

Abstract
Block of neurotransmitter receptors at the neuromuscular junction (NMJ) has been shown to trigger upregulation of the number of synaptic vesicles released (quantal content, QC), a response termed homeostatic synaptic plasticity. The mechanism underlying this plasticity is not known. Here, we used selective toxins to demonstrate that block of α1-containing nicotinic acetylcholine receptors (nAChRs) at the NMJ of male and female mice triggers the upregulation of QC. Reduction of current flow through nAChRs, induced by drugs with antagonist activity, demonstrated that reduction in synaptic current per se does not trigger upregulation of QC. These data led to the remarkable conclusion that disruption of synaptic transmission is not sensed to trigger upregulation of QC. During studies of the effect of partial block of nAChRs on QC, we observed a small but reproducible increase in the decay kinetics of miniature synaptic currents. The change in kinetics was correlated with the increase in QC and raises the possibility that a change in postsynaptic nAChR conformation may be associated with the presynaptic increase in QC. We propose that, in addition to functioning in synaptic transmission, ionotropic muscle nicotonic nAChRs may serve as signaling molecules that participate in synaptic plasticity. Because nAChRs have been implicated in a number of disease states, the finding that nAChRs may be involved in triggering synaptic plasticity could have wide-reaching implications.
SIGNIFICANCE STATEMENT The signals that initiate synaptic plasticity of the nervous system are still incompletely understood. Using the mouse neuromuscular junction as a model synapse, we studied how block of neurotransmitter receptors is sensed to trigger synaptic plasticity. Our studies led to the surprising conclusion that neither changes in synaptic current nor spiking of the presynaptic or postsynaptic cell are sensed to initiate synaptic plasticity. Instead, postsynaptic nicotinic acetylcholine receptors (nAChRs), in addition to functioning in synaptic transmission, may serve as signaling molecules that trigger synaptic plasticity. Because nAChRs have been implicated in a number of disease states, the finding that they may mediate synaptic plasticity has broad implications.
Keywords: endplate, homeostatic, neurotransmitter, plasticity, synaptic, transmission
Introduction
The signals that initiate synaptic plasticity of the nervous system are still incompletely understood. The neuromuscular junction (NMJ) is a synaptic preparation ideally suited to studies of synaptic plasticity because there is only one neurotransmitter and neurotransmitter receptor type present. At the adult mouse NMJ, release of acetylcholine (ACh) from the presynaptic terminal binds and opens a single type of nicotinic acetylcholine receptor (nAChR) containing two identical α subunits and a β, a δ, and an ε subunit (Auerbach, 2015). The presence of a uniform population of nAChRs simplifies interpretation of experiments in which plasticity of the synapse is studied in response to partial block of receptors.
Block of synaptic transmission at the NMJ triggers rapid upregulation of presynaptic transmitter release that serves to counteract the block of transmission and is thus termed homeostatic synaptic plasticity (Rich and Wenner, 2007; Turrigiano, 2012; Davis and Müller, 2015). A number of different blockers of nAChRs have been found to trigger increases of the number of synaptic vesicles released (quantal content, QC) to homeostatically regulate synaptic current at the NMJ of multiple species including mouse and human (Katz and Miledi, 1978; Miledi et al., 1978; Molenaar et al., 1979, 1991; Cull-Candy et al., 1980; Wilson, 1982; Harborne et al., 1988; Plomp et al., 1992, 1994; Tian et al., 1994; Plomp et al., 1995; Wang et al., 2010, 2016). Homeostatic regulation of synaptic current also occurs at the glutamatergic Drosophila NMJ after block of glutamate receptors (Frank et al., 2006). These studies suggest homeostatic regulation of QC at the NMJ is an evolutionarily conserved phenomenon.
There are at least two distinct forms of homeostatic upregulation of QC at the mouse NMJ. The first is triggered by block of neurotransmitter receptors and the mechanism is an increase in the number of releasable vesicles (the binomial parameter n); the second is triggered by block of evoked release and the mechanism is an increase in the probability of release (the binomial parameter p) (Wang et al., 2010). These pathways can be triggered independently and are additive (Wang et al., 2010).
It is not clear how disruption of synaptic activity is sensed to trigger homeostatic plasticity. Possibilities include spiking of the postsynaptic cell and block of synaptic transmission. At the Drosophila NMJ, the increase in QC occurs in the absence of evoked activity (Frank et al., 2006), suggesting that neither loss of spiking of muscle fibers nor block of evoked release of neurotransmitter is the signal that triggers upregulation of QC. Similarly, it has been found that homeostatic regulation of quantal amplitude in neurons occurs independently of spiking activity (Garcia-Bereguiain et al., 2016). It is assumed that current flow or depolarization occurring during spontaneous release of neurotransmitter is what is sensed to trigger the homeostatic response.
We probed how partial block of nAChRs is sensed to trigger homeostatic synaptic plasticity at the mouse NMJ. Our data suggest that block of postsynaptic α1-containing nAChRs triggers presynaptic plasticity and led to the surprising conclusion that neither evoked nor spontaneous synaptic transmission is sensed. Our data raise the possibility that binding of toxin to nAChRs triggers trans-synaptic signaling and the presynaptic increase in QC.
Materials and Methods
Ethical approval.
All procedures involving animals were approved by the Wright State institutional animal care and use committee.
Mice.
To easily visualize NMJs, mice expressing the YFP transgene driven by the Thy-1 promoter were used [B6.Cg-Tg(Thy1-YFP)16Jrs/J; The Jackson Laboratory). Mice with α7 subunit of nicotinic nAChRs knock-out were also obtained from The Jackson Laboratory (Chrna7). Both male and female mice were used for all experiments. No differences were noted between male and female mice.
Electrophysiological recording.
The experimental procedures used to measure QC in the mouse tibialis anterior muscle have been described previously (Wang et al., 2004, 2016). Briefly, 2- to 3-month-old mice were killed using CO2 inhalation and the tibialis anterior muscle was removed, pinned in a Sylgard plated dish and stained with 10 μm 4-(4-diethylaminostyryl)-N-methylpyridinium iodide (4-Di-2ASP; Invitrogen) to visualize NMJs using an upright epifluorescence microscope. Muscle strips were perfused at a speed of 3–6 ml per minute with external solution containing the following (in mm): 118 NaCl, 0.7 MgCl2, 2 CaSO4, 3.5 KCl, 26.2 NaHCO3, 1.7 NaH2PO4, and 5.5 glucose, equilibrated with 95% O2 and 5% CO2, pH 7.3–7.4, 20–22°C. For experiments in which temperature was increased to 32°C, an automatic temperature controller (Warner Instruments) was used.
Endplates were imaged and muscle fibers were impaled within 100 μm of the NMJ to ensure good space clamp of the NMJ region. Endplate currents (EPCs) were recorded using two-electrode voltage clamp while the nerve branch to the tibialis anterior muscle was stimulated via a tungsten bipolar electrode (FHC).
Unless otherwise indicated, muscle fibers were crushed on both ends away from the NMJ band to eliminate contractions upon nerve stimulation and the holding potential was set at −45 mV. In some experiments, a more negative potential was required to detect miniature EPCs (mEPCs) due to block of postsynaptic nAChRs. In these experiments, muscle fibers were not crushed and contraction was prevented by addition of μ-conotoxin GIIIB (μ-Ctx; Alomone Labs) to the external solution (final toxin concentration was 1–3 μm) to inhibit muscle Na+ channels. We have shown that QC obtained from both preparations is identical (Wang et al., 2004, 2005).
The QC corresponding to a given EPC was calculated from the ratio of the amplitude the EPC to the average of amplitude of at least 20 mEPCs obtained during the 20 s before and the 20 s after the EPC. For bars graphs of QC before and after block of nAChRs, EPC amplitude was determined from an average of 10 EPCs evoked at 0.5 Hz, whereas mEPC amplitude was determined from an average obtained from at least 30 mEPCs recorded over a 1 min period.
Drugs.
Drugs were applied either by adding into the bathing solution or by pressure puff delivery with a picospritzer II (20 psi) as described previously (Wang et al., 2016). α-bungarotoxin (BTX), D-tubocurarine (D-TC), carbachol (CCh), vecuronium bromide, succinylcholine chloride, gallamine, mecamylamine hydrochloride, atropine, and acetylcholine chloride were all from Sigma-Aldrich. α-conotoxin MI (α-CtxMI) and α-conotoxin ArIB[V11L;V16D] (α-CtxArIB) were provided by Dr. McIntosh.
Experimental design and statistical analysis.
