

Abstract
Downstream processing of monoclonal antibodies (mAbs) has evolved to allow the specific process for a new product to be developed largely by empirical specialization of a platform process that enables removal of impurities of different kinds. A more complete characterization of impurities and the product itself would provide insights into the rational design of efficient downstream processes. This work identifies and characterizes host cell protein (HCP) product associated impurities, i.e., HCP species carried through the downstream processes via direct interactions with the mAb. Interactions between HCP and mAbs are characterized using cross interaction chromatography under solution conditions typical of those used in downstream processing. The interacting species are then identified by two dimensional gel electrophoresis and mass spectrometry. This methodology has been applied to identify product associated impurities in one particular purification step, namely protein A affinity chromatography, for four therapeutic mAbs as well as the Fab and Fc domains of one of these mAbs. The results show both the differences in HCP-mAb interactions among different mAbs, and the relative importance of product association compared to co-elution in protein A affinity chromatography.
Keywords: Host cell protein impurities, mAb process development, protein A affinity chromatography, protein-protein interactions
1. Introduction
Monoclonal antibodies (mAbs) are currently the fastest growing sector of the pharmaceutical industry (Li and Zhu, 2010). There are 26 mAbs approved by the FDA for human therapeutic use currently marketed in the United States and 27 in the European Union (Reichert, 2012). The increase in use of mAbs has been accompanied by extensive improvements in upstream technology and productivity, with much higher titers than in the past (Shukla and Thommes, 2010; Wurm, 2004). Because of the very high titers and demanding purity standards – typically 1–100 ppm host cell protein (HCP) in final formulations (Champion et al., 2005) – downstream processes generally account for the majority of production costs. Downstream purification has been estimated as representing 50–80% of total manufacturing costs (Guiochon and Beaver, 2011).
Downstream purification of mAbs most commonly utilizes a platform process, which is largely similar across the pharmaceutical industry. The platform process features a series of orthogonal chromatographic steps (Gottschalk, 2008; Strube et al., 2011), the first of which is most often a protein A affinity capture step (Huse et al., 2002; Nogal et al., 2011; Shukla and Hinckley, 2008), followed by two to three polishing steps (Gottschalk, 2008; Strube et al., 2011). Due to the extensive similarities among mAb products, the downstream platform parameters generally require only empirical tuning to optimize product recovery and purity for each new pipeline molecule (Guiochon and Beaver, 2011).
The generic platform process is well known and has been extensively studied, but with an emphasis on characterizing product retention and recovery in each of the purification steps. This study focuses instead on the impurities to be removed in mAb downstream processing, specifically on HCPs. HCPs are one of the major classes of impurities that must be removed during downstream purification as they have the potential to cause antigenic effects in human patients (Singh, 2011). Mammalian cells secrete hundreds of HCP species along with the desired product molecules. Typical industrial processes monitor the total HCP content using ELISA, but the specific identity of HCP impurities is generally unknown. Previous studies have used proteomic analysis, such as two-dimensional gel electrophoresis (2-DE) and/or mass spectrometry (MS), to identify the HCP impurities remaining after various downstream purification steps (Doneanu et al., 2012; Jin et al., 2010; Tait et al., 2012) or to determine the chromatographic properties of identified HCP species (Nfor et al., 2012).
The present study aims to provide a better understanding of how HCP impurities interact with both chromatographic adsorbents and with the mAb product molecule in a typical protein A affinity chromatography step, and examines the impact of these interactions on impurity clearance. The HCP behavior in the presence of IgG1 or IgG2 products is of particular interest as they are the two most abundant subclasses among FDA-approved mAbs (Chames et al., 2009). Protein A affinity chromatography is the workhorse of mAb platform purification processes in that it serves as an efficient capture step and removes the majority of HCP impurities as well as most other impurities. However, not all HCP impurities are removed by the protein A step, and identification of persistent HCPs in the protein A step and more importantly the reasons that these particular HCP impurities are retained can be valuable in future efforts to optimize downstream process design.
There are two major mechanisms by which HCP can enter the product fraction of the protein A affinity step, or any bind-and-elute type chromatographic purification. The first mechanism is product association (Luhrs et al., 2009; Shukla and Hinkley, 2008; Tarrant et al., 2012), which refers to strongly attractive interactions that certain HCPs have with the mAb product molecule, resulting in binding to the mAb. The HCP species is then carried through the process in association with the mAb, both in binding to the protein A or other ligand and in elution into the product fraction. The second mechanism for HCP retention in a chromatographic operation is co-elution, which refers to HCP species that bind to either the chromatographic ligand or the resin backbone and are then eluted into the product fraction as impurities. A more complete understanding of the relative importance of co-elution and product association and identification of HCPs that follow each of the two mechanisms can provide much needed mechanistic knowledge of HCP impurity retention, which can aid in future process synthesis efforts.
2. Materials and Methods
2.1. Materials and Solutions
Trichloroacetic acid, glutaraldehyde, ethanolamine, EDTA and recombinant human insulin were purchased from Sigma-Aldrich Chemical Co. (St. Louis, MO). Sodium chloride, sodium phosphate, sodium carbonate and sodium acetate were purchased from Fisher Scientific (Pittsburgh, PA). Tris was purchased from ICN Biomedicals Inc. (Arora, OH). Solutions were made using DI water further purified using a Millipore Milli-Q® system (Billerica, MA) and pH-adjusted using small amounts of concentrated sodium hydroxide or hydrochloric acid solutions. The protein A loading buffer was 20 mM sodium phosphate, 150 mM sodium chloride, pH 7.4, and the protein A elution buffer and low-pH wash buffer for cross-interaction chromatography was 20 mM acetic acid. The high-salt wash for cross-interaction chromatography was 20 mM sodium phosphate, 2 M sodium chloride, pH 7.4.
