brazina J, Svadlenka J, Macurek L, DNA damage-induced regulatory interplay between DAXX, p53, ATM kinase and Wip1 phosphatase. Cell Cycle. 2015;14:375–387. doi: 10.4161/15384101.2014.988019. PMID:25659035 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.