

Supplemental Digital Content is Available in the Text.
Key Words: fostemsavir, temsavir, attachment inhibitor, resistance, phase 2b
Abstract
Background:
Fostemsavir is a prodrug of temsavir, an attachment inhibitor that binds to HIV-1 gp120, blocking viral attachment to host CD4+ T-cells. The phase 2b trial AI438011 investigated the safety, efficacy, and dose–response of fostemsavir vs ritonavir-boosted atazanavir (ATV/r) in treatment-experienced, HIV-1–infected subjects.
Methods:
Two hundred fifty-one treatment-experienced subjects with baseline (BL) susceptibility to study drugs [temsavir half-maximal inhibitory concentration (IC50) <100 nM, PhenoSense Entry assay] received fostemsavir or ATV/r, each with tenofovir disoproxil fumarate + raltegravir. Subjects meeting resistance-testing criteria were assessed for emergent viral drug resistance. Changes in temsavir IC50 from BL was given a conservative technical cutoff (>3-fold increase).
Results:
66/200 fostemsavir and 14/51 ATV/r subjects had resistance testing performed; 44/66 and 9/14 were successfully tested using the PhenoSense GT assay. No subjects had emergent tenofovir disoproxil fumarate or ATV resistance. Six fostemsavir-treated subjects developed emergent raltegravir resistance. 29/66 fostemsavir-treated subjects had an evaluable phenotype using PhenoSense Entry (which tests for viral susceptibility to temsavir) and 13/29 exhibited >3-fold increase in temsavir IC50 from BL. gp120 population sequencing was successful in 11/13 subjects and 7 had emergent substitutions in gp120 associated with reduced temsavir susceptibility (S375, M426, or M434). However, 5/13 fostemsavir-treated subjects achieved subsequent suppression to <50 copies/mL before the week 48 database lock, regardless of key gp120 substitutions.
Conclusions:
Response rates remained similar across study arms regardless of BL nucleoside reverse transcriptase inhibitor, nonnucleoside reverse transcriptase inhibitor, and protease inhibitor resistance-associated mutations. Emergent changes in viral susceptibility occurred more frequently with fostemsavir compared with ATV/r. However, the full impact of temsavir IC50 changes and emergent HIV-1 gp120 substitutions, and thus appropriate clinical cutoffs, requires further study. Fostemsavir is being evaluated in a phase 3 trial in heavily treatment-experienced subjects.
INTRODUCTION
Fostemsavir (formerly BMS-663068 or GSK3684934) is an oral prodrug of temsavir (formerly BMS-626529 or GSK2616713), a first-in-class HIV-1 attachment inhibitor that is currently being investigated in a phase 3 study in heavily treatment-experienced, HIV-1–infected patients with limited remaining treatment options. Temsavir binds directly to the viral envelope protein gp120, preventing initial viral attachment to CD4+ T cells.1 Binding is believed to occur within the structurally conserved outer domain of HIV-1 gp120, under the antiparallel β20–β21 sheet, and adjacent to the CD4 binding loop, which blocks formation of the 4-stranded bridging sheet and subsequent exposure and formation of the coreceptor binding site.1 Temsavir acts prior to the action of CCR5 antagonists and fusion inhibitors and is active against CCR5-, CXCR4- and dual-tropic (R5X4) strains of HIV-1.1–5
Findings from a previous phase 2a proof-of-concept study in treatment-naive and treatment-experienced patients showed that 8 days of fostemsavir monotherapy resulted in maximum median decreases in HIV-1 RNA from baseline (BL) of 1.21 to 1.73 log10 copies/mL.6 The majority (42/48) completing the study achieved a viral load decline of >1.0 log10 copies/mL, with the remaining 6 patients exhibiting a viral load decline of <1.0 log10 copies/mL (maximum change in HIV-1 RNA from BL 0.04 to −0.99 log10 copies/mL).6 Suboptimal efficacy was associated with a BL temsavir half-maximal inhibitory concentration (IC50) of >100 nM and the presence of the HIV-1 gp120 M426L substitution, although there was no strict correspondence with either parameter.4,7 Subsequent analyses also linked reduced viral susceptibility to BL substitutions at positions S375, M434, and M475.5 No emergent gp120 substitutions were observed using standard population genotypic and phenotypic approaches.4
Temsavir has a unique resistance profile and no in vitro cross-resistance has been observed with other classes of antiretrovirals, including nucleoside reverse transcriptase inhibitors (NRTIs), non-NRTIs (NNRTIs), protease inhibitors (PIs), integrase inhibitors, CCR5 antagonists, and fusion inhibitors (Bristol-Myers Squibb, unpublished data).2,3 Interestingly, considerable variability in viral susceptibility to temsavir has been observed in vitro,3,5 which may be linked to the significant diversity in HIV-1 gp120. A >6 log10 variation between viral isolates was reported, despite the majority having a temsavir IC50 of <10 nM,3 and a 2–3 log10 variation has been observed in envelope clones isolated from a single subject.5
AI438011 was a randomized, phase 2b, dose-finding study in HIV-1–infected, treatment-experienced subjects.8 A 7-day lead-in fostemsavir monotherapy substudy, with subjects receiving fostemsavir at doses of 400 mg twice daily (BID), 800 mg BID, 600 mg once daily (QD), and 1200 mg QD, showed median declines in HIV-1 RNA of 0.69–1.4 log10 copies/mL,8 and after 24 and 48 weeks of combination antiretroviral therapy (cART) using raltegravir (RAL) and tenofovir disoproxil fumarate (TDF), fostemsavir showed similar efficacy (proportion of subjects achieving <50 copies/mL) to an active ritonavir-boosted atazanavir (ATV/r) reference arm.8,9 The AI438011 study is in the process of closing, with participants receiving at least 4.2 years of study medication before study termination. In this study, we report a key secondary endpoint of treatment-emergent phenotypic and genotypic changes in viral drug susceptibility in AI438011 through 48 weeks of cART. In addition, we explore the effect of BL genotypic and phenotypic resistance on treatment response.