MAbs A and B, provided by Amgen Inc. (Seattle, WA), are closely related IgG2s that differ only in that two Arg residues near the CDRs of mAb A are an Ala and a Thr in mAb B. They were provided in formulation buffer at 150 and 32.2 mg/mL, respectively. MAb C is an IgG1 that was donated by Biogen Idec (San Diego, CA); it is a human antibody with variable regions derived from primates. MAb C was delivered in a formulation buffer at 49 mg/mL. MAb D (EMD Millipore, Bedford, MA) is an IgG1-κ that was provided in buffer at 60 mg/mL.
2.2. Chinese Hamster Ovary (CHO) Cell Culture for Null Supernatant
A null CHO-K1 cell line (ATCC, Manassas, VA) was adapted to adherent serum free growth in SFM4CHO-A media (Hyclone Laboratories Inc., Logan, UT) as described previously (Kumar et al., 2008). Cultures were seeded at 5·104 cells/mL in T-75 culture flasks and incubated for 3 – 4 days in a 37 °C cell culture incubator with 5% CO2 and 80% relative humidity until the final cell density reached 1·106 cells/mL at 97 – 99% viability. HCP for interaction measurements with Fc fragments were obtained from CHO cells that had been adapted to suspension culture in 125 mL shake flasks containing SFM4CHO suspension media (Hyclone Laboratories Inc.). The extracellular CHO HCPs were harvested by decanting the cell supernatant, separated from the residual cells by centrifugation (180 g, 5 min), and stored at −20 °C until further use.
2.3. Cell Culture Null Supernatant Preparation
CHO cell culture supernatant was buffer exchanged into protein A loading buffer using Slide-A-Lyzer cassettes from Pierce Biotechnology (Rockford, IL) by dialyzing three times against the final buffer for at least 4 hours each at 4 °C. The supernatant was then concentrated using Amicon® ultracentrifugal filter units (10 kDa nMWCO) from EMD Millipore (Bedford, MA) and was filtered using Millex 0.22 μm syringe filters from EMD Millipore. The same procedure was used to prepare mAbs for immobilization. Concentrations of mAbs were determined from UV absorbance at 280 nm, measured using a Perkin-Elmer Lambda 4B spectrophotometer (Waltham, MA). The extinction coefficients used were provided by the manufacturers or calculated from dilution curves of a solution of known concentration.
2.4. Protein A Purification
A series of protein A affinity chromatography purifications were completed similarly to previous work (Shukla and Hinckley, 2008; Tarrant et al., 2012), but excluding the use of ELISA, to determine the total amount of HCP that associates with various mAbs. An AP-Minicolumn (5 mm i.d.) was packed to a height of about 5 cm with 1 mL of rProtein A Sepharose Fast Flow resin from GE Healthcare (Piscataway, NJ) according to the manufacturer’s specifications. Three different materials were prepared for loading. CHO supernatant was prepared in protein A loading buffer as described in Section 2.3. The second mixture was an equal volume of buffer-exchanged supernatant spiked with purified mAb and equilibrated overnight. The third mixture contained only buffer and the same amount of mAb as the spiked sample.
Identical purification procedures were followed with these three starting materials, using an ÄKTA Purifier 900 using Unicorn 5.11 software (GE Healthcare, Piscataway, NJ). The sample was loaded onto the prepared protein A column using the sample pump, followed by a five column-volume wash and elution in low pH buffer. The entire low pH eluate was collected for all protein A affinity purifications to represent the product fraction. Product fractions from the three purifications were analyzed using a BCA assay (Pierce Biotechnology, Rockford, IL) to determine total protein concentrations. Each chromatographic experiment was run in triplicate. The known amount of mAb and the protein contribution from the HCP-only sample were subtracted from the results for the spiked sample to determine the total amount of HCP in the product fraction due to product association.
2.5. MAb Fragment Preparation
Fab and Fc fragments were generated using immobilized papain from Pierce Biotechnology (Rockford, IL). MAb D was digested by immobilized papain for 6 hours. Fragments were purified first using protein A affinity chromatography; the flow-through, which contained pure Fab fragments, was collected. The eluate produced using low-pH buffer was collected and buffer exchanged into 20 mM Tris, pH 8.5 with Amicon® ultracentrifugal filter units (10 kDa nMWCO). The buffer-exchanged eluate was then injected into a 1 mL AP-Minicolumn packed with Toyopearl QAE-550 (Tosoh Bioscience, King of Prussia, PA), washed with 15 column volumes (CVs) pH 8.5 buffer and eluted with a linear salt gradient from 0 to 400 mM NaCl over 15 CVs to separate Fc fragments from undigested mAb.
2.6. Cross-interaction Chromatography (CIC)
Cross interaction chromatography (CIC) (Cheng et al., 2008; Teske et al., 2004; Tessier et al., 2004a; Tessier et al., 2004b;) was used to probe the interactions of a complex mixture of HCP with a single mAb. It was also used to measure osmotic second virial cross-coefficients (B23) of insulin with immobilized mAbs and mAb fragments.
All four mAbs (A-D), Fab D and Fc D were immobilized separately onto Toyopearl AF-Amino-650 chromatographic particles (Tosoh Bioscience, King of Prussia, PA) using glutaraldehyde chemistry as previously described (Lewus et al., 2010). The immobilization density was measured using a Micro BCA™ Protein Assay Kit (Pierce Biotechnology, Rockford, IL). Immobilized mAb resin was flow packed into a 1 mL AP-Minicolumn with a flowrate of 2 mL/min water for 1 hour. The packing quality was verified by measuring the plate number using an acetone pulse.