Data were recorded from the same muscle (often the same NMJ) before and after drug treatment. Nested ANOVA (SYSTAT; Systat Software) was used for comparing the effect of drugs and other experimental manipulations. Details of the statistical tests used and the values obtained are listed in Table 1. Plots and curve fittings were made using Sigmaplot software (Systat Software). Averaged results are expressed as mean ± SE. p < 0.05 and p < 0.01 are denoted by one and two asterisks, respectively.
Table 1.
Data statistics
| Reference | Test | Result | Method |
|---|---|---|---|
| Temperature | Raise to 32oC on QC | t(7) = 1.626, p = 0.148 | Paired t test |
| D-TC on QC at 32oC | t(8) = −4.765, p = 0.00142 | Paired t test | |
| Fig. 1D | Holding at −40 mV and −90 mV on QC | t(9) = −0.635, p = 0.541 | Paired t test |
| Fig. 2A | Baseline QC (QC) | F(3,15) = 0.805, p = 0.499 | ANOVA |
| Fig. 2A | D-TC on QC | Paired t test | |
| Control | t(21) = −11.467, p = 1.67E-10 | ||
| αCtxArIB | t(8) = −10.814, p = 4.72E-6 | ||
| Atropine | t(4) = −8.102, p = 0.00126 | ||
| α7KO | t(19) = −7.316, p = 6.15E-7 | ||
| Fig. 2B | α-Ctx MI on QC | F(2,6) = 9.787, p < 0.0001 | ANOVA |
| Fig. 3C | D-TC on QC with CCh puff | t(9) = −15.213, p = 9.98E-8 | ANOVA |
| Fig. 3G | Effect of ACh | ANOVA | |
| EPC | F(1,8) = 72.463, p < 0.0001 | ||
| mEPC | F(1,8) = 184.117, p < 0.0001 | ||
| QC | F(1,8) = 0.267, p = 0.667 | ||
| Fig. 3H | Ctrl vs +ACh + atropine | ANOVA | |
| EPC | F(1,6) = 25.143 p < 0.0001 | ||
| mEPC | F(1,6) = 31.179, p ≤ 0.0001 | ||
| QC | F(1,6) = 0.026, p = 0.873 | ||
| Fig. 3H | Ctrl vs ACh + atropine + D-TC | ANOVA | |
| EPC | F(1,6) = 70.508, p ≤ 0.0001 | ||
| mEPC | F(1,6) = 269.432, p ≤ 0.0001 | ||
| QC | F(1,6) = 25.644, p ≤ 0.0001 | ||
| Fig. 3H | ACh + atropine vs ACh + atropine + D-TC | ANOVA | |
| EPC | F(1,6) = 10.924, p = 0.001 | ||
| mEPC | F(1,6) = 250.029, p ≤ 0.0001 | ||
| QC | F(1,6) = 25.644, p ≤ 0.0001 | ||
| Fig. 4A | Before vs vecuronium | ANOVA | |
| EPC | F(1,10) = 126.367, p < 0.0001 | ||
| mEPC | F(1,10) = 805.135, p < 0.0001 | ||
| QC | F(1,10) = 42.313, p < 0.0001 | ||
| Fig. 4A | Vecuronium vs washout | ANOVA | |
| EPC | F(1,10) = 54.36, p < 0.0001 | ||
| mEPC | F(1,10) = 596.527, p < 0.0001 | ||
| QC | F(1,10) = 27.124, p < 0.0001 | ||
| Fig. 4B | Before vs mecamylamine | ANOVA | |
| EPC | F(1,4) = 7.92, p = 0.006 | ||
| mEPC | F(1,4) = 338.71, p < 0.0001 | ||
| QC | F(1,4) = 54.659, p < 0.0001 | ||
| Fig. 4B | Mecamylamine vs washout | ANOVA | |
| EPC | F(1,4) = 10.835, p = 0.002 | ||
| mEPC | F(1,4) = 602.623, p < 0.0001 | ||
| QC | F(1,4) = 39.211, p < 0.0001 | ||
| Fig. 4D | Before vs succinylcholine | ANOVA | |
| EPC | F(1,8) = 273.314, p < 0.0001 | ||
| mEPC | F(1,8) = 493.958, p < 0.0001 | ||
| QC | F(1,8) = 0.528, p = 0.498 | ||
| Fig. 4D | Succinylcholine vs washout | ANOVA | |
| EPC | F(1,8) = 110.816, p < 0.0001 | ||
| mEPC | F(1,8) = 172.376, p < 0.0001 | ||
| QC | F(1,8) = 0.486, p < 0.487 | ||
| Fig. 5B | mEPC decay tau change by | Paired t test | |
| BTX | t(18) = 6.458, p = 4.48E-6 | ||
| D-TC | t(17) = 12.355, p = 1.35E-9 | ||
| Gallamine (GT) | t(9) = 5.825, p = 2.52E-4 | ||
| Mecamylamine | t(4) = 7.263, p = 0.00191 | ||
| Vecuronium | t(12) = 5.048, p = 2.85E-4 | ||
| Succinylcholine | t(11) = −1.956, p = 0.0790 | ||
| ACh/CCh | t(21) = 3.705, p = 0.0014 | ||
| BTX vs ACh/CCh on decay tau | t(38) = 4.197, p = 1.57E-4 | t test |
Results
Block of nAChRs triggers upregulation of QC independently of evoked release, membrane potential, and synaptic current
We and others found previously that block of nAChRs at the mammalian NMJ at room temperature triggers an increase in QC (Katz and Miledi, 1978; Miledi et al., 1978; Molenaar et al., 1979, 1991; Cull-Candy et al., 1980; Wilson, 1982; Harborne et al., 1988; Plomp et al., 1992, 1994; Tian et al., 1994; Plomp et al., 1995; Wang et al., 2010, 2016). To confirm that the finding is physiologically relevant, we examined whether it occurred at the more physiologic temperature of 32°C. Increasing temperature to 32°C increased mean mEPC amplitude from 1.44 ± 0.14 to 1.90 ± 0.08 nA (n = 8 NMJs) and accelerated the rate of decay from 1.04 ± 0.07 to 0.59 ± 0.04 ms. There was no effect on QC (56.6 ± 2.6 to 55.8 ± 2.5, t(7) = 1.6, p = 0.15). These results are similar to temperature-dependent changes reported previously at a central synapse (Kushmerick et al., 2006). The finding that QC does not change with increased temperature suggests that neither the number of releasable vesicles nor the probability of release has significant temperature dependence. When the temperature was 32°C, the addition of 0.1 μm D-TC triggered an increase in QC (45.1%, n = 9 NMJs, t(8) = −4.765, p = 0.001) that was similar to the increase occurring at room temperature. These data suggest that, at both room temperature and at physiologic temperature, partial block of nAChRs triggers an increase in QC.
There are several ways that the postsynaptic cell might sense reduction in synaptic activity. One is through reduction in postsynaptic spiking activity. This has been the mechanism proposed for detection of reduced network activity in neuronal systems (Turrigiano, 2012). In our system, this was not a possibility because postsynaptic spiking was prevented by crushing muscle fibers to depolarize the membrane potential and inactivate sodium channels. Despite the loss of postsynaptic spiking, partial block of nAChRs by D-TC triggered rapid upregulation of QC (Fig. 1A,B).
Figure 1.
