

ABSTRACT
The main pathways for the repair of DNA double strand breaks (DSBs) are non-homologous end-joining (NHEJ) and homologous recombination directed repair (HDR). These operate mutually exclusive and are activated by 53BP1 and BRCA1, respectively. As HDR can only succeed in the presence of an intact copy of replicated DNA, cells employ several mechanisms to inactivate HDR in the G1 phase of cell cycle. As cells enter S-phase, these inhibitory mechanisms are released and HDR becomes active. However, during DNA replication, NHEJ and HDR pathways are both functional and non-replicated and replicated DNA regions co-exist, with the risk of aberrant HDR activity at DSBs in non-replicated DNA. It has become clear that DNA repair pathway choice depends on inhibition of DNA end-resection by 53BP1 and its downstream factors RIF1 and MAD2L2. However, it is unknown how MAD2L2 accumulates at DSBs to participate in DNA repair pathway control and how the NHEJ and HDR repair pathways are appropriately activated at DSBs with respect to the replication status of the DNA, such that NHEJ acts at DSBs in pre-replicative DNA and HDR acts on DSBs in post-replicative DNA. Here we show that MAD2L2 is recruited to DSBs in H4K20 dimethylated chromatin by forming a protein complex with 53BP1 and RIF1 and that MAD2L2, similar to 53BP1 and RIF1, suppresses DSB accumulation of BRCA1. Furthermore, we show that the replication status of the DNA locally ensures the engagement of the correct DNA repair pathway, through epigenetics. In non-replicated DNA, saturating levels of the 53BP1 binding site, di-methylated lysine 20 of histone 4 (H4K20me2), lead to robust 53BP1-RIF1-MAD2L2 recruitment at DSBs, with consequent exclusion of BRCA1. Conversely, replication-associated 2-fold dilution of H4K20me2 promotes the release of the 53BP1-RIF1-MAD2L2 complex and favours the access of BRCA1. Thus, the differential H4K20 methylation status between pre-replicative and post-replicative DNA represents an intrinsic mechanism that locally ensures appropriate recruitment of the 53BP1-RIF1-MAD2L2 complex at DNA DSBs, to engage the correct DNA repair pathway.
KEYWORDS: DNA repair pathway choice, DNA replication, 53BP1, H4K20 dimethylation, chromatin, HDR, NHEJ
Introduction
In the absence of exogenous genotoxic factors, it is calculated that a mammalian cell encounters up to 105 spontaneous DNA lesions every day, caused by normal DNA metabolism1. Luckily, most of these are lesions in a single strand of the DNA helix and do not constitute a danger for genome integrity, as they are efficiently repaired by error-free pathways. A common strategy to repair these lesions is an endonuclease-dependent resection of a portion of the single strand containing the damage, followed by use of the intact complementary strand as a template to repair the resected portion of the DNA. Double strand breaks (DSBs) of DNA molecules, such as caused by reactive oxygen species produced by metabolic processes or ionizing radiation, are rare. However, these lesions constitute a high risk to genome integrity and cell survival due to the complexity of the repair mechanisms that, first, must keep the two ends of the broken DNA in close proximity and, second, recover the genetic material in the absence of an intact single DNA strand to use as template. However, when the cells are in S-phase, DSBs in a replicated DNA region can be faithfully repaired by homologous recombination directed repair (HDR), which uses the intact sister chromatid as a template. HDR is activated by an initial step of limited 5′-3′ end-resection catalysed by the MRN complex bound to CTIP, which is then extended by the exonuclease activity of both Exo1 and the BLM-DNA2 complex, which may work in a redundant manner2. S-phase specific activity of CDK-Cyclin A is required for initial steps of HDR and, in particular, for the end-resection that requires CDK-dependent phosphorylation of CTIP and NBS13–7. The long 3′ single stranded stretch of DNA is then used to invade the sister chromatid by annealing to its complementary strand, forming D-loop structures in a process dependent on the sequential recruitment of RPA, BRCA2, and RAD51. In the D-loops, DNA synthesis occurs, leading to Holliday junctions that are unknotted in the final step by a class of enzymes called resolvases.
Upstream of this entire process, the recruitment of BRCA1 is required to actively displace 53BP1 from DSBs in order to release the inhibitory effect of 53BP1 on 5′-3′ end-resection8,9. Conversely, the inhibition of 5′-3′ end-resection by 53BP1 is strictly necessary to activate non-homologous end-joining (NHEJ), the other major repair pathway of DSBs. End-resection inhibition is needed to allow recruitment of the KU70-KU80 dimer, that recognizes unprocessed DSBs, and subsequently DNA-PK, that is required for the final steps of NHEJ ligation, mediated by Ligase IV10. Thus, 53BP1 and BRCA1 compete to occupy DSBs, making NHEJ and HDR mutually exclusive DNA repair pathways. To carry out its inhibition on 5′-3′ end-resection, 53BP1 is phosphorylated by ATM, which leads to the binding of RIF16,11. This, in turn, is required for the recruitment of MAD2L2 at DSBs12,13. The mechanism by which the 53BP1-RIF1 complex leads to MAD2L2 accumulation at DSBs is unknown, while it is well established that 53BP1, RIF1, and MAD2L2 inhibit end-resection in an epistatic manner12,13.
Cells rely on NHEJ for DNA repair when HDR is not possible due to the absence of a sister chromatid, such as in the G1–phase of the cell cycle. Accordingly, 53BP1 accumulates at DSBs of cells in G1. When cells enter into S-phase, accumulation of 53BP1 at DSBs becomes less efficient, with a portion of DSBs characterized by the recruitment of BRCA1, which actively removes 53BP1 through the ubiquitination of lysine 125, 127, and 129 of histone H2A9. The exact contribution of each BRCA1-dependent ubiquitination and the exact mechanism behind this process are not known. The accumulation of 53BP1 at DSBs is dependent on two protein domains: the ubiquitin dependent recruitment domain (UDR) that recognizes mono-ubiquitin conjugated to lysine 15 of histone H2A (H2AK15ub) and the tandem tudor domain that binds di-methylated lysine 20 of histone H4 (H4K20me2). Both histone modifications must be present at DSBs to activate NHEJ, as the single disruption of one of the two binding sites completely abolishes 53BP1 foci formation14-16. Upstream of the recruitment of 53BP1 and BRCA1 at DSBs, a complex network of ubiquitination events is locally activated and is mediated by the ubiquitin ligases RNF8 and RNF168. Although this network is not yet completely understood, one of the endpoints is the ubiquitination of H2AK15 by RNF16817.
While H2AK15ub is induced at DSBs, for H4K20me2 this seems less evident. Indeed, in G1 about 90% of the genome contains H4K20me2, indicating that upon the formation of a DSB, this binding site for 53BP1 is already present to large extent. As cells enter into S-phase, new nucleosomes are synthesized and incorporated into replicated DNA, leading to a twofold dilution of histone posttranslational modifications, which are then re-established on the newly synthesized nucleosomes within one cell cycle to maintain the epigenetic code from generation to generation18–22. In particular, H4K20me2 is re-established at the end of G2 in a process that involves three methyl-transferases: SETD8, which mediates mono-methylation, and SUV4-20H1 and SUV4-20H2, which redundantly catalyse the addition of a second methyl group. SETD8 is degraded before entry into S-phase through ubiquitination, which is mediated by the ubiquitin ligase CRL4Cdt2. Specifically, the PIP domain of SETD8 is bound by PCNA, which interacts with CRL4Cdt2. SETD8 is re-expressed in late G2, driving the re-establishment of H4K20me2. The fact that the expression of SETD8 mirrors the levels of H4K20me2 throughout the cell cycle and the overexpression of a non-degradable version that carries a single mutation in the PIP domain (SETD8ΔPIP) increases the global level of H4K20me2 strongly indicates that SETD8 is the limiting factor for the control of H4K20me2 levels19,22.
Here we show that MAD2L2 can be found in complex with RIF1 and 53BP1 on H4K20me2 containing chromatin and can suppress BRCA1 foci at DSBs in S/G2 when ectopically present. Moreover, we show that H4K20me2 drives the pathway choice for the repair of DSBs depending on the cell cycle phase and replication status of the DNA. The twofold dilution of H4K20me2 in replicated DNA regions abolishes accumulation of 53BP1-RIF1 complexes at DSBs, while the presence of H4K20me2 in the majority of nucleosomes in G1 and late G2 leads to the re-establishment of 53BP1-RIF1 foci at DSBs, that assisted by the recruitment of MAD2L2 obstructs the loading of BRCA1 to DSBs. We postulate that upon replication, H4K20me2 nucleosomes are flanked by newly incorporated H4K20me0 nucleosomes that cannot be bound by 53BP1, and thus grant access to BRCA1, which then activates the removal of 53BP1 from the proximal nucleosomes through ubiquitination, resulting in DNA end-resection.