METHODS
Study Design and Participants
AI438011 was a phase 2b, multinational, randomized, active-controlled, partially blinded trial that has been previously described in detail.8 HIV-1–infected, treatment-experienced (defined as current or previous exposure to ≥1 week of ≥1 antiretroviral drug) adults were randomized 1:1:1:1:1 to one of 4 fostemsavir treatment arms (400 mg BID, 800 mg BID, 600 mg QD, or 1200 mg QD) and an active comparator arm (ATV/r 300/100 mg QD). After an initial elective 7-day fostemsavir monotherapy substudy (up to 10 subjects per study arm), subjects were treated with fostemsavir or ATV/r on a backbone of RAL 400 mg BID and TDF 300 mg QD for 48 weeks, with a primary analysis at 24 weeks and long-term follow-up (96 weeks).
Eligibility criteria included plasma HIV-1 RNA ≥1000 copies/mL, a CD4+ T-cell count >50 cells/mm3, and an HIV-1 genotype and phenotype indicating susceptibility to ATV, RAL, and TDF. Based on the results of the phase 2a study, a temsavir IC50 cutoff of <100 nM was also applied.
Protocol-Defined Virologic Failure
Plasma HIV-1 RNA levels were quantified as previously described.8 The criteria for protocol-defined virologic failure were a confirmed (second measurement within 2–4 weeks of original sample) plasma HIV-1 RNA measurement of ≥50 copies/mL at week 24 or later, or virologic rebound [confirmed HIV-1 RNA measurement of ≥50 copies/mL at any time after previous confirmed suppression to <50 copies/mL, or a confirmed increase in HIV-1 RNA of >1 log10 copies/mL above the nadir level (where the nadir was ≥50 copies/mL)].8
Resistance-Testing Criteria
Plasma samples for viral drug resistance testing were collected at each visit. Screening samples were used for analysis of BL genotypic resistance profiles. Resistance testing was uniformly performed at screening and subsequently conducted on patients who qualified for resistance testing in the event of protocol-defined virologic failure, or at a minimum in the event of confirmed plasma HIV-1 RNA ≥400 copies/mL at any time during the study (having previously achieved viral suppression to <50 copies/mL) or discontinuation before achieving viral suppression (to <50 copies/mL) after week 8 with a last plasma HIV-1 RNA measurement of ≥400 copies/mL.
Key substitutions conferring resistance to NRTI, NNRTI, PI, and integrase inhibitors were derived from the IAS-USA 2014 guidelines.10 Genotypic and phenotypic NRTI, NNRTI, PI, and integrase resistance were determined using the PhenoSense GT and Integrase (GeneSeq and PhenoSense) assays, respectively (Monogram Biosciences, LabCorp, South San Francisco, CA) and are defined in Supplemental Digital Content Table S1, http://links.lww.com/QAI/B94. Viral susceptibility to temsavir was determined using the PhenoSense Entry assay (Monogram Biosciences). Values were reported as fold change in IC50 (FC-IC50) compared with a reference virus used for normalization as previously reported.4 A technical cutoff (>3-fold increase) was used to assess potentially significant changes in temsavir FC-IC50 from BL because ≥95% of replicate measurements in the PhenoSense Entry assay are reported to be within 3-fold of each other.4
Population Sequencing of the Envelope Precursor HIV-1 gp160 env
Genotyping of gp160 in screening samples was performed retrospectively. Extraction of HIV-1 RNA from plasma samples, synthesis of first-strand cDNA, and polymerase chain reaction amplification of HIV-1 gp160 env were as previously described.5 In most cases, uncloned purified polymerase chain reaction products were used for population sequencing of gp160 env using a library of envelope-specific primers (Supplemental Digital Content Table S2, http://links.lww.com/QAI/B94).
HIV-1 env sequences were aligned to the HIV-1 subtype B consensus sequence available in the Los Alamos National Laboratories HIV sequence database (http://www.hiv.lanl.gov) using the AlignX software in the Vector NTI Advance package (version 11.5; Invitrogen, Carlsbad, CA). BL sequences were deposited in GenBank with the accession numbers MF990378 to MF990556. Amino acid positions were numbered per the HIV-1 HXB2 strain sequence. Amino acid substitutions in gp120 were visualized using GeneDoc software11 in difference display mode, and specific changes at gp120 positions S375, M434, M426, and M475 were assessed. If the nucleotide sequence had more than one possible base, all possibilities were expanded within the codon and amino acids were assigned as previously described.4 In addition, changes at positions L116 and A204, previously linked to reduced in vitro viral susceptibility to temsavir,5 were assessed. In the case of a novel polymorphism, the mutations were introduced into clinical isolates or the wild-type HIV-1 LAI strain by site-directed mutagenesis, and viral susceptibility to temsavir was assessed using a cell–cell fusion assay as described previously.5,12 All data were reported as FC in temsavir half-maximal effective concentration (EC50), normalized to an internal control.5
RESULTS
BL Characteristics and Genotypic Resistance Profile
A total of 581 subjects were screened, 254 were randomly assigned to treatment groups in the study, and 251 received treatment (200 subjects received fostemsavir).8 BL characteristics were generally well balanced between treatment arms.8 Most subjects had HIV-1 subtype B (65.7%) or C (19.9%); the remainder had HIV-1 subtype A (0.4%), A1 (4.0%), BF (0.4%), complex (6.8%), F1 (2.4%), or G (0.4%). The median BL viral load was 4.85 log10 copies/mL (43% ≥100,000 copies/mL), median CD4+ T-cell count was 229.5 cells/μL (38% <200 cells/μL), and median BL temsavir IC50 was 0.67 nM (range: 0.05–161 nM). One subject with a BL temsavir IC50 of 161 nM, which was higher than the IC50 cutoff of <100 nM specified in the entry criteria, was randomized to the study but achieved the primary efficacy endpoint (HIV-1 RNA <50 copies/mL at week 24) and remained on study through week 48.
