

Abstract
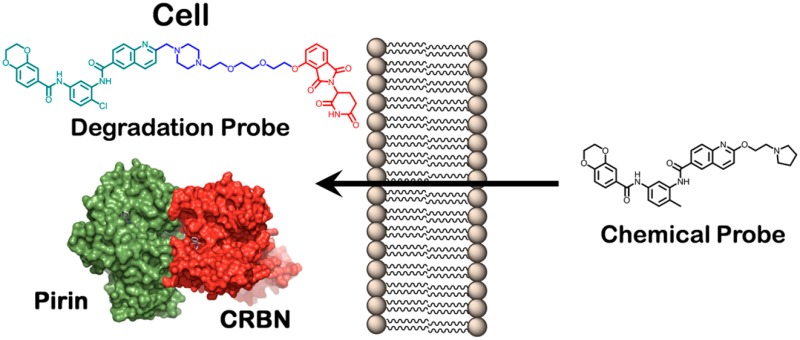
Demonstrating intracellular protein target engagement is an essential step in the development and progression of new chemical probes and potential small molecule therapeutics. However, this can be particularly challenging for poorly studied and noncatalytic proteins, as robust proximal biomarkers are rarely known. To confirm that our recently discovered chemical probe 1 (CCT251236) binds the putative transcription factor regulator pirin in living cells, we developed a heterobifunctional protein degradation probe. Focusing on linker design and physicochemical properties, we generated a highly active probe 16 (CCT367766) in only three iterations, validating our efficient strategy for degradation probe design against nonvalidated protein targets.
Introduction
Drug discovery is reliant on recombinant proteins and biochemical screens to develop structure–activity relationships (SAR) and progress compounds.1 However, the conditions in biochemical assays often display little relevance to the intracellular environment, which can result in a failure to translate high target affinity to activity within a living cell.2 Target-proximal biomarker modulation is the most important confirmation of intracellular target engagement.3 Unfortunately, this is often not possible in early stage chemical probe or drug discovery projects, especially against novel biological targets, where poorly understood biology and pharmacology make it difficult to discover and robustly validate biomarkers, a process that is particularly challenging for noncatalytic proteins.
Owing to the importance of confirming intracellular target engagement, several techniques have been developed. The overexpression of fusion proteins allows for a direct readout of target occupancy.4 However, the engineered cell lines are often difficult to generate and the protein label can impact compound binding. The cellular thermal shift assay (CETSA) is a label-free technique for intracellular target engagement.5 It exploits compound-induced stabilization of protein melting temperatures, but CETSA cannot be applied to all targets.6 Activity-based protein profiling (ABPP) methods utilize nonselective irreversible covalent ligands, or intracellular fluorescence polarization probes, combined with intracellular reversible ligand competition, to study target engagement.7 Proteolysis targeting chimeras (PROTACs)8,15 and specific and nongenetic IAP-dependent protein erasers (SNIPERs)9 are heterobifunctional molecules that induce rapid and selective protein degradation, via the proteasome, within living cells. One portion of the molecule engages the target protein, while the other, attached via a flexible linker, recruits an E3 ligase to ubiquitinate the target, marking it for degradation as part of the cullin–RING finger machinery.10,15
We recently reported the development of a high affinity pirin chemical probe 1 (CCT251236, SPR KD = 44 nM) discovered through a cell-based phenotypic screen for inhibitors of the heat shock transcription factor 1 (HSF1) stress pathway.11 Pirin is an iron-binding member of the cupin super family of proteins and has been reported as a putative transcription factor regulator.12,13 It has no known enzymatic function in mammalian cells, no endogenous ligands have been reported, and no validated proximal biomarkers have been described.14 This makes demonstrating intracellular target engagement in living cells very challenging.
We hypothesized that we could demonstrate chemical probe 1 binding to pirin within living cells by developing a pirin-targeting protein degradation probe (PDP).
Results and Discussion
Protein Degradation Probe Design
PDPs have been described against various proteins,15 with the bromodomain epigenetic target, BRD4, the most extensively studied.16 These heterobifunctional PDP molecules have utilized several ligands that bind to the E3 ligases: VHL,17 the IAP proteins,18 and CRBN.19 Despite the rapid expansion in PDP research, there remains no clear methodology to determine which proteins are amenable to PDP-mediated degradation,20 which E3 ligase ligand should be exploited or the optimal features of probe design. The structure of the linker, its length, and physicochemical properties have all been demonstrated to be important for PDP activity.21 The linker controls the formation of the essential ternary complex,22 with evidence that it may stabilize the protein–protein interaction (PPI) between the target and the E3 ligase rather than forming a detached linear ternary complex (Figure 1), although it is unclear whether the ternary complex is part of the multiprotein cullin–RING finger complex.23−25
Figure 1.

(top) Pirin (5JCT)/CRBN (5CI1)/PDP ternary complex design model. The PDP can either stabilize a PPI or simply bring the proteins in close proximity depending on the role of the linker. (bottom left) Cartoon representation of the chemical probe 1 (yellow) bound to recombinant pirin (5JCT). The cloud represents the shape of the binding pocket with key residues shown in black and the metal in orange. Red = oxygen, blue = nitrogen. Hydrogens and solvent are omitted for clarity except the water coordinated to the metal, shown as a red sphere. Both representations were generated using the PyMOL Molecular Graphics System, version 1.8; Schrödinger, LLC. (bottom right) Key residues in the binding site and the clear solvent exposed vector for the chemical probe 1 binding to pirin are shown adapted from an analysis using MOE 2014.09. The ethyl pyrrolidine solubilizing group of chemical probe 1 was not resolved in the crystal structure and therefore is not drawn in the analysis.
Although we had discovered a high affinity ligand for pirin, there was no evidence that this protein would be a suitable substrate for ubiquitination by a PDP-recruited E3 ligase, without which extensive linker optimization could be futile. Therefore, we initially designed a synthetically tractable 15-atom linker that we predicted would not affect the affinity of the PDP for the isolated target proteins.26 Analysis of the crystal structure of the chemical probe 1 bound to pirin (Figure 1, PDB 5JCT)11 suggested that the solvent-exposed solubilizing group vector should be amenable to linker attachment. We selected a CRBN-targeting thalidomide ligand as the basis of our E3 ligase binding motif due to its low molecular weight. The CRBN-targeting ligand would be attached via the solvent exposed hydroxyl group of the 4-hydroxythalidomide analogue 2.27 The linker would then attach the pirin- and CRBN-targeting specific binding groups via two amide moieties to give our first generation pirin-targeting PDP 3 (Scheme 1).
Scheme 1. Design and Synthesis of the First Generation PDP 3 Based on the Chemical Probe 1.

Reagents and conditions: (a) (i) PPh3, tbutyl 2-hydroxyacetate, DTBAD, THF, 0 °C → RT, 16 h, 75%, (ii) HCO2H, DCM, 40 °C, 16 h, 54%, (iii) HATU, DIPEA, DMF, tbutyl (2-(2-aminoethoxy)ethyl)carbamate, RT, 16 h, 81%, (iv) 4 M HCl in dioxane, 0.5 h, 100%; (b) ethyl 4-bromobutanoate, K2CO3, DMF, RT, 16 h, 37%; (c) Herrmann’s palladacycle,29tBu3PHBF4, MoCO6, DBU, 2-(trimethylsilyl)ethan-1-ol, 130 °C, 62%; (d) TBAF, THF, RT, 16 h; (e) (i) HATU, DIPEA, DMF, RT, 38% (over 2 steps), (ii) LiOH·H2O, MeOH/THF/H2O, RT, 48 h, 18%; (f) HATU, DIPEA, DMF, 74%. For the synthesis of hydroxythalidomide 2, see ref (28).
The CRBN-targeting motif 4 of PDP 3 was synthesized from 4-hydroxythalidomide 2 in two steps and 84% yield, in a similar manner to that previously described.28 The pirin-binding motif of the PDP 3 was synthesized from 6-bromoquinoline 5 via a palladium-mediated carbonylation reaction on the ether derivative 6. Trapping the carbonylation intermediate with 2-(trimethylsilyl)ethan-1-ol gave the (trimethylsilyl)ethyl ester 7,29 which facilitated TBAF-selective hydrolysis. Amide coupling to the previously described bis-aniline derivative 8(11) was followed by hydrolysis of the aliphatic linker ester to give acid 9. Final amide coupling to the CRBN-targeting derivative 4 gave the first-generation pirin-targeting PDP 3 in 10 steps and 0.4% overall yield.
The First Generation PDP
Analysis of the heterobifunctional PDP 3 revealed that it possessed good affinity for recombinant pirin (Table 1, entry 1) when measured using SPR, confirming the success of the rationally designed attachment vector.30 The affinity of PDP 3 was then assessed against the CRBN-DDB1 complex, with DDB1 acting as a scaffolding protein, using an FP-assay similar to that previously described (Supporting Information, Figure S6).31 PDP 3 displayed moderate affinity for CRBN-DDB1, with Ki = 230 nM,32 comparable to the affinities of the parent CRBN ligands, thalidomide and lenalidomide (Supporting Information, Figure S7).
Table 1. Physicochemical Properties and Affinities for Recombinant Protein Targets of the Three Generations of PDPsI.


HBD = hydrogen bond donor count.
ALogP was calculated using Biovia Pipeline Pilot, version 9.5, 2 SF.
Log D7.4 measured using a HPLC-based method, n = 1, 2 SF.
tPSA was calculated using ChemDraw (16.0.1.4) based on the O- and N-count, 3 SF.
KS = kinetic solubility in pH 7.4 phosphate buffer at room temperature, n = 1, 1 SF.
KD values are reported to 2 SF and are calculated by equilibrium analysis using a one site specific binding model from SPR sensorgrams at equilibrium where possible, pKD = −log(KD (M) × 10–9) and represents the geometric mean of n = 3 independent biological repeats.
IC50 values are reported to 2 SF and are calculated from an FP-assay dose–response curve to displace a thalidomide derived fluorescent probe using a log[inhibitor] vs response – variable slope (four parameters) model, pIC50 = −log(IC50 (M) × 10–9) and represents the geometric mean of n = 3 independent biological repeats, also see ref (31).
Ki values are calculated from the geometric mean CRBN-DDB1 complex IC50 and the FP-probe KD using methods described in ref (32).
SF = significant figure. All data was reprocessed using GraphPad Prism 7.01. See Supporting Information, Figures S8–S16. SEM = standard error of the mean.
Following confirmation that both binding motifs of the PDP 3 retained high affinity for their respective targets, we then investigated its activity against pirin in human cancer cell lines. Several cell lines were assessed for CRBN expression by quantitative capillary electrophoresis (Supporting Information, Figure S17).33 The SK-OV-3 ovarian carcinoma cell line34 displayed good basal CRBN and also pirin expression, and there was no observable depletion of pirin in these cells when treated with chemical probe 1 (1 μM, data not shown), so this line was selected for further study. Unfortunately, treatment of SK-OV-3 cells with PDP 3, at high concentrations (>1 μM) and for extended time periods (>48 h), resulted in no measurable effects on the cancer cells (data not shown).
The Second Generation PDP
We speculated several causes for the failure of the first generation PDP 3. Pirin may simply be incompatible with this methodology, and if so, no further improvements could be made.35 Alternatively, CRBN could be the wrong E3 ligase target to deplete pirin,36 or the linker length could be inconsistent with formation of the ternary complex.26 Finally, the physicochemical properties of PDP 3 could be limiting its intracellular free concentration.37 Given these variables, designing a PDP against a protein target that has not previously been validated as being susceptible to E3 ligase-directed degradation is challenging, as optimization cycles can appear lengthy and the difficult and low yielding synthesis discourages the generation of multiple analogues. It was also unclear how strict and narrow the requirements for optimal PDP design would be. We hypothesized that the physicochemical properties of the PDP were the primary cause of the failure of the first generation probe, and so began a redesign process that should increase cell membrane flux while maintaining the same linker length and CRBN-targeting ligand (Table 1). By carrying out multiple changes to the PDP, we aimed to minimize the number of iterative design cycles, so we could more rapidly validate this approach.
The design of heterobifunctional molecules unavoidably results in high molecular weight compounds, making it difficult to balance their physicochemical properties in a manner consistent with acceptable permeability and solubility.38 In the case of the first generation pirin-targeting PDP 3, although the Log D7.4 was acceptable (Table 1, entry 1),39 the linker had introduced two new hydrogen bond donors (HBD), which can have a negative impact on permeability. The calculated tPSA was very high (258 Å2), although it is inevitable that tPSA will be high for large molecules and outside the standard cut-offs for cell permeability unless the compounds are highly lipophilic.40 In designing a second generation PDP, we followed standard medicinal chemistry principles to reduce the tPSA and HBD count while maintaining an acceptable Log D7.4. To achieve this, we redesigned the ether linker in the first generation PDP 3 to include a methylene piperazine that would project into solvent based on analysis of the crystal structure of the chemical probe 1 bound to pirin. The resulting tertiary amide would remove one HBD. We also sought to mask the quinoline amide HBD, using a dipole–dipole interaction, via a bioisosteric replacement with fluorine.41 These changes resulted in the design of the second generation pirin-targeting PDP 10 (Scheme 2) with a modestly reduced tPSA (244 Å2).
Scheme 2. Synthesis of Second (10) and Third (16) Generation Probes and Control Compounds.

Reagents and conditions: (a) (i) X = F 12, 2-methylquinoline carboxylic acid, oxalyl chloride, DMF, DCM, RT, 3 h, then pyridine, 18 h quant, (ii) Fe(0), NH4Cl, EtOH/H2O, 90 °C, 1 h, quant; (b) 2,3-dihydrobenzo-[b][1,4]-dioxine-6-carboxylic acid, oxalyl chloride, DMF, DCM, RT, 3 h, then pyridine, RT, 2 h, 85%; (c) (i) SeO2, 1,4-dioxane/DMF, reflux, 1 h, (ii) N-Boc-piperazine, DCM, RT, 12 h then NaBH(OAc)3, DCM, RT, 2 h, 94% (over 2 steps), (iii) TFA, DCM, RT, 2 h, 69%; (d) (i) 2, PPh3, tbutyl 2-hydroxyacetate, DTBAD, THF, 0 °C → RT, 16 h, 75%, (ii) HCO2H, DCM, 40 °C, 16 h, 54%, (iii) HATU, DIPEA, DMF, tbutyl 3-(2-aminoethoxy)propanoate, RT, 16 h, 72%, (iv) HCO2H, DCM, 40 °C, 6 h, 93%; (e) HATU, DIPEA, DMF, RT, 16 h, 52%; (f) (i) X = Cl 17, 2-methylquinoline carboxylic acid, oxalyl chloride, DMF, DCM, RT, 3 h, then pyridine 2 h, 88%, (ii) Fe(0), NH4Cl, EtOH/H2O, 90 °C, 1 h, quant; (g) 2,3-dihydrobenzo-[b][1,4]-dioxine-6-carboxylic acid, oxalyl chloride, DMF, DCM, RT, 3 h, then pyridine 2 h, 61%; (h) (i) SeO2, DMF, 1,4-dioxane, 50 °C, 16 h, (ii) N-Boc-piperazine, NaBH3CN, AcOH, DMF, 0 °C → RT, 16 h, (iii) 4 M HCl in dioxane, MeOH 0 °C → RT, 16 h, 32% over 3 steps; (i) 2-[2-(2-hydroxyethoxy)ethoxy]ethyl 4-methylbenzenesulfonate, K2CO3, DMF, RT, 16 h, 48%; (j) PPh3, DTBAD, THF, RT, 2 h, 27%; (k) (i) 23, 2,3-dihydro-1,4-benzodioxine-5-carboxylic acid, oxalyl chloride, DMF, DCM, RT, 2 h, then pyridine 48 h, 84%, (ii) Pd/C, EtOH/DCM, H2 (1 atm), 77%, (iii) 2-methylquinoline carboxylic acid, oxalyl chloride, DMF, DCM, RT, 2 h, then pyridine 16 h, 58%, then same procedure as from 18, 6% yield over 5 steps. (l) (i) SeO2, 1,4-dioxane/DMF, 50 °C, 5.5 h, (ii) N-ethylpiperazine, DCM, RT, 20 h, then NaBH(OAc)3, DCM, RT, 2 h, 35% (over 2 steps).