Results
53BP1. and RIF1 foci formation is independent of CDK activity
Since the formation of 53BP1 and RIF1 protein complexes at DNA DSBs is the key upstream event that shifts the DNA repair pathway choice towards NHEJ2, we decided to investigate the mechanisms that regulate 53BP1-RIF1 foci recruitment. One candidate is CDK-Cyclin A activity, which activates DNA end-resection in S-phase by CTIP phosphorylation3. CTIP is required at DSBs for the removal of 53BP1 and consequent end-resection, thus we reasoned that CDK-dependent phosphorylation of CTIP might have a role in this function. To address this hypothesis, we analysed RIF1 foci formation 2 hours after irradiation in both G1 and S/G2 cells, respectively detected as negative and positive for Cyclin A expression. As previously shown23, the number of RIF1 foci is higher in G1 than in S/G2 cells. However, both G1 and S/G2 cells do not show differences in foci formation upon inhibition of CDK activity (Fig. 1A, B), indicating that RIF1 foci formation does not depend on CDK activity. In addition, we measured DNA end-resection upon induction of DSBs by RPA phosphorylation (Ser-4/8) and by RPA foci formation. As previously shown12, depletion of MAD2L2 increases the levels of RPA phosphorylation as result of the release of the inhibitory control of MAD2L2 on end-resection. Interestingly, CDK inhibition reduced the increased RPA phosphorylation in MAD2L2 depleted cells (Fig. 1C), and also reduced IR-induced RPA foci in both control and MAD2L2 depleted cells (Fig. 1D). Altogether these results indicate that CDK activity promotes end-resection downstream and independently of 53BP1, RIF1 and MAD2L2 recruitment. Strikingly, the analysis of RIF1 foci formation throughout the cell cycle showed that a subpopulation of cells, in a cell cycle phase displaying high levels of cyclin A above a certain threshold, forms a number of foci even higher than observed in G1 cells, both in the absence and presence of CDK inhibitor (Fig. 1A, B). This further indicates that 53BP1-RIF1 accumulation at DSBs is independent of CDK-Cyclin A activity and prompted us to investigate the mechanism that leads to reactivation of 53BP1-driven repair in cells with high levels of cyclin A.
Figure 1.
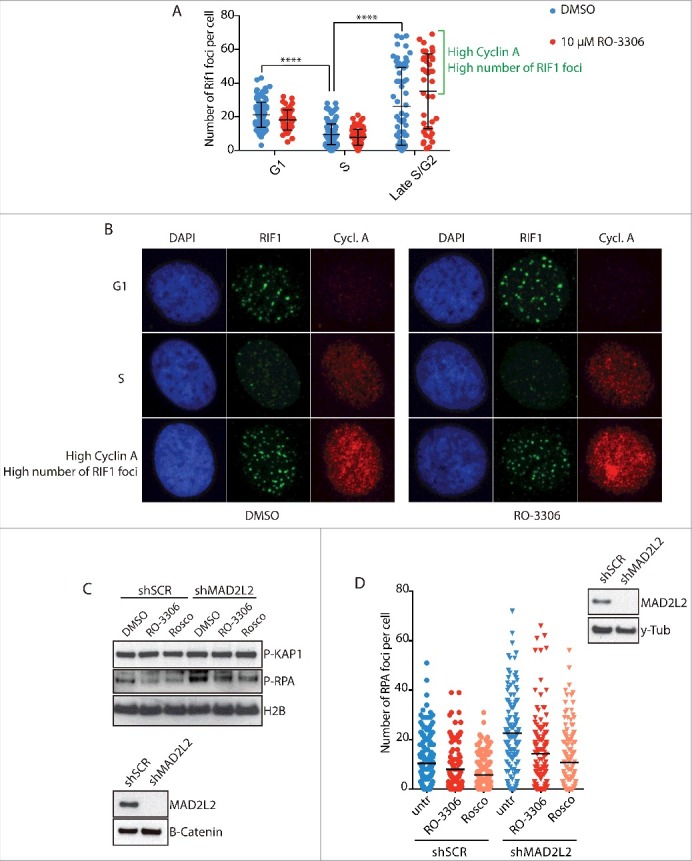
CDK activity regulates end resection downstream of RIF1 and MAD2L2. a) Analysis of RIF1 foci formation upon inhibition of CDK activity. HeLa cells were irradiated with 5Gy, fixed and stained for Cyclin A and RIF1 after 2h of recovery. 2 different thresholds of cyclin A levels were used to distinguish between G1/early S-phase (low cyclin A), S-phase (intermediate cyclin A), late S/G2 (high cyclin A). CDK inhibitor RO-3306 (10µM) was added to the medium 15 min before cell irradiation. The bracket highlights a subpopulation of cells in late S/G2 with a high number of foci, **** indicates p-value ≤ 0.0001. Statistical analysis is included in Material and Methods. b) Representative images of the experiment in a). c) Inhibition of CDK activity greatly reduced end-resection in MAD2L2 depleted U2OS cells. Cells were treated with 10µM RO-3306 or 20µM roscovitine for 15 minutes prior to adding neocarzinostatin (NCS) at 250ng/ml for 1 hour and western blotting of RPA phosphorylation (Ser-4/8) in whole cell extracts was used as a measure of end-resection. d) U2OS cells were treated with RO-3306 (10µM) or roscovitine (25µM) and irradiated with 5Gy. After 3 h, to allow for end-resection to take place, cells were pre-extracted to remove all chromatin-unbound RPA, fixed and stained for RPA foci. For each condition > 100 cells were quantified. Corresponding immunoblot shows the knockdown of MAD2L2 achieved.
53BP1. and RIF1 form foci at DSBs in late G2
First, we addressed whether the high number of RIF1 foci observed in the subpopulation of cells with highest levels of cyclin A reflect reactivation of RIF1 foci formation during the G2-phase of the cell cycle, as previous reports indicated that NHEJ becomes the predominant mechanism of DNA repair in G2-phase24,25. To distinguish between S- and G2-phase cells, asynchronous cell populations were pulsed with EdU for 45 min before irradiation. Based on the levels of EdU incorporation and Cyclin A, we distinguished early S-phase from late S-phase/early G2 and cells that already spent at least 45 min in G2 at the time of irradiation (late G2-phase) (Fig. 2A). We noted that RIF1 and 53BP1 accumulation at DSBs is progressively inhibited from G1/early S-phase to late S-phase, and then subsequently reactivated in late G2-phase. Consistently, inhibition of CDK activity before irradiation, did not affect either 53BP1 or RIF1 foci formation in any of the cell cycle phases analysed, confirming our previous results (Fig. 2B, C). Importantly, we observed 100% co-localization of 53BP1 and RIF1 in both Edu positive and negative cells, which indicates that the cell cycle dependent choice between NHEJ and HDR does not rely on the regulation of RIF1 binding to 53BP1 (Fig. 2D).
Figure 2.