BL genotypic resistance to PIs and NRTIs was described previously8 and is expanded in Supplemental Digital Content Table S3, http://links.lww.com/QAI/B94. Overall, 49% of subjects had ≥1 major PI, NRTI, or NNRTI mutation, and across all treatment arms, 22%–34% had ≥1 NRTI and NNRTI mutation at BL. The most common BL mutations (other than minor PI substitutions) were M184V (22%–40% of samples across all treatment arms), K103N (20%–39%), and thymidine analogue mutations (8%–16%). In line with study-entry criteria, no subject had virus with integrase resistance-associated mutations (RAMs) at BL. Three subjects had virus with the K70E substitution; however, this was not associated with reduced susceptibility to TDF.
Association of BL NRTI, NNRTI, and PI RAMs With Antiviral Response
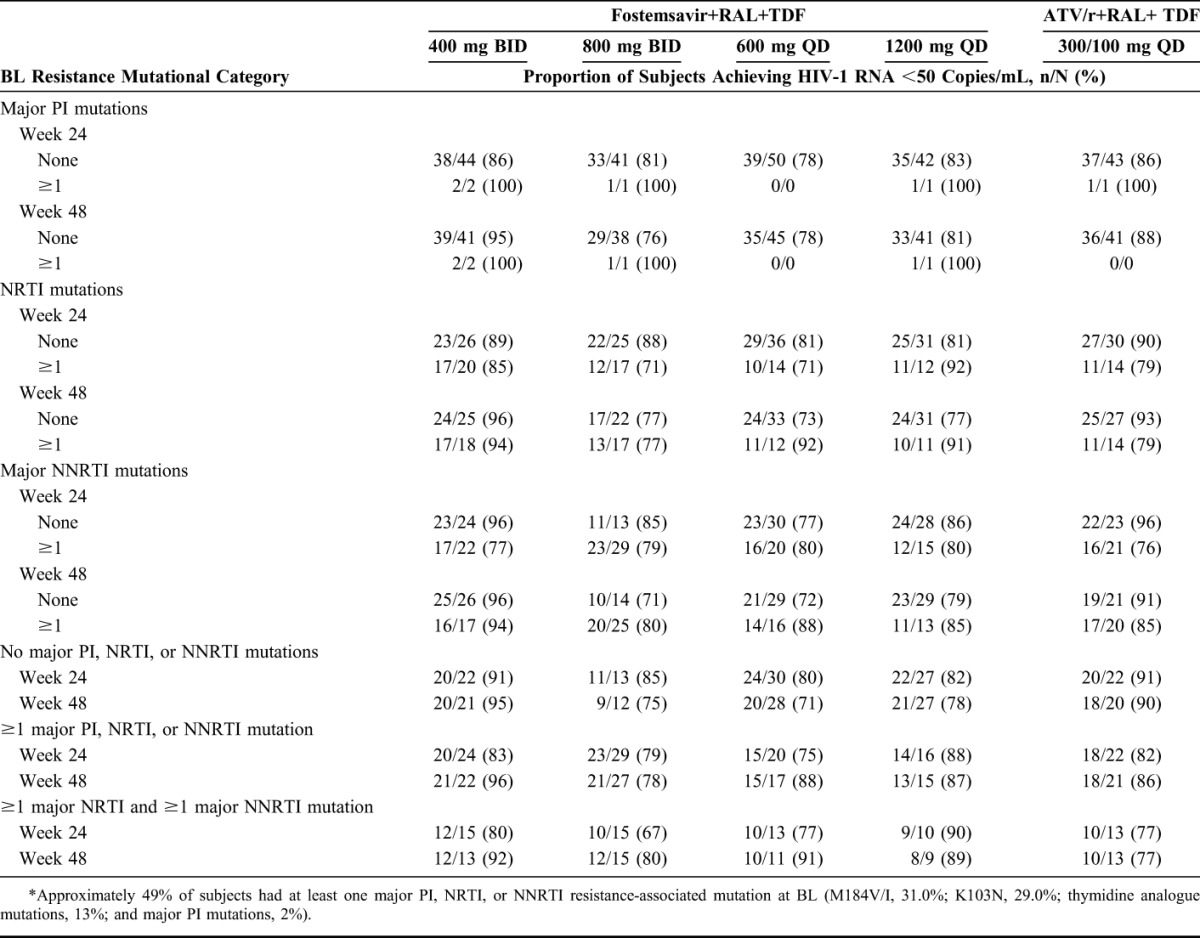
Through weeks 24 and 48, the proportion of subjects achieving HIV-1 RNA <50 copies/mL was similar across the fostemsavir and ATV/r arms, irrespective of BL NRTI, NNRTI, or major PI RAMs (Table 1).
TABLE 1.
Proportion of Subjects Achieving HIV-1 RNA <50 Copies/mL by BL PI, NRTI, and NNRTI Resistance Mutations Through Weeks 24 and 48 (Observed Analysis)*
Association of BL env Genotypes With Resistance Testing
To determine whether BL substitutions in HIV-1 gp120 at key positions S375, M426, M434, or M475, among others, were associated with the likelihood of subjects experiencing virologic failure, population sequencing of gp160 env was performed. Genotyping was successful in BL samples from 179/200 fostemsavir-treated subjects. Of these, 75/179 (42%) subjects had virus with a substitution at ≥1 of the 4 positions; the remainder had virus with no changes at these positions compared with the HXB2 reference sequence. Seventeen of 75 (23%) subjects had ≥2 polymorphisms at these positions.
