

NEDD4-2 is a ubiquitin ligase that catalyses the ubiquitination of specific membrane proteins to promote their endocytosis and turnover1,2. The best known target of NEDD4-2 is the amiloride-sensitive epithelial Na+ channel (ENaC). ENaC is comprised of three subunits (α, β and γ) and plays a critical role in Na+ homeostasis in multiple organs including the kidney, lung and distal colon by transporting Na+ from the lumen across the plasma membrane into lining epithelial cells2–4. By controlling the transport of Na+ from the extracellular fluid, ENaC plays an important role in maintaining fluid volume in the lung, as well as blood pressure in the renal and cardiovascular systems. Indeed, increased ENaC activity caused by gain of function mutations which inhibit NEDD4-2 binding cause Liddle's syndrome, an inherited form of salt-sensitive hypertension.
All three ENaC subunits consist of two transmembrane domains between cytosolic N-and C-termini (Figure 1). These subunits undergo proteolytic processing and glycosylation to generate the mature form of ENaC. In response to high cytosolic Na+, PPxY motifs located within the C-termini of ENaC bind to WW domains of NEDD4-2 to facilitate their ubiquitination. The physiological importance of NEDD4-2-dependent regulation of ENaC is evident from knockout (KO) studies showing that global deficiency of Nedd4-2 results in perinatal lethality due to respiratory distress and increased ENaC expression in the lung5. In addition, Nedd4-2-deficiency has been linked to salt-sensitive hypertension in different mouse KO lines4.
Figure 1.
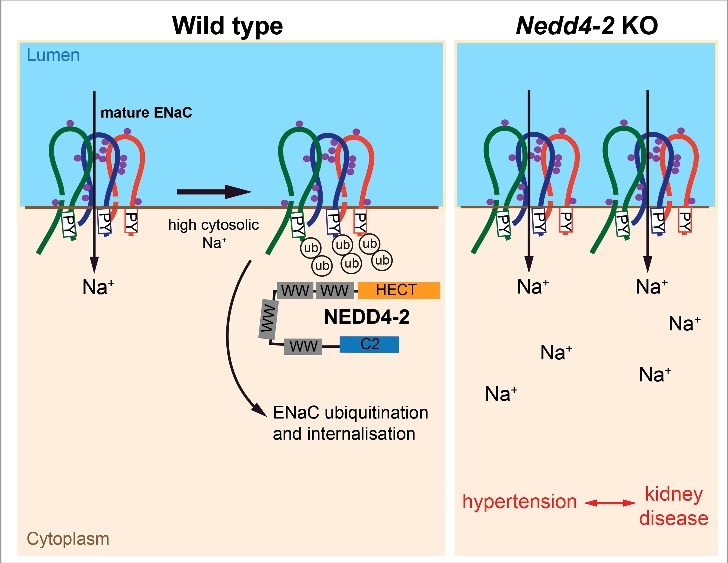
Model of ENaC maturation and regulation by NEDD4-2. Immature ENaC subunits undergo processing to mature forms by proteolytic cleavage and glycosylation (purple dots: processed N-glycans). Under high cytosolic Na+ conditions, PPxY (PY) motifs on ENaC subunits bind to WW domains of NEDD4-2. This results in ubiquitination of ENaC and subsequent removal from the membrane. In Nedd4-2 KO mice, mature ENaC is retained on the membrane leading to increased Na+ reabsorption and ultimately resulting in hypertension and kidney disease.
Our recent results show that both global and kidney-tubule specific Nedd4-2 KO in mice cause progressive kidney disease, largely due to increased retention of functional ENaC at the apical membrane in the tubular epithelia6. In global Nedd4-2 knockout mice that survive birth, kidney damage characterised by cortical cysts, cellular debris, mesenchymal infiltration and fibrosis becomes apparent within a few days after birth, and progresses with time until the animals die from respiratory distress at three weeks of age. A kidney-tubule specific Nedd4-2 KO mouse model was then generated (Nedd4-2Ksp1.3), where Nedd4-2 is deleted in renal epithelial cells, enabling further investigation of the kidney pathology in older animals. We found that these KO mice also develop kidney pathology with a slightly delayed onset (about 20 days) and by six months of age, display hydronephrosis, polydipsia and polyuria. As expected, these mice also exhibit high blood pressure on a standard salt diet, as well as an increase in blood Na+ concentration, decreased serum K+ and low aldosterone levels. This discovery that Nedd4-2-defiency causes a phenotype similar to chronic kidney disease (CKD), suggests that human Nedd4-2 (NEDD4L) variants linked to familial hypertension may also contribute to kidney disease.