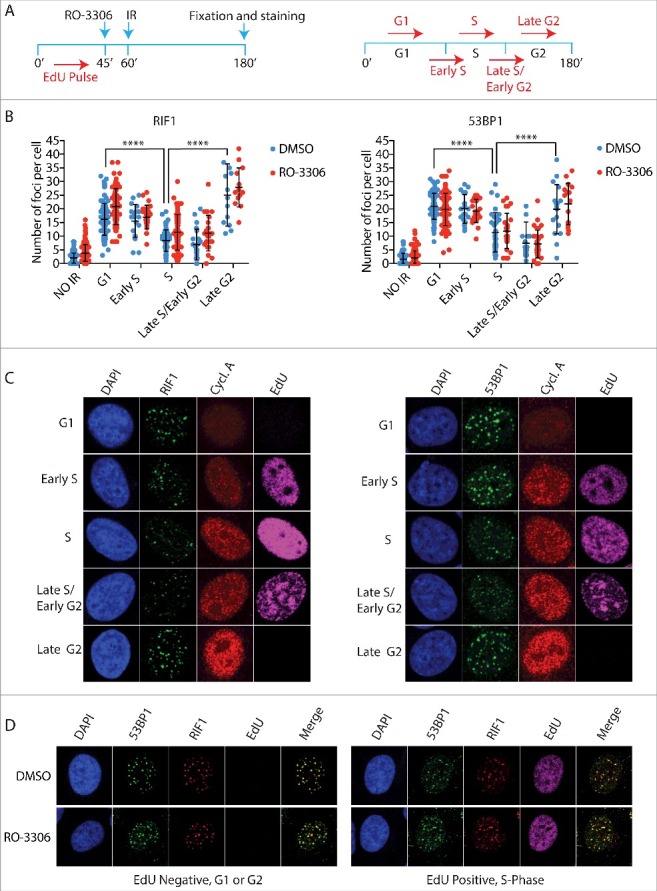
The 53BP1-RIF1 complex forms foci in late G2 cells. a) Schematic representation of experimental design (left) and how cell cycle phases were categorized (right). Asynchronous HeLa cells were pulsed for 45 min with 10 µM EdU, a thymidine analogue, to mark replicated DNA. After EdU wash, 10 µM RO-3306 was added to the medium 15 min before irradiation with 5Gy. 2 h after irradiation cells were stained for EdU, cyclin A and either 53BP1 or RIF1. G1 cells are EdU and cyclin A negative. Early S phase cells show low levels of cyclin A and EdU staining. High levels of EdU and intermediate cyclin A levels identify S-phase cells, as they spent the EdU incubation time entirely in S-phase. Late S/Early G2 cells have low levels of EdU and high levels of cyclin A, as the low levels of EdU indicate that they spent part of the EdU incubation in G2. Late G2 cells show high levels of cyclin A and are EdU negative, which indicate that they were already in G2 at the time of irradiation for at least 45 min. b) Quantification of RIF1 and 53BP1 foci during the different phases of the cell cycle schematized in a). Statistical analysis is included in Material and Methods. c) Representative images of experiment in b). d) Representative images that show 100% co-localization between RIF1 and 53BP1. Cells were pulsed with EdU for 45 min, irradiated and co-stained for RIF1 and 53BP1 after a recovery of 2 h.
H4K20me2 levels control DNA repair pathway choice
Because of its cell cycle phase related changes, the level of di-methylation of the lysine 20 of histone 4 (H4K20me2) could potentially serve as a key regulator at the crossroads of NHEJ and HDR. H4K20me2 levels are high in G1, reduced by a factor of two in S-phase due to the incorporation of unmodified newly synthesized H4K20me0 nucleosomes behind the replication fork, and re-established in late G2 through the re-expression of SETD8. As shown above, the efficiency of 53BP1-RIF1 foci formation follows the exact same pattern (Fig. 1A, B). H4K20me2 is essential for the binding of 53BP1 to the nucleosomes around DSBs, thus we hypothesized that the high concentration of H4K20me2 nucleosomes at DSBs in G1 is determining the formation of 53BP1-RIF1 foci and, conversely, the two-fold dilution in H4K20me2 on the replicated DNA of S-phase cells drives 53BP1-RIF1 dissociation from chromatin, in favour of BRCA1 recruitment. Supporting this hypothesis, we observed that, in our cells, H4K20me2 levels fluctuate from G1 to G2 (Fig. 3A, B). Detection of H4K20me2 by immunofluorescence shows that G1 cells have high H4K20me2 levels that drop progressively with the increasing levels of Cyclin A as cells progress to G2. In contrast, a subpopulation of cells in late G2-phase, with high levels of Cyclin A, shows re-establishment of H4K20me2, presumably on the newly incorporated nucleosomes. Furthermore, the number of 53BP1 foci is proportional to the levels of H4K20me2 (Fig. 3C).
Figure 3.
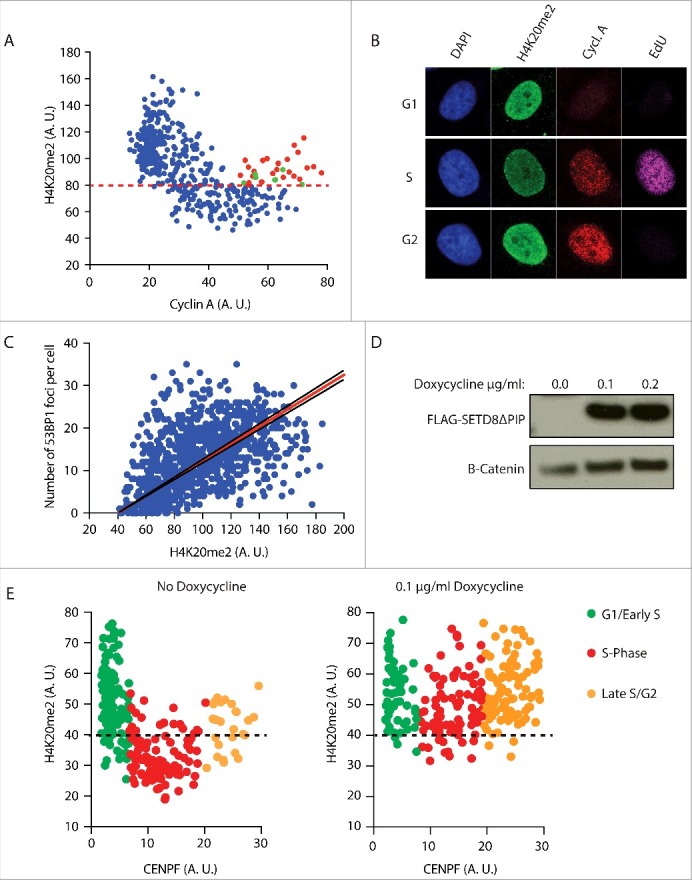
The number of 53BP1 foci correlates with the level of H4K20me2. a) Fluctuation of H4K20me2 throughout the cell cycle. Asynchronous HeLa cells were pulsed for 45 min with 10 µM EdU and stained for Cyclin A, EdU and H4K20me2. Levels of H4K20me2 of each cell were plotted against the levels of cyclin A. Levels are represented as mean fluorescence intensity (arbitrary units). Red dots indicate late G2 cells (EdU negative and high Cyclin A) that show high levels of H4K20me2. Green dots indicate late G2 cells that haven't re-established H4K20me2 levels yet. Red dashed line mark differentiates high levels of H4K20me2 typical of G1/Early S-phase cells and low levels of H4K20me2 typical of S-phase cells. b) Representative images of experiment in a). c) 53BP1 foci formation is proportional to the levels of H4K20me2. Cells were irradiated with 5 Gy and stained for 53BP1 and H4K20me2 after 2 h recovery. The number of 53BP1 foci was plotted against the levels of H4K20me2 represented as mean fluorescence intensity (arbitrary units). Calculated correlation index is 0.49. d) Western blot analysis of flag-tagged SetD8ΔPIP overexpression in U2OS cells incubated with 0.1 µg/ml and 0.2 µg/ml of doxycycline for 16 h. Anti-flag antibody was used to detect flag-tagged SetD8ΔPIP. e) In parallel, cells were stained for H4K20me2 and CENPF, which mirrors cyclin A expression from G1 to G2 phase of the cell cycle. Cells not treated with doxycycline show fluctuation of H4K20me2 (left). Induction of SETD8ΔPIP overexpression with 0.1 µg/ml doxycycline for 16 h increases H4K20me2 levels in S-phase (right). G1/Early S, S-phase and Late S/G2 cells are characterized using 2 thresholds of CENPF levels. Black dashed line differentiates high levels of H4K20me2 typical of G1/Early S-phase cells and low levels of H4K20me2 typical of S-phase cells. Levels are represented as mean fluorescence intensity (arbitrary units).