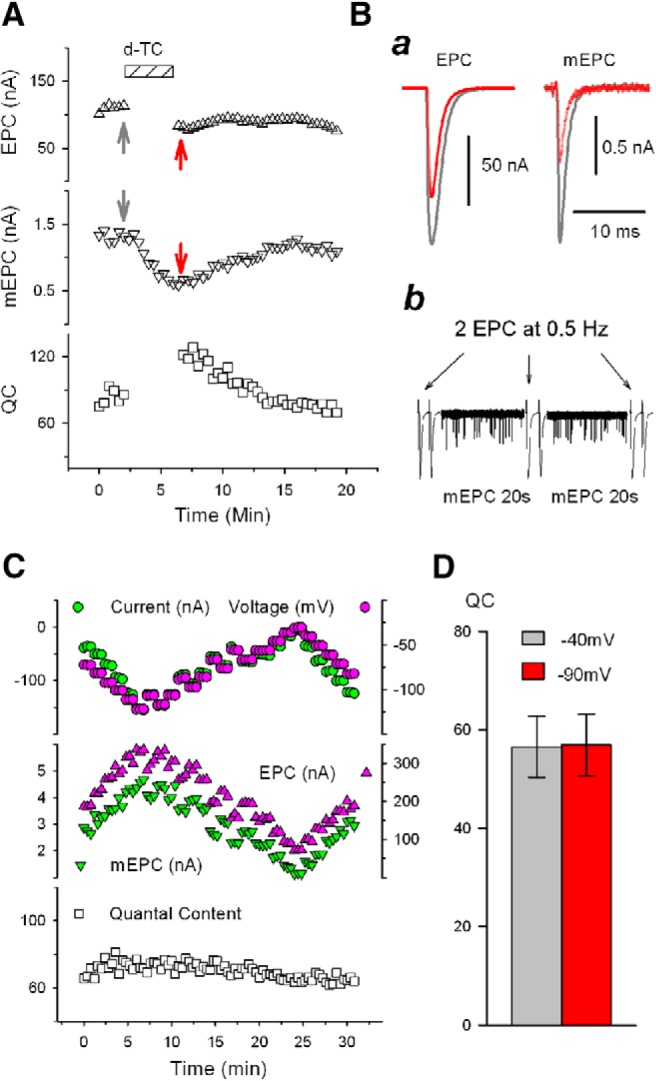
Rapidly reversible increase in QC is triggered independent of synaptic current and evoked release. A, Increase in QC triggered by blocking nAChRs is not dependent on evoked ACh release. EPC (top) and mEPC (middle) were recorded alternately by repeating the protocol shown in the bottom row of B. The corresponding QC was calculated from the ratio of the EPC amplitude/average mEPC (see Materials and Methods) and plotted in the bottom trace. The bathing solution contained 0.1 μΜ D-TC as indicated by striped bar. Nerve stimulation/EPC collection was paused during D-TC infusion. Ba, Representative traces of EPC and mEPC, from the experiment shown in A before (gray) and during (red) infusion of D-TC at the times indicated by the correspondingly colored arrows in A. Bb, Recording protocol used for experiments in which QC was followed in individual fibers during infusion of nAChR blockers. Two EPCs at 0.5 Hz and 20 s of mEPCs were alternately recorded and the averaged values were used to calculate corresponding QC and construct the plot (e.g., A and C). EPC and mEPC amplitudes are scaled differently so that both fit on the same plot. C, QC is not affected by varying EPC and mEPC amplitude by changing the muscle fiber holding potential. Using 2-electrode voltage clamp, the muscle voltage in the region of the endplate was varied by 5 mV every minute (top, green circle). Muscle action potentials were prevented by the presence of 2 μΜ μ-conotoxin in the bathing solution. The holding current (top, purple circle) is also plotted. EPCs and mEPCs (middle) were acquired alternately by repeating the protocol shown in Bb. The calculated QC is shown at the bottom and was unvarying despite a >2-fold change in EPC and mEPC amplitude. D, EPC and mEPC were collected at holding potentials of both −40 mV and −90 mV on 10 NMJs and QC was compared. Holding potential exerted no significant effect on QC (t(9) = −0.635, p = 0.541).
A second possibility is that depolarization of the postsynaptic muscle fiber during evoked release is sensed to trigger the homeostatic upregulation of QC. We tested this possibility in two ways. First, we voltage clamped individual muscle fibers such that the membrane potential in the region of the NMJ was constant before, during, and after the infusion of D-TC. Despite a constant membrane potential, QC was still upregulated by infusion of D-TC (Fig. 1A). Second, we measured QC in individual NMJs and then infused D-TC in the absence of presynaptic spikes triggered by nerve stimulation. The increase in QC was evident in the EPC evoked by the very first nerve stimulation after infusion of D-TC (Fig. 1A. n = 6 NMJs), but not when D-TC was omitted (n = 4 NMJs). These data indicate that the increase in QC can occur in the absence of changes in muscle membrane potential and in the absence of prior evoked release.
The upregulation of QC could be triggered by interruption of nAChRs activation during spontaneous release of ACh. Spontaneous release of vesicles containing ACh occurs at the NMJ at a rate of ∼1 Hz and triggers mEPCs. One way that mEPCs might signal is through depolarization of the membrane potential; however, this was ruled out by the voltage-clamp experiments described above. The next possibility that we considered is that reduction in mEPC current (rather than depolarization) is sensed to trigger the upregulation of QC. We altered the holding potential of the postsynaptic muscle fiber to alter driving force for current flow through nAChRs (Fig. 1C,D). Increasing and decreasing mEPC amplitude by twofold by altering holding potential had no effect on QC (Fig. 1C,D). These data suggest that reduction in postsynaptic current is not the trigger of increases in QC.
Block of nicotinic α1 containing nAChRs triggers the increase in QC
The finding that reduction in postsynaptic current does not trigger upregulation of presynaptic QC caused us to consider that the nAChRs triggering the upregulation of QC might be on either the presynaptic terminal or the Schwann cell. Evidence for the presence of AChRs on terminal Schwann cells at the mouse NMJ comes from a study showing that Schwann cell Ca2+ transients are triggered by nerve stimulation or application of ACh (Rochon et al., 2001). Muscarinic AChRs appear to be involved as the Schwann cell Ca2+ transients were blocked by atropine and were unaffected by application of BTX (Rochon et al., 2001). We applied atropine to muscles and found this triggered no change in QC (Fig. 2A). Application of D-TC after application of atropine triggered the normal increase in QC (Fig. 2A). These data indicate that muscarinic AChRs are not involved in D-TC-induced increase in QC at the NMJ.
Figure 2.
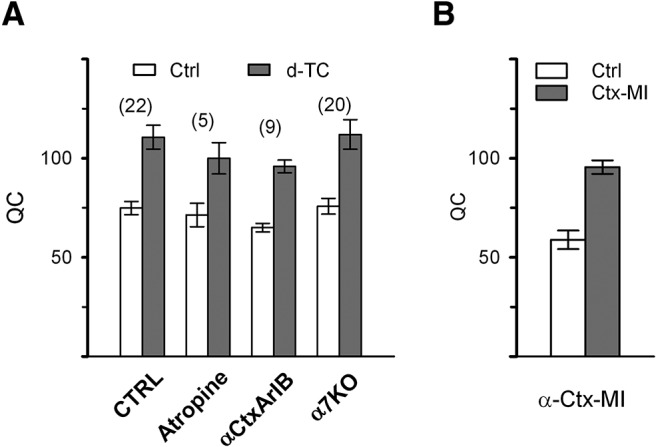
Increase in QC is triggered by block of nAChRs containing α1 subunits. A, QC was measured before and in the presence of D-TC (0.1 μm) in control muscle (n = 8 mice, 22 NMJs), in muscle pretreated with atropine (10 μm) to block muscarinic AChRs (2 mice, 5 NMJs), in muscle pretreated with α-CtxArIB (17 nm) to block α7 containing nAChRs (3 mice, 9 NMJs), and in muscle from mice lacking nAChRs composed of α7 subunits (6 mice, 20 NMJs). There was no significant difference in baseline QC among the 4 groups (F(3,15) = 0.805, p = 0.499, ANOVA) and D-TC triggered a significant increase in QC in all 4 groups (Table 1). B, QC is significantly increased by α-Ctx MI (40 nm), which blocks nAChRs containing the α1 subunit. Control, n = 3 mice, 30 NMJs; in the presence of α-Ctx MI, n = 3 mice, 50 NMJs. ANOVA, F(2,6) = 9.787, p = 0.0001.
Due to the rapidity of the upregulation of QC after D-TC, it has been hypothesized that block of presynaptic nAChRs triggers the upregulation (Bowman et al., 1990; MacDermott et al., 1999). D-TC-sensitive α7-subunit-containing nAChRs are found on many presynaptic terminals in the CNS (Albuquerque et al., 2009) such that block of this type of nAChR could be involved in the D-TC-induced increase in QC. α-CtxArIB is a rapid-acting blocker of α7 containing nAChRs (Whiteaker et al., 2007; Innocent et al., 2008) and α-CtxArIB triggered no increase in QC (Fig. 2A). Furthermore, the increase in QC triggered by application of D-TC was unaffected by prior block of α7 nAChRs with α-CtxArIB. We also applied D-TC to NMJs from mice lacking the α7 AChR subunit (α7 KO mice) and found an increase in QC that was similar to the increase present in wild-type siblings (Fig. 2A). These data suggest block of presynaptic α7 containing nAChRs is not the trigger for the increase in QC.
α1-containing nAChRs are present in high concentration on the postsynaptic muscle fiber at the NMJ and, to our knowledge, have not been shown to be expressed by either motor neurons or Schwann cells. α-CtxMI is a selective blocker of α1-containing nAChRs (Johnson et al., 1995; Luo and McIntosh, 2004) and it triggered an increase in QC similar to that seen with D-TC (Fig. 2B). These data strongly suggest that block of α1-containing nAChRs on muscle is the trigger for upregulation of QC.