HCPs were prepared for CIC by ultrafiltration in Amicon® centrifugal ultrafiltration filters, 10 kDa nMWCO, to 100 times the original concentration, and buffer exchange into protein A loading buffer. Adherent CHO cell culture supernatant was used for all CIC work except that for Fc CIC, where suspension cell culture supernatant was used. A 200 μL injection into the immobilized mAb column was followed by 10 CV of isocratic elution at the solution conditions being tested. This work emphasized typical protein A loading conditions, but the method can be adapted to seek interacting HCP at any solution condition of interest. The procedure was terminated with a high-salt elution (2 M NaCl) and a low-pH (pH 3.0) elution to remove any HCP impurities very strongly bound to the immobilized mAb.
2.7. Proteomic Analysis
Proteomic analysis consisted of 2 DE and mass spectrometry. Various fractions from CIC experiments were analyzed using 2-DE and the HCP populations of these fractions were compared to the total HCP that was injected into the immobilized mAb column.
2.7.1. Sample Preparation
Approximately 1 mg of each CHO HCP containing sample was precipitated with trichloroacetic acid on ice for a minimum of 1 hr, followed by centrifugation (14,000 g for 5 minutes) and two acetone (Fisher Scientific, Fair Lawn, NJ) wash centrifugation cycles (14,000 g for 5 minutes). The protein pellet was dried under argon.
2.7.2. 2-DE
2-DE was performed as described previously (Valente et al., 2012). Materials and equipment were obtained from Bio Rad Laboratories (Hercules, CA) unless otherwise specified. Briefly, precipitated proteins were resolubilized in rehydration solution (8 mM tris, 8 M urea, 30 mM DTT, 2% CHAPS (Sigma-Aldrich Chemical Co.), 0.4% BioLytes, and trace bromophenol blue), which was used to rehydrate 18 cm, pH 3–10 nonlinear Immobiline DryStrips (GE Healthcare). Isoelectric focusing (IEF) was performed using a PROTEAN IEF Cell for 100,000 Vh, after which IEF gels were sequentially equilibrated with DTT and iodoacetamide (Sigma-Aldrich Chemical Co.). SDS-PAGE was performed using 12 %T, 2.6 %C polyacrylamide slab gels measuring 18 cm × 16 cm × 1.5 mm. Gels were stained with SYPRO Ruby (Molecular Probes, Eugene, OR) and imaged on an FLA-3000 Fluorescent Image Analyzer (Fujifilm Corp., Tokyo, Japan). Gel images were analyzed and compared using ImageMaster 2D Platinum Software v5.0 (GE Healthcare). Spots were detected using the auto-detect feature and manually edited to remove artifacts. Spot matching was manually completed by comparing images of the CIC fractions to those of the total HCP supernatant that was loaded onto the CIC column. Spots of interest were either manually excised and stored at −80 °C or compared to a reference gel with previously identified spots using ImageMaster software.
2.7.3. Mass Spectrometry
Gel plugs containing spots of interest were subjected to in-gel digestion with trypsin (Promega Corporation, Madison, WI). The resulting peptides were desalted and concentrated with ZipTips (EMD Millipore) and spotted onto stainless steel target plates with α-cyano-4-hydroxycinnamic acid (Sigma-Aldrich Chemical Co.) matrix. Analysis by matrix-assisted laser desorption/ionization tandem time-of-flight (MALDI-TOF/TOF) MS was performed as described previously (Hayduk et al., 2004) on an Applied Biosystems 4800 MALDI TOF/TOF Analyzer (Framingham, MA). Data were acquired in positive ion MS reflector mode and MS/MS, then submitted for Mascot v2.2 (Matrix Science Ltd., London, UK) database searches through GPS Explorer software v3.6 (Applied Biosystems). Spectra were searched against translations of the CHO genome (Xu et al., 2011) and the NCBInr database (downloaded July 15, 2009), and identifications with 95% confidence or greater were accepted.
3. Results and Discussion
The results presented in this section show the relative importance and abundance of product association in a typical protein A affinity purification step for three different mAbs. Further characterization of the product association was obtained using CIC followed by proteomic analysis to identify HCPs that associate with four different mAbs. The same analysis was completed for the Fc and Fab domains from one of the four mAbs.
3.1. Protein A Affinity Purification
The total HCP content in the product fraction of protein A affinity purifications with and without mAb spiked into the starting material is shown in Figure 1 for three different mAbs. These results are consistent with those reported by Shukla and Hinckley (Shukla and Hinckley, 2008) as they show an increase in HCP in the product fraction when a mAb is included; the observed increase in HCP is presumably due to HCP mAb association.
FIGURE 1.
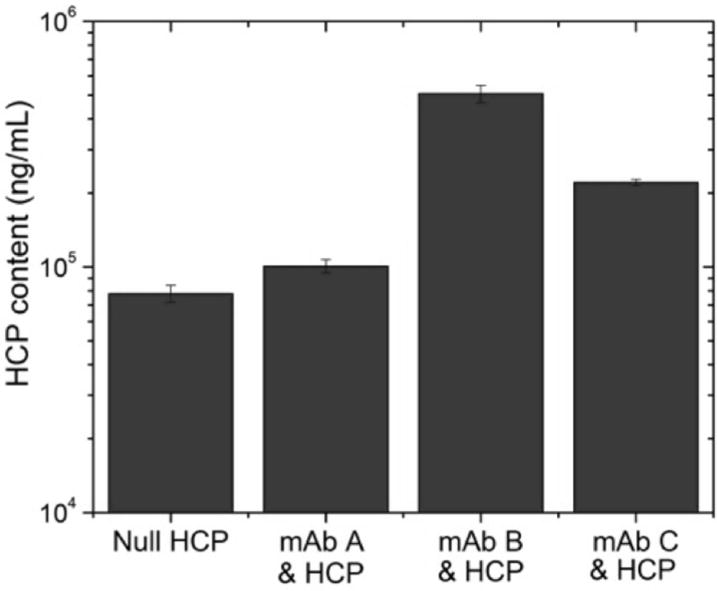
HCP content in protein A product fraction for purification of null CHO supernatant compared to purification of null CHO supernatant spiked with 3 different mAbs (A-C). Error bars represent chromatography replicates.