Next, we tested whether enforced methylation of H4K20 in newly synthesized nucleosomes would restore 53BP1-RIF1 foci formation in S-phase. Overexpression of SETD8 has been shown to increase the levels of H4K20me2 in whole cell extracts22. We reasoned that the observed increase of H4K20me2 is due to a forced methylation of newly synthesized nucleosomes in S-phase, before cells progress to late G2. To increase the levels of H4K20me2 in S-phase, we overexpressed a SETD8 version with a single mutation in the PIP domain that inhibits its degradation in S-phase (SETD8ΔPIP) (Fig. 3D). As we aimed for, this prematurely increased the levels of H4K20me2 in S-phase cells (Fig. 3E). To investigate whether the increased levels of H4K20me2 favoured NHEJ over HDR at DSBs of S-phase cells, we synchronized cells at the G1/S boundary of the cell cycle and overexpressed SETD8ΔPIP before releasing cells into S-phase. Three hours after the release, cells were irradiated and after 4 hours of recovery they were co-stained for 53BP1 and BRCA1. In the absence of SETD8ΔPIP in S-phase and, thus, with replication-associated reduced H4K20me2, we observed a lower number of 53BP1 foci than BRCA1 foci, indicating that HDR is the mechanism of preference for the repair of DSBs in S-phase (Fig. 4A, B). Importantly, there was virtually no co-localization between 53BP1 and BRCA1 foci, indicating that at 4 hours post irradiation the choice between NHEJ and HDR at each DSB has been completed (Fig. 4C: no doxycycline, cell 1 and cell 2). This is different from earlier time points when co-localization between 53BP1 and BRCA1 can still be observed9. This indicates that DNA repair pathway choice is a dynamic process that entails 53BP1 and BRCA1 antagonizing each other at DSBs until one prevails and promotes its own repair pathway. Strikingly, expression of SETD8ΔPIP, thereby overriding the PCNA/PIP-box dependent degradation of SETD8 in S-phase and elevating H4K20me2 in S-phase nucleosomes, leads to increased numbers of 53BP1 foci at the cost of BRCA1 foci (Fig. 4B and 4C: doxycycline panels, cell 1). This strongly indicates that the concentration of H4K20me2 is critical for repair pathway choice. Moreover, a subpopulation of cells overexpressing SETD8ΔPIP show co-localization between large 53BP1 foci and small BRCA1 foci, suggesting that, at these particular DSBs, H4K20me2 was not at saturation, allowing a limited access of BRCA1 to DSBs, most likely in the regions with low H4K20me2 (Fig. 4C: doxycycline panels, cell 2).
Figure 4.
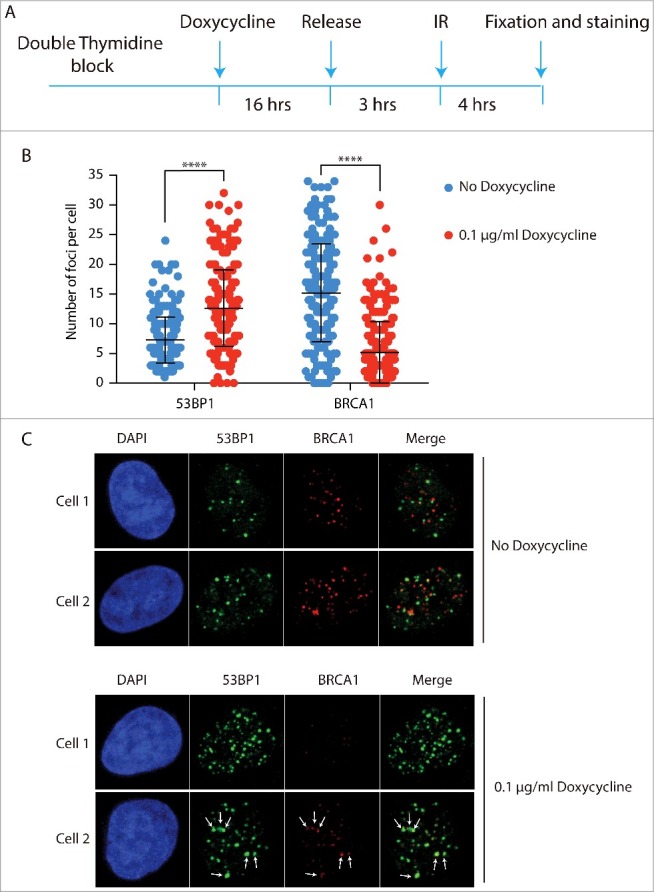
H4K20me2 levels control DNA repair pathway choice between NHEJ and HR. a) Schematic representation of experimental design. U2OS cells were synchronized with a double thymidine block and SETD8ΔPIP overexpression was induced for 16 h before the release in S-phase by incubation with 0.1 µg/ml Doxycycline. Cells were irradiated after 3 h from the release and, after 4 h recovery, co-stained for 53BP1 and BRCA1. b) Quantification of 53BP1 and BRCA1 foci in the experiment described in a). A 2 tailed t-test was run to compare cells treated either with buffer or 0.1 µg/ml doxycycline for both 53BP1 and BRCA1 foci with resulting p values < 0.0001. c) 2 representative cells for each condition, i.e. no doxycycline and 16 h incubation with 0.1 µg/ml doxycycline. White arrows in Cell 2 incubated with doxycycline indicate co-localization between big 53BP1 foci and small BRCA1 foci.
MAD2L2 forms a complex with 53BP1 and RIF1 that suppresses BRCA1 loading to DSBs
How the 53BP1-RIF1 complex prohibits the accumulation of BRCA1 at DSBs remains poorly understood. One insight in the mechanism came from the discovery that 53BP1-RIF1 accumulation at DSBs leads to the recruitment of MAD2L2, which is necessary to inhibit end-resection and thereby promote recruitment of downstream NHEJ factors12,13. Abolishing MAD2L2 localisation to DSBs allows initiation of end-resection, despite the presence of 53BP1 and RIF1 at DSBs. How the 53BP1-RIF1 complex promotes the recruitment of MAD2L2 is unknown. To address this, we performed immunoprecipitation of 53BP1 after cell irradiation. In this setting, MAD2L2 specifically co-purified from chromatin extracts of irradiated cells, together with H4K20me2, indicating that MAD2L2 forms a protein complex with 53BP1 and RIF1 bound to nucleosomes containing H4K20me2 (Fig. 5A). Furthermore, mass spectrometry analysis of affinity-purified Flag-tagged MAD2L2 and its interacting proteins, confirmed previously reported interactions as well as an interaction between MAD2L2 and 53BP1 that was enhanced upon irradiation (Fig. 5B, C). Within the same experiments MAD2L2 also interacted with RIF, in a manner that appears less affected by irradiation, suggesting that RIF1 and MAD2L2 might constitutively interact with each other and accumulate at DSBs through the ATM-phosphorylation dependent binding of RIF1 to 53BP1. Interestingly, overexpression of MAD2L2 suppressed BRCA1 localisation to DSBs in S/G2, suggesting that MAD2L2 could be a limiting factor in DNA repair pathway choice (Fig. 5D). While we succeeded in detecting 53BP1-RIF1-MAD2L2 interactions, we did not reproducibly identify any enzymes in our purifications, besides ATM involved in 53BP1 phosphorylation, that can be directly linked to the mechanism of inhibition of end-resection as we currently understand it. Such an enzymatic activity still remains to be discovered or alternatively, with all the nucleosomes containing H4K20me2, 53BP1-RIF1-MAD2L2 might simply physically obstruct the access of BRCA1 to DSBs (Fig. 6). Conversely, the presence of proximal H4K20me0 nucleosomes in replicated DNA will grant access of BRCA1 to DSBs, which will then through enzymatic activity, i.e. ubiquitination9, actively remove 53BP1 and its interactors to release the inhibition of end-resection and finally initiate HDR.
Figure 5.

MAD2L2 is recruited at DSBs by protein interaction with the 53BP1-RIF1 complex and suppresses BRCA1 accumulation. a) HeLa cells were irradiated with 10 Gy and 53BP1 was immuno-precipitated after 2 h recovery. H4K20me2 and MAD2L2 were co-purified with 53BP1 exclusively upon irradiation. b) Cells expressing FLAG-tagged MAD2L2 were irradiated and FLAG immunoprecipitation was performed after 2 h recovery. Western blot analysis of inputs, supernatant and elution shows that FLAG-MAD2L2 is efficiently and equally purified from both non-irradiated and irradiated cells. c) 53BP1, RIF1 and ATM were identified by mass spectrometry analysis as novel protein interactors of MAD2L2 together with 7 previously known MAD2L2 protein interactors. PSM (peptide spectrum matches) counts are shown for 2 independent experiments. Specificity of the interaction is addressed in a smaller scale FLAG-MAD2L2 immunoprecipitation (Supplementary Figure 1). d) U2OS cells transfected with control GFP vector (EV) or GFP-MAD2L2 where irradiated with 10 Gy at 48 h after transfection, fixed at 1 h post IR and stained for Cyclin A (serving as S/G2 marker) and BRCA1 to assess BRCA1 accumulation to DSBs in S/G2 (n = 2 independent replicates).
Figure 6.