Upregulation of QC is independent of synaptic transmission
Synaptic current through nAChRs is primarily carried by Na+ and K+ ions, but there is significant Ca2+ current as well (Bregestovski et al., 1979; Adams et al., 1980; Vernino et al., 1994). At the mouse, frog, and Drosophila NMJ, it has been proposed that Ca2+ flow through postsynaptic receptors is sensed to trigger homeostatic plasticity (Frank et al., 2006; Ouanounou et al., 2016). Changes in Ca2+ entry through nAChRs will be relatively insensitive to changes in holding potential becauase the driving force for Ca2+ entry is large. Therefore, the lack of effect of changing holding potential does not rule out a role for Ca2+ current through nAChRs.
To examine the possibility that reduced Ca2+ entry through nAChRs is sensed, we increased synaptic current by directly applying agonist during infusion of D-TC. Carbachol (CCh) was selected as the agonist as it causes less desensitization than ACh. The duration and amplitude of the CCh-gated current in response to intermittent application of CCh was far greater than the amplitude of currents carried by mEPCs and EPCs, yet pulsatile application of CCh did not prevent the upregulation of QC triggered by D-TC (Fig. 3A–C, n = 11 NMJs from 6 mice). These data demonstrate that D-TC can still trigger an increase in QC, even when Ca2+ entry is greatly increased relative to baseline by puffs of CCh. A caveat is that the large increases in Ca2+ entry through nAChRs triggered by CCh do not mimic the small pulsatile Ca2+ entry occurring during mEPCs.
Figure 3.

Upregulation of QC is independent of synaptic function. A, Pulsatile application of the agonist CCh that produces large CCh-gated currents does not prevent the D-TC-induced increase in QC. EPC, puffed CCh-evoked current, and mEPC were acquired alternately by the protocol shown in Cb. Average EPC (up triangle, open, average mEPC (down triangle), and QC (square, open) were plotted against time. For comparison, the value of CCh-evoked current (up triangle, filled) was normalized to that of EPC recorded before D-TC infusion, and the ratio of CCh-evoked current/mEPC were also plotted (square, filled) along-side QC. The bathing solution was switched to a solution containing 0.1 μm D-TC as indicated by horizontal bars. B, Representative traces of EPC, CCh-evoked current, and mEPC were taken at times denoted by the numbers in A. Ca, Comparison of QC increases (percentage of control) by D-TC infusion with (n = 22, same data as in Fig. 2A control) /without (n = 11) puffing of CCh (t(9)=−15.213, p = 9.98E-8, paired t). Cb, Protocol used in A. It is a variation of protocol shown in Figure 1Bb. with addition of pressure puffing of agonist onto the NMJ being recorded after acquisition of EPC and recording of 25 s mEPC in each cycle. D, Total postsynaptic current for a given time period of the recording shown in A was measured by taking the integral of the various currents over each 25 s epoch. The top panel is a plot of the summed integrals of EPCs and mEPCs before and after D-TC infusion. The bottom panel is a plot of the current in response to intermittent application of carbachol. Despite the CCh induced currents being 200 times larger than the combined total EPC and mEPC currents in the same recording cycle, the D-TC-induced increase in QC is robust. E, Desensitization of nAChRs does not trigger an increase in QC. Plots of average EPC, mEPC, and QC obtained using the protocol shown in Figure 1Bb. Despite a 40% reduction in EPC and mEPC amplitudes (measured at the time indicated by the red symbols), there was no increase in QC. F, Averaged EPC and mEPC traces before (gray) and in the presence of ACh (red) taken from times corresponding to colored symbols on the plots shown in D. G, Mean data from 10 muscles treated with ACh: n = 43 NMJs before and 102 NMJs during ACh infusion (Table 1). H, Mean data from 8 muscles before (white, n = 38 NMJs) treated with ACh 0.1 mm and atropine 10 μm (gray, n = 43 NMJs) and with addition of 0.1 μm D-TC (red, n = 56 NMJs). Nested analysis showed significant reductions in EPC and mEPC amplitude after application of ACh and further significant reductions after addition of D-TC. **p < .01. QC was statistically increased after D-TC infusion, whereas application of ACh and atropine alone had no effect (Table 1).
We wished to determine whether we could reduce Ca2+ entry during mEPCs without triggering an increase in QC. We reduced the amplitude of mEPCs by application of ACh to trigger desensitization. To further confirm that upregulation of QC is independent of the reduction in synaptic current/Ca2+ entry, we reduced the amplitudes of synaptic currents by application of ACh to trigger desensitization. We applied ACh to the bath at a dose (0.3–0.5 mm) that reduced mEPC amplitude by >30%. This degree of reduction in mEPC amplitude normally reliably triggers an increase in QC after application of D-TC. However, when mEPC amplitude was reduced by desensitization of nAChRs, there was no increase in QC (Fig. 3D–F). Similar results were obtained with 10 to 30 μm CCh (n = 10 NMJs, data not shown). These results are consistent with the possibility that reduction in Ca2+ entry/current during mEPCs is not what is sensed to trigger the upregulation of QC.
We next considered whether an aspect of synaptic function that we did not measure might be involved in signaling the increase in QC. Several studies have suggested there is nonvesicular release of ACh that is continuous (Katz and Miledi, 1977; Vyskocil et al., 2009). Such nonvesicular release is undetectable in our recording conditions. The amount of continuous ACh release has been hypothesized to be two orders of magnitude greater than that released during mEPCs (Katz and Miledi, 1977; Vyskocil et al., 2009). Perhaps block of this continuous neurotransmission underlies the induction of the increase in QC. We tested whether increasing continuous ACh signaling could prevent the D-TC effect by adding ACh to the bath. We used 0.1 mm ACh, a dose that triggers relatively mild desensitization of nAChRs and thus would be expected to greatly increase continuous ACh-mediated current. To avoid the possibility of activation of muscarinic nAChRs by the addition of ACh, 10 μm atropine was also added. The increase in QC after application of D-TC was unaffected by the addition of ACh to the bath (Fig. 3H). These data suggest that increasing continuous activation of nAChRs by application of agonist has no effect on the signaling triggered by application of D-TC.
Efficacy of different blocker of nAChRs
We wished to determine whether all blockers of nicotinic nAChRs had the same efficacy in triggering the increase in QC. It has been reported that vecuronium blocks nAChRs, but does not trigger an increase in QC (Tian et al., 1994). However, when we applied vecuronium, the QC increase was similar to other blockers of nAChRs (Fig. 4A). We measured the efficacy of both competitive and noncompetitive blockers of nAChRs. Both D-TC and gallamine are reversible competitive antagonists (Rang et al., 2003; Ostroumov et al., 2008). Mecamylamine is a noncompetitive antagonist that does not block binding of ACh to the nAChR but instead is an open-channel blocker (Varanda et al., 1985). We compared the efficacies of gallamine and mecamylamine with those of D-TC, vecuronium, and BTX and found that all of these blockers had similar efficacy in increasing QC (Fig. 4B). In contrast, succinylcholine blocked nAChRs, as indicated by the decrease of mEPC amplitude, but has partial agonist activity (Marshall et al., 1990; Baraka, 2007) and did not trigger QC upregulation (Fig. 4C,D).
Figure 4.

Upregulation of QC occurs only in blockers with no agonist activity. A, Application of vecuronium triggers upregulation of QC. Left, Plot of average EPC and mEPC amplitude and QC before and after infusion of vecuronium (0.25 μm) at the time indicated. Right, Averaged EPC and mEPC traces before (gray) and in the presence of vecuronium (red) taken from times corresponding to colored symbols on the plot on the left. Below are plots of mean EPC, mEPC, QC, and mEPC amplitude before (n = 10 muscle, 35 NMJs), during (n = 10 muscles, 99 NMJs) and after washout of vecuronium (n = 10 muscles, 45 NMJs. For these experiments, muscle was held at −70 mV and contraction was prevented by addition of 2 μm μ-Ctx (Table 1). B, Application of mecamylamine triggers upregulation of QC. The same experimental protocol was used as described in A for vecuronium. Mecamylamine (12 μm) triggered similar changes in EPC and mEPC amplitude and a similar increase in QC. Bottom summarizes data from 6 muscles before mecamylamine (n = 31 NMJs) and during (n = 32 NMJs) and after washout (n = 43 NMJs) (Table 1). C, Application of succinylcholine fails to trigger upregulation of QC. Left, Plot of average EPC and mEPC amplitude and QC before and after infusion of succinylcholine (1 μm) at the time indicated. D, Averaged EPC and mEPC traces before (gray) and in the presence of succinylcholine (red), taken from times corresponding to colored symbols on the plot shown in C. Bottom, mean data from 10 muscles treated with succinylcholine (500 nm): n = 61 NMJs before, n = 87 NMJs during succinylcholine infusion, and 76 NMJs after washout (Table 1). E, Comparison of changes in mEPC amplitude (gray triangle) and QC (red circle) induced by infusion of ACh (left) or by infusion of mecamylamine (right). All the values shown were normalized to the average value (as 100%) from the beginning of the plots, respectively. Although mEPC amplitude was reduced by a similar proportion, QC increased during mecamylamine infusion, but not ACh infusion. F, Summary of changes in mEPC amplitude and QC induced by BTX (control, 51 NMJs and BTX, 78 NMJs), D-TC (29 and 25), gallamine (33 and 55), mecamylamine (Meca, 31 and 43), vecuronium (33 and 99), succinylcholine (suc, 61 and 87), and ACh (43 and 102). **p < .01. Averaged values were scaled to controls, which were set at 100%.
