

Infection by microbial pathogens modulates apoptotic cell death of the host cells as one of the host immune defense mechanisms. Counteracting the host apoptotic cell death could be directed by the nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB), which is induced upon infection and triggers the host inflammatory response against pathogens. The NF-κB family of five transcription factors directs nuclear responses leading to many cellular processes including inflammation, proliferation or survival. Apoptotic cell death of infected cells benefits the host as it limits the infection through the elimination of infected cells, in the form of apoptotic bodies, by professional phagocytes and thereby makes microbial antigens available for presentation by major histocompatibility complex class I or II molecules to induce a protective immune response. Bacterial pathogens however tend to suppress host cell death during the initial stages of infection for colonization and later, induce host cell death which is necessary not only as a source of nutrients from the cell debris, but also for the further propagation of the bacteria. For these reasons in fact, bacteria have evolved sophisticated strategies to manipulate the host cell death pathways.1 Notably, these evasion strategies originate from the synthesis and translocation of bacterial effector proteins to interfere with the NF-κB activation.2 An early report from Yanai and colleagues3 showed the involvement of NF-κB in promoting the survival of cells infected with the human gastric pathogen Helicobacter pylori, opening up the possibility that rather than utilizing self-synthesized effector molecules, bacteria can benefit from infection-activated NF-κB. Our latest report showed specifically the upregulation of NF-κB-dependent A20 after infections by H. pylori and Campylobacter jejuni.4 A20 (also known as Tumor Necrosis Factor Alpha-Induced Protein 3, TNFAIP3) is a unique zinc finger protein possessing a deubiquitinylase activity as well as an E3 ubiquitin ligase activity. Both activities are required to terminate NF-κB activation in a negative feedback loop, explaining the transient nature of an inflammatory response. In H. pylori-infected gastric epithelial cells, A20 inhibits not only prosurvival NF-κB signaling, preventing the expression of proinflammatory IL-8, but also apoptotic cell death. Specifically for the latter, we provided evidence demonstrating that A20 interferes with the activation of procaspase-8 and interestingly, another NF-κB target p62 (sequestosome-1) is important for this interaction of A20 with procaspase-84 (Fig. 1). Our observations suggest that H. pylori induction of the NF-κB target gene TNFAIP3 can be advantageous for pathogen infection by specifically restricting host apoptotic cell death. This ‘pro-survival’ function of A20 can also be extrapolated to other pathogens, although different mechanisms are at work. In T-cells infected by human T-cell leukemia virus type I, A20 interacts directly with procaspase-8 and thus blocks the activation of procaspase-8 by preventing its recruitment to FADD.5 The parasite Leishmania donovani also benefits from infection-elicited NF-κB activation and the consecutive upregulation of A20. Here however, A20 functions in a different manner to support pathogen infection, namely by deubiquitinylation of TRAF6 to inhibit the Toll-like receptor 2 response and thereby circumvent the host immune response.6 For the intracellular pathogen Mycobacterium tuberculosis however, an effector molecule called ESAT-6 is secreted that downregulates miR-let-7f leading to the increase in expression of A207. Collectively, these findings increase our appreciation of what is likely hitherto an undervalued feature of infection-induced NF-κB activity which plays to the benefit of the pathogen. With this recognition also comes the awareness that plausibly, with the multitude of host signaling pathways that are activated upon infection, there are still proteins to be discovered which have this ‘secondary action’ of enhancing the infection by pathogens. This should also be taken into consideration when evaluating the suitability of host proteins as therapeutic targets.
Figure 1.
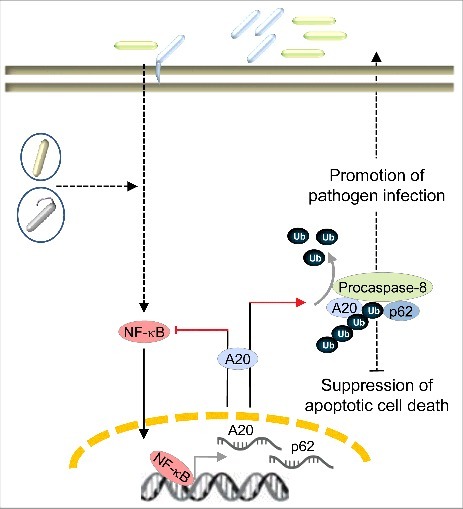
Schematic presentation of pathogen-induced A20 and its role in promoting H. pylori infection. In response to infection by extra- and intracellular pathogens (different colored rods), the host activates multiple signaling cascades, one of which is the NF-κB signaling pathway. NF-κB-induced A20 can be advantageous for pathogen infection. In H. pylori-infected gastric epithelial cells, A20 participates in the apoptotic cell death pathway by deubiquitinylating procaspase-8. Interestingly, the scaffold protein p62 is important for this interaction of A20 with procaspase-8. These events suppress host apoptotic cell death, which is beneficial for the colonization and persistence of H. pylori.
Funding Statement
Deutsche Forschungsgemeinschaft
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
References
- [1].Robinson KS, Aw R. The Commonalities in bacterial effector inhibition of apoptosis. Trends Microbiol. 2016;24:665-80. doi: 10.1016/j.tim.2016.04.002. [DOI] [PubMed] [Google Scholar]
- [2].Neish A, Naumann M. Microbial-induced immunomodulation by targeting the NF-κB system. Trends Microbiol. 2011;19:596-605. doi: 10.1016/j.tim.2011.08.004. [DOI] [PubMed] [Google Scholar]
- [3].Yanai A, Hirata Y, Mitsuno Y, Maeda S, Shibata W, Akanuma M, Yoshida H, Kawabe T, Omata M. Helicobacter pylori induces antiapoptosis through nuclear factor–κB activation. J Infect Dis. 2003;188:1741-51. doi: 10.1086/379629. [DOI] [PubMed] [Google Scholar]
- [4].Lim MCC, Maubach G, Sokolova O, Feige MH, Diezko R, Buchbinder J, Backert S, Schluter D, Lavrik IN, Naumann M. Pathogen-induced ubiquitin-editing enzyme A20 bifunctionally shuts off NF-[kappa]B and caspase-8-dependent apoptotic cell death. Cell Death Differ. 2017;24:1621-31. doi: 10.1038/cdd.2017.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Saitoh Y, Hamano A, Mochida K, Kakeya A, Uno M, Tsuruyama E, Ichikawa H, Tokunaga F, Utsunomiya A, Watanabe T, et al.. A20 targets caspase-8 and FADD to protect HTLV-I-infected cells. Leukemia. 2016;30:716-27. doi: 10.1038/leu.2015.267. [DOI] [PubMed] [Google Scholar]
- [6].Srivastav S, Kar S, Chande AG, Mukhopadhyaya R, Das PK. Leishmania donovani exploits host deubiquitinating enzyme A20, a negative regulator of TLR signaling, to subvert host immune response. J Immunol. 2012;189:924-34. doi: 10.4049/jimmunol.1102845. [DOI] [PubMed] [Google Scholar]
- [7].Kumar M, Sahu Sanjaya K, Kumar R, Subuddhi A, Maji Ranjan K, Jana K, Gupta P, Raffetseder J, Lerm M, Ghosh Z, et al.. MicroRNA let-7 Modulates the immune response to Mycobacterium tuberculosis infection via control of A20, an inhibitor of the NF-κB pathway. Cell Host Microbe. 2015;17:345-56. doi: 10.1016/j.chom.2015.01.007. [DOI] [PubMed] [Google Scholar]