These results also demonstrate the variability in total product association with different mAbs. As shown in Figure 1, mAb B gives rise to the largest amount of HCP in the eluate whereas the increase in the presence of mAb A is very small. Although mAbs A and B differ by only 2 charged residues near the CDR, this relatively minor difference has a very large impact on total product association. This considerable difference in total associating HCP could be caused by a single abundant species that binds appreciably more strongly to the mutated molecule or it could be due to binding of a number of different HCP species. These alternatives are not easily distinguished because the method used in developing Figure 1 indicates only total product association as opposed to determining specific HCP species responsible for the interaction.
2-DE was used to monitor HCPs that persist through a typical protein A affinity purification process and appear in the product fraction. A representative example (Figure 2) compares 2-DE images for product fractions of protein A affinity purifications of null CHO supernatant spiked with mAb A (Figure 2A) and an equivalent quantity of only mAb A (Figure 2B). Given the inherent order-of-magnitude difference between the HCP and mAb concentrations, the heavy and light chains of the mAb obstruct most of the gel, hindering the ability to observe HCP impurities. It is therefore difficult to monitor both the mAb product and HCP impurities simultaneously using a method such as 2-DE. Despite this uncertainty and the low HCP concentrations compared to those of the mAb, the HCP concentration is still likely unacceptable in terms of final purity. Sypro Ruby has been reported to enable detection of protein amounts as low as 0.3–1.0 ng (Tscheliessnig et al., 2013), but it has been shown that for in-process samples, where HCP is in the ppm range, 2DE is unlikely to provide significant information about HCP populations (Jin et al., 2010).
FIGURE 2.
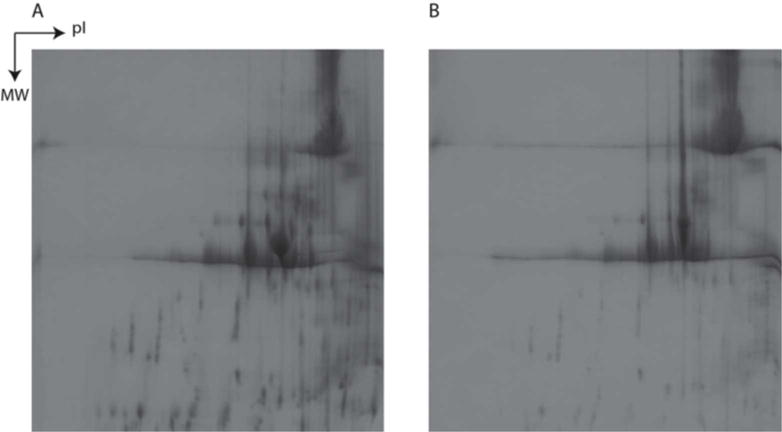
2-DE images of product fraction of protein A affinity purification of (A) mAb A spiked into null CHO supernatant and (B) 2-DE image of only mAb A.
3.2. HCP CIC
Measuring and identifying the complete set of HCP impurities in the product fraction of protein A affinity purification is informative and has been done previously for a similar system (Doneanu et al., 2012). However, this previous work did not provide information about the mechanisms by which specific HCP impurities are retained in the product fraction. CIC was used here to measure interactions between CHO HCP and various mAb products, and was coupled to 2-DE and MS to obtain identities of the specific strongly associating HCP impurities for each mAb. Procedures for each of the 4 mAbs, and the Fc and Fab fragments of mAb D were run in duplicate. A typical CIC chromatogram is shown in Figure 3. The majority of HCPs are found in the flow-through fraction, and the column wash yields a secondary peak containing strongly interacting HCP.
FIGURE 3.
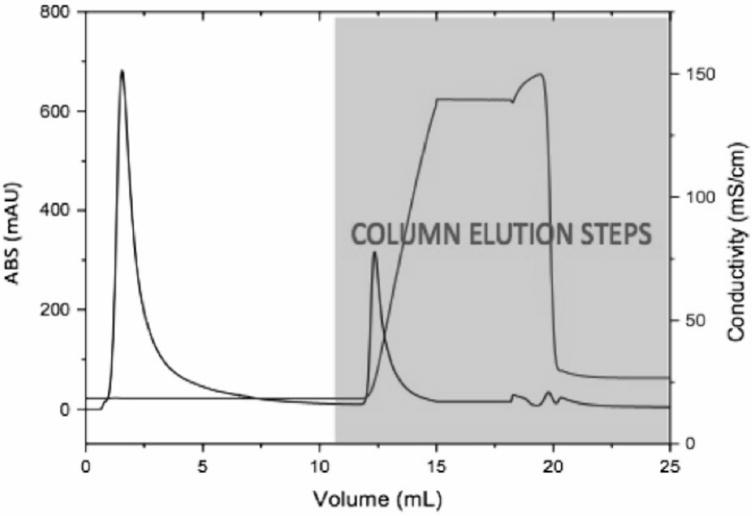
Typical chromatogram from HCP-mAb CIC run.