It is possible that because of the partial agonist activity, mEPC amplitude was not reduced to the same extent as after application of blockers with no agonist activity and this was why QC was not increased after application of ACh and succinylcholine. To investigate this, we compared the magnitude of reduction of mEPC amplitude with the percentage increase in QC for all blockers tested and found that the reduction in mEPC amplitude was similar after drugs that did and did not trigger increased QC (Fig. 4E,F). Therefore, the lack of increase in QC could not be explained by a less severe block of synaptic transmission.
Increase in QC is associated with a speeding of mEPC decay
If block of nAChRs does not trigger increased QC by reducing synaptic current, what possible explanations remain? We considered the possibility that nAChRs undergo a conformational change after application of blocker. Evidence consistent with a conformational change in nAChRs came from analysis of the rate of decay of mEPCs. After partial block of nAChRs with blockers with no agonist activity, there was a small, but statistically significant increase in the rate of decay of mEPCs (Fig. 5). At CNS synapses, the presence of multiple receptor types made up of different mixes of subunits means that changes in the kinetics of currents could be due to preferential block of certain receptor subtypes. This is not the case at the mouse NMJ, where there is only one type of nAChR at the adult NMJ that is made up of an identical mix of subunits (Unwin, 2013; Auerbach, 2015). Therefore, the more rapid mEPC decay could indicate a change in behavior of the adult α1-containing muscle AChR.
Figure 5.

Upregulation of QC correlates with an increase in decay rate of mEPC. A, Plots of averaged mEPC amplitude (triangle), decay tau* (circle), and QC (square) against time. Data were acquired using a protocol shown in Figure 1A. BTX (200 μg/ml) was pressure puffed onto the endplate at the time indicated by the arrow. Left top, Superimposed mEPC traces (left) and peak normalized traces (right) shown were averaged from the time points indicated by the gray and red filled symbols on the mEPC plot (middle). For reference, same colorations apply to symbols in QC plot (bottom). B, Decay constant of mEPCs in the presence of drug relative to that of control. The same endplate was used to obtain mEPC values in the presence and absence of drug and the number of endplates in each case is indicated in parentheses. Statistically significant decrease in normalized decay time constant was seen in all pairs (except for an increase in succinylcholine) (Table 1). Comparison of mEPC decay time constant reduction between the ACh/CCh group and the BTX group yielded significance (t(38) = 4.197, p = 1.57E-4, t test). GT, Gallamine. C, Scattered plots of mEPC decay tau before and in the presence of nAChR antagonists. Two measurements of each endplate are connected by a colored line. Data are from the same experiments shown in B (Table 1). *The rate of decay was measured as the time elapsed from the peak of the averaged mEPC to peak/e.
It is known that the time constant of mEPC and EPC decay and the mean open time of ACh-gated channels is voltage dependent (Magleby and Stevens, 1972); however, we voltage clamped individual NMJs at the same potential before and after toxin application and therefore voltage-dependent differences in nAChRs open time does not explain the differences in mEPC decay. Another possible explanation for the increase in rate of mEPC decay is that blockers decrease the density of ACh-binding sites and thereby accelerate the diffusion of ACh from the synaptic cleft, which is manifested by more rapid decay of the synaptic current (Katz and Miledi, 1973).
If a conformational change in nAChRs is involved in signaling the increase in QC, then blockers that trigger the increase in QC should trigger an increase in rate of decay even if they have different mechanisms of action. Consistent with this possibility, blockers that triggered increased QC had significant shortening of mEPC decay time (Fig. 5B). In the case of mecamylamine, single-channel recordings have shown that the increase rate of decay is likely secondary to a shorter channel open time (Varanda et al., 1985). Succinylcholine, the only nAChR blocker that did not trigger an increase in QC, triggered prolongation in mEPC decay rather than a shortening. This increase in decay time has been shown to be due to a longer open time of ACh-activated currents (Nojima et al., 1992). Desensitization of nAChRs with ACh or CCh triggered a slight, but statistically significant, speeding of mEPC decay. The increase in decay rate after desensitization was statistically significantly less than the increase in decay rate induced by blockers that triggered the increase in QC (Fig. 5). Because desensitization of nAChRs with ACh or CCh decreases the number of unoccupied ACh-binding sites, there should have been a similar increase in diffusion of ACh such that mEPC decay should have been similarly shortened. The finding that mEPC shortening was less after desensitization is consistent with the possibility that part of the shortening is due to a conformational change in unblocked nAChRs.
Discussion
It has been proposed that disruption of synaptic transmission is sensed to trigger homeostatic upregulation of QC at the NMJ (Rich and Wenner, 2007; Frank, 2014; Davis and Müller, 2015). Studies performed to determine how disruption of synaptic transmission is sensed led us to propose that interruption of synaptic transmission may not be what is sensed to trigger synaptic plasticity at the mouse NMJ. We instead suggest that muscle α1-containing nicotinic nAChRs may serve as signaling molecules that trigger synaptic plasticity at the NMJ.
Technical considerations
Given the surprising nature of our hypothesis that block of postsynaptic AChRs triggers directly an increase in presynaptic release of ACh within seconds, it is worth considering the possibility that our findings are due to an artifact of the way that we performed the experiments. For example, the change in QC could be due to different ACh concentrations during the EPC and mEPC; because of the much higher concentration of ACh in the synaptic cleft during the EPC than in mEPC, antagonist blockade could more efficiently decrease the mEPC compared with the EPC, thus altering the EPC/mEPC ratio. This issue would be compounded if there was focal multivesicular release, as has been demonstrated at CNS synapses (Wadiche and Jahr, 2001; Taschenberger et al., 2002). Alternatively, there could be an issue with voltage clamp of the larger EPC and better clamp is achieved after partial block of nAChRs. Both of these possibilities would lead to the incorrect conclusion that QC was increased after partial block of nAChRs. Supporting both of these possibilities is the finding that, when QC is lowered by decreasing external Ca2+, the increase in QC is no longer seen (Tian et al., 1994; Wang et al., 2016).
What then, is our evidence that the increase in QC is real? One finding suggesting the increase is real is that block with BTX triggers the same increase as D-TC. BTX binds nAChRs with high affinity (Lukas et al., 1981) such that a higher concentration of ACh in the cleft during evoked release will not overcome block. Furthermore, the increase can be prevented by two manipulations that have minimal effect on EPC and mEPC amplitude. The first manipulation is block of vesicle refilling with vesamicol. Although this eventually leads to reduction of EPC amplitude, before causing a significant reduction in EPC amplitude it eliminates the increase in QC after infusion of D-TC (Wang et al., 2016). Second, mice that have Ca2+ indicator dye expressed presynaptically (perhaps buffering presynaptic Ca2+) have normal EPC amplitude, but a significantly blunted increase in QC after infusion of D-TC (Wang et al., 2016). Finally, mEPC and EPC amplitudes were reduced after application of ACh and succinylcholine without triggering an increase in QC. The finding that synaptic current can be reduced without triggering an increase in QC rules out technical artifacts such as poor voltage clamp of large currents. Our interpretation is that the increase in QC is real and is due to mobilization of a unique pool of synaptic vesicles in a manner that is dependent on elevation of presynaptic Ca2+ (Wang et al., 2016).
Triggers of homeostatic synaptic plasticity at different synapses
It has been proposed that spiking activity is sensed by CNS neurons to trigger homeostatic upregulation of quantal amplitude (synaptic scaling) (Turrigiano et al., 1998; Burrone et al., 2002; Stellwagen and Malenka, 2006). If this is the case, then it would suggest that the way in which disruption of synaptic activity is sensed at central synapses is fundamentally different from the way that disruption is sensed at the NMJ. However, it was recently demonstrated that restoring network activity to baseline levels in the presence of ongoing block of neurotransmission had no effect on synaptic scaling (Fong et al., 2015; Garcia-Bereguiain et al., 2016). These findings strongly suggest blocking neurotransmitter receptors is sufficient to trigger homeostatic plasticity independent of network activity in the CNS and raise the possibility that the signals triggering homeostatic regulation are similar at CNS synapses and the NMJ.