Several novel polymorphisms were observed at positions M426 and M434; so, these substitutions were examined in a cell–cell fusion assay to determine their effect on viral susceptibility to temsavir in vitro. Each polymorphism was introduced into a functional LAI envelope clone by site-directed mutagenesis.5 Only the M426P substitution resulted in a >3-fold increase in viral susceptibility to temsavir compared with the wild-type LAI virus (Supplemental Digital Content Table 4, http://links.lww.com/QAI/B94).
There was no correlation between substitutions at gp120 positions S375, M426, M434, and M475 and the number of subjects qualifying for resistance testing through week 48, regardless of whether the substitutions were linked to a <3-fold or >3-fold reduction in viral susceptibility to temsavir (or a previous attachment inhibitor, BMS-488043) in this study or previous in vitro studies (Fig. 1).5,13
FIGURE 1.

Association of BL substitutions in HIV-1 gp120 at positions S375, M434, M426, and M475, with the number of subjects undergoing resistance testing through week 48 (N = 179). (Left) Susceptibility to temsavir was grouped by BL sequence into subjects without polymorphisms in any of the 4 targeted amino acids (None), subjects with a polymorphism in 1 or more of the 4 targeted amino acids that results in an FC > 3 (includes 4 subjects with 2 polymorphisms; Any FC > 3) and subjects with a polymorphism in 1 of the 4 targeted amino acids that results in a FC < 3; (Any FC ≤ 3). The blue line signifies the number of subjects who met criteria for resistance testing and the orange line signifies the number of subjects who did not meet criteria for resistance testing. Actual numbers with percentages of subjects meeting criteria for resistance testing in each group are shown to the right. Only subjects who had virus with a determinable genotype at gp120 positions 375, 426, 434, and 475 were included in the analysis. (Right) The Any FC > 3 cohort was broken out by individual polymorphisms and blue and orange lines were as before. This analysis includes 17 subjects who had virus with multiple polymorphisms. Ranges of observed temsavir susceptibility in each cohort are noted in both figures.
Analysis of Treatment-Emergent Resistance Through Week 48
Through week 48, 66/200 (33%) fostemsavir-treated subjects and 14/51 (28%) ATV/r-treated subjects met criteria for resistance testing. Of these, 37/66 fostemsavir-treated and 9/14 ATV/r-treated subjects had available genotype/phenotype data using the Integrase assay, 44/66 fostemsavir-treated and 9/14 ATV/r-treated subjects had available genotype/phenotype data using PhenoSense GT assay, and 29/66 fostemsavir-treated subjects had a successful PhenoSense Entry assay measurement (Table 2). Common causes for assay failure listed by the manufacturer included low viral load (HIV-1 RNA <500 copies/mL) and primer incompatibility.
TABLE 2.
Treatment-Emergent Genotypic and Phenotypic Resistance Profile Through Week 48
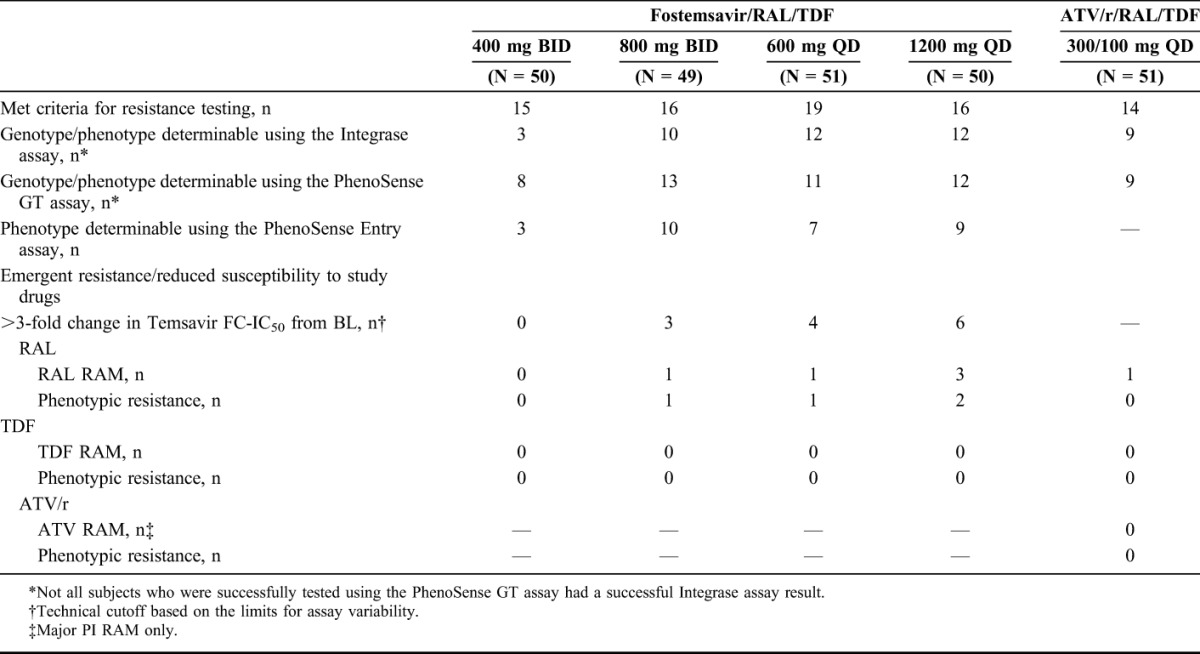
Through week 48, no emergent TDF resistance was detected in any study arm and no subjects developed phenotypic resistance to any study drug in the ATV/r arm. Four subjects across the fostemsavir arms developed genotypic and phenotypic resistance to RAL and 1 fostemsavir-treated and 1 ATV/r-treated subject had emergent RAL RAM but did not develop phenotypic resistance (<1.5-fold change in FC-IC50). Across the fostemsavir arms, 13/29 subjects with an evaluable result in the PhenoSense Entry assay had virus with a >3-fold increase in temsavir FC-IC50 from BL.