Direct identification of the strongly interacting HCPs was not possible as the relatively low concentration of HCP in the secondary peak was not sufficient for detection by 2-DE. In addition, very strongly interacting HCP species may be retained following elution and not appear in the secondary peak at all. The interacting HCP impurities were therefore instead identified by comparing 2-DE images of the CIC flow-through, containing non-interacting HCPs, to those of the CIC feed, containing all CHO HCPs. Spots that were detected on the feed image and absent from the flow-through image correspond to HCPs that associate strongly with the mAb being tested and thus remain in the column during isocratic elution. Missing spots in the flow-through were determined either by a 50% decrease in integrated spot volume using Image Master software or by qualitative changes based on visual inspection; further details are provided in Supplementary Material. These identified spots are marked on the feed gel and were either excised for further identification or compared to a reference gel with previously identified spots, as can be seen in Figures 4 and 5. The proteins identified as strongly associating HCPs are listed for each mAb and the Fab and Fc domains of mAb D in Table 1. As noted above, mAbs A and B differ by only 2 residues near the CDR, while mAbs C and D are unrelated products. Not surprisingly, mAbs A and B share 9 strongly interacting HCPs but each also has strong associations with a number of HCPs that do not interact with the other. As was shown in Figure 1, the total amount of HCP association was dramatically changed by the mutation from mAb A to B. Here it can be seen that there is a significant change in the population of interacting HCP species due to the two-residue mutation.
FIGURE 4.
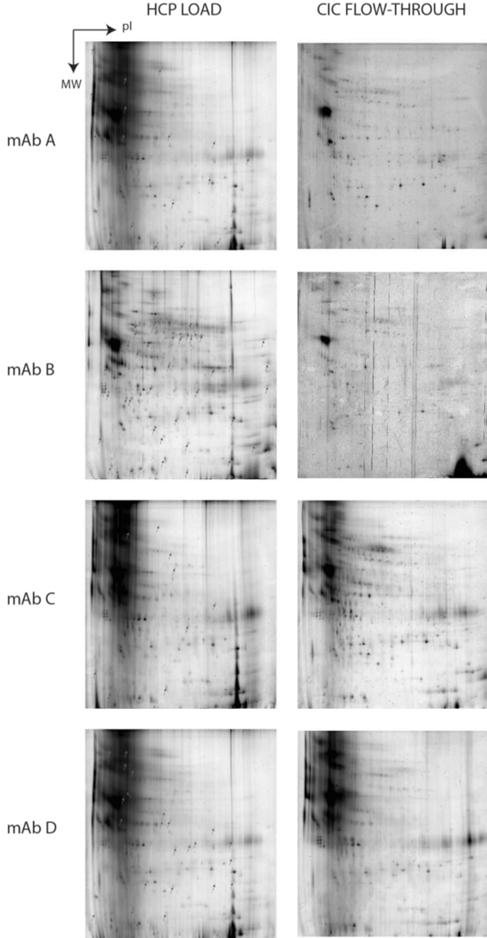
Analyzed 2-DE load (left column) and flow-through (right column) gels for HCP CIC of mAbs A-D. Indicated spots on ‘load’ gels are absent from corresponding flow-through gels and represent strongly interacting HCPs.
FIGURE 5.
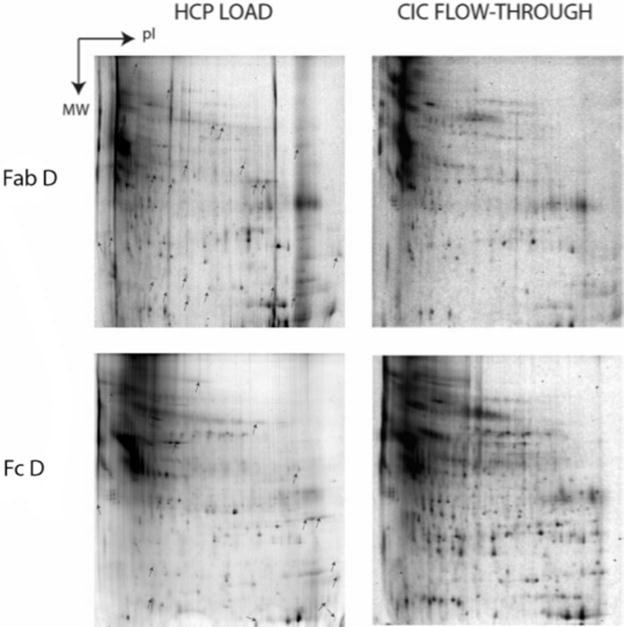
Analyzed 2-DE load (left column) and flow-through (right column) gels for HCP CIC of Fab D and Fc D. Indicated spots on ‘load’ gels are absent from corresponding flowthrough gels and represent strongly interacting HCPs.
Table I.
CHO HCP impurities with attractive interactions to mAbs A–D, Fc D and Fab D at 150 mM NaCl, pH 7.4.