At CNS synapses, homeostatic synaptic plasticity can also be triggered by blocking Na channels rather than neurotransmitter receptors (Turrigiano et al., 1998; Burrone et al., 2002; Stellwagen and Malenka, 2006). Do similar forms of homeostatic plasticity exist at the NMJ? At the mouse NMJ, block of evoked activity triggers an increase in probability of vesicle release and an increase in quantal amplitude that is independent of nAChR block (Wang et al., 2004, 2005, 2010) and, at the Drosophila NMJ, postsynaptic overexpression of K channels triggers an increase in QC despite normal activation of glutamate receptors (Paradis et al., 2001). These forms of synaptic plasticity appear to be triggered by disruption of activation of the presynaptic nerve terminal or the postsynaptic muscle fiber rather than block of neurotransmitter receptors and thus represent homeostatic synaptic plasticity that is triggered by disruption of synaptic transmission. This suggests that at both the NMJ and central synapses there are at least two different sensors that can independently trigger homeostatic synaptic plasticity. Some sensors monitor synaptic/network activity, whereas others may sense factors that are independent of synaptic/network activity. At the NMJ, each sensor engages different mechanisms to homeostatically regulate synaptic strength (Wang et al., 2010).
Differences between homeostatic regulation at the mouse and Drosophila NMJ
The system in which the molecular mechanisms underlying homeostatic upregulation of QC has been studied in the greatest detail is the Drosophila NMJ. Most relevant to the current study, two different retrograde signaling molecules have been identified (Wang et al., 2014; Orr et al., 2017). The retrograde molecular signals act to modify presynaptic actin to potentiate the readily releasable pool, increase insertion of DegENaC channels into the presynaptic membrane, and increase Ca2+ entry through presynaptic Cav2.1 channels (Müller and Davis, 2012; Younger et al., 2013; Orr et al., 2017). All of these changes appear to contribute to the increase in QC.
Could the same molecular signals underlie the retrograde homeostatic signaling occurring at the mouse NMJ? In both systems, block of postsynaptic neurotransmitter receptors triggers an upregulation in presynaptic QC that occurs relatively rapidly. Although the similarities suggest that the answer certainly could be yes, there are two differences that suggest caution should be used when extrapolating results from Drosophila to the mammalian NMJ. First, in the mouse, upregulation of QC occurs as rapidly as it can be measured (Wang et al., 2016). In Drosophila, there is a several minute lag between block of receptors and upregulation of QC (Frank et al., 2006). Second, reduction of external Ca2+ concentration eliminates the upregulation of QC at the mouse NMJ (Tian et al., 1994; Wilson et al., 1995; Wang et al., 2016). In Drosophila, the increase in QC occurs at all levels of extracellular Ca2+ (Frank et al., 2006; Müller et al., 2015; Orr et al., 2017). These dissimilarities suggest fundamental differences in homeostatic mechanisms between the mouse and Drosophila NMJ.
Type and location of nAChRs for which block triggers the increase in QC
Two assumptions underlying our interpretation of the data are that the nAChRs blocked are located on the postsynaptic muscle fiber and that there is only a single type of nAChR present on the muscle fiber. The data for and against our assumptions are considered below.
It has been hypothesized that block of α7-containing nicotinic nAChRs present on the presynaptic nerve terminal at the NMJ is responsible for the rapid upregulation of QC after block of nAChRs (Bowman et al., 1990; MacDermott et al., 1999). We ruled out involvement of presynaptic α7 nAChRs as the trigger using α7 knock-out mice and toxins selective for the α1 versus α7 AChR subunits (Luo and McIntosh, 2004; Whiteaker et al., 2007). Our data strongly suggest that block of α1-containing nAChRs triggers the upregulation of QC. Because we are aware of no evidence that either Schwann cells or the presynaptic motor terminal express α-1-containing nAChRs, we favor the possibility that block of α-1-containing nAChRs on the postsynaptic muscle fiber triggers the upregulation of QC.
A second possibility that would complicate interpretation is that there might be two different types of nAChRs at the NMJ with differential sensitivity to block. If spontaneous and evoked release differentially activated the two nAChR subtypes, then this could lead to erroneous conclusions about changes in QC after block. For example, if the nAChRs preferentially activated by spontaneous release were more sensitive to block, one could erroneously conclude that QC is increased after block. Support for this possibility comes from studies at a number of different synapses, which suggest the vesicle pools responsible for evoked and spontaneous release are distinct (for review, see Kavalali, 2015). However, there is no evidence for multiple nAChR subtypes at the adult, innervated NMJ (Unwin, 2013; Auerbach, 2015). Nevertheless, it remains possible that two populations exist and this could account for our findings. The following data argue against this possibility: when extracellular Ca2+ is lowered, partial block of nAChRs triggers no increase in QC (Tian et al., 1994; Wang et al., 2016). Similarly, there is no nAChR block-induced upregulation of QC when vesamicol is added to prevent refilling of vesicles (Wang et al., 2016). If there were two types of nAChRs differentially activated by spontaneous and evoked release and the nAChR types had differential sensitivity to blockers, then one would not expect there to be situations in which block of nAChRs would not cause the apparent upregulation of QC.
How block of nAChRs is sensed
The nAChR-block-induced increase in QC did not require prior evoked release and could be triggered in NMJs of fibers that were voltage clamped. These data indicate that neither evoked release nor membrane potential is sensed to trigger the increase in QC. Another possibility is that Ca2+/ion flow through nAChRs during spontaneous release of ACh is sensed (Bregestovski et al., 1979; Vernino et al., 1994). Block of Ca2+ flow through postsynaptic receptors has been proposed as the sensor for homeostatic plasticity at the frog, mouse, and Drosophila NMJ (Frank et al., 2006; Ouanounou et al., 2016). We addressed this possibility by applying agonist and found no relationship between reduction of current flow through nAChRs and upregulation of QC. These data raise the possibility that reduction of neither Ca2+ entry nor synaptic current is the signal sensed. Further experiments directly measuring Ca2+ entry would be required to confirm this conjecture.
One way that nAChRs could initiate signaling is by undergoing a conformational change. Evidence consistent with unblocked nAChRs undergoing a conformational change comes from the finding that block of nAChRs triggers a small, but statistically significant increase in the rate of decay of mEPCs. The increase in rate of decay cannot be explained by an effect of membrane potential on decay rate (Magleby and Stevens, 1972) or competition between ligand and blocker (Wadiche and Jahr, 2001) because it occurs after block with an essentially irreversible blocker (BTX). It is possible that the increase in decay rate is due to occupation of ACh-binding sites, which leads to an increase in the rate of diffusion of ACh out of the synaptic cleft (Katz and Miledi, 1973). However, desensitization of nAChRs with ACh or CCh also causes a decrease in ACh-binding sites available, but does not trigger as big an increase in decay rate. Reduction of mEPC amplitude by application of ACh and succinylcholine, two treatments that did not trigger the same increase in decay rate, did not trigger an increase in QC. These data raise the possibility that there is a conformational change in unblocked nAChRs that may be responsible for the increase in mEPC decay.
The signaling role that we are proposing for nAChRs at the NMJ has been termed noncanonical signaling (Valbuena and Lerma, 2016). In noncanonical signaling, ionotropic neurotransmitter receptors also serve as signaling molecules. Noncanoncial signaling involving nAChRs has been shown previously in leukocytes (Razani-Boroujerdi et al., 2007; Hecker et al., 2015; Richter et al., 2016). Ours is the first study to suggest that postsynaptic nAChRs may be involved in noncanonical signaling that regulates synaptic function and that noncanonical signaling may be involved in triggering homeostatic synaptic plasticity. Because nAChRs have been implicated in a number of disease states involving the nervous system (Del Bufalo et al., 2014; Deutsch et al., 2015; Lombardo and Maskos, 2015), the hypothesis that nAChRs may trigger synaptic plasticity directly has wide-reaching implications.
Footnotes
This work was supported by National Institutes of Health (Grant P01NS057228 to M.M.R. and Grants GM49677 and GM103801 to J.M.M.). We thank Drs. Peter Wenner and Doju Yoshikami for helpful comments.
The authors declare no competing financial interests.