The synthesis of PDP 10 began from the fluoroaniline carboxamide 11, synthesized in a similar manner to that previously described from 2-fluoro-5-nitroaniline 12 in three steps and 85% yield. Amide coupling to give bisamide 13 and benzylic oxidation using selenium dioxide42 was followed by reductive amination of the resulting aldehyde with N-Boc-piperazine, and subsequent deprotection, to give 14. The CRBN-targeting thalidomide derivative precursor 15 was prepared in four steps and 27% yield from 4-hydroxythalidomide 2. Final amide coupling with monosubstituted piperazine 14 gave the second generation pirin-targeting PDP 10 in 11 steps and 8% overall yield (Scheme 2).
PDP 10 displayed a similar affinity for recombinant pirin and CRBN-DDB1 to our first generation probe 3 (Table 1, entry 2), so it was progressed to cellular assessment of pirin degradation. Pleasingly, treatment of SK-OV-3 ovarian cancer cells with 10 at 3.0 μM total concentration for up to 48 h revealed a clear and time-dependent depletion of intracellular pirin expression (Supporting Information, Figure S18).43 This confirmed, for the first time, that pirin is amenable to modulation using a PDP and that the bisamide chemotype not only binds recombinant pirin with high affinity but also binds pirin within living cells.
Although demonstrating pirin depletion with the second generation probe 10 was a very encouraging result, we found the effects were poorly reproducible. The concentrations of PDP 10 needed to observe pirin degradation were high and close to its kinetic solubility (KS). Furthermore, at least 24 h of compound exposure was needed before pirin degradation was observed (Supporting Information, Figure S18). To explore the cause of the variable results obtained with PDP 10, we assessed its chemical stability. At room temperature, PDP 10 was stable as a solid and in DMSO stock solution (>1 month, data not shown), but at 37 °C in pH 7.4 phosphate buffer, consistent with the cell assay conditions, it underwent rapid decomposition (Supporting Information, Table S1), displaying a half-life of only ∼4 h. This poor chemical stability was consistent with the known decomposition of the parent CRBN-targeting thalidomide ligand under these conditions, where multiple hydrolysis products of the imide and glutaramide moieties are observed.44 The chemical probe 1 displayed no instability under these conditions; therefore, we concluded that the facile hydrolysis of the CRBN-targeting motif limited the reproducibility of our slow-acting second generation pirin-targeting PDP 10.
The Third Generation PDP
In the design of a third generation probe, we aimed to increase permeability further. This should result in higher intracellular free concentrations more quickly, mitigating the poor stability of the CRBN-targeting motif. We decided to carry out a bioisosteric replacement of the central ring fluorine for the larger and more sterically hindering chlorine substituent.45 However, we were concerned that the increase in lipophilicity from this exchange would negatively impact both the solubility and permeability of the probe. Large lipophilic heterobifunctional molecules are particularly susceptible to aggregation,46 and this decreases the free concentration that drives cell membrane flux.47 To balance the lipophilicity, we removed the tertiary amide bond to the piperazine, introducing a cationic amine, which would be substantially charged at pH 7.4 (MoKa, version 2.5.2, pKa = 8.0).48 The second amide in the linker was also removed, reducing the HBD count further and increasing the overall flexibility of the ligand, consistent with the formation of the crucial PDP ternary protein complex. The linker length was reduced by one atom to accommodate these changes and resulted in the design of the third generation pirin-targeting PDP 16 (CCT367766), which now displayed a notably reduced, but still high, tPSA (207 Å2).
The synthesis of the pirin-targeting motif of the third generation PDP 16 was carried out in a similar manner to that previously described starting from 2-chloro-5-nitroaniline 17 to give chlorobisamide 18 in three steps and 54% yield (Scheme 2). Oxidation of the methylquinoline moiety with SeO2 and reductive amination of the resulting aldehyde with N-Boc-piperazine and N-Boc deprotection gave 19 in 32% yield. Following SN2 alkylation with the ether linker to give 20, selective Mitsunobu alkylation with 4-hydroxythalidomide 2 gave the third generation PDP 16 in eight steps and 2% overall yield. A nonpirin-binding negative control matched pair compound, PDP 21, based on our negative control pirin chemical probe,11 was synthesized from regioisomer 23 in a similar manner in 2% overall yield. A non-CRBN binding control 22(49) was also synthesized from 18, utilizing reductive amination with N-ethylpiperazine in 35% yield (Scheme 2).
Analysis of the third generation PDP 16 confirmed that it retained acceptable lipophilicity (Table 1, entry 3) but also displayed a 4.2-fold increase in affinity for recombinant pirin compared to the second generation PDP 10 (Figure 2A) and comparable affinity for CRBN (Figure 2B). SK-OV-3 cells were then treated with PDP 16 at concentrations from 50 to 1500 nM for up to 24 h (Figure 3).
Figure 2.

(A) Representative SPR sensorgram of the third generation PDP 16 and recombinant pirin. (B) Representative binding curve of PDP 16 in the CRBN-DDB1 FP-assay.
Figure 3.

(A) Immunoblot of SK-OV-3 human ovarian cancer cells demonstrating the depletion of pirin protein using the third generation PDP 16 and the time-dependent hook-effect. (B) Immunoblot demonstrating the concentration-dependent depletion of pirin protein after 2 h exposure in SK-OV-3 cells. (C) Capillary electrophoresis and immunoassay were used to quantify the pirin protein expression after 2 h exposure with PDP 16, and all values are normalized to vinculin loading control and relative to the measured basal pirin protein expression, all bars represent the arithmetic mean of n = 3 independent biological repeats, error bars are SEM. (D) Proteomics analysis of the third generation PDP 16 (50 nM) exposure (4 h) in SK-OV-3 cells compared to vehicle control, using a tandem mass tagging (TMT) MS2 protocol on the cell lysate, 8547 quantifiable proteins were identified; each blue dot represents a single quantifiable protein, pirin is marked in red (adjusted p value = 1.4 × 10–4), p values were calculated using a linear modeling based t test and corrected for multiple comparisons using the Benjamini–Hochberg method to give the p(adj) values shown, dotted lines represent 2-fold depletion of the protein and a p(adj) = 0.05.
In contrast to our earlier generation PDPs, near complete pirin degradation was now observed with just 50 nM treatment and only 2 h exposure (Figure 3A). Increasing the total initial concentration of pirin-targeting PDP 16 resulted in a clear hook effect. This bell-shaped concentration–response is consistent with the formation of a ternary complex.22 Interestingly the hook effect was seen to decrease over time (24 h), possibly either due to the slower degradation at high concentration or from the depletion of PDP 16 due to thalidomide hydrolysis (half-life = ∼3 h, Supporting Information, Figure S1), which would reduce the effective free concentration below the negative cooperativity threshold of the ternary complex.22 Pirin degradation was subsequently shown to be concentration-responsive with activity at concentrations as low as 0.5 nM (Figure 3B), which was confirmed with a quantitative capillary electrophoresis-based immunoassay (Figure 3C). The negative control benzodioxane regioisomer PDP 21 (Table 1, entry 4) displayed no pirin depletion at equimolar concentrations (Supporting Information, Figure S19),50 and degradation was also confirmed to be proteasome-dependent by rescue following preincubation with the proteosome inhibitor, MG132 (500 nM, Supporting Information, Figure S19).51 We then carried out whole proteome mass spectrometry to estimate the cellular selectivity of the pirin-targeting PDP 16 in an unbiased manner, quantified using tandem mass tagging (TMT) (Figure 3D, www.proteomics.com). After treating SK-OV-3 cells with 50 nM PDP 16 for 4 h, and comparing to vehicle treated cells using Benjamini–Hochberg corrected p values, we found that from 8547 quantifiable proteins identified, only pirin (2.3-fold reduction, p(adj) = 1.4 × 10–4) displayed a statistically significant (p(adj) < 0.05)52 difference in protein expression.
To confirm that our chemical probe 1 also bound pirin within SK-OV-3 cancer cells, we carried out competition experiments designed to rescue pirin depletion by PDP 16. The concentrations required for a mutually exclusive binding ligand to displace a probe molecule are dependent on the affinity of the ligand relative to the ratio of the free concentration of the probe and its affinity for the protein target complex.53 Because the depletion of pirin is a nonequilibrium event that does not necessarily require complete target occupancy, it is more difficult to observe competition at later time points owing to continued protein turnover.54 Pretreating SK-OV-3 cells with 10 μM thalidomide, as a CRBN-binding competitive ligand, demonstrated that after 2 h treatment with PDP 16 (5 nM), we successfully rescued pirin depletion (Supporting Information, Figure S19). The non-CRBN-binding control chlorobisamide 22 displayed high affinity for recombinant pirin (SPR, KD = 21 nM, pKD = 7.68 ± 0.03, n = 3). Pretreatment of SK-OV-3 cells with chlorobisamide 22 displayed concentration-dependent rescue of pirin expression following treatment with the PDP 16 (Figure 4, top), with complete rescue observed at 1 μM. Finally, pretreatment with the chemical probe 1 demonstrated clear rescue of pirin depletion, confirming intracellular target engagement (Figure 4, bottom).
Figure 4.

Intracellular competition studies with PDP 16 and the chemical probes. SK-OV-3 cells were pretreated with increasing concentrations of chemical probe for 4 h before exposing to PDP 16 for 2 h at the concentrations shown. Cells were then lysed and protein expression analyzed using immunoblot. For clarity, gel images have been cropped where appropriate.
Conclusion
Exploiting cell-based phenotypic screens to identify new disease-associated therapeutic targets is an increasingly frequent strategy in drug discovery. While this approach can identify novel targets with unique mechanisms of action, these proteins are often poorly characterized and can lack identifiable enzymatic activity, ligands, and biomarkers of target engagement. The main focus of research into PDPs has been as potential therapeutics with novel mechanisms of action. We designed a PDP as an intracellular probe against the poorly understood and noncatalytic molecular target, pirin. Developing PDPs to confirm intracellular target engagement, and potentially develop intracellular SAR, against challenging proteins, is an important addition to the current methods for compound profiling.
For PDPs to be used as target engagement probes, their rapid development and validation is crucial. The ideal strategies for efficient and successful PDP design are still under investigation and will clearly improve as more protein targets are modulated and additional crystallographic evidence of the target protein/E3 ligase/PDP ternary complexes are discovered. The number of variables involved in PDP design against nonvalidated target proteins can make the process daunting. By focusing on the physicochemical properties of our probe molecules, in only three iterations, we developed a selective degradation probe that eliminates pirin at low concentration and in a short time period. This confirmed our chemical probe 1 does bind pirin in an intracellular environment, and PDP 16 provides another chemical tool to study a largely unexplored protein.
Experimental Section
General Experimental
Unless otherwise stated, reactions were conducted in oven-dried glassware under an atmosphere of nitrogen or argon using anhydrous solvents. All commercially obtained reagents and solvents were used as received. Thin layer chromatography (TLC) was performed on precoated aluminum sheets of silica (60 F254 nm, Merck) and visualized using short-wave UV light. Flash column chromatography was carried out on Merck silica gel 60 (partial size 40–65 μm). Column chromatography was also performed on a Biotage SP1 or Biotage Isolera Four purification system using Biotage Flash silica cartridges (SNAP KP-Sil) or for reverse phase purifications SNAP Ultra C18 cartridges. Ion exchange chromatography was performed using acidic Isolute Flash SCX-II columns. Semipreparative HPLC was performed on an Agilent 6120 system, flow 20 mL/min, eluents 0.1% acetic acid in water and 0.1% acetic acid in methanol, gradient of 10–100% organic phase. Lipophilic method: Chromatographic separation at room temperature was carried out using a 1200 series preparative HPLC (Agilent, Santa Clara, USA) over a 15 min gradient elution (gradient 15 min, 20 mL) from 60:40 to 0:100 water:methanol (both modified with 0.1% formic acid) at a flow rate of 20 mL/min. 1H NMR spectra were recorded on Bruker AMX500 (500 MHz) spectrometers using an internal deuterium lock. Chemical shifts are quoted in parts per million (ppm) using the following internal references: CDCl3 (δH 7.26), MeOD (δH 3.31), and DMSO-d6 (δH 2.50). Signal multiplicities are recorded as singlet (s), doublet (d), triplet (t), quartet (q), multiplet (m), doublet of doublets (dd), doublet of doublet of doublets (ddd), broad (br), or obscured (obs). Coupling constants, J, are measured to the nearest 0.1 Hz. 13C NMR spectra were recorded on Bruker AMX500 spectrometers at 126 MHz using an internal deuterium lock. Chemical shifts are quoted to 0.01 ppm, unless greater accuracy was required, using the following internal references: CDCl3 (δC 77.0), MeOD (δC 49.0), and DMSO-d6 (δC 39.5). High resolution mass spectra were recorded on an Agilent 1200 series HPLC and diode array detector coupled to a 6210 time-of-flight mass spectrometer with dual multimode APCI/ESI source or on a Waters Acquity UPLC and diode array detector coupled to a Waters G2 QToF mass spectrometer fitted with a multimode ESI/APCI source. All compounds were >95% purity by LCMS analysis unless otherwise stated.
Thalidomide (http://www.sigmaaldrich.com/catalog/product/sigma/t144?lang=en®ion=GB, accessed August 29, 2017), Lenalidomide (http://www.sigmaaldrich.com/catalog/product/aldrich/cds022536?lang=en®ion=GB, accessed August 29, 2017) and MG132 (http://www.sigmaaldrich.com/catalog/product/sigma/m8699?lang=en®ion=GB, accessed August 29, 2017) were purchased from Sigma-Aldrich and used without further purification.