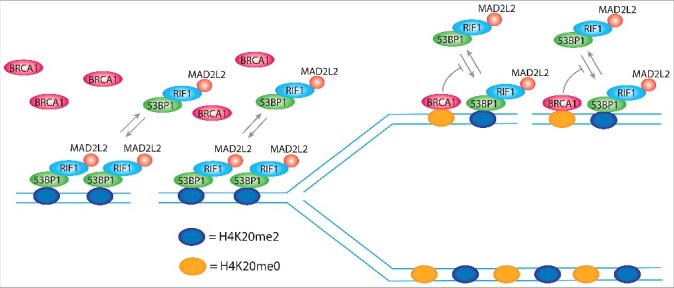
H4K20me2 levels control DNA repair pathway choice. 53BP1 forms foci at DSBs by simultaneous binding to ubiquitinated lysine 15 of histone H2A (H2AK15ub; not represented in the model) and di-methylated lysine of histone H4 (H4K20me2), using respectively a ubiquitin dependent recruitment domain (UDR) and a tandem tudor domain. The single disruption of one of the 2 binding sites completely abolishes 53BP1 foci formation. H2AK15ub is specifically induced at DSBs, ensuring the binding of 53BP1 exclusively at sites of damage, while H4K20me2 is present in more than 90% of the nucleosomes of non-replicated DNA, which indicates that it is not induced for the formation of 53BP1 foci. However, H4K20me2 plays a critical role for the choice of the correct DNA repair pathway depending on the replication state of DNA. In non-replicated DNA, all the nucleosomes bear H4K20me2 and thereby present the binding site for 53BP1, which in complex with RIF1 and MAD2L2, leaves no access points for BRCA1. FRAP experiments show that 80% of 53BP1 molecules at foci dynamically exchange with the nucleoplasmic pool within 30 min after photo-bleaching. In replicated DNA, H4K20me2 nucleosomes are flanked by newly synthesized nucleosomes with unmodified lysine 20 of histone H4 (H4K20me0). This opens breaches for the access of BRCA1 to the chromatin that by ubiquitination of lysine 125, 127, and 129 of histone H2A modifies the binding site for 53BP1, inhibiting the rebinding of 53BP1 from the nucleoplasmic pool and, thus, leading to its consequent release.
Discussion
DSBs are particularly dangerous lesions that require highly complex repair mechanisms to avoid cells going through mitosis with broken chromosomes. NHEJ and HDR are the major pathways of DSB repair, both of which are characterized by multiple and intricate networks that not only reflect the complexity of the repair process, but also the importance of choosing the correct pathway depending on the cell cycle phase. Multiple mechanisms operate to suppress inappropriate DNA repair activities that would have detrimental consequences. For instance, in mitosis CDK-cyclin B inhibits the recruitment of RNF8 and 53BP1 to avoid the activation of DNA repair at telomeres while they are temporarily uncapped and at risk of forming telomere fusions26–28. In S-phase, CDK-cyclin A is required for DNA end-resection, excluding the possibility of HDR activation in G1 when CDK-cyclin A is absent or low. Additional CDK-independent mechanisms are present in G1 to prevent HDR at DSBs, such as reduced expression of CTIP and USP11 in G1, which are required in S-phase to initiate DNA end-resection and formation of the BRCA1-PALB2-BRCA2 complex, respectively4,29,30. The presence of multiple pathways of HDR inhibition in G1 emphasizes the detrimental consequences of unwanted activation of resection and HDR at DSBs in non-replicated DNA. Indeed, activation of DNA end-resection at DSBs in non-replicated DNA would preclude the possibility to repair DSBs with both HDR and NHEJ. This because HDR cannot succeed in the absence of an intact template while NHEJ relies on minimal processing of DSB ends to allow the re-joining reaction performed by Ligase IV10.
In S-phase, the replication of the genome represents a particularly interesting challenge to the control of DNA repair pathways, as before the completion of DNA replication both non-replicated and replicated DNA co-exist, each requiring a different form of DNA repair to maintain genome integrity.
Mechanisms of HDR inhibition acting in G1 phase do not suffice to prevent unwanted HDR on non-replicated DNA in S-phase as both the NHEJ and HDR machineries need to be capable to perform repair on the correct type of chromatin. Relying on a matter of probability to perform HDR, which would increase proportionally to the concentration of a functional HDR machinery in S-phase, seems an unlikely successful method to safeguard genome integrity. Furthermore, NHEJ needs to be suppressed to prevent replication intermediates from forming NHEJ dependent chromosomal fusions. Thus, maintenance of genome integrity upon DSB formation in S-phase appears to require local control of DNA repair pathway choice. Our results indicate that the DNA repair network tackles this issue by using the epigenetic mark H4K20me2 to distinguish between non-replicated and replicated DNA to safely activate the correct repair pathway depending on the DNA replication state. 53BP1 forms foci by direct binding to H4K20me2 through its tandem tudor domain. We show that a twofold dilution of H4K20me2 in replicated DNA, due to incorporation of H4K20me0, is sufficient to inhibit 53BP1 foci formation, since overexpression of SETD8ΔPIP in S-phase and consequent di-methylation of newly synthesized nucleosomes, leads to a drastic increase of 53BP1 foci at the disadvantage of BRCA1 foci. A possible mechanistic explanation is that saturation levels of H4K20me2, i.e. 90% of the nucleosomes of non-replicated DNA under physiological conditions, lead 53BP1, and its interacting proteins, to fully occupy the area surrounding DSBs of non-replicated DNA, leaving insufficient opportunity for access by BRCA1. Once into the breaches, BRCA1 can promote the release of 53BP1 through ubiquitination of K125, K127 and K128 of H2A (Fig. 6).
Recent reports have also identified an important role for H4K20 methylation as an epigenetic mark that allows the recognition of the DNA replication state to recruit the correct DNA repair pathway. First of all, data, highly similar and in complete agreement to what we present here, were recently reported while this manuscript was in preparation, supporting the same conclusion that H4K20 methylation modulates the cell cycle dependent response of 53BP1 to DSBs and in particular that replication-associated dilution of H4K20me2 directs the 53BP1-driven repair pathway to pre-replicative chromatin31. In addition, other recent work has reported that the TONSL-MMS22L homologous recombination complex is recruited specifically to replicated DNA through direct binding of the TONSL ankyrin repeat domain (ARD) to newly synthesized H4K20me0 histones. Under replication stress conditions, this interaction protects stalled replication forks and promotes RAD51-dependent repair of collapsed replication forks32.
Despite that BRCA1-dependent ubiquitination is required for the removal of 53BP1, the exact mechanism of 53BP1 release is not known. FRAP experiments show that the binding of 53BP1 to the nucleosomes of DSBs is dynamic, with an average of 80% of 53BP1 molecules at foci that exchange nucleosome-bound 53BP1 with the nucleoplasmic pool of unbound 53BP1 within 30 min after photo-bleaching33. One possibility is that BRCA1-mediated ubiquitination might affect the rebinding of nucleoplasmic 53BP1 during this dynamic exchange by promoting modification of 53BP1 binding sites (Fig. 6), e. g. either changing the relative orientation of H2K15ub and H4K20me2 or by their removal. One candidate for the first mechanism could be SMARCAD1, an ATP-dependent nucleosome-remodeler, recently characterized as an effector of BRCA1 mediated antagonization of 53BP19. Alternatively, BRCA1 could promote the access of either a deubiquitinating enzyme or a demethylase to H4K20me2 nucleosomes to remove either H2AK15ub or H4K20me2.
Why cells reactivate NHEJ in late G2 by re-establishing high levels of H4K20me2 that promote 53BP1-RIF1 binding (Figs. 2,3), is unclear. H4K20me2, together with H3K79me2, is linked to proper replication licensing34–36. The reason why both are re-established in late G2 instead of in G1, is not completely understood. Since both H4K20me2 and H3K79me2 are strongly linked to chromatin compaction20,21, one possibility is that both histone modifications prepare the ground for chromatin condensation in the next phase of the cell cycle, i.e. prophase of mitosis. HDR steps, such as DNA end-resection and strand invasion, are not feasible on compacted chromatin, so the reactivation of NHEJ at the end of G2 might be a necessity, rather than a matter of probability, which increases with the re-establishment of H4K20me2.