References
- Adams DJ, Dwyer TM, Hille B (1980) The permeability of endplate channels to monovalent and divalent metal cations. J Gen Physiol 75:493–510. 10.1085/jgp.75.5.493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albuquerque EX, Pereira EF, Alkondon M, Rogers SW (2009) Mammalian nicotinic acetylcholine receptors: from structure to function. Physiol Rev 89:73–120. 10.1152/physrev.00015.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auerbach A. (2015) Activation of endplate nicotinic acetylcholine receptors by agonists. Biochem Pharmacol 97:601–608. 10.1016/j.bcp.2015.06.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baraka A. depolarizing block is an endplate-muscular block, not a neuromuscular block. Anesthesiology 106:399–400, 2007; author reply 400. [DOI] [PubMed] [Google Scholar]
- Bowman WC, Prior C, Marshall IG (1990) Presynaptic receptors in the neuromuscular junction. Ann N Y Acad Sci 604:69–81. 10.1111/j.1749-6632.1990.tb31983.x [DOI] [PubMed] [Google Scholar]
- Bregestovski PD, Miledi R, Parker I (1979) Calcium conductance of acetylcholine-induced endplate channels. Nature 279:638–639. 10.1038/279638a0 [DOI] [PubMed] [Google Scholar]
- Burrone J, O'Byrne M, Murthy VN (2002) Multiple forms of synaptic plasticity triggered by selective suppression of activity in individual neurons. Nature 420:414–418. 10.1038/nature01242 [DOI] [PubMed] [Google Scholar]
- Cull-Candy SG, Miledi R, Trautmann A, Uchitel OD (1980) On the release of transmitter at normal, myasthenia gravis and myasthenic syndrome affected human end-plates. J Physiol 299:621–638. 10.1113/jphysiol.1980.sp013145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis GW, Müller M (2015) Homeostatic control of presynaptic neurotransmitter release. Annu Rev Physiol 77:251–270. 10.1146/annurev-physiol-021014-071740 [DOI] [PubMed] [Google Scholar]
- Del Bufalo A, Cesario A, Salinaro G, Fini M, Russo P (2014) Alpha9 alpha10 nicotinic acetylcholine receptors as target for the treatment of chronic pain. Curr Pharm Des 20:6042–6047. 10.2174/1381612820666140314150634 [DOI] [PubMed] [Google Scholar]
- Deutsch SI, Burket JA, Urbano MR, Benson AD (2015) The alpha7 nicotinic acetylcholine receptor: a mediator of pathogenesis and therapeutic target in autism spectrum disorders and down syndrome. Biochem Pharmacol 97:363–377. 10.1016/j.bcp.2015.06.005 [DOI] [PubMed] [Google Scholar]
- Fong MF, Newman JP, Potter SM, Wenner P (2015) Upward synaptic scaling is dependent on neurotransmission rather than spiking. Nat Commun 6:6339. 10.1038/ncomms7339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank CA. (2014) Homeostatic plasticity at the Drosophila neuromuscular junction. Neuropharmacology 78:63–74. 10.1016/j.neuropharm.2013.06.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank CA, Kennedy MJ, Goold CP, Marek KW, Davis GW (2006) Mechanisms underlying the rapid induction and sustained expression of synaptic homeostasis. Neuron 52:663–677. 10.1016/j.neuron.2006.09.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Bereguiain MA, Gonzalez-Islas C, Lindsly C, Wenner P (2016) Spontaneous release regulates synaptic scaling in the embryonic spinal network in vivo. J Neurosci 36:7268–7282. 10.1523/JNEUROSCI.4066-15.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harborne AJ, Bowman WC, Marshall IG (1988) Effects of tubocurarine on end-plate current rundown and quantal content during rapid nerve stimulation in the snake. Clin Exp Pharmacol Physiol 15:479–490. 10.1111/j.1440-1681.1988.tb01104.x [DOI] [PubMed] [Google Scholar]
- Hecker A, Küllmar M, Wilker S, Richter K, Zakrzewicz A, Atanasova S, Mathes V, Timm T, Lerner S, Klein J, Kaufmann A, Bauer S, Padberg W, Kummer W, Janciauskiene S, Fronius M, Schweda EK, Lochnit G, Grau V (2015) Phosphocholine-modified macromolecules and canonical nicotinic agonists inhibit ATP-induced IL-1beta release. J Immunol 195:2325–2334. 10.4049/jimmunol.1400974 [DOI] [PubMed] [Google Scholar]
- Innocent N, Livingstone PD, Hone A, Kimura A, Young T, Whiteaker P, McIntosh JM, Wonnacott S (2008) Alpha-conotoxin arenatus IB [V11L, V16D] [corrected] is a potent and selective antagonist at rat and human native alpha7 nicotinic acetylcholine receptors J Pharmacol Exp Ther 327:529–537. 10.1124/jpet.108.142943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson DS, Martinez J, Elgoyhen AB, Heinemann SF, McIntosh JM (1995) alpha-conotoxin ImI exhibits subtype-specific nicotinic acetylcholine receptor blockade: preferential inhibition of homomeric alpha 7 and alpha 9 receptors. Mol Pharmacol 48:194–199. [PubMed] [Google Scholar]
- Katz B, Miledi R (1973) The binding of acetylcholine to receptors and its removal from the synaptic cleft. J Physiol 231:549–574. 10.1113/jphysiol.1973.sp010248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz B, Miledi R (1977) Transmitter leakage from motor nerve endings. Proc R Soc Lond B Biol Sci 196:59–72. 10.1098/rspb.1977.0029 [DOI] [PubMed] [Google Scholar]
- Katz B, Miledi R (1978) A re-examination of curare action at the motor endplate. Proc R Soc Lond B Biol Sci 203:119–133. 10.1098/rspb.1978.0096 [DOI] [PubMed] [Google Scholar]
- Kavalali ET. (2015) The mechanisms and functions of spontaneous neurotransmitter release. Nat Rev Neurosci 16:5–16. 10.1038/nrn3875 [DOI] [PubMed] [Google Scholar]
- Kushmerick C, Renden R, von Gersdorff H (2006) Physiological temperatures reduce the rate of vesicle pool depletion and short-term depression via an acceleration of vesicle recruitment. J Neurosci 26:1366–1377. 10.1523/JNEUROSCI.3889-05.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lombardo S, Maskos U (2015) Role of the nicotinic acetylcholine receptor in Alzheimer's disease pathology and treatment. Neuropharmacology 96:255–262. 10.1016/j.neuropharm.2014.11.018 [DOI] [PubMed] [Google Scholar]
- Lukas RJ, Morimoto H, Hanley MR, Bennett EL (1981) Radiolabeled alpha-bungarotoxin derivatives: kinetic interaction with nicotinic acetylcholine receptors. Biochemistry 20:7373–7378. 10.1021/bi00529a008 [DOI] [PubMed] [Google Scholar]
- Luo S, McIntosh JM (2004) Iodo-alpha-conotoxin MI selectively binds the alpha/delta subunit interface of muscle nicotinic acetylcholine receptors. Biochemistry 43:6656–6662. 10.1021/bi049906y [DOI] [PubMed] [Google Scholar]
- MacDermott AB, Role LW, Siegelbaum SA (1999) Presynaptic ionotropic receptors and the control of transmitter release. Annu Rev Neurosci 22:443–485. 10.1146/annurev.neuro.22.1.443 [DOI] [PubMed] [Google Scholar]
- Magleby KL, Stevens CF (1972) The effect of voltage on the time course of end-plate currents. J Physiol 223:151–171. 10.1113/jphysiol.1972.sp009839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall CG, Ogden DC, Colquhoun D (1990) The actions of suxamethonium (succinyldicholine) as an agonist and channel blocker at the nicotinic receptor of frog muscle. J Physiol 428:155–174. 10.1113/jphysiol.1990.sp018205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miledi R, Molenaar PC, Polak RL (1978) Alpha-bungarotoxin enhances transmitter “released” at the neuromuscular junction. Nature 272:641–643. 10.1038/272641a0 [DOI] [PubMed] [Google Scholar]
- Molenaar PC, Polak RL, Miledi R, Alema S, Vincent A, Newsom-Davis J (1979) Acetylcholine in intercostal muscle from myasthenia gravis patients and in rat diaphragm after blockade of acetylcholine receptors. Prog Brain Res 49:449–458. 10.1016/S0079-6123(08)64657-9 [DOI] [PubMed] [Google Scholar]
- Molenaar PC, Oen BS, Plomp JJ, Van Kempen GT, Jennekens FG, Hesselmans LF (1991) A non-immunogenic myasthenia gravis model and its application in a study of transsynaptic regulation at the neuromuscular junction. Eur J Pharmacol 196:93–101. 10.1016/0014-2999(91)90413-K [DOI] [PubMed] [Google Scholar]