The pirin chemical probe 1 CCT251236 was synthesized using the previously described procedure.11
2-(2,6-Dioxopiperidin-3-yl)-4-hydroxyisoindoline-1,3-dione 2
3-Aminopiperidine-2,6-dione hydrochloride (300 mg, 1.82 mmol) was added to a solution of 4-hydroxyisobenzofuran-1,3-dione (299 mg, 1.82 mmol) and triethylamine (0.38 mL, 2.73 mmol) in anhydrous THF (36.5 mL) and the reaction heated to reflux for 24 h under inert atmosphere. The reaction mixture was allowed to cool to room temperature, then 1-ethyl-3-(3-(dimethylamino)propyl)carbodiimide (384 mg, 2.00 mmol) and DMAP (22 mg, 0.18 mmol) were added. The reaction was heated to reflux for 24 h. Then, the reaction mixture was allowed to cool, and the solvents were removed under reduced pressure. The resulting residue was taken up in methanol and passed sequentially through two ion exchange columns (Isolute SCX-II), eluting with methanol to afford the title compound as an amorphous off-white solid (422 mg, 84%). 1H NMR (500 MHz, DMSO-d6) δ 11.18 (s, 1H), 11.09 (s, 1H), 7.65 (dd, J = 8.4, 7.2 Hz, 1H), 7.32 (dd, J = 7.1, 0.7 Hz, 1H), 7.25 (dd, J = 8.4, 0.7 Hz, 1H), 5.07 (dd, J = 12.8, 5.5 Hz, 1H), 2.88 (ddd, J = 17.0, 13.9, 5.5 Hz, 1H), 2.64–2.46 (m, 2H), 2.02 (dtd, J = 13.0, 5.3, 2.2 Hz, 1H). LCMS (ESI+) RT = 0.77 min, 100%, M + H+ 275, M + Na+ 297.28 This compound is also commercially available from several suppliers.
tert-Butyl 2-((2-(2,6-Dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)oxy)acetate
Triphenylphosphine (0.261 g, 0.996 mmol) and tert-butyl 2-hydroxyacetate (101 mg, 0.77 mmol) were dissolved in anhydrous THF (3 mL) while stirring at 0 °C. Then a solution of DTBAD (229 mg, 0.10 mmol) in anhydrous THF (2 mL) was added dropwise. Finally, a solution of (2-(2,6-dioxopiperidin-3-yl)-4-hydroxyisoindoline-1,3-dione) 2 in anhydrous THF (3 mL) was added. The mixture was stirred for 1 h at 0 °C then allowed to warm to room temperature and stirred overnight. Then the solvent was removed under reduced pressure. The crude product was purified by Biotage chromatography using a gradient of 0–50% EtOAc in cyclohexane to afford the title compound as an amorphous white solid (224 mg, 0.577 mmol, 75% yield). 1H NMR (500 MHz, DMSO-d6) δ 11.11 (s, 1H), 7.80 (dd, J = 8.5, 7.3 Hz, 1H), 7.48 (d, J = 7.2 Hz, 1H), 7.38 (d, J = 8.5 Hz, 1H), 5.10 (dd, J = 12.8, 5.5 Hz, 1H), 4.97 (s, 2H), 2.89 (ddd, J = 17.0, 13.9, 5.4 Hz, 1H), 2.64–2.52 (m, 2H), 2.07–2.00 (m, 1H), 1.43 (s, 9H). 13C NMR (126 MHz, DMSO-d6) δ 172.78, 169.89, 167.14, 166.71, 165.12, 155.03, 136.76, 133.25, 119.96, 116.44, 115.90, 81.92, 65.50, 48.80, 30.95, 27.68, 21.97. HRMS (ESI+): calcd for C15H13N2O7 (M + H, −tBu)+ 333.0717, found 333.0722.
2-((2-(2,6-Dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)oxy)acetic Acid
tert-Butyl 2-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)oxy)acetate (1.30 g, 3.35 mmol) was dissolved in anhydrous DCM, then formic acid (12.8 mL, 335 mmol) was added while stirring at room temperature. The reaction was stirred overnight at 40 °C. The solvents were removed under reduced pressure and the resulting residue purified by reverse phase Biotage chromatography using a gradient of 0–50% MeOH in water + 0.1% formic acid to afford the title compound as an amorphous white solid (600 mg, 1.81 mmol, 54% yield). 1H NMR (500 MHz, DMSO-d6) δ 13.29 (s, 1H), 11.11 (s, 1H), 7.79 (dd, J = 8.5, 7.3 Hz, 1H), 7.47 (d, J = 7.2 Hz, 1H), 7.39 (d, J = 8.6 Hz, 1H), 5.10 (dd, J = 12.8, 5.4 Hz, 1H), 4.98 (s, 2H), 2.89 (ddd, J = 16.9, 13.9, 5.4 Hz, 1H), 2.64–2.52 (m, 2H), 2.04 (dtd, J = 13.0, 5.4, 2.3 Hz, 1H). LCMS (ESI+): RT = 0.71 min, 100%, M + Na+ 355.28
tert-Butyl 3-(2-(2-((2-(2,6-Dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)oxy)acetamido)ethoxy)propanoate
2-((2-(2,6-Dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)oxy)acetic acid (217 mg, 0.65 mmol), tert-butyl 3-(2-aminoethoxy)propanoate (124 mg, 0.65 mmol), and DIPEA (0.34 mL, 1.96 mmol) were dissolved in anhydrous DMF (3.27 mL) at room temperature under inert atmosphere. HATU (230 mg, 0.98 mmol) was added and the reaction stirred overnight. The solvents were removed under reduced pressure, and the resulting residue was purified by reverse phase Biotage chromatography using a gradient of 10–90% MeOH in water + 0.1% formic acid to afford the title compound as an amorphous white solid (236 mg, 72%). 1H NMR (500 MHz, MeOD) δ 7.82 (dd, J = 8.4, 7.4 Hz, 1H), 7.55 (dd, J = 7.3, 0.5 Hz, 1H), 7.46–7.43 (m, 1H), 5.16 (dd, J = 12.6, 5.5 Hz, 1H), 4.78 (s, 2H), 3.71 (t, J = 6.2 Hz, 2H), 3.62–3.56 (m, 2H), 3.52–3.48 (m, 2H), 2.90 (ddd, J = 18.0, 14.4, 5.2 Hz, 1H), 2.82–2.70 (m, 2H), 2.50 (td, J = 6.2, 3.8 Hz, 2H), 2.16 (dddd, J = 10.8, 7.9, 5.1, 2.4 Hz, 1H), 1.42 (s, 9H). 13C NMR (126 MHz, MeOD) δ 174.55, 172.81, 171.33, 170.00, 168.31, 167.59, 156.21, 138.16, 134.96, 121.60, 119.30, 117.89, 81.75, 70.01, 69.26, 67.75, 50.56, 40.15, 37.22, 32.20, 28.34, 23.66. HRMS (ESI+): calcd for C24H30N3O9 (M + H)+ 504.1982, found 504.1981.
tert-Butyl (2-(2-(2-((2-(2,6-Dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)oxy)acetamido)ethoxy)ethyl)carbamate
2-((2-(2,6-Dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)oxy)acetic acid (0.19 g, 0.56 mmol), tert-butyl (2-(2-aminoethoxy)ethyl)carbamate (0.11 g, 0.56 mmol), and DIPEA (0.29 mL, 1.68 mmol) were dissolved in anhydrous DMF (2.80 mL) under inert atmosphere at room temperature. HATU (198 mg, 0.84 mmol) was added and the reaction stirred for 16 h. The reaction mixture was concentrated in vacuo and the resulting residue purified by Biotage chromatography using a gradient of 0–2% methanol in ethyl acetate. However, this material was not pure, therefore the material was purified by further Biotage chromatography using a gradient of 1–10% EtOH in DCM to afford the title compound as an amorphous white solid (243 mg, 81%). 1H NMR (500 MHz, DMSO-d6) δ 11.11 (s, 1H), 8.02 (t, J = 5.4 Hz, 1H), 7.81 (dd, J = 8.5, 7.3 Hz, 1H), 7.49 (d, J = 7.2 Hz, 1H), 7.39 (d, J = 8.5 Hz, 1H), 6.76 (t, J = 4.7 Hz, 1H), 5.12 (dd, J = 12.8, 5.4 Hz, 1H), 4.79 (s, 2H), 3.44 (t, J = 5.6 Hz, 2H), 3.38 (t, J = 6.1 Hz, 2H), 3.31 (d, J = 5.5 Hz, 2H), 3.08 (q, J = 5.6 Hz, 2H), 2.89 (ddd, J = 16.6, 13.8, 5.3 Hz, 1H), 2.65–2.51 (m, 2H), 2.08–1.98 (m, 1H), 1.36 (s, 9H). 13C NMR (126 MHz, DMSO-d6) δ 172.73, 169.85, 166.88, 166.80, 166.72, 165.45, 155.59, 155.00, 136.92, 133.03, 120.34, 116.75, 116.02, 77.63, 69.02, 68.55, 67.48, 48.80, 38.38, 30.95, 28.22, 21.99. HRMS (ESI+): calcd for C24H30N4O9Na (M + Na)+ 541.1911, found 541.1915.
N-(5-(2,3-Dihydrobenzo[b][1,4]dioxine-6-carboxamido)-2-methylphenyl)-2-(4-((2-(2-(2-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)oxy)acetamido)ethoxy)ethyl)amino)-4-oxobutoxy)quinoline-6-carboxamide 3
N-(2-(2-Aminoethoxy)ethyl)-2-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)oxy)acetamide hydrochloride 4 (21.0 mg, 0.046 mmol), 4-((6-((5-(2,3-dihydrobenzo[b][1,4]dioxine-6-carboxamido)-2-methylphenyl)carbamoyl)quinolin-2-yl)oxy)butanoic acid (25.0 mg, 0.046 mmol), and DIPEA (0.032 mL, 0.184 mmol) were dissolved in anhydrous DMF (0.23 mL) under inert atmosphere. HATU (16.0 mg, 0.069 mmol) was added and the reaction left to stir at room temperature for 2 h. The reaction was concentrated in vacuo, then purified by reverse phase Biotage chromatography using a gradient of 20–100% MeOH in water + 0.1% formic acid to afford the title compound as a beige solid (32 mg, 74%). 1H NMR (500 MHz, DMSO-d6) δ 11.09 (s, 1H), 10.07 (s, 1H), 10.06 (s, 1H), 8.56 (d, J = 1.9 Hz, 1H), 8.37 (d, J = 8.8 Hz, 1H), 8.21 (dd, J = 8.8, 2.1 Hz, 1H), 8.01 (t, J = 5.5 Hz, 1H), 7.91 (t, J = 5.5 Hz, 1H), 7.87–7.83 (m, 2H), 7.80 (dd, J = 8.5, 7.3 Hz, 1H), 7.58 (dd, J = 8.3, 2.1 Hz, 1H), 7.54 (d, J = 2.1 Hz, 1H), 7.51 (dd, J = 8.4, 2.2 Hz, 1H), 7.48 (d, J = 7.2 Hz, 1H), 7.39 (d, J = 8.5 Hz, 1H), 7.24 (d, J = 8.5 Hz, 1H), 7.08 (d, J = 8.8 Hz, 1H), 6.98 (d, J = 8.4 Hz, 1H), 5.12 (dd, J = 12.8, 5.4 Hz, 1H), 4.79 (s, 2H), 4.42 (t, J = 6.5 Hz, 2H), 4.33–4.26 (m, 4H), 3.45 (t, J = 5.7 Hz, 2H), 3.42 (t, J = 5.9 Hz, 2H), 3.36–3.28 (m, J = 5.5 Hz, 2H, obscured by water peak), 3.24 (q, J = 5.8 Hz, 2H), 2.89 (ddd, J = 16.3, 13.6, 5.1 Hz, 1H), 2.64–2.52 (m, 2H), 2.29 (t, J = 7.3 Hz, 2H), 2.23 (s, 3H), 2.11–1.93 (m, 3H). 13C NMR (126 MHz, DMSO-d6) δ 172.74, 171.66, 169.87, 166.90, 166.71, 165.44, 164.91, 164.34, 162.79, 154.96, 147.58, 146.33, 142.92, 140.16, 137.28, 136.90, 136.33, 133.01, 130.10, 130.00, 128.75, 128.37, 128.10, 127.68, 126.72, 123.99, 121.19, 120.32, 118.61, 118.11, 116.83, 116.74, 116.65, 116.02, 113.94, 68.91, 68.58, 67.48, 65.38, 64.39, 64.02, 48.80, 38.44, 38.33, 31.78, 30.94, 24.62, 21.99, 17.48. HRMS: calcd for C49H48N7O13 (M + H)+ 942.3305, found 942.3322.
N-(2-(2-Aminoethoxy)ethyl)-2-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)oxy)acetamide 4
tert-Butyl (2-(2-(2-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)oxy)acetamido)ethoxy)ethyl)carbamate (24 mg, 0.05 mmol) was stirred in 4 M HCl in dioxane (1.16 mL, 4.63 mmol) at room temperature. After 30 min, the solution was concentrated and used as the hydrochloride salt without further purification. LCMS (ESI+): RT = 0.63 min, 100%, M + H+ 419.
Ethyl 4-((6-Bromoquinolin-2-yl)oxy)butanoate 6
6-Bromoquinolin-2(1H)-one 5 (200 mg, 0.89 mmol) was dissolved in anhydrous DMF (4.5 mL) at room temperature under nitrogen, and potassium carbonate (185 mg, 1.34 mmol) was added. The reaction mixture was allowed to stir for 1.5 h, then ethyl 4-bromobutanoate (0.26 mL, 1.79 mmol) was added dropwise and the reaction stirred overnight. The reaction mixture was poured into water (50 mL) and the aqueous layer extracted with DCM (3 × 15 mL). The combined organic layer was dried (Na2SO4) and concentrated in vacuo to afford the crude product as an orange oil. This material was purified by Biotage chromatography using a gradient of 0–100% EtOAc in cyclohexane to afford the required O-linked isomer as a white solid (111 mg, 37%) and the undesired N-linked isomer as a yellow oil (164 mg, 55%). The regioisomers were distinguishable by NOE spectroscopy. 1H NMR (500 MHz, CDCl3) δ 7.91 (d, J = 8.9 Hz, 1H), 7.88–7.87 (br m, 1H), 7.71–7.69 (m, 2H), 6.92 (d, J = 8.8 Hz, 1H), 4.52 (t, J = 6.3 Hz, 2H), 4.18 (q, J = 7.1 Hz, 2H), 2.55 (t, J = 7.5 Hz, 2H), 2.19 (ddd, J = 13.7, 7.4, 6.3 Hz, 2H), 1.28 (t, J = 7.1 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 173.39, 162.35, 145.41, 137.80, 132.82, 129.61, 129.11, 126.42, 117.29, 114.30, 65.17, 60.59, 31.27, 24.60, 14.39. HRMS (ESI+): calcd for C15H1779BrNO3 (M + H)+ 339.0419, found 339.0403.