| Protein ID | mAb A | mAb B | mAb C | mAb D | Fab D | Fc D | Location | MW (kDa) | pI |
|---|---|---|---|---|---|---|---|---|---|
| Neural cell adhesion molecule | − | + | + | + | − | − | Cell membrane | 17.9 | 4.7 |
| Renin receptor | − | + | − | + | + | − | Cell membrane | 33.9 | 5.2 |
| Lipoprotein lipase | + | + | + | − | − | + | Cell membrane, secreted | 50.5 | 7.9 |
| Chondroitin sulfate protoglycan 4 | + | − | + | + | + | − | Cell membrane, cell projection, membrane | 153.8 | 6.1 |
| Alpha-enolase | − | + | − | − | − | − | Cytoplasm, cell membrane | 15.5 | 5 |
| Galectin-3-binding protein | + | − | − | + | − | + | Membrane | 63.8 | 5.1 |
| G-protein coupled receptor 56 | + | − | + | + | − | + | Membrane | 77.4 | 9.1 |
| V-type proton ATPase subunit S1 | − | + | − | − | − | − | Membrane, vacuole | 28.1 | 6.5 |
| Nidogen-1 | + | + | + | + | − | + | Basement membrane, extracellular matrix | 30.1 | 8.1 |
| ATP synthase subunit beta, mitochondrial | + | + | − | − | − | + | Mitochondrion inner membrane | 56.3 | 5.2 |
| Vimentin | + | + | − | − | − | − | Cytoplasm | 53.7 | 5.1 |
| Heat shock protein | + | + | − | − | − | − | Cytoplasm | 32.6 | 5.7 |
| Actin | + | + | − | + | − | + | Cytoplasm | 41.5 | 5.2 |
| Peroxirodoxin 1 | − | − | − | − | − | + | Cytoplasm, melanosome | 22.3 | 8.2 |
| SPARC | + | + | + | + | + | − | Secreted | 28.2 | 4.9 |
| Clusterin | + | + | + | + | + | − | Secreted | 51.8 | 6.1 |
| Complement C1r-a sub-component | + | − | − | − | − | − | Extracellular | 80.1 | 6.1 |
| Metalloproteinase inhibitor 1 | + | − | − | − | − | − | Secreted | 22.4 | 8.8 |
| Insulin | + | + | + | + | − | − | Secreted | 28.1 | 5.5 |
| Cathepsin D | − | + | − | − | − | − | Lysosome, melanosome | 44.9 | 6.7 |
| Sulfated glycoprotein 1 | + | − | − | − | − | − | Lysosome | 27.4 | 5.4 |
| Lysosomal protective protein | − | − | − | − | − | + | Lysosome | 54 | 6.1 |
MAbs C and D, which are unrelated products, differ considerably more with each other and with mAbs A and B. However, there were 4 HCP impurities that associated strongly with all 4 mAbs, and 5 additional species associated with 3 of the 4 mAbs tested. These results show a baseline set of HCP impurities that bind strongly to all mAbs and additional HCPs that bind to smaller subsets of mAbs or only single mAbs. The smaller subsets are likely due to HCPs with high affinity for the hyper-variable region or different IgG subtypes or specific constructs. The 9 HCP that bind to at least 3 of the mAbs tested are likely interacting with one of the constant domains of these mAbs. Although the IgG subtype may be a significant factor determining HCP-mAb interactions, the set of IgG1 and IgG2 molecules studied here is not large enough to enable specific conclusions.
Lipoprotein lipase, nidogen-1, heat shock protein, actin and clusterin were all found in this study to bind at least 2 mAbs, and they were also previously identified as HCP impurities found in protein A product fractions for mAb purification (Doneanu et al., 2012). The results here, using mAbs unrelated to those used by Doneanu et al., are further evidence that this group of HCP binds to one of the constant regions of mAbs.
CIC experiments were completed for Fab D and Fc D, the Fab and Fc domains from mAb D. The analyzed 2-DE feed and flow-through images can be found in Figure 5. Since the Fab domain contains the variable CDR region as well as many constant regions, it was anticipated that the associating HCP for Fab D would be similar to those for mAb D. The results from the 2- DE and MS analysis showed only 4 associated HCP that were also identified for the full mAb. For Fc D, 4 out of 8 interacting HCP were identified as also interacting strongly with mAb D. It is plausible that the additional interacting HCP are associating to an epitope on the Fc that is not accessible in the full molecule.
It was previously shown that clusterin binds multivalently to the Fab and Fc domains of IgGs (Wilson, 1992). Therefore the finding here that clusterin binds to all mAbs and the Fab that were tested is consistent with previous observations, however clusterin was not identified as strongly attracted to the Fc D. Insulin was also found to bind to all mAbs tested, but did not interact with Fab D or Fc D. It is possible that the inconsistencies for clusterin and insulin are a result of using suspension cell culture for Fc CIC that may have contained less clusterin and insulin than the adherent cell culture, making them difficult to detect. Nidogen-1 binds strongly to mAbs A-D and Fc D, but does not bind Fab D. This suggests that nidogen-1 binds to the Fc region of IgG molecules.
The majority of interacting HCPs were slightly acidic proteins. About a third of the interacting proteins have previously been reported as being secreted, with the remaining two-thirds all characterized as intracellular proteins (The UniProt Consortium, 2012). Abundant intracellular proteins, such as actin, are likely released into the extracellular space from a small amount of cell lysis during culture. The mechanism of release of low-abundance intracellular proteins, such as V-type proton ATPase subunit S1, is unknown; however, it is possible that these proteins may be actively secreted by a non-classical secretory pathway, as has been reported for heat shock protein 70 (Mambula et al., 2007).
3.3. Insulin-mAb CIC
Insulin was found to bind strongly to all the mAbs tested. The insulin-mAb interaction was probed further using CIC to measure interaction strengths quantitatively in terms of B23 values at sodium chloride concentrations greater than or equal to the protein A loading conditions used in HCP CIC. Figure 6 shows the B23 values for all 4 mAbs and the Fc and Fab domains from mAb D with insulin as the mobile phase protein. The results for each mAb and fragment are consistent: salt concentrations close to the protein A loading conditions result in very negative B23 values, indicating highly attractive interactions, and as salt concentrations are increased the attractions between insulin and all mAbs become weaker. At the highest salt concentrations the interactions become slightly repulsive for mAbs C and D, Fab D and Fc D. The results from HCP CIC identified insulin as a likely product-associated impurity in a protein A affinity purification, but this more detailed analysis shows that if a high salt wash is applied, the insulin will be easily removed prior to product elution. These results are unique to insulin; CIC with additional product-associated impurities is necessary to assess the effectiveness of high salt washes.
FIGURE 6.