- Müller M, Davis GW (2012) Transsynaptic control of presynaptic Ca(2)(+) influx achieves homeostatic potentiation of neurotransmitter release. Curr Biol 22:1102–1108. 10.1016/j.cub.2012.04.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller M, Genç Ö, Davis GW (2015) RIM-binding protein links synaptic homeostasis to the stabilization and replenishment of high release probability vesicles. Neuron 85:1056–1069. 10.1016/j.neuron.2015.01.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nojima H, Muroi M, Kimura I, Kimura M (1992) Indirect inhibitory effect of succinylcholine on acetylcholine-activated channel activities and its modulation by external Ca2+ in mouse skeletal muscles. Br J Pharmacol 105:23–26. 10.1111/j.1476-5381.1992.tb14205.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orr BO, Fetter RD, Davis GW (2017) Retrograde semaphorin-plexin signalling drives homeostatic synaptic plasticity. Nature 550:109–113. 10.1038/nature24017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostroumov K, Shaikhutdinova A, Skorinkin A (2008) Modeling study of mecamylamine block of muscle type acetylcholine receptors. Eur Biophys J 37:393–402. 10.1007/s00249-007-0224-5 [DOI] [PubMed] [Google Scholar]
- Ouanounou G, Baux G, Bal T (2016) A novel synaptic plasticity rule explains homeostasis of neuromuscular transmission. eLife 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paradis S, Sweeney ST, Davis GW (2001) Homeostatic control of presynaptic release is triggered by postsynaptic membrane depolarization. Neuron 30:737–749. 10.1016/S0896-6273(01)00326-9 [DOI] [PubMed] [Google Scholar]
- Plomp JJ, van Kempen GT, Molenaar PC (1992) Adaptation of quantal content to decreased postsynaptic sensitivity at single endplates in alpha-bungarotoxin-treated rats. J Physiol 458:487–499. 10.1113/jphysiol.1992.sp019429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plomp JJ, van Kempen GT, Molenaar PC (1994) The upregulation of acetylcholine release at endplates of alpha-bungarotoxin-treated rats: its dependency on calcium. J Physiol 478:125–136. 10.1113/jphysiol.1994.sp020236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plomp JJ, Van Kempen GT, De Baets MB, Graus YM, Kuks JB, Molenaar PC (1995) Acetylcholine release in myasthenia gravis: regulation at single end-plate level. Ann Neurol 37:627–636. 10.1002/ana.410370513 [DOI] [PubMed] [Google Scholar]
- Rang HP, Dale MM, Ritter JM, Moore PK (2003) Pharmacology, Ed 5 New York: Churchill Livingstone. [Google Scholar]
- Razani-Boroujerdi S, Boyd RT, Dávila-García MI, Nandi JS, Mishra NC, Singh SP, Pena-Philippides JC, Langley R, Sopori ML (2007) T cells express alpha7-nicotinic acetylcholine receptor subunits that require a functional TCR and leukocyte-specific protein tyrosine kinase for nicotine-induced Ca2+ response. J Immunol 179:2889–2898. 10.4049/jimmunol.179.5.2889 [DOI] [PubMed] [Google Scholar]
- Rich MM, Wenner P (2007) Sensing and expressing homeostatic synaptic plasticity. Trends Neurosci 30:119–125. 10.1016/j.tins.2007.01.004 [DOI] [PubMed] [Google Scholar]
- Richter K, Mathes V, Fronius M, Althaus M, Hecker A, Krasteva-Christ G, Padberg W, Hone AJ, McIntosh JM, Zakrzewicz A, Grau V (2016) Phosphocholine: an agonist of metabotropic but not of ionotropic functions of alpha9-containing nicotinic acetylcholine receptors. Sci Rep 6:28660. 10.1038/srep28660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rochon D, Rousse I, Robitaille R (2001) Synapse-glia interactions at the mammalian neuromuscular junction. J Neurosci 21:3819–3829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stellwagen D, Malenka RC (2006) Synaptic scaling mediated by glial TNF-alpha. Nature 440:1054–1059. 10.1038/nature04671 [DOI] [PubMed] [Google Scholar]
- Taschenberger H, Leão RM, Rowland KC, Spirou GA, von Gersdorff H (2002) Optimizing synaptic architecture and efficiency for high-frequency transmission. Neuron 36:1127–1143. 10.1016/S0896-6273(02)01137-6 [DOI] [PubMed] [Google Scholar]
- Tian L, Prior C, Dempster J, Marshall IG (1994) Nicotinic antagonist-produced frequency-dependent changes in acetylcholine release from rat motor nerve terminals. J Physiol 476:517–529. 10.1113/jphysiol.1994.sp020151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turrigiano G. (2012) Homeostatic synaptic plasticity: local and global mechanisms for stabilizing neuronal function. Cold Spring Harb Perspect Biol 4:a005736. 10.1101/cshperspect.a005736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turrigiano GG, Leslie KR, Desai NS, Rutherford LC, Nelson SB (1998) Activity-dependent scaling of quantal amplitude in neocortical neurons. Nature 391:892–896. 10.1038/36103 [DOI] [PubMed] [Google Scholar]
- Unwin N. (2013) Nicotinic acetylcholine receptor and the structural basis of neuromuscular transmission: insights from torpedo postsynaptic membranes. Q Rev Biophys 46:283–322. 10.1017/S0033583513000061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valbuena S, Lerma J (2016) Non-canonical signaling, the hidden life of ligand-gated ion channels. Neuron 92:316–329. 10.1016/j.neuron.2016.10.016 [DOI] [PubMed] [Google Scholar]
- Varanda WA, Aracava Y, Sherby SM, VanMeter WG, Eldefrawi ME, Albuquerque EX (1985) The acetylcholine receptor of the neuromuscular junction recognizes mecamylamine as a noncompetitive antagonist. Mol Pharmacol 28:128–137. [PubMed] [Google Scholar]
- Vernino S, Rogers M, Radcliffe KA, Dani JA (1994) Quantitative measurement of calcium flux through muscle and neuronal nicotinic acetylcholine receptors. J Neurosci 14:5514–5524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vyskocil F, Malomouzh AI, Nikolsky EE (2009) Non-quantal acetylcholine release at the neuromuscular junction. Physiol Res 58:763–784. [DOI] [PubMed] [Google Scholar]
- Wadiche JI, Jahr CE (2001) Multivesicular release at climbing fiber-purkinje cell synapses. Neuron 32:301–313. 10.1016/S0896-6273(01)00488-3 [DOI] [PubMed] [Google Scholar]
- Wang T, Hauswirth AG, Tong A, Dickman DK, Davis GW (2014) Endostatin is a trans-synaptic signal for homeostatic synaptic plasticity. Neuron 83:616–629. 10.1016/j.neuron.2014.07.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Engisch KL, Li Y, Pinter MJ, Cope TC, Rich MM (2004) Decreased synaptic activity shifts the calcium dependence of release at the mammalian neuromuscular junction in vivo. J Neurosci 24:10687–10692. 10.1523/JNEUROSCI.2755-04.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Li Y, Engisch KL, Nakanishi ST, Dodson SE, Miller GW, Cope TC, Pinter MJ, Rich MM (2005) Activity-dependent presynaptic regulation of quantal size at the mammalian neuromuscular junction in vivo. J Neurosci 25:343–351. 10.1523/JNEUROSCI.3252-04.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Wang Q, Engisch KL, Rich MM (2010) Activity-dependent regulation of the binomial parameters p and n at the mouse neuromuscular junction in vivo. J Neurophysiol 104:2352–2358. 10.1152/jn.00460.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Pinter MJ, Rich MM (2016) Reversible recruitment of a homeostatic reserve pool of synaptic vesicles underlies rapid homeostatic plasticity of quantal content. J Neurosci 36:828–836. 10.1523/JNEUROSCI.3786-15.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whiteaker P, Christensen S, Yoshikami D, Dowell C, Watkins M, Gulyas J, Rivier J, Olivera BM, McIntosh JM (2007) Discovery, synthesis, and structure activity of a highly selective alpha7 nicotinic acetylcholine receptor antagonist. Biochemistry 46:6628–6638. 10.1021/bi7004202 [DOI] [PubMed] [Google Scholar]
- Wilson DF. (1982) Influence of presynaptic receptors on neuromuscular transmission in rat. Am J Physiol 242:C366–372. 10.1152/ajpcell.1982.242.5.C366 [DOI] [PubMed] [Google Scholar]
- Wilson DF, West AE, Lin Y (1995) Inhibitory action of nicotinic antagonists on transmitter release at the neuromuscular junction of the rat. Neurosci Lett 186:29–32. [DOI] [PubMed] [Google Scholar]
- Younger MA, Müller M, Tong A, Pym EC, Davis GW (2013) A presynaptic ENaC channel drives homeostatic plasticity. Neuron 79:1183–1196. 10.1016/j.neuron.2013.06.048 [DOI] [PMC free article] [PubMed] [Google Scholar]