2-(Trimethylsilyl)ethyl 2-(4-ethoxy-4-oxobutoxy)quinoline-6-carboxylate 7
Ethyl 4-((6-bromoquinolin-2-yl)oxy)butanoate 6 (554 mg, 1.64 mmol), tri-tert-butylphosphonium tetrafluoroborate (95 mg, 0.33 mmol), and Herrmann’s palladacyle (77 mg, 0.08 mmol) were added to a microwave vial and suspended in 2-(trimethylsilyl)ethanol (15 mL). Molybdenum hexacarbonyl (865 mg, 3.28 mmol) followed by DBU 1.0 M in THF (4.91 mL, 4.91 mmol) were then added and the vial promptly sealed. The reaction was heated to 130 °C for 1 h in a microwave. The reaction mixture was diluted with DCM and filtered to remove solids. The filtrate was concentrated in vacuo. This residue was diluted with water (20 mL) and extracted with DCM (3 × 20 mL). The combined organic layer was washed with brine and concentrated under reduced pressure. The resulting crude was purified by Biotage chromatography using a gradient of 0–100% EtOAc in cyclohexane to give 472 mg of product (88% purity by LCMS, 62%) as a brown oil. 1H NMR (500 MHz, CDCl3) δ 8.47 (d, J = 1.9 Hz, 1H), 8.23 (dd, J = 8.7, 1.9 Hz, 1H), 8.06 (d, J = 8.8 Hz, 1H), 7.84 (d, J = 8.7 Hz, 1H), 6.94 (d, J = 8.8 Hz, 1H), 4.56 (t, J = 6.3 Hz, 2H), 4.51–4.43 (m, 2H), 4.17 (q, J = 7.1 Hz, 2H), 2.55 (t, J = 7.4 Hz, 2H), 2.19 (p, J = 7.0, 6.5 Hz, 2H), 1.27 (t, J = 7.1 Hz, 3H), 1.23–1.15 (m, 2H), 0.12 (s, 9H). LCMS (ESI+): RT = 1.79 min, 88%, (M + H)+ 404.
Ethyl 4-((6-((5-(2,3-Dihydrobenzo[b][1,4]dioxine-6-carboxamido)-2-methylphenyl)carbamoyl)quinolin-2-yl)oxy)butanoate
To a stirring solution of 2-(trimethylsilyl)ethyl 2-(4-ethoxy-4-oxobutoxy)quinoline-6-carboxylate 7 (463 mg, 1.15 mmol) in anhydrous THF (12 mL) at room temperature under nitrogen was dropwise added TBAF 1.0 M in THF (1.72 mL, 1.72 mmol). The reaction was allowed to stir at RT overnight, after which time the starting material had been consumed as indicated by LCMS. The reaction mixture was diluted with water and concentrated in vacuo to remove the THF. The aqueous layer was acidified to ∼ pH 2 with 1 M HCl aq and extracted with DCM (3 × 15 mL). The combined organic layer was washed with brine (20 mL) and dried (Na2SO4) to afford the crude product as a yellow oil. This material was used directly in the next step without further purification. LCMS (ESI+): RT = 1.57 min, 84%, (M + H)+ 304.
2-(4-Ethoxy-4-oxobutoxy)quinoline-6-carboxylic acid (74 mg, 0.24 mmol) was dissolved in anhydrous DMF (2 mL), and DIPEA (0.12 mL, 0.67 mmol) was added, followed by HATU (105 mg, 0.28 mmol). The reaction was allowed to stir for 5 min, and then N-(3-amino-4-methylphenyl)-2,3-dihydrobenzo[b][1,4]dioxine-6-carboxamide (63 mg, 0.22 mmol) was added. The reaction was stirred at room temperature overnight. The reaction mixture was poured into water and the resulting precipitate collected by filtration. The precipitate was dissolved in DCM/MeOH and preabsorbed onto silica. The crude material was purified by Biotage chromatography using a gradient of 0–5% MeOH in DCM and then by preparative HPLC (20 mL, lipophilic method) to afford the title compound as an amorphous beige solid (48 mg, 38% over two steps). 1H NMR (500 MHz, CDCl3) δ 8.30 (d, J = 1.9 Hz, 1H), 8.13–8.05 (m, 3H), 7.93–7.87 (m, 3H),7.68 (dd, J = 8.3, 2.1 Hz, 1H), 7.44 (d, J = 2.1 Hz, 1H), 7.38 (dd, J = 8.4, 2.2 Hz, 1H), 7.23 (d, J = 8.4 Hz, 1H), 6.97 (d, J = 8.8 Hz, 1H), 6.94 (d, J = 8.4 Hz, 1H), 4.57 (t, J = 6.3 Hz, 2H), 4.31 (ddd, J = 11.1, 3.7, 1.8 Hz, 4H), 4.18 (q, J = 7.1 Hz, 2H), 2.56 (t, J = 7.4 Hz, 2H), 2.35 (s, 3H), 2.21 (p, J = 6.7 Hz, 2H), 1.28 (t, J = 7.1 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 173.37, 165.49, 165.09, 163.48, 148.69, 146.89, 143.67, 139.54, 136.92, 136.11, 131.17, 130.22, 128.23, 128.05, 127.62, 127.30, 125.17, 124.61, 120.59, 117.63, 117.52, 116.89, 114.86, 114.58, 65.40, 64.71, 64.34, 60.62, 31.26, 24.58, 17.52, 14.39. HRMS (ESI+): calcd for C32H32N3O7 (M + H)+ 570.2253, found 570.2222.
4-((6-((5-(2,3-Dihydrobenzo[b][1,4]dioxine-6-carboxamido)-2-methylphenyl)carbamoyl)quinolin-2-yl)oxy)butanoic Acid 9
To a stirring solution of ethyl 4-((6-((5-(2,3-dihydrobenzo[b][1,4]dioxine-6-carboxamido)-2-methylphenyl)carbamoyl)quinolin-2-yl)oxy)butanoate (37 mg, 0.065 mmol) in THF (0.5 mL) and MeOH (0.25 mL) was added a solution of LiOH.H2O (13.6 mg, 0.325 mmol) in water (0.25 mL). After 2 days, the reaction mixture was diluted with water (30 mL) and the aqueous layer acidified to pH ∼2 with 1 M aq HCl. The aqueous layer was extracted with DCM (3 × 15 mL). The combined organic layer was washed with 1 M aq HCl (20 mL) and brine (20 mL), and dried (Na2SO4) then concentrated in vacuo to afford the product as an amorphous pale-yellow solid (6.5 mg, 18%). This material was used directly in the next reaction without further purification. 1H NMR (500 MHz, DMSO-d6) δ 12.16 (br s, 1H), 10.07 (s, 2H), 8.56 (d, J = 1.9 Hz, 1H), 8.38 (d, J = 8.8 Hz, 1H), 8.22 (dd, J = 8.7, 2.1 Hz, 1H), 7.89–7.83 (m, 2H), 7.58 (dd, J = 8.3, 2.1 Hz, 1H), 7.54 (d, J = 2.1 Hz, 1H), 7.51 (dd, J = 8.4, 2.2 Hz, 1H), 7.24 (d, J = 8.6 Hz, 1H), 7.11 (d, J = 8.8 Hz, 1H), 6.99 (s, 1H), 4.47 (t, J = 6.5 Hz, 2H), 4.34–4.21 (m, 4H), 2.43 (t, J = 7.4 Hz, 2H), 2.24 (s, 3H), 2.03 (p, J = 7.0 Hz, 2H). 13C NMR (126 MHz, DMSO-d6) δ 174.11, 164.94, 164.35, 162.78, 147.57, 146.34, 142.93, 140.23, 137.29, 136.33, 130.11, 130.05, 128.76, 128.40, 128.12, 127.68, 126.75, 124.03, 121.20, 118.61, 118.11, 116.84, 116.66, 113.94, 65.06, 64.39, 64.02, 30.30, 24.05, 17.49. HRMS (ESI+): calcd for C30H28N3O7 (M + H)+ 542.1922, found 542.1905.
N-(2-Fluoro-5-nitrophenyl)-2-methylquinoline-6-carboxamide
Oxalyl chloride (3.25 mL, 38.4 mmol) was added dropwise to a solution of 2-methylquinoline-6-carboxylic acid (6.59 g, 35.2 mmol) and DMF (0.0062 mL, 0.080 mmol) in anhydrous DCM (80 mL). The reaction mixture was stirred at room temperature for 3 h and then concentrated under reduced pressure. The residue was dissolved in DCM (30 mL) and concentrated again under reduced pressure. The resulting dry residue was dissolved in pyridine (80 mL), and 2-fluoro-5-nitroaniline 12 (5.00 g, 32.0 mmol) was added in one portion. The reaction mixture was stirred at room temperature for 18 h and then poured onto water (100 mL). The green precipitate was filtered and washed with water (3 × 20 mL), diethyl ether (3 × 20 mL), and DCM (10 mL), to afford the title compound as a light-green solid, which was carried onto the next step without further purification (10.4 g, quant). 1H NMR (500 MHz, DMSO-d6) δ 10.71 (s, 1H), 8.72 (dd, J = 6.5, 2.9 Hz, 1H), 8.63 (d, J = 2.0 Hz, 1H), 8.43 (d, J = 8.5 Hz, 1H), 8.23 (dd, J = 8.8, 2.0 Hz, 1H), 8.21–8.16 (m, 1H), 8.05 (d, J = 8.8 Hz, 1H), 7.65 (app t, J = 9.5 Hz, 1H), 7.55 (d, J = 8.4 Hz, 1H), 2.71 (s, 3H). 13C NMR (126 MHz, DMSO-d6) δ 165.53, 161.22, 158.67 (d, J = 258.2 Hz), 148.65, 143.72, 137.32, 130.36, 128.88, 128.48, 128.00, 127.08 (d, J = 13.9 Hz), 125.33, 123.18, 122.14 (d, J = 9.6 Hz), 121.25 (d, J = 3.8 Hz), 117.19 (d, J = 22.8 Hz), 25.07. 19F NMR (470 MHz, DMSO-d6) δ −110.20. HRMS (ESI+): calcd for C17H13FN3O3 (M + H)+ 326.0935, found 326.0931.
N-(5-(2,3-Dihydrobenzo[b][1,4]dioxine-6-carboxamido)-2-fluorophenyl)-2-((4-(3-(2-(2-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)oxy)acetamido)ethoxy)propanoyl)piperazin-1-yl)methyl)quinoline-6-carboxamide 10
3-(2-(2-((2-(2,6-Dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)oxy)acetamido)ethoxy)propanoic acid 15 (15 mg, 0.033 mmol), N,N-(5-(2,3-dihydrobenzo[b][1,4]dioxine-6-carboxamido)-2-fluorophenyl)-2-(piperazin-1-ylmethyl)quinoline-6-carboxamide 14 (18 mg, 0.033 mmol), and HATU (19 mg, 0.050 mmol) were dissolved in anhydrous DMF (0.34 mL) at room temperature under inert atmosphere. DIPEA (23.4 μL, 0.134 mmol) was added and the reaction stirred for 16 h. The reaction mixture was concentrated in vacuo and purified by reverse phase Biotage chromatography (10–80% MeOH in water + 0.1% formic acid), however, the material obtained was not pure. Further purification by Biotage chromatography using a gradient of 2–50% EtOH in DCM afforded the product as an off-white amorphous solid (17 mg, 52%). 1H NMR (500 MHz, DMSO-d6) δ 11.12 (s, 1H), 10.39 (s, 1H), 10.18 (s, 1H), 8.65 (d, J = 2.0 Hz, 1H), 8.50 (d, J = 8.4 Hz, 1H), 8.25 (dd, J = 8.8, 2.0 Hz, 1H), 8.14 (dd, J = 7.1, 2.6 Hz, 1H), 8.08 (d, J = 8.8 Hz, 1H), 7.99 (t, J = 5.6 Hz, 1H), 7.80 (dd, J = 8.5, 7.3 Hz, 1H), 7.74 (d, J = 8.5 Hz, 1H), 7.65 (ddd, J = 9.0, 4.4, 2.7 Hz, 1H), 7.56–7.46 (m, 3H), 7.39 (d, J = 8.5 Hz, 1H), 7.30 (dd, J = 10.1, 9.0 Hz, 1H), 6.99 (d, J = 8.4 Hz, 1H), 5.12 (dd, J = 12.8, 5.4 Hz, 1H), 4.79 (s, 2H), 4.31 (td, J = 5.3, 3.6 Hz, 4H), 3.82 (s, 2H), 3.62 (t, J = 6.6 Hz, 2H), 3.52–3.41 (m, 6H), 3.33 (s, 2H), 2.89 (ddd, J = 16.7, 13.7, 5.4 Hz, 1H), 2.62–2.52 (m, 4H), 2.43 (d, J = 21.7 Hz, 4H), 2.09–1.99 (m, 1H). 13C NMR (126 MHz, CDCl3) δ 171.71, 169.89, 168.79, 167.12, 166.76, 166.06, 165.15, 165.08, 161.23, 154.62, 149.43 (d, J = 240.9 Hz), 149.10, 146.95, 143.59, 137.71, 137.06, 134.97, 133.71, 131.94, 129.99, 127.99, 127.94 (d, J = 9.5 Hz), 127.28, 126.81, 126.42 (d, J = 11.2 Hz), 122.32, 120.74, 119.68, 118.21, 117.48, 117.46 (d, J = 6.5 Hz), 117.05 (d, J = 7.0 Hz), 116.93, 115.30 (d, J = 20.0 Hz), 113.74, 69.20, 68.22, 67.52, 64.74, 64.70, 64.32, 53.39, 53.15, 49.42, 45.80, 41.68, 39.47, 33.60, 31.52, 23.00. HRMS (ESI+): calcd for C50H48FN8O12 (M + H)+ 971.3370, found 971.3343.
N-(5-Amino-2-fluorophenyl)-2-methylquinoline-6-carboxamide 11
To a solution of N-(2-fluoro-5-nitrophenyl)-2-methylquinoline-6-carboxamide (10.4 g, 32.0 mmol) in ethanol (120 mL) and water (40 mL), ammonium chloride (12.0 g, 224 mmol) and iron powder (12.5 g, 224 mmol) were added in one portion, and the resulting suspension was allowed to stir at 90 °C for 1 h. The reaction mixture was allowed to cool to room temperature, diluted with MeOH (20 mL) and DCM (20 mL), and filtered through a pad of Celite. The resulting filtrate was concentrated under vacuum to afford a light-brown solid which was redissolved in a mixture of DCM:MeOH (9:1, 150 mL) and washed with saturated aqueous NaHCO3 (150 mL). The organic phase was dried over Na2SO4, filtered, and concentrated under reduced pressure to afford a yellow solid as crude product, which was taken directly onto the next step without further purification (9.46 g, quant). 1H NMR (500 MHz, DMSO-d6) δ 10.05 (s, 1H), 8.57 (d, J = 1.8 Hz, 1H), 8.39 (d, J = 8.5 Hz, 1H), 8.19 (dd, J = 8.8, 2.0 Hz, 1H), 8.01 (d, J = 8.8 Hz, 1H), 7.52 (d, J = 8.4 Hz, 1H), 6.94 (dd, J = 10.3, 8.8 Hz, 1H), 6.89 (dd, J = 6.6, 2.7 Hz, 1H), 6.46–6.39 (m, 1H), 5.05 (br s, 2H), 2.70 (s, 3H). 13C NMR (126 MHz, DMSO-d6) δ 164.93, 160.84, 148.49, 147.72 (d, J = 233.9 Hz), 145.08 (d, J = 1.9 Hz), 137.19, 131.19, 128.44, 128.33, 127.95, 125.50 (d, J = 13.1 Hz), 125.34, 122.99, 115.54 (d, J = 20.6 Hz), 111.52, 111.39 (d, J = 6.6 Hz), 25.05. 19F NMR (470 MHz, DMSO-d6) δ −138.12. HRMS (ESI+): calcd for C17H15FN3O (M + H)+ 296.1194, found 296.1191.