Previous participation in the 7-day monotherapy substudy did not seem to affect the number of subjects who met resistance-testing criteria. Overall, 10/32 (31%) monotherapy participants met resistance-testing criteria through week 48. This is compared with 56/168 (33.3%) subjects who did not receive monotherapy who met resistance-testing criteria. Of the 10 monotherapy participants who met minimum resistance-testing criteria, 2 had virus with a >3-fold increase in temsavir FC-IC50 from BL, in line with the overall fostemsavir-treated population.
Analysis of Emergent Substitutions in HIV-1 gp120 Through Week 48
Population sequencing of gp160 env was performed for viral samples obtained from the 66 fostemsavir-treated subjects who met resistance-testing criteria. Given the considerable heterogeneity observed within HIV-1 env between individuals and the heterogeneity observed in BL susceptibility to temsavir, this analysis concentrated on emergent changes at gp120 positions L116, A204, S375, M434, M426, and M475 compared with the HXB2 reference strain. These amino acids were previously shown to be important for temasvir susceptibility.5
Population sequencing was successful for 11/13 fostemsavir-treated subjects who had virus with a >3-fold increase in temsavir FC-IC50 from BL (Table 3). Of these, 7 had virus with emergent changes at ≥1 of the 4 key gp120 positions associated with reduced susceptibility to temsavir (S375, M426, M434, and M475) in the proof-of-concept study.4,5 The most common changes occurred at position M426; 5 subjects had virus with an emergent M426L substitution (2 of these had a mixed M426M/L genotype). In addition, 2 subjects with wild-type S375 at baseline had emergent S375S/N substitutions, while 1 had virus with emergent substitutions at M434 (M434K). However, a consensus amino acid could not be determined for all gp120 positions because of variability in env.
TABLE 3.
Emergent Substitutions in HIV-1 gp120 for Subjects With a >3-fold Increase in Temsavir FC-IC50 From BL Through Week 48*
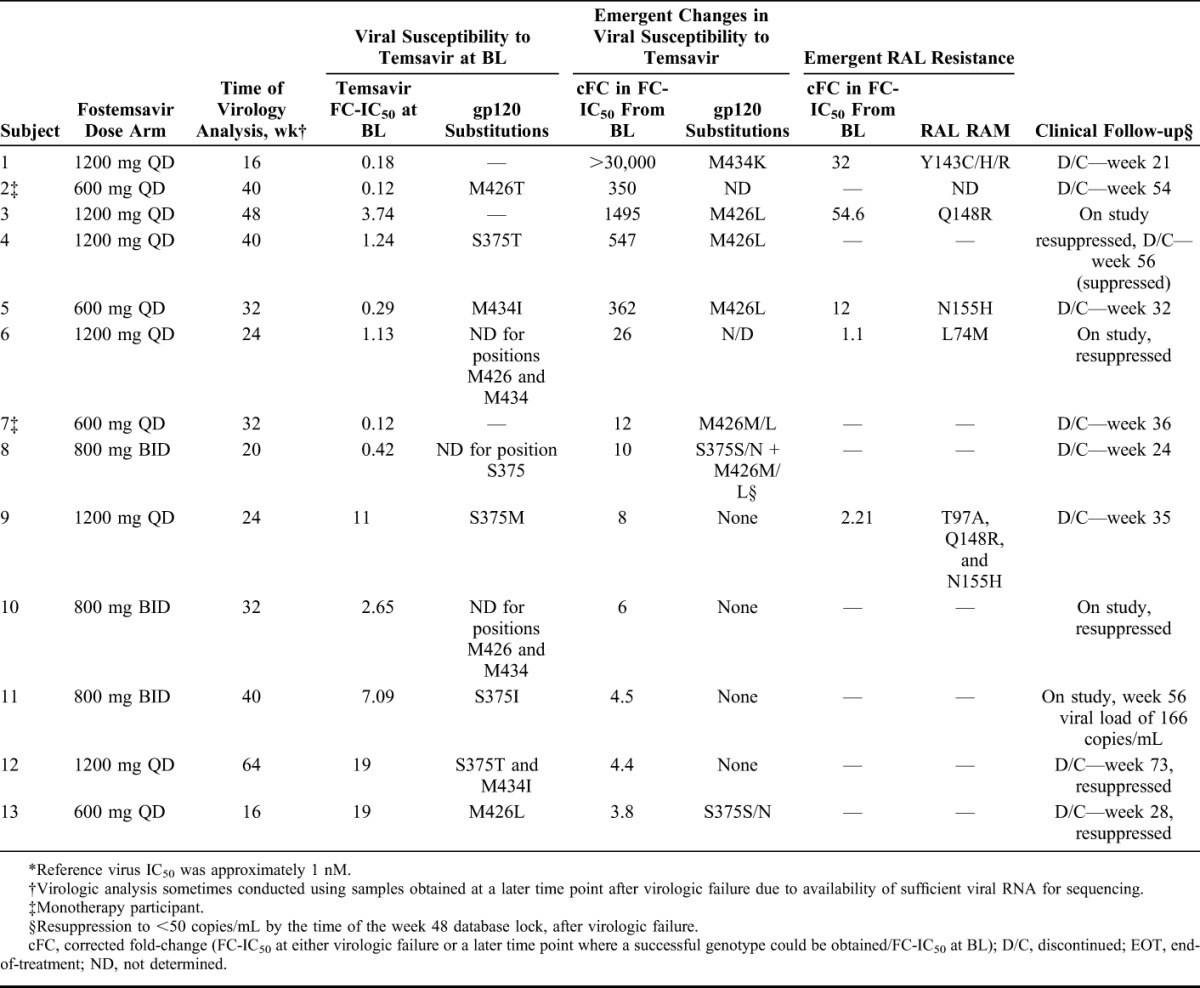
Because the M434K substitution has not been observed previously, it was analyzed further. A single functional gp120 viral envelope clone was isolated from subject 1 and site-directed mutagenesis was used to create a clone containing the HXB2 consensus amino acid (M434M). After analysis in a cell–cell fusion assay, the clone containing M434K showed a large decrease in susceptibility to temsavir (EC50 >20,000) relative to the clone with M434M (EC50 210 nM).