In Nedd4-2Ksp1.3 animals the expression of the mature, active forms of all three ENaC subunits is increased6. Importantly, the administration of amiloride to these mice reduces the renal damage, suggesting that the elevated levels of active ENaC are, at least in part, responsible for the observed kidney disease. In contrast, although we also observe an increase in the levels of the Na+Cl− cotransporter (NCC) protein, treatment of mice with an inhibitor of NCC (hydrochlorothiazide) does not ameliorate the kidney disease, suggesting that ENaC, and not NCC, is the major contributor to the pathology6.
Our data suggest that in addition to hypertension caused by an upregulation of mature membrane-localized ENaC (and also elevated NCC expression7), the increased Na+ reabsorption via ENaC in mice lacking Nedd4-2 also causes progressive kidney injury6. However, the mechanism by which this elevated Na+ reabsorption in renal tubules causes cell death and kidney damage remains to be fully understood. NEDD4-2 has been shown to interact with and regulate a number of other membrane proteins, including ion channels and transporters. Although amiloride feeding experiments suggest that increased ENaC expression and activity are the primary drivers of the kidney disease, it is possible that other NEDD4-2 substrates contribute to elements of the pathology. Hence, further exploration into the mechanisms of the renal disease in Nedd4-2-deficient animals and its potential clinical significance is warranted. Our kidney tubule-specific Nedd4-2 KO mouse may provide a useful model for better understanding the molecular basis of CKD-like diseases and to evaluate the extent to which lowering Na+ intake or reabsorption can reduce renal injury.
Disclosure of potential conflicts of interest
Authors declare no conflicts of interest.
References
- [1].Foot N, Henshall T, Kumar S. Ubiquitination and the regulation of membrane proteins. Physiol Rev. 2017;97:253–281. doi: 10.1152/physrev.00012.2016. PMID:27932395 [DOI] [PubMed] [Google Scholar]
- [2].Goel P, Manning J, Kumar S. NEDD4-2 (NEDD4L): The ubiquitin ligase for multiple membrane proteins. Gene. 2015;557:1–10. doi: 10.1016/j.gene.2014.11.051. PMID:25433090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Harvey KF, Dinudom A, Cook DI, Kumar S. The Nedd4-like protein KIAA0439 is a potential regulator of the epithelial sodium channel. J Biol Chem. 2001;276:8597–8601. doi: 10.1074/jbc.C000906200. PMID:11244092 [DOI] [PubMed] [Google Scholar]
- [4].Rizzo F, Staub O. NEDD4-2 and salt-sensitive hypertension. Curr Opin Nephrol Hypertens. 2015;24:111–116. doi: 10.1097/MNH.0000000000000097. PMID:25602517 [DOI] [PubMed] [Google Scholar]
- [5].Boase NA, Rychkov GY, Townley SL, Dinudom A, Candi E, Voss AK, Tsoutsman T, Semsarian C, Melino G, Koentgen F, et al.. Respiratory distress and perinatal lethality in Nedd4-2-deficient mice. Nat Commun. 2011;2:287. doi: 10.1038/ncomms1284. PMID:21505443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Henshall TL, Manning JA, Alfassy OS, Goel P, Boase NA, Kawabe H, Kumar S. Deletion of Nedd4-2 results in progressive kidney disease in mice. Cell Death Differ. 2017;24(12):2150–2160. doi: 10.1038/cdd.2017.137. PMID:28862701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Ronzaud C, Loffing-Cueni D, Hausel P, Debonneville A, Malsure SR, Fowler-Jaeger N, Boase NA, Perrier R, Maillard M, Yang B, et al.. Renal tubular NEDD4-2 deficiency causes NCC-mediated salt-dependent hypertension. J Clin Invest. 2013;123:657–665. PMID:23348737 [DOI] [PMC free article] [PubMed] [Google Scholar]