N-(5-(2,3-Dihydrobenzo[b][1,4]dioxine-6-carboxamido)-2-fluorophenyl)-2-methylquinoline-6-carboxamide 13
To a suspension of 2,3-dihydrobenzo[b][1,4]dioxine-6-carboxylic acid (12.7 g, 70.5 mmol) in anhydrous DCM (100 mL), a catalytic amount of anhydrous DMF (6.16 μL, 0.080 mmol) and oxalyl chloride (6.51 mL, 77.0 mmol) were added dropwise, and the resulting green solution was allowed to stir at room temperature for 3 h, after which time, the reaction mixture was concentrated in vacuo to afford a dry pale-green solid. The solid was dissolved in pyridine (100 mL), and N-(5-amino-2-fluorophenyl)-2-methylquinoline-6-carboxamide (9.46 g, 32.0 mmol) was added in one portion. The resulting dark-yellow suspension was allowed to stir for 2 h and was then poured onto water (100 mL). The yellow precipitate was filtered and washed with water (3 × 20 mL), diethyl ether (3 × 20 mL), and DCM (10 mL) to afford the crude product as a pale-yellow solid, which was taken directly onto the next step without purification (12.5 g, 85%). 1H NMR (500 MHz, DMSO-d6): δ 10.38 (s, 1H), 10.19 (s, 1H), 8.62 (d, J = 1.7 Hz, 1H), 8.41 (d, J = 8.8 Hz, 1H), 8.23 (dd, J = 8.8, 1.9 Hz, 1H), 8.13 (dd, J = 7.0, 2.5 Hz, 1H), 8.03 (d, J = 8.8 Hz, 1H), 7.68–7.62 (m, 1H), 7.57–7.46 (m, 3H), 7.29 (app t, J = 9.5, 1H), 6.99 (d, J = 8.4 Hz, 1H), 4.34–4.28 (m, 4H), 2.71 (s, 3H). 13C NMR (126 MHz, DMSO-d6) δ 165.09, 164.48, 160.95, 151.83 (d, J = 243.4 Hz), 148.57, 146.45, 142.95, 137.22, 135.42 (d, J = 2.0 Hz), 130.87, 128.50, 128.41, 127.94, 127.47, 125.39 (d, J = 9.5 Hz), 125.35, 123.05, 121.25, 118.81, 118.75 (d, J = 13.0 Hz), 116.89, 116.69, 115.56 (d, J = 21.2 Hz), 64.41, 64.03, 25.06. 19F NMR (470 MHz, DMSO-d6) δ −126.65. HRMS (ESI+): calcd for C26H21FN3O4 (M + H)+ 458.1511, found 458.1499.
N-(5-(2,3-Dihydrobenzo[b][1,4]dioxine-6-carboxamido)-2-fluorophenyl)-2-(piperazin-1-ylmethyl)quinoline-6-carboxamide 14
A solution of N-(5-(2,3-dihydrobenzo[b][1,4]dioxine-6-carboxamido)-2-fluorophenyl)-2-methylquinoline-6-carboxamide 13 (5.00 g, 10.9 mmol) and selenium dioxide (1.33 g, 12.0 mmol) in anhydrous DMF (40 mL) and 1,4-dioxane (120 mL) was heated at reflux for 1 h. Then the reaction mixture was allowed to cool to room temperature, diluted with DCM (20 mL), and filtered through a pad of Celite. The filtrate was concentrated under vacuum (using a heptane/EtOAc azeotrope to remove DMF) to afford the crude product as a yellow solid, which was carried onto the next step without further purification (5.15 g).
A solution of N-(5-(2,3-dihydrobenzo[b][1,4]dioxine-6-carboxamido)-2-fluorophenyl)-2-formylquinoline-6-carboxamide (255 mg, 0.541 mmol) and tert-butyl piperazine-1-carboxylate (302 mg, 1.62 mmol) in anhydrous DCM (5 mL) was allowed to stir at 20 °C for 12 h, after which time sodium triacetoxyborohydride (344 mg, 1.62 mmol) was added in one portion and the resulting mixture was allowed to stir at 20 °C for 2 h. The reaction was quenched with NaHCO3 saturated aqueous solution (5 mL) and extracted with a DCM:MeOH, 9:1 mixture (3 × 5 mL). The crude product (pale-yellow solid) was purified by column chromatography on silica gel using a gradient of 0–6% MeOH in DCM, followed by trituration in diethyl ether to afford the desired product as a pale-beige solid (325 mg, 94%).
To a suspension of tert-butyl 4-((6-((5-(2,3-dihydrobenzo[b][1,4]dioxine-6-carboxamido)-2-fluorophenyl)carbamoyl)quinolin-2-yl)methyl)piperazine-1-carboxylate (300 mg, 0.468 mmol) in anhydrous DCM (5 mL), TFA (0.179 mL, 2.33 mmol) was added dropwise, and the resulting mixture was allowed to stir at 20 °C for 3 h. The reaction mixture was concentrated under reduced pressure to afford the crude product as a light-brown oil. The crude was purified by column chromatography on silica gel using a gradient of 0–15% MeOH in DCM, followed by trituration in diethyl ether to afford the title compound as a white solid (174 mg, 69%). 1H NMR (500 MHz, DMSO-d6): δ 10.42 (s, 1H), 10.20 (s, 1H), 8.73 (br s, 1H), 8.67 (d, J = 1.66 Hz, 1H), 8.52 (d, J = 8.71 Hz, 1H), 8.27 (dd, J = 8.71, 1.66 Hz, 1H), 8.15 (dd, J = 7.08, 2.72 Hz, 1H), 8.09 (d, J = 8.71 Hz, 1H), 7.74 (d, J = 8.42 Hz, 1H), 7.66–7.61 (m, 1H), 7.54 (d, J = 2.11 Hz, 1H), 7.52 (dd, J = 8.42, 2.11 Hz, 1H), 7.30 (app t, J = 10.10 Hz, 1H), 6.99 (d, J = 8.42 Hz, 1H), 4.35–4.27 (m, 4H), 3.90 (s, 2H), 3.19–3.09 (m, 4H), 2.76–2.66 (m, 4H). 13C NMR (126 MHz, DMSO-d6) δ 164.98, 164.50, 160.59, 151.87 (d, J = 243.8 Hz), 148.27, 146.47, 142.96, 137.76, 135.45 (d, J = 2.7 Hz), 131.45, 128.79, 128.57, 128.13, 127.46, 126.25, 125.30 (d, J = 13.2 Hz), 121.95, 121.26, 118.86, 118.78 (d, J = 7.8 Hz), 116.90, 116.70, 115.59 (d, J = 20.9 Hz), 64.42, 64.04, 63.67, 49.45, 43.03. HRMS (ESI+): calcd for C30H29FN5O4 (M + H)+ 542.2198, found 542.2190.
3-(2-(2-((2-(2,6-Dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)oxy)acetamido)ethoxy)propanoic Acid 15
tert-Butyl 3-(2-(2-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)oxy)acetamido)ethoxy)propanoate (29 mg, 0.06 mmol) and formic acid (0.22 mL, 5.76 mmol) were dissolved in anhydrous DCM (0.29 mL) at 40 °C for 6 h. The reaction mixture was concentrated in vacuo and the resulting residue purified by reverse phase Biotage chromatography using a gradient of 10–80% MeOH in water + 0.1% formic acid to afford the title compound as an amorphous white solid (24 mg, 93%). 1H NMR (500 MHz, DMSO-d6) δ 12.21 (br s, 1H), 11.12 (s, 1H), 8.01 (t, J = 5.6 Hz, 1H), 7.81 (dd, J = 8.5, 7.3 Hz, 1H), 7.50 (d, J = 7.2 Hz, 1H), 7.39 (d, J = 8.4 Hz, 1H), 5.11 (dd, J = 12.9, 5.4 Hz, 1H), 4.79 (s, 2H), 3.60 (t, J = 6.4 Hz, 2H), 3.44 (t, J = 5.7 Hz, 2H), 3.30 (q, J = 5.8 Hz, 2H, partially obscured by water peak), 2.94–2.82 (m, 1H), 2.64–2.51 (m, 2H), 2.48–2.41 (m, 2H), 2.08–1.99 (m, 1H). 13C NMR (126 MHz, DMSO-d6) δ 172.80, 172.69, 169.91, 166.92, 166.77, 165.48, 155.00, 136.98, 133.06, 120.36, 116.78, 116.07, 68.58, 67.49, 66.06, 48.84, 38.32, 34.67, 30.98, 22.01. HRMS (ESI+): calcd for C20H21N3O9 (M + H)+ 448.1356, found 448.1351.
N-(2-Chloro-5-nitrophenyl)-2-methylquinoline-6-carboxamide
2-Methylquinoline-6-carboxylic acid (1.50 g, 8.01 mmol) was suspended in anhydrous DCM (40 mL) under inert atmosphere. DMF (1.40 μL, 0.018 mmol) and oxalyl chloride (0.74 mL, 8.74 mmol) were added dropwise, and the resulting green solution was allowed to stir at 20 °C for 3 h, after which time it was concentrated in vacuo to afford a dry pale-green solid. The solid was dissolved in pyridine (40.0 mL) and 2-chloro-5-nitroaniline 17 (1.26 g, 7.28 mmol) was added in one portion. The resulting dark-yellow suspension was allowed to stir for 2 h, then it was poured onto water and the yellow precipitate was filtered and washed several times with water, Et2O, and finally with a minimum amount of DCM to afford the crude product as a yellow amorphous solid which was used without further purification (2.20 g, 88%). 1H NMR (500 MHz, DMSO-d6): δ 10.59 (s, 1H), 8.65 (d, J = 2.0 Hz, 1H), 8.60 (d, J = 2.7 Hz, 1H), 8.44 (d, J = 8.6 Hz, 1H), 8.25 (dd, J = 8.6, 2.0 Hz, 1H), 8.15 (dd, J = 8.6, 2.7 Hz, 1H), 8.06 (d, J = 8.4 Hz, 1H), 7.91 (d, J = 8.6 Hz, 1H), 7.55 (d, J = 8.0 Hz, 1H), 2.71 (s, 3H). HRMS (ESI+): found [M + H]+ 342.0646, C17H1335ClN3O3 requires 342.0640.
N-(2-Chloro-5-(2,3-dihydrobenzo[b][1,4]dioxine-6-carboxamido)phenyl)-2-((4-(2-(2-(2-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)oxy)ethoxy)ethoxy)ethyl)piperazin-1-yl)methyl)quinoline-6-carboxamide 16 (CCT367766)
2-(2,6-Dioxo-3-piperidyl)-4-hydroxy-isoindoline-1,3-dione 2 (29 mg, 0.100 mmol) was dissolved in anhydrous THF (1 mL) under argon and then triphenylphosphine (29 mg, 0.110 mmol) was added and N-(2-chloro-5-(2,3-dihydro-1,4-benzodioxine-6-carbonylamino)phenyl)-2-((4-(2-(2-(2-hydroxyethoxy)ethoxy)ethyl)piperazin-1-yl)methyl)quinoline-6-carboxamide 20 (72.00 mg, 0.100 mmol) were added, followed by tert-butyl (NE)-N-tert-butoxycarbonyliminocarbamate (25 mg, 0.110 mmol). The reaction was stirred at room temperature for 2 h. The crude product was purified by Biotage chromatography using a gradient of 0–30% MeOH in DCM to afford the product as a pale-yellow amorphous solid (27 mg, 27%). 1H NMR (500 MHz, DMSO-d6) δ 11.13 (s, 1H), 10.34 (s, 1H), 10.28 (s, 1H), 8.67 (d, J = 2.1 Hz, 1H), 8.50 (d, J = 8.5 Hz, 1H), 8.27 (dd, J = 8.7, 2.0 Hz, 1H), 8.16 (d, J = 2.5 Hz, 1H), 8.10 (d, J = 8.8 Hz, 1H), 7.80 (dd, J = 8.5, 7.3 Hz, 1H), 7.75 (dd, J = 8.8, 2.5 Hz, 1H), 7.72 (d, J = 8.5 Hz, 1H), 7.57–7.52 (m, 4H), 7.45 (d, J = 7.2 Hz, 1H), 7.01 (d, J = 8.4 Hz, 1H), 5.10 (dd, J = 12.8, 5.4 Hz, 1H), 4.38–4.28 (m, 6H), 3.87–3.76 (m, 4H), 3.65 (d, J = 5.0 Hz, 2H), 3.54 (s, 4H), 2.89 (ddd, J = 16.9, 13.8, 5.4 Hz, 1H), 2.69–2.32 (m, 12H), 2.08–1.99 (m, 1H). 13C NMR (126 MHz, DMSO-d6) δ 172.78, 169.93, 166.79, 165.27, 164.98, 164.65, 155.83, 148.27, 146.58, 142.97, 138.59, 137.56, 136.97, 134.88, 133.23, 131.37, 129.37, 128.83, 128.42, 127.90, 127.30, 126.23, 123.55, 121.82, 121.33, 119.99, 119.82, 119.24, 116.93, 116.75, 116.29, 115.39, 79.18, 70.10, 69.70, 68.90, 68.66, 64.42, 64.03, 57.22, 53.03, 48.75, 30.96, 22.02 (1 signal not observed/overlapping signals). HRMS (ESI+): calcd for C49H4935ClN7O11 (M + H)+ 947.3173, found 947.3173.
N-(2-Chloro-5-(2,3-dihydrobenzo[b][1,4]dioxine-6-carboxamido)phenyl)-2-methylquinoline-6-carboxamide 18
2,3-Dihydrobenzo[b][1,4]dioxine-6-carboxylic acid (1.27 g, 7.06 mmol) was suspended in anhydrous DCM (20 mL) under inert atmosphere. DMF (1.23 μL, 0.016 mmol) and oxalyl chloride (0.65 mL, 7.70 mmol) were added dropwise, and the resulting green solution was allowed to stir at 20 °C for 3 h, after which time it was concentrated under vacuum to afford a dry pale-green solid. The solid was dissolved in pyridine (20 mL), and N-(5-amino-2-chlorophenyl)-2-methylquinoline-6-carboxamide 24 (2.00 g, 6.42 mmol) was added in one portion. The resulting dark-yellow suspension was allowed to stir for 2 h, then it was poured onto water and the yellow precipitate was filtered and washed several times with water, Et2O, and finally with a minimum amount of DCM to afford the crude product as a pale-yellow amorphous solid, which was carried onto the next step without purification (1.86 g, 61%). 1H NMR (500 MHz, DMSO-d6): δ 10.31 (s, 1H), 10.27 (s, 1H), 8.63 (d, J = 1.5 Hz, 1H), 8.43 (d, J = 8.8 Hz, 1H), 8.25 (dd, J = 8.8, 2.2 Hz, 1H), 8.14 (d, J = 2.2 Hz, 1H), 8.04 (d, J = 8.8 Hz, 1H), 7.75 (dd, J = 8.8, 2.9 Hz, 1H), 7.58–7.49 (m, 4H), 7.00 (d, J = 8.8 Hz, 1H), 4.37–4.26 (m, 4H), 2.71 (s, 3H). HRMS (ESI+): found [M + H]+ 474.1210, C26H2135ClN3O4 requires 474.1215.