There was a wide variation in temsavir susceptibility among the 13 fostemsavir-treated subjects who had virus with a >3-fold increase in temsavir FC-IC50 from BL (range: 3.8-fold increase with S375S/N to >30,000-fold increase with M434I; Table 3). Variability was observed regardless of substitutions at positions S375, M434, and M426.
Among the same 13 subjects, 9 discontinued from the study and 4 remained on study at the week 48 database lock. Five of the 13 subjects, who all had initial suppression to <50 copies/mL before week 24, qualified for resistance testing but achieved resuppression to <50 copies/mL while on study by the week 48 database lock (Table 3), despite a 547-fold increase in temsavir FC-IC50 from BL (subject 4) and the presence of the S375S/N substitution (subject 13; Table 3). All 5 subjects retained susceptibility to the optimized backbone therapy.
The remaining 53/66 fostemsavir-treated subjects, who met resistance-testing criteria, had virus either with a <3-fold increase in temsavir FC-IC50 from BL, or a nondeterminable temsavir susceptibility phenotype (data not shown). Seventeen of 53 subjects had successfully genotyped samples. Emergent changes were detected in 1/17 subject—a S375T substitution.
DISCUSSION
Fostemsavir showed similar efficacy to ATV/r when combined with RAL and TDF in treatment-experienced, HIV-1–infected subjects, regardless of BL viral load (<100,000 or ≥100,000 copies/mL) or temsavir IC50 category (<0.1 or ≥0.1 nM, <1 or ≥1 nM, and <10 or ≥10 nM; an IC50 cutoff of <100 nM was required for enrollment in the study).8,9 We have shown that the proportion of subjects achieving HIV-1 RNA virologic suppression through 24 and 48 weeks of cART remained similar across the fostemsavir and ATV/r arms regardless of the presence of NRTI, NNRTI, or PI RAMs at BL. This was expected, given the lack of cross-resistance between temsavir and other classes of antiretrovirals in vitro,2,3 and reported activity against a range of NRTI, NNRTI, PI, and integrase inhibitor–resistant isolates (Bristol-Myers Squibb, unpublished data).3
Substitutions at positions S375, M434, M426, and M475 in HIV-1 gp120 have been associated with reduced viral susceptibility to temsavir.4,5 These positions map in and around the modeled gp120 binding site for temsavir, adjacent to the region for CD4 binding.1 Interestingly, although BL substitutions at positions S375, M434, M426, and M475 were present in viral samples obtained from 75/179 evaluable subjects in this study, no correlation was found between these substitutions and the number of subjects who qualified for resistance testing. This suggests that despite the high variability in susceptibility to temsavir in this population (FC-IC50 range of 3.8-fold increase with S375S/N to >30,000-fold increase with M434I), virologic failure was not dependent on BL IC50 as long as subjects retained some susceptibility to temsavir. These findings are similar to those in the phase 2a study (AI438001), where although the M426L substitution was linked to nonresponse (decline in HIV-1 RNA of <1.0 log10 copies/mL) after 8 days of fostemsavir monotherapy, 2 subjects who had virus with this BL substitution still achieved a decline of >1.0 log10 copies/mL,4,5 suggesting that these substitutions may not fully predict antiviral response.
Through week 48, a similar proportion of fostemsavir-treated (33%) and ATV/r-treated (27.5%) subjects met resistance-testing criteria. Emergent changes in viral drug susceptibility were more frequent in the fostemsavir arms: 4/37 evaluable subjects had emergent RAL resistance and 13/29 evaluable subjects had virus with a >3-fold increase in temsavir FC-IC50 from BL. By contrast, no emergent resistance to study drugs was observed in the ATV/r arm. This may be due in part to the higher genetic barrier to resistance for ATV/r, a boosted PI, which may confer better backbone protection, and the relatively lower barrier to resistance for RAL, where a single mutation (such as N155H, Q148R, and Y143R) can result in a >10-fold reduction in viral susceptibility.14 Previous participation in the 7-day fostemsavir monotherapy substudy did not influence the likelihood of subjects who met criteria for resistance testing, suggesting that fostemsavir monotherapy did not compromise its subsequent use as part of cART.