MAD2L2 was identified as a NHEJ promoting factor participating in DNA repair pathway choice that localises to DSBs in a 53BP1, RIF1 and ATM kinase dependent manner12,13. How the 53BP1-RIF1 complex enables the recruitment of the downstream factor MAD2L2, was still an open question. Here, we show that MAD2L2 is recruited at DSBs by forming a protein complex with 53BP1 and RIF1, which is detected both by using a candidate approach and by mass spectrometry analysis (Fig. 5). Except for ATM, which is known to be required for the binding of RIF1 to 53BP1, our mass spectrometry analysis did not reproducibly detect enzymatic activities in association with the 53BP1-RIF1-MAD2L2 complex that with the current knowledge clarify its mechanism of end-resection control. We and others previously reported that depletion of MAD2L2 is sufficient to at least partially abrogate DNA end-resection inhibition while 53BP1 and RIF1 are still present at DSBs12,13. This suggests that, in DNA repair pathway choice, MAD2L2 is somehow important for the action of an enzymatic activity that still remains to be discovered. Alternatively, MAD2L2 in complex with 53BP1, RIF1 and potentially other proteins, when bound to DSBs in chromatin containing saturating H4K20me2, might help form a physical obstruction that excludes BRCA1 and end-resection at DSBs.
Materials and methods
Cell culture, cell synchronization, inhibitor treatment and generation of stable cell lines
All cells were grown as described before12,37. Cell synchronization at the G1/S boundary was performed by a double thymidine block. Day 1 at 2 pm, cells were incubated in medium containing 2.5 mM Thymidine (Sigma, T9250). Day 2 at 9 am, cells were washed twice with PBS and released in thymidine-free medium. Day 2 at 6 pm, cells were incubated in medium containing 2.5 mM Thymidine. Day 3 at 9 am, cells were washed twice with PBS and released in S-phase with thymidine-free medium. RO-3306 (Millipore, 217699) was used at 10µM, roscovitine (Sigma-Aldrich, R7772) at 20µM (western blot) or 25µM (IF) and neocarzinostatin (NCS, Sigma-Aldrich, N9162) at a final concentration of 250ng/ml for 1 hour. Doxycycline inducible U2OS cells were generated in a 2-step procedure. First, cells were transduced as before37 with pLKO-Tet-On lentivirus38 to express the Tet-repressor and selected with G418 (2 mg/ml). Selected cells were transfected according to the manufacturer (Mirus, TransIT-LT1, MIR 2300) with both pcDNA4/TO-Flag-SETD8WT and Flag-SETD8ΔPIP22 and selected with zeocin (0.4 mg/ml) for 3 weeks. Clones were isolated and tested for inducible expression of SETD8 constructs. Cells expressing SETD8WT were excluded from experimental procedures because of low expression levels due to PIP domain-dependent degradation. To produce cells expressing 3xFlag N-terminal tagged MAD2L2, endogenous MAD2L2 was depleted by transducing cells with PLKO-puro shRNA obtained from Mission library clones (Sigma; TRCN0000006573: 5′-CATCTTCCAGAAACGCAAGAA-3′) and puromycin selected cells were transduced with pMSCV-3xFlag-MAD2L2-blasticidin retroviruses as described before37. To obtain MAD2L2 depleted cells, U2OS or HeLa cells were infected with lentivirus containing an PLKO-puro shRNA targeting human MAD2L2 or a PLKO-puro scrambled control (Human MAD2L2 sh Sigma Mission library clone, TRCN0000006570: 5′-CCCGGAGCTGAATCAGTATAT-3′ and Scrambled control shRNA: 5′-CAACAAGATGAAGAGCACCAA-3′) and selected with puromycin.
Immunofluorescence
Immunofluorescence was performed on cells grown in 8-well chamber slides (Millicell EZ slide), essentially as described before12. Cells were washed twice with cold PBS, fixed with 2% paraformaldehyde for 10 min at RT, washed twice with PBS, permeabilized with 0.5% Triton/PBS for 10 min, washed 3 times with PBS, then stored at 4°C in PBS containing sodium-azide or used directly for the staining procedure. For RPA staining, cells were washed twice with PBS and pre-extracted for 5 minutes using 0.5% Triton/PBS prior to fixation. For staining, cells were incubated with blocking solution (0.02% Triton, 5% NGS, 5% FBS in PBS) for 30 min at RT, incubated with the primary antibody in blocking solution overnight at 4°C, washed 3 times for 5 min with 0.02% Triton/PBS, incubated for 1 h with secondary antibody in blocking solution and washed 3 times for 5 min with 0.02% Triton/PBS. Slides were then mounted in Vectashield containing DAPI (Vector Laboratories). Primary antibodies used were against 53BP1 (A300-272A, Bethyl, 1:2000; MAB3802, clone BP13, Millipore, 1:500), RIF1 (A300-569A, Bethyl, 1:1000), Cyclin A (MS-1061-S0, Ab-6, Thermo Fisher Scientific, 1:500; sc-751, H-432, Santa Cruz, 1:100), H4K20me2 (ab9052, Abcam, 1:500) and RPA34-20 (GTX16850, GeneTex, 1:500). Alexa Fluor 488 or 568 goat anti-mouse or anti-rabbit IgG (Invitrogen) were used as secondary antibodies. The EdU staining with Alexa 647 was performed according to the manufacturer's instructions (Click-it, Life Technologies). Images were acquired by taking z stacks of the nuclei on a Leica SP5 confocal system with a 63x NA 1.32 oil objective and LAS-AF software. Foci were counted on maximum-intensity projection using an automatic and objective analysis as described before39.
Immunoblotting
Whole-cell lysates were prepared as described before12. Primary antibodies used were against MAD2L2 (sc135977, Santa Cruz, 1:400), phospho-RPA32 S4/S8 (A300-245A, Bethyl, 1:1000; NBP1-23017, Novus Biologicals, 1:1000), H2B (07-371, Millipore, 1:1000), P-Kap1 S824 (A300-767A, Bethyl, 1:500), B-catenin (610154, BD Biosciences, 1:2000), 53BP1 (A300-272A, Bethyl, 1:2000), y-Tubulin (T6557, Sigma-Aldrich, 1:1000), Flag epitope (F7425, Sigma, 1:1000), H4K20me2 (ab9052, Abcam 1:2000) and γH2AX (p-histone H2AX S139, Cell Signaling 2577S, 1:1000).
53BP1. immunoprecipitation
About 50 × 10e6 HeLa cells were used for each sample, i.e. non-irradiated and irradiated cells (10Gy). Cell culture medium was completely removed while cells were kept on ice. Protein crosslinking was performed for 10 min directly in the petri-dishes with 2% paraformaldehyde/PBS at RT. Freshly prepared 0.125M Glycine/PBS was added and incubated for 5 min to quench the cross-linking reaction. Cells were placed back on ice and washed twice with ice cold PBS before being scraped and collected in 15 ml falcon tubes. Next, cells were centrifuged at 2000 rcf for 5 min and PBS was completely removed before freezing the pellets in liquid nitrogen. All following steps were performed in ice cold buffers and at 4°C, unless specified differently. Pellets were re-suspended and incubated for 10 min in 10 ml of buffer LB1 (50mM Hepes-KOH, pH7.5; 140 mM NaCl; 1 mM EDTA; 10% Glycerol; 0.5% Igepal CA-630; 0.25% Triton X-100) on a rotating wheel. Cells were pelleted at 2000 rcf for 5 min, re-suspended in 10 ml buffer LB2 (10 mM Tris-HCl, pH8.0; 100 mM NaCl; 1 mM EDTA; 0.5 mM EGTA), incubated for 10 min on a rotating wheel, pelleted at 2000 rcf for 5 min and resuspended in 3 ml buffer LB3 (10mM Tris-HCl, pH8.0; 100 mM NaCl; 1 mM EDTA; 0.5 mM EGTA; 0.1% Na-Deoxycholate; 0.5% N-lauroylsarcosine). Samples were sonicated for 7 cycles of 30” ON/30” OFF. Lysates were cleared by centrifugation at 14.000 rpm for 10 min using a benchtop centrifuge. Triton was added to a final concentration of 0.2% before the incubation with beads. A mix of 80% protein A and 20% protein G dynabeads (final volume 100 µl) was washed 3 times with 500 µl PBS/BSA buffer containing 0.25 mg/ml BSA (Sigma-aldrich, A4737), incubated with 10 µg of anti-53BP1 antibody (A300-272A, Bethyl) for 4 h in 300 µl PBS/BSA at 4°C, washed 3 times with 500 µl PBS/BSA buffer, re-suspended in 100 µl PBS/BSA and equally divided into 2 aliquots that were added to the lysate of non-irradiated and irradiated cells. After overnight incubation on a wheel at 4°C, beads were washed 3 times with 1 ml buffer LB3 supplemented with 0.2% triton. Elution and reversion of formaldehyde crosslinks were performed by incubating the beads in 75 µl SDS buffer (125 mM Tris-HCl, pH8.0; 20% glycerol; 4% SDS) using a benchtop thermomixer at 95°C and 750 rpm. 2ml round-bottom tubes were used to avoid sedimentation of beads during the elution process.