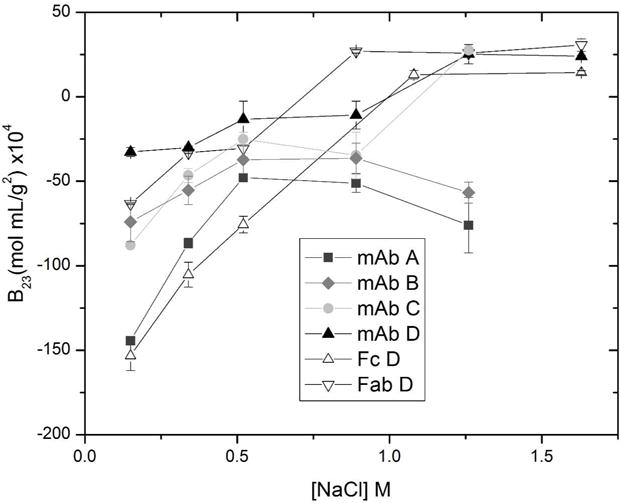
Second osmotic virial cross-coefficient (B23) measured by cross-interaction chromatography of insulin with mAbs A-D, Fab D and Fc D at pH 7.4 and varying sodium chloride concentration.
4. Conclusions
This work elucidates the characteristics of product association as a mechanism by which HCP impurities are retained in product fractions after a protein A purification. As was seen in previous work (Shukla and Hinckley, 2008; Tarrant et al., 2012), product association is as common as or more common than co-elution resulting from non-specific binding of HCP to chromatographic resins. We have extended these studies to show that the total amount of product association varies significantly for different mAb products. These differences can make some mAbs considerably more difficult to purify to acceptable HCP levels. The results also demonstrate that small changes in the mAb sequence can yield dramatic changes in HCP interactions; it is not reasonable to assume that related mAbs will interact with the same population of HCP impurities.
The work also showed the difficulties of directly monitoring HCP impurity content using 2-DE of downstream process product fractions. The nature of the mAb purification process results in HCP and mAb concentrations that differ by orders of magnitude, even though the relatively low HCP content is still important and would be considered unacceptable by regulatory agencies.
The CIC method developed here provided insight into the mechanism of HCP impurity retention while avoiding the issues of product and impurity concentration differences. For example, CIC with mAbs and mAb fragments showed the specific domains with which some HCPs associate, such as the consistent binding of nidogen-1 via the Fc domain. Overall, the number of HCP that were found to bind to each mAb (10–15) and the number of HCP that were found to bind to at least 3 of the 4 mAbs tested (9) are small compared to the total population of HCP. A relatively small subpopulation of CHO HCP therefore appears responsible for giving rise to product-associated impurities. While some strongly binding proteins, such as insulin, may bind strongly at loading conditions but be relatively easy to remove in a wash step, this is not necessarily true for other proteins, which would require further investigation.
Supplementary Material
Acknowledgments
We thank Amgen Inc., Biogen Idec and EMD Millipore for providing the antibodies used in this study. We are also grateful for financial support from the National Science Foundation (grant number CBET-0966644).
References
- Chames P, Van Regenmortel M, Weiss E, Baty D. Therapeutic antibodies: successes, limitations and hopes for the future. Br J of Pharmacol. 2009;157:220–233. doi: 10.1111/j.1476-5381.2009.00190.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Champion K, Madden H, Dougherty J, Shacter E. Defining Your Product Profile and Maintaining Control Over It, Part 2. BioProcess Int. 2005;3(8):52–57. [Google Scholar]
- Chen YC, Bianco CL, Sandler SI, Lenhoff AM. Salting-out of lysozyme and ovalbumin from mixtures: predicting precipitation performance from protein-protein interactions. Ind Eng Chem Res. 2008;47:5203–5213. [Google Scholar]
- Doneanu CE, Xenopoulos A, Fadgen K, Murphy J, Skilton SJ, Prentice H, Stapels M, Chen W. Analysis of host-cell proteins in biotherapeutic proteins by comprehensive online two-dimensional liquid chromatography / mass spectrometry. Commun Integr Biol. 2012;4:24–44. doi: 10.4161/mabs.4.1.18748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottschalk U. Bioseparation in antibody manufacturing: the good, the bad and the ugly. Biotechnol Prog. 2008;24:496–503. doi: 10.1021/bp070452g. [DOI] [PubMed] [Google Scholar]
- Guiochon G, Beaver LA. Separation science is the key to successful biopharmaceuticals. J Chromatogr, A. 2011;1218:8836–8858. doi: 10.1016/j.chroma.2011.09.008. [DOI] [PubMed] [Google Scholar]
- Hayduk EJ, Choe LH, Lee KH. A two-dimensional electrophoresis map of Chinese hamster ovary cell proteins based on fluorescence staining. Electrophoresis. 2004;25:2545–2556. doi: 10.1002/elps.200406010. [DOI] [PubMed] [Google Scholar]
- Huse K, Bohme HJ, Scholz GH. Purification of antibodies by affinity chromatography. J Biochem Biophys Methods. 2002;51:217–231. doi: 10.1016/s0165-022x(02)00017-9. [DOI] [PubMed] [Google Scholar]
- Jin M, Szapiel N, Zhang J, Hickey J, Ghose S. Profiling of host cell proteins by two-dimensional difference gel electrophoresis (2D-DIGE): Implications for downstream process development. Biotechnol Bioeng. 2010;105:306–316. doi: 10.1002/bit.22532. [DOI] [PubMed] [Google Scholar]
- Kumar N, Maurya P, Gammell P, Dowling P, Clynes M, Meleady P. Proteomic profiling of secreted proteins from CHO cells using surface-enhanced laser desorption ionization time-of-flight mass spectrometry. Biotechnol Prog. 2008;24:273–278. doi: 10.1021/bp070244o. [DOI] [PubMed] [Google Scholar]