N-(2-Chloro-5-(2,3-dihydrobenzo[b][1,4]dioxine-6-carboxamido)phenyl)-2-(piperazin-1-ylmethyl)quinoline-6-carboxamide 19
N-[2-Chloro-5-(2,3-dihydro-1,4-benzodioxine-6-carbonylamino)phenyl)-2-methyl-quinoline-6-carboxamide 18 (4.00 g, 8.44 mmol) was taken up in anhydrous DMF (20.00 mL) and anhydrous 1,4-dioxane (20 mL). Selenium dioxide (1.03 g, 9.28 mmol) was added, and the reaction was degassed via 3× vacuum/nitrogen cycles. The reaction mixture was heated to 50 °C and stirred for 16 h. Then, the reaction was filtered through Celite, eluting with 1:1 DCM/EtOAc and concentrated in vacuo. Remaining DMF was removed by azeotrope with EtOAc/heptane to afford N-(2-chloro-5-(2,3-dihydrobenzo[b][1,4]dioxine-6-carboxamido)phenyl)-2-formylquinoline-6-carboxamide as a brown solid. The crude material was used directly in the next step, assuming 100% yield, without further purification. Sodium cyanoborohydride (2.12 g, 33.8 mmol) was added to a stirring suspension of N-(2-chloro-5-(2,3-dihydro-1,4-benzodioxine-6-carbonylamino)phenyl)-2-formyl-quinoline-6-carboxamide (4.12 g, 8.44 mmol) and 1-Boc-piperazine (3.15 g, 16.89 mmol) in anhydrous DMF (60 mL) at 0 °C under nitrogen. Then acetic acid (0.53 mL, 9.29 mmol) was added. The reaction mixture was allowed to warm to room temperature and stirred overnight. A bleach bubbler was used to vent the reaction. The reaction was quenched by slow addition of an aqueous solution of 1 M NaOH (50 mL), and the reaction mixture concentrated in vacuo by using heptane for azeotropic removal of DMF. The resulting residue was taken up in a small amount of MeOH and poured into a large volume of water (300–400 mL). The precipitate formed was collected by filtration, washed with water, then Et2O, and dried under vacuum overnight to afford the crude tert-butyl 4-((6-((2-chloro-5-(2,3-dihydrobenzo[b][1,4]dioxine-6-carboxamido)phenyl)carbamoyl)quinolin-2-yl)methyl)piperazine-1-carboxylate as a pale-brown solid (4.57 g). This material was used directly in the next reaction without further purification.
To a solution of tert-butyl 4-((6-((2-chloro-5-(2,3-dihydrobenzo[b][1,4]dioxine-6-carboxamido)phenyl)carbamoyl)quinolin-2-yl)methyl)piperazine-1-carboxylate (4.57 g, 6.94 mmol) in anhydrous MeOH (60 mL) at 0 °C under argon was added 4 M HCl in dioxane (26.1 mL, 104.4 mmol). Upon addition of the acid, the reaction mixture became darker red/brown in color. The reaction was allowed to warm to room temperature and stirred overnight. Then the solvents were removed in vacuo. The resulting residue was suspended in a small amount of MeOH. The suspension was poured into a large volume of water (with stirring), and the precipitate formed was collected by filtration, washed well with water, then Et2O, and dried under vacuum to afford the crude product as a brown solid (3.42 g). Purification by Biotage column chromatography using a gradient of 0–10% MeOH in DCM + 1% 7N NH3 in MeOH afforded the title compound as a dark-yellow amorphous solid (1.53 g, 32% over 3 steps). 1H NMR (500 MHz, DMSO-d6) δ 10.32 (s, 1H), 10.27 (s, 1H), 8.65 (d, J = 2.0 Hz, 1H), 8.49 (d, J = 8.4 Hz, 1H), 8.26 (dd, J = 8.8, 2.0 Hz, 1H), 8.15 (d, J = 2.5 Hz, 1H), 8.09 (d, J = 8.8 Hz, 1H), 7.76–7.71 (m, 2H), 7.58–7.47 (m, 3H), 7.00 (d, J = 8.4 Hz, 1H), 4.40–4.23 (m, 4H), 3.76 (s, 2H), 2.74 (t, J = 4.8 Hz, 4H), 2.47–2.42 (br m, 4H) (1 proton missing). 13C NMR (126 MHz, DMSO-d6) δ 165.00, 164.64, 161.79, 148.29, 146.57, 142.97, 138.58, 137.45, 134.89, 131.32, 129.36, 128.81, 128.39, 127.85, 127.31, 126.21, 123.54, 121.85, 121.32, 119.81, 119.23, 116.92, 116.74, 65.11, 64.42, 64.03, 54.26, 45.54. HRMS (ESI+): calcd for C30H2935ClN5O4 (M + H)+ 558.1903, found 558.1885.
2-(2-(2-(4-((6-((2-Chloro-5-(2,3-dihydrobenzo[b][1,4]dioxine-6-carboxamido)phenyl)carbamoyl)quinolin-2-yl)methyl)piperazin-1-yl)ethoxy)ethoxy)ethyl 4-Methylbenzenesulfonate 20
N-(2-Chloro-5-(2,3-dihydrobenzo[b][1,4]dioxine-6-carboxamido)phenyl)-2-(piperazin-1-ylmethyl)quinoline-6-carboxamide 19 (500 mg, 0.90 mmol) was dissolved in anhydrous DMF (8 mL) under nitrogen at room temperature. K2CO3 (372 mg, 2.69 mmol) was added, followed by a solution of 2-(2-(2-hydroxyethoxy)ethoxy)ethyl 4-methylbenzenesulfonate (573 mg, 1.88 mmol) in anhydrous DMF (2 mL), and the reaction stirred at room temperature overnight. Further 2-(2-(2-hydroxyethoxy)ethoxy)ethyl 4-methylbenzenesulfonate (286 mg, 0.94 mmol) in anhydrous DMF (1 mL) was added and the reaction stirred overnight. The reaction mixture was poured into water and the aqueous layer extracted three times with 10% MeOH in DCM. The combined organic layer was washed with brine, dried (Na2SO4), and concentrated in vacuo. EtOAc/heptane was added to remove remaining traces of DMF by azeotrope. The crude oil was purified by Biotage chromatography using a gradient of 0–10% MeOH in DCM + 1% 7N NH3 in MeOH to afford the title compound as a yellow amorphous solid (297 mg, 48%). 1H NMR (500 MHz, CDCl3) δ 8.63 (s, 1H), 8.59 (d, J = 2.5 Hz, 1H), 8.42 (d, J = 1.9 Hz, 1H), 8.28 (d, J = 8.5 Hz, 1H), 8.21 (d, J = 8.8 Hz, 1H), 8.17 (dd, J = 8.8, 2.0 Hz, 1H), 7.98 (dd, J = 8.8, 2.5 Hz, 1H), 7.95 (s, 1H), 7.76 (d, J = 8.5 Hz, 1H), 7.47 (s, 1H), 7.45 (d, J = 6.6 Hz, 1H), 7.40 (dd, J = 8.4, 2.2 Hz, 1H), 6.97 (d, J = 8.4 Hz, 1H), 4.42–4.43 (m, 4H), 3.90 (s, 2H), 3.78–3.71 (m, 2H), 3.71–3.66 (m, 2H), 3.66–3.59 (m, 6H), 2.74–2.56 (m, 10H). 13C NMR (126 MHz, CDCl3) δ 165.20, 165.02, 162.36, 149.26, 147.08, 143.70, 138.10, 137.49, 134.77, 131.89, 130.26, 129.63, 127.91, 127.88, 126.89, 122.42, 120.64, 117.96, 117.57, 117.05, 116.93, 112.63, 72.76, 70.49, 70.44, 68.81, 65.16, 64.70, 64.32, 61.83, 57.90, 53.69, 53.39 (1 signal not observed/overlapping signals). HRMS (ESI+): calcd for C36H4135ClN5O7 (M + H)+ 690.2689, found 690.2692.
N-(4-Chloro-3-nitrophenyl)-2,3-dihydrobenzo[b][1,4]dioxine-5-carboxamide
2,3-Dihydro-1,4-benzodioxine-5-carboxylic acid (1.73 g, 9.61 mmol) was suspended in anhydrous DCM (45 mL), and 3 drops of anhydrous DMF were added. Oxalyl chloride (0.81 mL, 9.61 mmol) was added dropwise (effervescence observed) and the reaction stirred at room temperature under nitrogen for 2 h. After this time, the effervescence had ceased and the reaction mixture had become fully dissolved. Then the solvents were removed in vacuo. Anhydrous DCM (10 mL) was added and the reaction concentrated again. The residue was taken up in anhydrous DCM (5 mL, then 1 mL to rinse flask) and added slowly to a solution of 4-chloro-3-nitro-aniline 23 (1.11 g, 6.41 mmol) and pyridine (1.55 mL, 19.22 mmol) in anhydrous DCM (45 mL). The reaction was stirred at room temperature under nitrogen for 48 h. The solvent was removed in vacuo and the resulting residue partitioned between saturated aqueous NaHCO3 solution and 10% MeOH in DCM (30 mL). The aqueous layer was extracted with two further portions of 10% MeOH in DCM (30 mL), the combined organic layer was washed with water, brine, dried (Na2SO4) and concentrated in vacuo, and then dried under high vacuum to afford the crude product as a pale-brown solid (2.24 g).
However, this material was contaminated with 2,3-dihydro-1,4-benzodioxine-5-carboxylic acid, therefore the solid was suspended in a small volume of MeOH and diluted with saturated aqueous NaHCO3 solution. The suspension was vigorously stirred for 1 h, after which time the precipitate was collected by filtration. The precipitate was washed with copious amounts of water, followed by a small portion of Et2O, then dried under vacuum to afford the product as a beige amorphous solid (1.82 g, 84%). 1H NMR (500 MHz, DMSO-d6) δ 10.62 (s, 1H), 8.55 (d, J = 2.5 Hz, 1H), 7.95 (dd, J = 8.8, 2.5 Hz, 1H), 7.75 (d, J = 8.8 Hz, 1H), 7.15 (dd, J = 7.6, 1.6 Hz, 1H), 7.05 (dd, J = 8.1, 1.6 Hz, 1H), 6.94 (t, J = 7.8 Hz, 1H), 4.39–4.26 (m, 4H). 13C NMR (126 MHz, DMSO-d6) δ 164.71, 147.29, 143.70, 141.31, 138.82, 132.01, 124.79, 124.51, 121.25, 120.84, 119.59, 118.73, 115.87, 64.52, 63.77. HRMS (ESI+): calcd for C15H1235ClN2O5 (M + H)+ 335.0429, found 335.0413.
N-(3-Amino-4-chlorophenyl)-2,3-dihydrobenzo[b][1,4]dioxine-5-carboxamide
N-(4-Chloro-3-nitro-phenyl)-2,3-dihydro-1,4-benzodioxine-5-carboxamide (1.50 g, 4.48 mmol) was suspended in a mixture of DCM (18 mL) and EtOH (9 mL). Then 10% Pd/C (200 mg) was added and the reaction mixture stirred for 3 days under 1 atm H2. The reaction mixture was filtered through Celite, eluting with 10% MeOH in DCM. The filtrate was concentrated in vacuo and dried under high vacuum to afford the crude product as a pale-brown solid (1.13 g, 93% purity, 77%), which was used directly in the next reaction. A portion of this material (320 mg) was purified by Biotage chromatography using a gradient of 0–5% EtOAc in DCM to afford the title compound as a pale-yellow amorphous solid (198 mg, 52% based on starting material). 1H NMR (500 MHz, DMSO-d6) δ 9.93 (s, 1H), 7.35 (d, J = 2.2 Hz, 1H), 7.10 (d, J = 8.8 Hz, 2H), 6.99 (dd, J = 8.0, 1.5 Hz, 1H), 6.91 (t, J = 7.8 Hz, 1H), 6.81 (dd, J = 8.6, 2.3 Hz, 1H), 5.37 (s, 2H), 4.39–4.32 (m, 2H), 4.31–4.27 (m, 2H). 13C NMR (126 MHz, DMSO-d6) δ 163.77, 144.72, 143.63, 141.13, 138.48, 128.82, 125.80, 121.21, 120.70, 118.98, 111.77, 108.78, 106.29, 64.43, 63.74. HRMS (ESI+): calcd for C15H1435ClN2O3 (M + H)+ 305.0687, found 305.0688.
N-(2-Chloro-5-(2,3-dihydrobenzo[b][1,4]dioxine-5-carboxamido)phenyl)-2-methylquinoline-6-carboxamide
2-Methylquinoline-6-carboxylic acid (685 mg, 3.66 mmol) was dissolved in anhydrous DCM (24 mL) at room temperature under inert atmosphere and 1 drop anhydrous DMF added. Then oxalyl chloride (0.31 mL, 3.66 mmol) was added dropwise (effervescence observed) and the reaction stirred at room temperature for 2 h, over which time effervescence ceased and the solution became dark green. The solvent was removed in vacuo. The resulting residue was redissolved in anhydrous DCM (2 mL) and concentrated in vacuo (×2). The solid was taken up in anhydrous DCM (1 mL, then 1 mL to rinse flask) and slowly added to a stirring solution of N-(3-amino-4-chloro-phenyl)-2,3-dihydro-1,4-benzodioxine-5-carboxamide (800.00 mg, 2.44 mmol) in anhydrous pyridine (18 mL) at room temperature under inert atmosphere. The reaction was stirred overnight. A precipitate formed which was isolated by filtration, washed with water (×2) and ether, and then dried under vacuum to afford the product as a beige amorphous solid (672 mg, 58%). 1H NMR (500 MHz, DMSO-d6) δ 10.33 (s, 1H), 10.32 (s, 1H), 8.63 (d, J = 1.8 Hz, 1H), 8.42 (d, J = 8.5 Hz, 1H), 8.25 (dd, J = 8.8, 2.0 Hz, 1H), 8.07 (d, J = 2.3 Hz, 1H), 8.04 (d, J = 8.8 Hz, 1H), 7.68 (dd, J = 8.8, 2.4 Hz, 1H), 7.54 (dd, J = 8.6, 2.8 Hz, 2H), 7.15 (dd, J = 7.6, 1.5 Hz, 1H), 7.02 (dd, J = 8.0, 1.6 Hz, 1H), 6.93 (t, J = 7.8 Hz, 1H), 4.37 (dd, J = 5.1, 2.4 Hz, 2H), 4.31 (dd, J = 5.0, 2.7 Hz, 2H), 2.71 (s, 3H). 13C NMR (126 MHz, DMSO-d6) δ 165.07, 164.24, 160.97, 148.57, 143.67, 141.20, 138.27, 137.25, 135.12, 130.87, 129.53, 128.46, 128.44, 127.85, 125.51, 125.39, 123.79, 123.07, 121.19, 120.76, 119.31, 119.19, 118.67, 64.47, 63.77, 25.05. HRMS (ESI+): calcd for C26H2135ClN3O4 (M + H)+ 474.1215, found 474.1192.