Flag-immunoprecipitation of MAD2L2 and mass spectrometry analysis
U2OS cells depleted of endogenous MAD2L2 and complemented with endogenous levels of 3xFlag-tagged MAD2L2 were used for protein complex purification. About 150 × 10e6 cells were used for each sample, i.e. non-irradiated and irradiated cells (10 Gy). All steps, unless specified differently, were performed with ice cold buffers and on ice. Centrifugation steps were performed at 4°C. Cells were washed twice with PBS, scraped and centrifuged at 400 × g for 5 min. The packed cell volume (PCV) was estimated and kept as reference for all the following steps, i.e. not taking into account the changes in cell/nuclei pellet that occur in following steps. Pellets were washed twice with 3X PCV of buffer A (10 mM Hepes-KOH, pH7.5; 10 mM KAc; 1.5 mM MgCl2; 0.1 mM DTT; phosphatase inhibitor cocktail-PhosSTOP – Sigma Aldrich), re-suspended in 2X PCV of buffer A and centrifuged at higher speed, 1100 × g, to favour breakage of cytoplasmic membranes. The resulting mix of cells and nuclei were resuspended in 2X PCV of buffer M1 (20 mM Hepes-KOH, pH7.5; 60 mM KAc; 3 mM CaCL2; 0.05% Igepal CA-630; phosphatase inhibitor cocktail, PhosSTOP, Roche; EDTA-free protease inhibitor cocktail, Roche), shortly sonicated at low voltage for 4 cycles of 5” ON/30” OFF and incubated with Micrococcal nuclease (Thermo Scientific, 88216, 217 U/µl) for 3.5 min at 37°C using ∼80 units per 500 µl of lysate. The reaction was stopped by adding 20 mM EGTA and tubes were put back in ice. Lysates were cleared by centrifugation at 14.000 rpm for 10 min using a benchtop centrifuge. The slurry of magnetic anti-flag M2 beads (Sigma-Aldrich, M8823) was washed 4 times with 5X the volume of packed beads in buffer M1 and the initial slurry was reconstituted with buffer M1 before the incubation with lysates for 2 h at 4°C. 30 µl of packed beads were used per every 500 µl of lysate. After incubation, beads were washed 4 times with buffer M1, using for each wash the same volume of initial cleared lysate. A dilution of 0.5 mg/ml of 3xFlag peptide (Sigma-Aldrich, F4799) in buffer M1 was used for elution of protein complexes. Per sample 2 cycles of elution were performed for 20 min at RT in a thermomixer (750 rpm) using 3 times the volume of packed beads for each elution. 2ml round-bottom tubes were used to avoid sedimentation of beads during the elution process. Samples were then frozen in liquid nitrogen and stored at −80°C. 0.1% of input and immunoprecipitation supernatant and 1% of total elution were used for western blot analysis. Eluted proteins were concentrated with an Amicon Ultra-0.5 ml filter unit (Merk Millipore, UFC501008), separated on SDS-PAGE and silver-stained (Pierce, silver stain kit, 24600, before mass spectrometry analysis. Lanes were excised from silver stained gels and cut into 10 gel bands per IP sample. Bands were destained, proteins were reduced with DTT (1 hr at 600C) and subsequently alkylated using iodoacetamide (30 min at RT). In-gel digestion with 3ng/uL trypsin (Gold, Mass Spectrometry Grade, Promega) in 50 mM ammonium bicarbonate (pH 8.5) was performed overnight at 37 oC. Peptides were extracted with acetonitrile, dried down in a speed vacuum centrifuge and reconstituted in 10% formic acid prior to mass spectrometry analysis. Peptide mixtures were analyzed by nanoLC-MS/MS on an Orbitrap Fusion Tribrid mass spectrometer equipped with a Proxeon nLC1000 system (Thermo Scientific) employing a linear 45-min gradient, essentially as described previously40. Raw data files were processed with Proteome Discoverer (version 1.4.1.14, Thermo Fisher Scientific) searching against the Swissprot human database (april 2015, 20.205 entries) using Mascot (version 2.4.1, Matrix Science, UK). Carbamidomethylation of cysteines and oxidation of methionine were set as a fixed and variable modifications, respectively, and up to two trypsin miscleavages were allowed. Data filtering was performed using percolator, resulting in 1% false discovery rate (FDR). Additional filters were search engine rank 1 peptides and peptide ion score >20. Peptide spectrum matches (PSM) of proteins identified in multiple gel bands were summed up for each IP sample.
Statistics
For statistical analysis of the data represented in Figs. 1A, 2B and 4B 2-tailed t-tests were run to compare different cell populations. In each of the figures p-values ≤ 0.0001 are indicated with 4 asterisks. No consistent significant changes were observed between the DMSO and RO-3306 treated cell populations in Figs. 1A and 2B.
Funding Statement
This work was supported by the Netherlands Organization for Scientific Research (NWO) [grant number Proteins@Work 184.032.201] and EC European Research Council (ERC) [grant number ERC-StG 311565].
Acknowledgments
We thank Lee Zou's laboratory for SETD8 constructs, Bram van den Broek for help with imaging acquisition and analysis, members of the Jacqueline Jacobs and René Medema laboratories for discussion, Behzad Moumbeini and Ewald Van Dyk for statistical analysis, Piet Borst for critical reading of the manuscript. This work was supported by the European Research Council under Grant ERC-StG 311565 to J.J.L.J and by the Netherlands Organization for Scientific Research (NWO) as part of the National Roadmap Large-scale Research Facilities of the Netherlands, Proteins@Work (project number 184.032.201).
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
References
- [1].Ciccia A, Elledge SJ. The DNA damage response: making it safe to play with knives. Mol Cell. 2010;40:179–204. doi: 10.1016/j.molcel.2010.09.019. PMID:20965415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Panier S, Durocher D. Push back to respond better: regulatory inhibition of the DNA double-strand break response. Nat Rev Mol Cell Biol. 2013;14:661–672. doi: 10.1038/nrm3659. PMID:24002223 [DOI] [PubMed] [Google Scholar]
- [3].Jazayeri A, Falck J, Lukas C, et al.. ATM- and cell cycle-dependent regulation of ATR in response to DNA double-strand breaks. Nat Cell Biol. 2006;8:37–45. doi: 10.1038/ncb1337. PMID:16327781 [DOI] [PubMed] [Google Scholar]
- [4].Yu X, Chen J. DNA damage-induced cell cycle checkpoint control requires CtIP, a phosphorylation-dependent binding partner of BRCA1 C-terminal domains. Mol Cell Biol. 2004;24:9478–9486. doi: 10.1128/MCB.24.21.9478-9486.2004. PMID:15485915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Yata K, Esashi F. Dual role of CDKs in DNA repair: to be, or not to be. DNA Repair (Amst). 2009;8:6–18. doi: 10.1016/j.dnarep.2008.09.002. PMID:18832049 [DOI] [PubMed] [Google Scholar]
- [6].Escribano-Diaz C, Orthwein A, Fradet-Turcotte A, et al.. A cell cycle-dependent regulatory circuit composed of 53BP1-RIF1 and BRCA1-CtIP controls DNA repair pathway choice. Mol Cell. 2013;49:872–883. doi: 10.1016/j.molcel.2013.01.001. PMID:23333306 [DOI] [PubMed] [Google Scholar]