Among the 13 subjects who had virus with a >3-fold increase in temsavir FC-IC50 from BL, a wide variation in temsavir FC-IC50 from BL was observed (3.8 to >30,000-fold increase in temsavir FC-IC50). This is in line with in vitro data, which showed a considerable range in susceptibility of viral isolates to temsavir (>6 log10 variation), despite the majority having a temsavir half-maximal effective concentration of <10 nM.3 The reasons for this variability are not known, but may be linked to the considerable diversity found in HIV-1 gp120.15
Seven of 11 evaluable subjects with a >3-fold increase in temsavir FC-IC50 had emergent substitutions in HIV-1 gp120 at positions 375, 426, and 434. Treatment-emergent substitutions in this study were not observed at amino acid position 475, or at positions 116 or 204. Substitutions at positions 116 or 204 have only been observed thus far during in vitro selection experiments. Overall, viruses with emergent gp120 substitutions tended to have larger increases in temsavir FC-IC50 from BL compared with those who had virus with no changes at positions 375, 426, 434, and 475. M426L was the most common emergent substitution, and substitutions at positions S375 and M434 were also observed, including a novel M434K substitution that resulted in a large decline in susceptibility to temsavir in vitro. S375 and M426 lie in the modeled binding site for temsavir and substitutions at these positions may reduce binding of temsavir to gp120.1 M434 lies distal to the temsavir binding site, but is involved in bridging sheet packing and may shift gp120 away from an effective temsavir binding conformation.1
Among 13 subjects who had virus with a >3-fold increase in temsavir FC-IC50 from BL, 5 subsequently achieved viral resuppression on the same regimen. Trends suggested that viral resuppression may have been related to a smaller corrected FC in temsavir FC-IC50 from BL, absence of gp120 substitutions at positions S375, M426, and M434, and absence of emergent RAL resistance. However, 2/5 subjects achieved viral resuppression despite the presence of a gp120 M426L or S375S/N substitution, and 1 subject despite a 547-fold increase in temsavir FC-IC50. Similar results were observed in the proof-of-concept study, where 2 subjects who had virus with an M426L substitution at BL, including 1 subject with a very high BL temsavir IC50 (∼5300 nM) still achieved a decline in HIV-1 RNA of >1 log10 copies/mL after 8 days of monotherapy.4,5 Therefore, subjects may still respond to therapy despite BL or emergent gp120 substitutions. Because their effect seems to be context dependent, it is possible that any associated increase in temsavir FC-IC50 may only be relevant if it takes the overall temsavir IC50 value over a certain threshold.
The >3-fold cutoff for analysis of emergent changes in viral susceptibility to temsavir used in this study was a technical cutoff, based on the limits for assay variability. At present, there is no clinical cutoff for changes in viral susceptibility to temsavir and, in this study, 5 subjects with a 4–547-fold increase in temsavir FC-IC50 from BL still achieved viral resuppression after virologic failure. Furthermore, the 100-nM BL temsavir IC50 required for entry in this study was based on the results from the phase 2a study, and within the confines of this limit, no difference in response was seen after 24 or 48 weeks of cART with RAL and TDF when subjects were stratified by temsavir IC50 category.8 Further analysis will be required to determine an appropriate clinical cutoff for fostemsavir because the technical cutoff >3 does not seem to correlate with clinical response in this study.
This study had some limitations. The BL genotypic resistance profile for the study population was more in line with first-line or second-line virologic failure; so, the activity of fostemsavir in treatment-experienced populations with a range of different BL genotypic resistance profiles remains to be confirmed. In addition, the study was restricted to participants with a BL temsavir IC50 <100 nM. Because no clinical cutoff is currently available, the impact of higher BL temsavir IC50 values on antiviral activity and changes in viral susceptibility requires further evaluation. Amino acids could not be determined for all gp120 positions, mainly due to insertions and deletions in HIV-1 gp120; so, changes at the 4 key positions could not be determined for all samples. However, there was no increase in the proportion of sequence failures in the virologic failure samples compared with BL sequences. Finally, not all subjects who were successfully tested using the PhenoSense GT assay had a successful Integrase assay result.
In conclusion, we have shown that response rates remained similar across the fostemsavir and ATV/r arms through weeks 24 and 48, regardless of NRTI, NNRTI, and PI RAM at BL. Emergent changes in viral susceptibility occurred more frequently in the fostemsavir arms compared with the ATV/r arm; however, of the 13/200 fostemsavir-treated subjects who had virus with a >3-fold increase in temsavir FC-IC50 from BL, 5 achieved resuppression to <50 copies/mL during the study, regardless of S375S/N and M426L substitutions. The impact of changes in temsavir IC50 and emergent substitutions in HIV-1 gp120 will require more evaluation, and thus, an appropriate clinical cutoff requires further study. Because the greatest unmet medical need lies with patients who have few remaining treatment options, fostemsavir is being evaluated in a phase 3 trial in heavily treatment-experienced subjects who have limited options; this phase 3 trial does not include a cutoff for BL temsavir IC50. A retrospective analysis of the effect of BL susceptibility to temsavir will be conducted to potentially elucidate an appropriate clinical cutoff.
Supplementary Material
ACKNOWLEDGMENTS
The authors thank the AI438011 participants and their families, and the AI438011 investigators: JD Altclas, G Amaya-Tapia, JF Andrade-Villanueva, V Arama, AI Arango-Duque, K Arastéh, J Arribas, R Bardinas-Rodriguez, CM Benites-Villafane, J Bogner, C Brinson, PE Cahn, WM Casapia-Morales, LI Cassetti, B Clotet, E DeJesus, IG Diaconescu, JI Echevarría, R Elion, J Ernst, J Feinberg, J Fourie, J Galindez, JM Gatell, ER Granados-Reyes, S Hassler, C Hicks, D Johnson, R Kaplan, J Lalezari, JR Lama-Valdivia, AM La Rosa Rodriguez, GH Latiff, MY Leon, SH Lupo, F Marquez-Diaz, MD Martins, M Mckellar, FC Mendo-Urbina, Y Pinedo-Ramirez, LJ Prisacariu, J Rockstroh, S Rugina, MR Salazar-Castro, AR Scribner, J G Sierra-Madero, LM Sloan, A Stoehr, OA Sussmann-Pena, P Tebas, M Thompson, SC Treviño-Pérez, OA Tsybakova, EE Voronin, AA Yakovlev, and NG Zakharova. In addition, the authors thank the following people at Bristol-Myers Squibb: John Coumbis, Neelanjana Ray, Nancy Cusack, Carey Hwang, Todd Correll, and also Peter Lill and John Riefler at ICON CRO for their assistance with the study. ViiV Healthcare has acquired fostemsavir. During the conduct of this study, authors were affiliated with Bristol-Myers Squibb. Professional medical writing and editorial assistance were provided by Anna Shirazi and Sharmin Bovill, and funded by Bristol-Myers Squibb and ViiV Healthcare.