N-(2-Chloro-5-(2,3-dihydrobenzo[b][1,4]dioxine-5-carboxamido)phenyl)-2-(piperazin-1-ylmethyl)quinoline-6-carboxamide
N-(2-Chloro-5-(2,3-dihydro-1,4-benzodioxine-5-carbonylamino)phenyl)-2-methyl-quinoline-6-carboxamide (500 mg, 1.06 mmol) was taken up in anhydrous DMF (5 mL) and anhydrous 1,4-dioxane (5 mL). Selenium dioxide (128 mg, 1.16 mmol) was added, the reaction was degassed via 3× vacuum/nitrogen cycles, and stirred at 50 °C for 5 h. The reaction mixture was filtered through Celite (eluting with 1:1 DCM/EtOAc), then concentrated in vacuo, using EtOAc/heptane to remove traces of DMF. This material was used directly in the next reaction without further purification, assuming 100% yield.
N-[2-Chloro-5-(2,3-dihydro-1,4-benzodioxine-5-carbonylamino)phenyl]-2-formyl-quinoline-6-carboxamide (0.51 g, 1.06 mmol) and 1-Boc-piperazine (392 mg, 2.11 mmol) were dissolved in anhydrous DMF (10 mL) at 0 °C under nitrogen. Then sodium cyanoborohydride (265 mg, 4.22 mmol) was added in one portion, followed by acetic acid (0.07 mL, 1.16 mmol). A bleach bubbler was used to vent the reaction. The reaction was allowed to warm to room temperature and stirred overnight. The reaction was quenched by addition of 1 M NaOH (aq) and then concentrated in vacuo. The resulting residue was suspended in MeOH and diluted with water to afford a brown precipitate. The precipitate was collected by filtration and washed well with water, followed by Et2O, and dried under vacuum to afford the product as a pale-brown solid (520 mg). This material was used directly in the next reaction without further purification.
tert-Butyl 4-((6-((2-chloro-5-(2,3-dihydro-1,4-benzodioxine-5 carbonylamino)phenyl)carbamoyl)-2-quinolyl)methyl)piperazine-1-carboxylate (508 mg, 0.770 mmol) was dissolved in anhydrous MeOH (7 mL) at 0 °C under inert atmosphere. Then 4 M HCl in dioxane (2.90 mL, 11.61 mmol) was added and the reaction warmed to room temperature. After 1.5 h, a further 4 M HCl in dioxane (2.90 mL, 11.61 mmol) was added and the reaction stirred overnight. Then, the reaction mixture was concentrated in vacuo. The resulting residue was taken up in 1:1 MeOH/DCM and subjected to Isolute Flash SCX-II chromatography to afford the free salt, eluting first with MeOH followed by 10% 7N NH3 in MeOH. This material was further purified by Biotage chromatography using a gradient of 0–10% MeOH in DCM + 1% 7N NH3 in MeOH to afford the title compound as a pale-brown amorphous solid (253 mg, 43% over 3 steps). 1H NMR (500 MHz, DMSO-d6) δ 10.35 (s, 1H), 10.32 (s, 1H), 8.71–8.54 (m, 1H), 8.49 (d, J = 8.5 Hz, 1H), 8.26 (dd, J = 8.8, 1.8 Hz, 1H), 8.09 (app. d, J = 8.6 Hz, 2H), 7.74 (d, J = 8.5 Hz, 1H), 7.68 (dd, J = 8.8, 2.3 Hz, 1H), 7.54 (d, J = 8.8 Hz, 1H), 7.14 (dd, J = 7.6, 1.3 Hz, 1H), 7.02 (dd, J = 8.0, 1.5 Hz, 1H), 6.93 (t, J = 7.8 Hz, 1H), 4.42–4.34 (m, 2H), 4.33–4.29 (m, 2H), 3.77 (s, 2H), 2.76 (t, J = 4.5 Hz, 4H), 2.42 (br s, 4H) (1 proton missing). 13C NMR (126 MHz, DMSO-d6) δ 165.00, 164.24, 161.72, 148.30, 143.67, 141.20, 138.27, 137.48, 135.08, 131.28, 129.54, 128.81, 128.44, 127.88, 126.22, 125.50, 123.78, 121.86, 121.19, 120.76, 119.32, 119.19, 118.70, 65.02, 64.47, 63.77, 53.94, 48.60, 45.37 (1 missing/overlapping signal). HRMS (ESI+): calcd for C30H2935ClN5O4 (M + H)+ 558.1903, found 558.1890.
N-(2-Chloro-5-(2,3-dihydrobenzo[b][1,4]dioxine-5-carboxamido)phenyl)-2-((4-(2-(2-(2-hydroxyethoxy)ethoxy)ethyl)piperazin-1-yl)methyl)quinoline-6-carboxamide
N-(2-Chloro-5-(2,3-dihydro-1,4-benzodioxine-5-carbonylamino)phenyl)-2-(piperazin-1-ylmethyl)quinoline-6-carboxamide (248 mg, 0.440 mmol) was dissolved in anhydrous DMF (3 mL) at room temperature under inert atmosphere, and K2CO3 (371 mg, 2.69 mmol) was added. Then a solution of 2-(2-(2 hydroxyethoxy)ethoxy)ethyl 4-methylbenzenesulfonate (573 mg, 1.88 mmol) in anhydrous DMF (1 mL) was added and reaction stirred overnight. The reaction was diluted with water and the aqueous layer extracted (3 × 10% MeOH in DCM). The combined organic layer was washed with brine and dried (Na2SO4). Purification by Biotage chromatography using a gradient of 0–10% MeOH in DCM + 1% 7N NH3 in MeOH gave the title compound as a pale-yellow amorphous solid (108 mg, 35%). 1H NMR (500 MHz, CDCl3) δ 9.59 (s, 1H), 8.63 (s, 1H), 8.54 (d, J = 2.5 Hz, 1H), 8.42 (d, J = 1.6 Hz, 1H), 8.27 (d, J = 8.5 Hz, 1H), 8.21 (d, J = 8.8 Hz, 1H), 8.17 (dd, J = 8.8, 1.9 Hz, 1H), 8.00 (dd, J = 8.8, 2.5 Hz, 1H), 7.81 (dd, J = 7.8, 1.7 Hz, 1H), 7.75 (d, J = 8.5 Hz, 1H), 7.43 (d, J = 8.8 Hz, 1H), 7.07 (dd, J = 8.0, 1.7 Hz, 1H), 6.99 (t, J = 7.9 Hz, 1H), 4.56 (dd, J = 5.0, 3.1 Hz, 2H), 4.38 (dd, J = 5.0, 3.1 Hz, 2H), 3.89 (s, 2H), 3.77–3.71 (m, 2H), 3.70–3.58 (m, 8H), 2.73–2.53 (m, 10H) (1 proton missing). 13C NMR (126 MHz, CDCl3) δ 164.98, 163.11, 162.34, 149.27, 143.81, 141.99, 138.12, 137.44, 134.75, 131.96, 130.31, 129.55, 127.80, 126.89, 124.52, 122.41, 122.16, 121.76, 121.56, 118.01, 117.66, 113.13, 72.74, 70.48, 70.44, 68.82, 65.46, 65.14, 63.75, 61.82, 57.87, 53.67, 53.36 (1 overlapping/missing signal). HRMS (ESI+): calcd for C36H4135ClN5O7 (M + H)+ 690.2689, found 690.2696.
N-(2-Chloro-5-(2,3-dihydrobenzo[b][1,4]dioxine-5-carboxamido)phenyl)-2-((4-(2-(2-(2-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)oxy)ethoxy)ethoxy)ethyl)piperazin-1-yl)methyl)quinoline-6-carboxamide 21
2-(2,6-Dioxo-3-piperidyl)-4-hydroxy-isoindoline-1,3-dione (19.87 mg, 0.070 mmol) was dissolved in anhydrous THF (0.72 mL) under inert atmosphere, and then triphenylphosphine (19.95 mg, 0.080 mmol) and tert-butyl (NE)-N-tert-butoxycarbonyliminocarbamate (17.51 mg, 0.080 mmol) were added, followed by N-(2-chloro-5-(2,3-dihydro-1,4-benzodioxine-5-carbonylamino)phenyl)-2-((4-(2-(2-(2-hydroxyethoxy)ethoxy]ethyl)piperazin-1-yl)methyl)quinoline-6-carboxamide (50.00 mg, 0.070 mmol). The reaction was stirred at room temperature for 2 h. The reaction mixture was preabsorbed onto silica and purified by Biotage chromatography using a gradient of 0–30% MeOH in DCM to afford the product as a pale-yellow amorphous solid (28 mg, 41%). 1H NMR (500 MHz, DMSO-d6) δ 11.12 (s, 1H), 10.35 (s, 1H), 10.32 (s, 1H), 8.65 (d, J = 2.0 Hz, 1H), 8.49 (d, J = 8.6 Hz, 1H), 8.26 (dd, J = 8.8, 2.0 Hz, 1H), 8.13–8.06 (m, 2H), 7.79 (dd, J = 8.6, 7.2 Hz, 1H), 7.75–7.65 (m, 2H), 7.53 (t, J = 8.4 Hz, 2H), 7.44 (d, J = 7.2 Hz, 1H), 7.14 (dd, J = 7.6, 1.7 Hz, 1H), 7.02 (dd, J = 8.0, 1.7 Hz, 1H), 6.93 (t, J = 7.8 Hz, 1H), 5.09 (dd, J = 12.8, 5.5 Hz, 1H), 4.42–4.26 (m, 6H), 3.85–3.73 (m, 4H), 3.64 (dd, J = 5.9, 3.7 Hz, 2H), 3.54–3.46 (dt, J = 11.8, 5.7 Hz, 4H), 2.88 (ddd, J = 16.9, 13.8, 5.4 Hz, 1H), 2.65–2.23 (m, 12H), 2.09–1.96 (m, 1H). 13C NMR (126 MHz, DMSO-d6) δ 172.76, 169.90, 166.79, 165.24, 164.98, 164.23, 161.71, 155.84, 148.28, 143.66, 141.19, 138.26, 137.51, 136.95, 135.07, 133.23, 131.27, 129.53, 128.81, 128.43, 127.87, 126.21, 125.50, 123.76, 121.79, 121.18, 120.76, 120.01, 119.30, 119.18, 118.68, 116.31, 115.37, 70.12, 69.72, 68.90, 68.67, 68.21, 64.46, 64.35, 63.76, 57.22, 54.91, 53.13, 53.01, 48.74, 40.02, 30.95, 22.01. HRMS (ESI+): calcd for C49H4935ClN7O11 (M + H)+ 946.3173, found 946.3183.
N-(2-Chloro-5-(2,3-dihydrobenzo[b][1,4]dioxine-6-carboxamido)phenyl)-2-((4-ethylpiperazin-1-yl)methyl)quinoline-6-carboxamide 22
N-(2-Chloro-5-(2,3-dihydrobenzo[b][1,4]dioxine-6-carboxamido)phenyl)-2-methylquinoline-6-carboxamide 18 (1.00 g, 2.11 mmol) was taken up in anhydrous DMF (10 mL) and anhydrous 1,4-dioxane (10 mL). Selenium dioxide (280 mg, 2.53 mmol) was added, and the reaction was degassed via 3× vacuum/nitrogen cycles and stirred at 50 °C under nitrogen for 3.5 h. Further selenium dioxide (47 mg, 0.42 mmol) was added and the reaction stirred at 50 °C for an additional 2 h. The reaction mixture was cooled, filtered through Celite (eluting with DCM/EtOAc), and concentrated in vacuo, using EtOAc/heptane azeotrope to remove DMF. This material was used directly in the next reaction without further purification, assuming 100% yield.
N-(2-Chloro-5-(2,3-dihydrobenzo[b][1,4]dioxine-6-carboxamido)phenyl)-2-formylquinoline-6-carboxamide (1.03 g, 2.11 mmol) was suspended in anhydrous DCM (20 mL) at room temperature under nitrogen, and 1-ethylpiperazine (0.80 mL, 6.33 mmol) was added. The reaction was allowed to stir overnight, and then NaBH(OAc)3 (1.34 g, 6.33 mmol) was added and stirred for 3 h. The reaction mixture was quenched with a saturated aqueous solution of NaHCO3 and the aqueous layer extracted with 10% MeOH in DCM (×3). The combined organic layer was dried (Na2SO4) and concentrated in vacuo. The residue was purified by Biotage chromatography using a gradient of 0–10% MeOH in DCM. This material was further purified by Isolute Flash SCX-II chromatography, eluting with MeOH, followed by 10% 7N NH3 in MeOH to afford the product as an amorphous yellow solid (431 mg, 35%). 1H NMR (500 MHz, DMSO-d6) δ 10.32 (s, 1H), 10.27 (s, 1H), 8.65 (d, J = 1.9 Hz, 1H), 8.50 (d, J = 8.5 Hz, 1H), 8.26 (dd, J = 8.8, 2.0 Hz, 1H), 8.15 (d, J = 2.5 Hz, 1H), 8.09 (d, J = 8.8 Hz, 1H), 7.77–7.74 (m, 1H), 7.73 (d, J = 8.7 Hz, 1H), 7.56–7.50 (m, 3H), 7.00 (d, J = 8.4 Hz, 1H), 4.34–4.26 (m, 4H), 3.80 (s, 2H), 2.60–2.25 (m, 10H), 0.99 (t, J = 7.2 Hz, 3H). 13C NMR (126 MHz, DMSO-d6) δ 165.00, 164.64, 161.70, 148.28, 146.57, 142.97, 138.58, 137.52, 134.89, 131.35, 129.35, 128.82, 128.39, 127.87, 127.31, 126.23, 123.53, 121.82, 121.32, 119.80, 119.23, 116.92, 116.74, 64.42, 64.35, 64.03, 52.94, 52.34, 51.59, 11.92. HRMS (ESI+): calcd for C32H3335ClN5O4 (M + H)+ 586.2216, found 586.2239.
N-(5-Amino-2-chlorophenyl)-2-methylquinoline-6-carboxamide 24
N-(2-Chloro-5-nitrophenyl)-2-methylquinoline-6-carboxamide was suspended in water (7 mL) and EtOH (21 mL). Ammonium chloride (2.41 g, 45.1 mmol) and iron powder (2.52 g, 45.1 mmol) were added, and the resulting suspension was allowed to stir at 90 °C for 1 h. The reaction mixture was allowed to cool to room temperature, diluted with MeOH and DCM, and filtered through a pad of Celite. The resulting filtrate was concentrated under vacuum to afford a light-brown amorphous solid as crude product, which was used directly in the next step without any further purification (2.00 g, 100%). 1H NMR (500 MHz, DMSO-d6): δ 9.96 (s, 1H), 8.58 (d, J = 2.2 Hz, 1H), 8.41 (d, J = 8.7 Hz, 1H), 8.21 (dd, J = 8.7, 2.2 Hz, 1H), 8.02 (d, J = 8.7 Hz, 1H), 7.53 (d, J = 7.6 Hz, 1H), 7.15 (d, J = 8.7 Hz, 1H), 6.87 (d, J = 2.2 Hz, 1H), 6.50 (dd, J = 8.7, 2.2 Hz, 1H), 5.41 (bs, 2H), 2.70 (s, 3H). HRMS (ESI+): found [M + H]+ 312.0902, C17H1535ClN3O requires 312.0898.