- [7].Falck J, Forment JV, Coates J, et al.. CDK targeting of NBS1 promotes DNA-end resection, replication restart and homologous recombination. EMBO Rep. 2012;13:561–568. doi: 10.1038/embor.2012.58. PMID:22565321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Bouwman P, Aly A, Escandell JM, et al.. 53BP1 loss rescues BRCA1 deficiency and is associated with triple-negative and BRCA-mutated breast cancers. Nat Struct Mol Biol. 2010;17:688–695. doi:nsmb.1831[pii] 10.1038/nsmb.1831. PMID:20453858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Densham RM, Garvin AJ, Stone HR, et al.. Human BRCA1-BARD1 ubiquitin ligase activity counteracts chromatin barriers to DNA resection. Nat Struct Mol Biol. 2016;23:647–655. doi: 10.1038/nsmb.3236. PMID:27239795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Langerak P, Russell P. Regulatory networks integrating cell cycle control with DNA damage checkpoints and double-strand break repair. Philos Trans R Soc Lond B Biol Sci. 2011;366:3562–3571. doi: 10.1098/rstb.2011.0070. PMID:22084383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Callen E, Di Virgilio M, Kruhlak MJ, et al.. 53BP1 mediates productive and mutagenic DNA repair through distinct phosphoprotein interactions. Cell. 2013;153:1266–1280. doi: 10.1016/j.cell.2013.05.023. PMID:23727112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Boersma V, Moatti N, Segura-Bayona S, et al.. MAD2L2 controls DNA repair at telomeres and DNA breaks by inhibiting 5' end resection. Nature. 2015;521:537–540. doi: 10.1038/nature14216. PMID:25799990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Xu G, Chapman JR, Brandsma I, et al.. REV7 counteracts DNA double-strand break resection and affects PARP inhibition. Nature. 2015;521:541–544. doi: 10.1038/nature14328. PMID:25799992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Fradet-Turcotte A, Fradet-Turcotte A, Canny MD, et al.. 53BP1 is a reader of the DNA-damage-induced H2A Lys 15 ubiquitin mark. Nature. 2013;499:50–54. doi: 10.1038/nature12318. PMID:23760478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Botuyan MV, Lee J, Ward IM, et al.. Structural basis for the methylation state-specific recognition of histone H4-K20 by 53BP1 and Crb2 in DNA repair. Cell. 2006;127:1361–1373. doi: 10.1016/j.cell.2006.10.043. PMID:17190600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Wilson MD, Benlekbir S, Fradet-Turcotte A, et al.. The structural basis of modified nucleosome recognition by 53BP1. Nature. 2016;536:100–103. doi: 10.1038/nature18951. PMID:27462807 [DOI] [PubMed] [Google Scholar]
- [17].Mattiroli F, Vissers JH, van Dijk WJ, et al.. RNF168 ubiquitinates K13-15 on H2A/H2AX to drive DNA damage signaling. Cell. 2012;150:1182–1195. doi: 10.1016/j.cell.2012.08.005. PMID:22980979 [DOI] [PubMed] [Google Scholar]
- [18].Alabert C, Barth TK, Reverón-Gómez N, et al.. Two distinct modes for propagation of histone PTMs across the cell cycle. Genes Dev. 2015;29:585–590. doi: 10.1101/gad.256354.114. PMID:25792596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Rice JC, Nishioka K, Sarma K, et al.. Mitotic-specific methylation of histone H4 Lys 20 follows increased PR-Set7 expression and its localization to mitotic chromosomes. Genes Dev. 2002;16:2225–2230. doi: 10.1101/gad.1014902. PMID:12208845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].O'Sullivan RJ, Kubicek S, Schreiber SL, et al.. Reduced histone biosynthesis and chromatin changes arising from a damage signal at telomeres. Nat Struct Mol Biol. 2010;17:1218–1225. doi: 10.1038/nsmb.1897. PMID:20890289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Jorgensen S, Schotta G, Sorensen CS. Histone H4 lysine 20 methylation: key player in epigenetic regulation of genomic integrity. Nucleic Acids Res. 2013;41:2797–2806. doi: 10.1093/nar/gkt012. PMID:23345616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Centore RC, Havens CG, Manning AL, et al.. CRL4(Cdt2)-mediated destruction of the histone methyltransferase Set8 prevents premature chromatin compaction in S phase. Mol Cell. 2010;40:22–33. doi: 10.1016/j.molcel.2010.09.015. PMID:20932472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Chapman JR, Barral P, Vannier JB, et al.. RIF1 is essential for 53BP1-dependent nonhomologous end joining and suppression of DNA double-strand break resection. Mol Cell. 2013;49:858–871. doi: 10.1016/j.molcel.2013.01.002. PMID:23333305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Karanam K, Kafri R, Loewer A, et al.. Quantitative live cell imaging reveals a gradual shift between DNA repair mechanisms and a maximal use of HR in mid S phase. Mol Cell. 2012;47:320–329. doi: 10.1016/j.molcel.2012.05.052. PMID:22841003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Shibata A, Conrad S, Birraux J, et al.. Factors determining DNA double-strand break repair pathway choice in G2 phase. EMBO J. 2011;30:1079–1092. doi: 10.1038/emboj.2011.27. PMID:21317870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Denchi EL, Li J. Let it go: how to deal with a breakup in mitosis. Nat Struct Mol Biol. 2014;21:433–435. doi: 10.1038/nsmb.2825. PMID:24799037 [DOI] [PubMed] [Google Scholar]
- [27].Hayashi MT, Cesare AJ, Fitzpatrick JA, et al.. A telomere-dependent DNA damage checkpoint induced by prolonged mitotic arrest. Nat Struct Mol Biol. 2012;19:387–394. doi: 10.1038/nsmb.2245. PMID:22407014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Orthwein A, Fradet-Turcotte A, Noordermeer SM, et al.. Mitosis inhibits DNA double-strand break repair to guard against telomere fusions. Science. 2014;344:189–193. doi: 10.1126/science.1248024. PMID:24652939 [DOI] [PubMed] [Google Scholar]
- [29].Yu X, Baer R. Nuclear localization and cell cycle-specific expression of CtIP, a protein that associates with the BRCA1 tumor suppressor. J Biol Chem. 2000;275:18541–18549. doi: 10.1074/jbc.M909494199. PMID:10764811 [DOI] [PubMed] [Google Scholar]
- [30].Orthwein A, Noordermeer SM, Wilson MD, et al.. A mechanism for the suppression of homologous recombination in G1 cells. Nature. 2015;528:422–426. doi: 10.1038/nature16142. PMID:26649820 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [31].Pellegrino S, Michelena J, Teloni F, et al.. Replication-Coupled Dilution of H4K20me2 Guides 53BP1 to Pre-replicative chromatin. Cell Rep. 2017;19:1819–1831. doi: 10.1016/j.celrep.2017.05.016. PMID:28564601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Saredi G, Huang H, Hammond CM, et al.. H4K20me0 marks post-replicative chromatin and recruits the TONSL-MMS22L DNA repair complex. Nature. 2016;534:714–718. doi: 10.1038/nature18312. PMID:27338793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Pryde F, Khalili S, Robertson K, et al.. 53BP1 exchanges slowly at the sites of DNA damage and appears to require RNA for its association with chromatin. J Cell Sci. 2005;118:2043–2055. doi: 10.1242/jcs.02336. PMID:15840649 [DOI] [PubMed] [Google Scholar]
- [34].Kuo AJ, Song J, Cheung P, et al.. The BAH domain of ORC1 links H4K20me2 to DNA replication licensing and Meier-Gorlin syndrome. Nature. 2012;484:115–119. doi: 10.1038/nature10956. PMID:22398447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Beck DB, Burton A, Oda H, et al.. The role of PR-Set7 in replication licensing depends on Suv4-20h. Genes Dev. 2012;26:2580–2589. doi: 10.1101/gad.195636.112. PMID:23152447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Fu H, Maunakea AK, Martin MM, et al.. Methylation of histone H3 on lysine 79 associates with a group of replication origins and helps limit DNA replication once per cell cycle. PLoS Genet. 2013;9:e1003542. doi: 10.1371/journal.pgen.1003542. PMID:23754963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Peuscher MH, Jacobs JJ. DNA-damage response and repair activities at uncapped telomeres depend on RNF8. Nat Cell Biol. 2011;13:1139–1145. doi: 10.1038/ncb2326. PMID:21857671 [DOI] [PubMed] [Google Scholar]
- [38].Wiederschain D, Wee S, Chen L, et al.. Single-vector inducible lentiviral RNAi system for oncology target validation. Cell Cycle. 2009;8:498–504. doi: 10.4161/cc.8.3.7701. PMID:19177017 [DOI] [PubMed] [Google Scholar]
- [39].Typas D, Luijsterburg MS, Wiegant WW, et al.. The de-ubiquitylating enzymes USP26 and USP37 regulate homologous recombination by counteracting RAP80. Nucleic Acids Res. 2016;44:2976. doi: 10.1093/nar/gkv1480. PMID:26673702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Ameziane N, May P, Haitjema A, et al.. A novel Fanconi anaemia subtype associated with a dominant-negative mutation in RAD51. Nat Commun. 2015;6:8829. doi: 10.1038/ncomms9829. PMID:26681308 [DOI] [PMC free article] [PubMed] [Google Scholar]