Footnotes
This study was funded by Bristol-Myers Squibb.
Presented in part at the International Workshop on Antiretroviral Drug Resistance; June 3–7, 2014; Berlin, Germany and at the 8th IAS Conference on HIV Pathogenesis, Treatment and Prevention; July 19–22, 2015; Vancouver, Canada.
M.L., S.R.J., S.L., D.A.S., G.J.H., and M.K. were employees and held stock or stock options at Bristol-Myers Squibb during the conduct of the study. M.L., S.R.J., and M.K. are current employees at ViiV Healthcare and hold stock or stock options at GSK. N.Z. is an employee and holds stock or stock options at Bristol-Myers Squibb. G.J.H. is a current employee at Merck Research Laboratories.
Supplemental digital content is available for this article. Direct URL citations appear in the printed text and are provided in the HTML and PDF versions of this article on the journal's Web site (www.jaids.com).
REFERENCES
- 1.Langley DR, Kimura SR, Sivaprakasam P, et al. Homology models of the attachment inhibitor BMS-626529 bound to gp120 suggest a unique mechanism of action. Proteins. 2015;83:331–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Li Z, Zhou N, Sun Y, et al. Activity of the HIV-1 attachment inhibitor BMS-626529, the active component of the prodrug BMS-663068, against CD4-independent viruses and HIV-1 envelopes resistant to other entry inhibitors. Antimicrob Agents Chemother. 2013;57:4172–4180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nowicka-Sans B, Gong YF, McAuliffe B, et al. In vitro antiviral characteristics of HIV-1 attachment inhibitor BMS-626529, the active component of the prodrug BMS-663068. Antimicrob Agents Chemother. 2012;56:3498–3507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ray N, Hwang C, Healy MD, et al. Prediction of virological response and assessment of resistance emergence to the HIV-1 attachment inhibitor BMS-626529 during 8-day monotherapy with its prodrug BMS-663068. J Acquir Immune Defic Syndr. 2013;64:7–15. [DOI] [PubMed] [Google Scholar]
- 5.Zhou N, Nowicka-Sans B, McAuliffe B, et al. Genotypic correlates of susceptibility to HIV-1 attachment inhibitor BMS-626529, the active agent of the prodrug BMS-663068. J Antimicrob Chemother. 2014;69:573–581. [DOI] [PubMed] [Google Scholar]
- 6.Nettles R, Schurmann D, Zhu L, et al. Pharmacodynamics, safety, and pharmacokinetics of BMS-663068, an oral HIV-1 attachment inhibitor in HIV-1-infected subjects. J Infect Dis. 2012;206:1002–1011. [DOI] [PubMed] [Google Scholar]
- 7.Zhou N, Ray N, Healy M, et al. Genotypic and phenotypic correlates of virologic response to the attachment inhibitor BMS-626529 in a short-term monotherapy study with its prodrug BMS-663068. Poster presented at: International Workshop on HIV & Hepatitis Virus Drug Resistance; June 5–9, 2012; Sitges, Spain. Poster presentation 6.
- 8.Lalezari JP, Latiff GH, Brinson C, et al. Safety and efficacy of the HIV-1 attachment inhibitor prodrug BMS-663068 in treatment-experienced individuals: 24 week results of AI438011, a phase 2b, randomised controlled trial. Lancet HIV. 2015;2:e427–e437. [DOI] [PubMed] [Google Scholar]
- 9.Thompson M, Lalezari J, Kaplan R, et al. Safety and efficacy of the HIV-1 attachment inhibitor prodrug fostemsavir in antiretroviral-experienced subjects: week 48 analysis of AI438011, a phase IIb, randomized controlled trial. Antivir Ther. 2017;22:215–223. [DOI] [PubMed] [Google Scholar]
- 10.Wensing AM, Calvez V, Gunthard HF, et al. 2014 update of the drug resistance mutations in HIV-1. Top Antivir Med. 2014;22:642–650. [PMC free article] [PubMed] [Google Scholar]
- 11.Nicholas KB, Nicholas HBJ, Deerfield DWI. GeneDoc: analysis and visualization of genetic variation. Embnet News. 1997;4:1–4. [Google Scholar]
- 12.Lin PF, Blair W, Wang T, et al. A small molecule HIV-1 inhibitor that targets the HIV-1 envelope and inhibits CD4 receptor binding. Proc Natl Acad Sci U S A. 2003;100:11013–11018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhou N, Nowicka-Sans B, Zhang S, et al. In vivo patterns of resistance to the HIV attachment inhibitor BMS-488043. Antimicrob Agents Chemother. 2011;55:729–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tang MW, Shafer RW. HIV-1 antiretroviral resistance: scientific principles and clinical applications. Drugs. 2012;72:e1–e25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brower ET, Schon A, Freire E. Naturally occurring variability in the envelope glycoprotein of HIV-1 and development of cell entry inhibitors. Biochemistry. 2010;49:2359–2367. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.