- Lewus RA, Darcy PA, Lenhoff AM, Sandler SI. Interactions and phase behavior of a monoclonal antibody. Biotechnol Prog. 2010;27:280–289. doi: 10.1002/btpr.536. [DOI] [PubMed] [Google Scholar]
- Li J, Zhu Z. Research and development of next generation of antibody-based therapeutics. Acta Pharmacol Sin. 2010;31:1198–1207. doi: 10.1038/aps.2010.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luhrs KA, Harris DA, Summers S, Parseghian MH. Evicting hitchhiker antigens from purified antibodies. J Chromatogr B. 2009;877:1543–1552. doi: 10.1016/j.jchromb.2009.03.042. [DOI] [PubMed] [Google Scholar]
- Mambula SS, Stevenson MA, Ogawa K, Calderwood SK. Mechanisms for HSP secretion: crossing membranes without a leader. Methods. 2007;43:168–175. doi: 10.1016/j.ymeth.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nfor BK, Ahamed T, Pinkse MW, van der Wielen LA, Verhaert PD, van Dedem GW, Eppink MH, van de Sandt EJ, Ottens M. Multi-dimensional fractionation and characterization of crude protein mixtures: Toward establishment of a database of protein purification process development parameters. Biotechnol Bioeng. 2012;109:3070–3083. doi: 10.1002/bit.24576. [DOI] [PubMed] [Google Scholar]
- Nogal B, Chhiba K, Emery JC. Select host cell proteins coelute with monoclonal antibodies in protein a chromatography. Biotechnol Prog. 2011;28:454–458. doi: 10.1002/btpr.1514. [DOI] [PubMed] [Google Scholar]
- Reichert JM. Marketed therapeutic antibodies compendium. mAbs. 2012;4:413–415. doi: 10.4161/mabs.19931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shukla AA, Hinckley P. Host cell protein clearance during protein A chromatography: development of an improved column wash step. Biotechnol Prog. 2008;24:1115–1121. doi: 10.1002/btpr.50. [DOI] [PubMed] [Google Scholar]
- Shukla AA, Thommes J. Recent advances in large-scale production of monoclonal antibodies and related proteins. Trends Biotechnol. 2010;28:253–261. doi: 10.1016/j.tibtech.2010.02.001. [DOI] [PubMed] [Google Scholar]
- Singh SK. Impact of product-related factors on immunogenicity of biotherapeutics. J Pharm Sci. 2011;100:354–387. doi: 10.1002/jps.22276. [DOI] [PubMed] [Google Scholar]
- Strube J, Grote F, Josch J, Ditz R. Process Development and Design of Downstream Processes. Chem Ing Tech. 2011;83:1044–1065. [Google Scholar]
- Tait AS, Hogwood CEM, Smales CM, Bracewell DG. Host cell protein dynamics in the supernatant of a mAb producing CHO cell line. Biotechnol Bioeng. 2012;109:971–982. doi: 10.1002/bit.24383. [DOI] [PubMed] [Google Scholar]
- Tarrant RDR, Velez-Suberbie ML, Tait AS, Smales CM, Bracewell DG. Host cell protein adsorption characteristics during protein a chromatography. Biotechnol Prog. 2012;28:10371044. doi: 10.1002/btpr.1581. [DOI] [PubMed] [Google Scholar]
- Teske CA, Blanch HW, Prausnitz JM. Chromatographic measurement of interactions between unlike proteins. Fluid Phase Equilib. 2004;219:139–148. [Google Scholar]
- Tessier PM, Sandler SI, Lenhoff AM. Direct measurement of protein osmotic second virial cross coefficients by cross-interaction chromatography. Protein Sci. 2004a:1379–1390. doi: 10.1110/ps.03419204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tessier PM, Verruto VJ, Sandler SI, Lenhoff AM. Correlation of diafiltration sieving behavior of lysozyme-BSA mixtures with osmotic second virial cross-coefficients. Biotechnol Bioeng. 2004b;87:303–310. doi: 10.1002/bit.20115. [DOI] [PubMed] [Google Scholar]
- The UniProt Consortium. Reorganizing the protein space at the universal protein resource (UniProt) Nucleic Acids Res. 2012;40:D71–D75. doi: 10.1093/nar/gkr981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tscheliessnig AL, Konrath J, Bates R, Jungbauer A. Host cell protein analysis in therapeutic protein bioprocessing-methods and applications. Biotechnol J. 2013;8:655–670. doi: 10.1002/biot.201200018. [DOI] [PubMed] [Google Scholar]
- Valente KN, Choe LC, Lenhoff AM, Lee KH. Optimization of sample preparation for two-dimensional electrophoresis. Electrophoresis. 2012;33:1947–1957. doi: 10.1002/elps.201100659. [DOI] [PubMed] [Google Scholar]
- Wilson M. Clusterin binds by a multivalent mechanism to the Fc and Fab regions of IgG. Biochim Biophys Acta. 1992;1159:319–326. doi: 10.1016/0167-4838(92)90062-i. [DOI] [PubMed] [Google Scholar]
- Wurm FM. Production of recombinant protein therapeutics in cultivated mammalian cells. Nat Biotechnol. 2004;22:1393–1398. doi: 10.1038/nbt1026. [DOI] [PubMed] [Google Scholar]
- Xu X, Nagarajan H, Lewis NE, Pan S, Liu X, Chen W, Xie M, Wang W, Hammond S, Andersen M, Neff N, Passarelli B, Koh W, Lee KH, Betenbaugh MJ, Quake SR, Famili I, Palsson BO, Wang J. The genomic sequence of the Chinese hamster ovary (CHO)-K1cell line. Nat Biotechnol. 2011;29:735–741. doi: 10.1038/nbt.1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.