1-(6-((2-(2-(2-((2-(2,6-Dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)oxy)acetamido)ethoxy)ethyl)amino)-6-oxohexyl)-3,3-dimethyl-2-((1E,3E)-5-((E)-1,3,3-trimethyl-5-sulfoindolin-2-ylidene)penta-1,3-dien-1-yl)-3H-indol-1-ium-5-sulfonate
In the absence of light, N-(2-(2-aminoethoxy)ethyl)-2-(2-(2,6-dioxo-3-piperidyl)-1,3-dioxo-isoindolin-4-yl)oxy-acetamide hydrochloride 4 (0.60 mg, 0.0013 mmol) was added to a solution of sodium (2E)-2-((2E,4E)-5-(1-(6-(2,5-dioxopyrrolidin-1-yl)oxy-6-oxo-hexyl)-3,3-dimethyl-5-sulfonato-indol-1-ium-2-yl)penta-2,4-dienylidene)-1,3,3-trimethyl-indoline-5-sulfonate (1.00 mg, 0.0013 mmol) dissolved in 20 μL of triethylamine and 300 μL of DMF, and the deep-blue solution was left to stand for for 16 h. The crude product was then purified by semipreparative HPLC to give the desired product. LCMS (ESI+) RT = 2.59 min, 77%, M + H+ 1043.
Acknowledgments
This work was supported by Cancer Research UK grant numbers C309/A8274 and C309/A11566. We acknowledge CRUK Centre funding and NHS funding to the NIHR Biomedical Research Centre at The Institute of Cancer Research, the Royal Marsden Hospital, and funding from Cancer Research Technology Pioneer Fund and Battle Against Cancer Investment Trust. Paul Workman is Cancer Research UK Life Fellow. We would like to thank Dr. Bissan Al-Lazikani for her helpful discussions and Nicky Evans for editorial assistance.
Glossary
Abbreviations Used
- SAR
structure–activity relationships
- CETSA
cellular thermal shift assay
- ABPP
activity-based protein profiling
- PROTAC
proteolysis targeting chimera
- SNIPER
specific and nongenetic IAP-dependent protein eraser
- PDP
protein degradation probe
- SPR
surface plasmon resonance spectroscopy
- SEM
standard error of the mean
- HPLC
high-performance liquid chromatography
- KS
kinetic solubility
- tPSA
topological polar surface area
- DMSO
dimethyl sulfoxide
- HSF1
heat shock transcription factor 1
- BRD4
bromodomain-containing protein 4
- VHL
Von Hippel–Lindau disease tumor suppressor
- IAP
inhibitors of apoptosis proteins
- CRBN
protein cereblon
- PPI
protein–protein interaction
- TBAF
tetra-n-butylammonium fluoride
- HBD
hydrogen bond donor
- TMT
tandem mass tagging
- MS2
MS/MS mass spectrometry
- DTBAD
di-tert-butylazocarboxylate
- THF
tetrahydrofuran
- DCM
dichloromethane
- RT
room temperature
- HATU
1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxid hexafluorophosphate
- DIPEA
diisopropylethylamine
- DMF
dimethylformamide
- DBU
1,8-diazabicyclo(5.4.0)undec-7-ene
- SF
significant figures.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jmedchem.7b01406.
Author Contributions
N.E.A.C. and S.Y.S. contributed equally. S.Y.S. carried out in vitro cellular biology experiments. N.E.A.C and J.J.C. synthesized compounds. N.E.A.C., J.J.C., A.E.P., B.W., G.C., I.C., M.D.C., and K.J. contributed to the design of compounds. M.R. ran and designed the physicochemical and stability analysis of compounds. P.C.M. expressed and purified the recombinant proteins. B.O. analyzed proteomics data. M.R., M.S., and R.B. designed and carried out biochemical experiments. N.E.A.C., S.Y.S., P.C., P.W., M.D.C., and K.J. designed studies and interpreted results. N.E.A.C. and M.D.C. wrote the manuscript. All authors have given approval to the final version of the manuscript.
The authors declare the following competing financial interest(s): The Institute of Cancer Research has a potential financial interest in ligands of pirin and operates a Rewards to Discoverers scheme.
Supplementary Material
References
- Swinney D. C. Biochemical mechanisms of drug action: What does it take for success?. Nat. Rev. Drug Discovery 2004, 3, 801–808. 10.1038/nrd1500. [DOI] [PubMed] [Google Scholar]
- a Schürmann M.; Janning P.; Ziegler S.; Waldmann H. Small-molecule target engagement in cells. Cell Chem. Biol. 2016, 23, 435–441. 10.1016/j.chembiol.2016.03.008. [DOI] [PubMed] [Google Scholar]; b Simon G. M.; Niphakis M. J.; Cravatt B. F. Determining target engagement in living systems. Nat. Chem. Biol. 2013, 9, 200–205. 10.1038/nchembio.1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durham T. B.; Blanco M. Target engagement in lead generation. Bioorg. Med. Chem. Lett. 2015, 25, 998–1008. 10.1016/j.bmcl.2014.12.076. [DOI] [PubMed] [Google Scholar]
- a Sun Y. S.; Hays N. M.; Periasamy A.; Davidson M. W.; Day R. N. Monitoring protein interactions in living cells with fluorescence lifetime imaging microscopy. Methods Enzymol. 2012, 504, 371–391. 10.1016/B978-0-12-391857-4.00019-7. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Robers M. B.; Dart M. L.; Woodroofe C. C.; Zimprich C. A.; Kirkland T. A.; Machleidt T.; Kupcho K. R.; Levin S.; Hartnett J. R.; Zimmerman K.; Niles A. L.; Ohana R. F.; Daniels D. L.; Slater M.; Wood M. G.; Cong M.; Cheng Y.; Wood K. V. Target engagement and drug residence time can be observed in living cells with BRET. Nat. Commun. 2015, 6, 10091. 10.1038/ncomms10091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Franken H.; Mathieson T.; Childs D.; Sweetman G. M. A.; Werner T.; Togel I.; Doce C.; Gade S.; Bantscheff M.; Drewes G.; Reinhard F. B.; Huber W.; Savitski M. M. Thermal proteome profiling for unbiased identification of direct and indirect drug targets using multiplexed quantitative mass spectrometry. Nat. Protoc. 2015, 10, 1567–1593. 10.1038/nprot.2015.101. [DOI] [PubMed] [Google Scholar]; b Savitski M. M.; Reinhard F. B. M.; Franken H.; Werner T.; Savitski M. F.; Eberhard D.; Molina D. M.; Jafari R.; Dovega R. B.; Klaeger S.; Kuster B.; Nordlund P.; Bantscheff M.; Drewes G. Tracking cancer drugs in living cells by thermal profiling of the proteome. Science 2014, 346, 1255784. 10.1126/science.1255784. [DOI] [PubMed] [Google Scholar]
- a Huber K. V. M.; Olek K. M.; Müller A. C.; Tan C. S. H.; Bennett K. L.; Colinge J.; Superti-Furga G. Proteome-wide drug and metabolite interaction mapping by thermal-stability profiling. Nat. Methods 2015, 12, 1055–1057. 10.1038/nmeth.3590. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Mateus A.; Määttä T. A.; Savitski M. M. Thermal proteome profiling: unbiased assessment of protein state through heat-induced stability changes. Proteome Sci. 2017, 15, 13. 10.1186/s12953-017-0122-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Cravatt B. F.; Wright A. T.; Kozarich J. W. Activity-based protein profiling: from enzyme chemistry to proteomic chemistry. Annu. Rev. Biochem. 2008, 77, 383–414. 10.1146/annurev.biochem.75.101304.124125. [DOI] [PubMed] [Google Scholar]; b Lanning B. R.; Whitby L. R.; Dix M. M.; Douhan J.; Gilbert A. M.; Hett E. C.; Johnson T. O.; Joslyn C.; Kath J. C.; Niessen S.; Roberts L. R.; Schnute M. E.; Wang C.; Hulce J. J.; Wei B.; Whiteley L. O.; Hayward M. M.; Cravatt B. F. A road map to evaluate the proteome-wide selectivity of covalent kinase inhibitors. Nat. Chem. Biol. 2014, 10, 760–769. 10.1038/nchembio.1582. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Dubach J. M.; Kim E.; Yang K.; Cuccarese M.; Giedt R. J.; Meimetis L. G.; Vinegoni C.; Weissleder R. Quantitating drug-target engagement in single cells in vitro and in vivo. Nat. Chem. Biol. 2016, 13, 168–173. 10.1038/nchembio.2248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bondeson D. P.; Mares A.; Smith I. E. D.; Ko E.; Campos S.; Miah A. H.; Mulholland K. E.; Routly N.; Buckley D. L.; Gustafson J. L.; Zinn N.; Grandi P.; Shimamura S.; Bergamini G.; Faelth-Savitski M.; Bantscheff M.; Cox C.; Gordon D. A.; Willard R. R.; Flanagan J. J.; Casillas L. N.; Votta B. J.; den Besten W.; Famm K.; Kruidenier L.; Carter P. S.; Harling J. D.; Churcher I.; Crews C. M. Catalytic in vivo protein knockdown by small-molecule PROTACs. Nat. Chem. Biol. 2015, 11, 611–617. 10.1038/nchembio.1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohoka N.; Okuhira K.; Ito M.; Nagai K.; Shibata N.; Hattori T.; Ujikawa O.; Shimokawa K.; Sano O.; Koyama R.; Fujita H.; Teratani M.; Matsumoto H.; Imaeda Y.; Nara H.; Cho N.; Naito M. In vivo knockdown of pathogenic proteins via specific and nongenetic inhibitor of apoptosis protein (IAP)-dependent protein erasers (SNIPERs). J. Biol. Chem. 2017, 292, 4556–4570. 10.1074/jbc.M116.768853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J.; Zhou P. DCAFs, the missing link of the CUL4-DDB1 ubiquitin ligase. Mol. Cell 2007, 26, 775–780. 10.1016/j.molcel.2007.06.001. [DOI] [PubMed] [Google Scholar]
- Cheeseman M. D.; Chessum N. E.; Rye C. S.; Pasqua A. E.; Tucker M. J.; Wilding B.; Evans L. E.; Lepri S.; Richards M.; Sharp S. Y.; Ali S.; Rowlands M.; O’Fee L.; Miah A.; Hayes A.; Henley A. T.; Powers M.; Te Poele R.; De Billy E.; Pellegrino L.; Raynaud F.; Burke R.; van Montfort R. L.; Eccles S. A.; Workman P.; Jones K. Discovery of a chemical probe bisamide (CCT251236): an orally bioavailable efficacious pirin ligand from a heat shock transcription factor 1 (HSF1) phenotypic screen. J. Med. Chem. 2017, 60, 180–201. 10.1021/acs.jmedchem.6b01055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F.; Rehmani I.; Esaki S.; Fu R.; Chen L.; de Serrano V.; Liu A. Pirin is an iron-dependent redox regulator of NF-κB. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 9722–9727. 10.1073/pnas.1221743110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunwell J. M.; Purvis A.; Khuri S. Cupins: the most functionally diverse protein superfamily?. Phytochemistry 2004, 65, 7–17. 10.1016/j.phytochem.2003.08.016. [DOI] [PubMed] [Google Scholar]
- Komai K.; Niwa Y.; Sasazawa Y.; Simizu S. Pirin regulates epithelial to mesenchymal transition independently of BCL3-SLUG signaling. FEBS Lett. 2015, 589, 738–743. 10.1016/j.febslet.2015.01.040. [DOI] [PubMed] [Google Scholar]
- a Ohoka N.; Shibata N.; Hattori T.; Naito M. Protein knockdown technology: Application of ubiquitin ligase to cancer therapy. Curr. Cancer Drug Targets 2016, 16, 136–146. 10.2174/1568009616666151112122502. [DOI] [PubMed] [Google Scholar]; b Collins I.; Wang H.; Caldwell J. J.; Chopra R. Chemical approaches to targeted protein degradation through modulation of the ubiquitin-proteasome pathway. Biochem. J. 2017, 474, 1127–1147. 10.1042/BCJ20160762. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Toure M.; Crews C. M. Small-molecule PROTACS: New approaches to protein degradation. Angew. Chem., Int. Ed. 2016, 55, 1966–1973. 10.1002/anie.201507978. [DOI] [PubMed] [Google Scholar]; d Lai A. C.; Crews C. M. Induced protein degradation: An emerging drug discovery paradigm. Nat. Rev. Drug Discovery 2017, 16, 101–114. 10.1038/nrd.2016.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For example,; a Zengerle M.; Chan K.; Ciulli A. Selective small molecule induced degradation of the BET bromodomain protein BRD4. ACS Chem. Biol. 2015, 10, 1770–1777. 10.1021/acschembio.5b00216. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Lu J.; Qian Y.; Altieri M.; Dong H.; Wang J.; Raina K.; Hines J.; Winkler J. D.; Crew A. P.; Coleman K.; Crews C. M. Hijacking the E3 ubiquitin ligase cereblon to efficiently target BRD4. Chem. Biol. 2015, 22, 755–763. 10.1016/j.chembiol.2015.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galdeano C.; Gadd M. S.; Soares P.; Scaffidi S.; Van Molle I.; Birced I.; Hewitt S.; Dias D. M.; Ciulli A. Structure guided design and optimization of small molecules targeting the protein-protein interaction between the von hippel-lindau (VHL) E3 ubiquitin ligase and the hypoxia inducible factor (HIF) alpha subunit with in vitro nanomolar affinities. J. Med. Chem. 2014, 57, 8657–8663. 10.1021/jm5011258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demizu Y.; Shibata N.; Hattori T.; Ohoka N.; Motoi H.; Misawa T.; Shoda T.; Naito M.; Kurihara M. Development of BCR-ABL degradation inducers via the conjugation of an imatinib derivative and a cIAP1 ligand. Bioorg. Med. Chem. Lett. 2016, 26, 4865–4869. 10.1016/j.bmcl.2016.09.041. [DOI] [PubMed] [Google Scholar]
- Winter G. E.; Buckley D. L.; Paulk J.; Roberts J. M.; Souza A.; Dhe-Paganon S.; Bradner J. E. Phthalimide conjugation as a strategy for in vivo target protein degradation. Science 2015, 348, 1376–1381. 10.1126/science.aab1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- A recent study demonstrated a method to determine which E3 ligase can be manipulated for protein degradation:Ottis P.; Toure M.; Cromm P. M.; Ko E.; Gustafson J. L.; Crews C. M. Assessing different E3 ligases for small molecule induced protein ubiquitination and degradation. ACS Chem. Biol. 2017, 12, 2570–2578. 10.1021/acschembio.7b00485. [DOI] [PubMed] [Google Scholar]
- Cyrus K.; Wehenkel M.; Choi E.; Han H.; Lee H.; Swanson H.; Kim K. Impact of linker length on the activity of PROTACs. Mol. BioSyst. 2011, 7, 359–364. 10.1039/C0MB00074D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douglass E. F.; Miller C. J.; Sparer G.; Shapiro H.; Spiegel D. A. A comprehensive mathematical model for three-body binding equilibria. J. Am. Chem. Soc. 2013, 135, 6092–6099. 10.1021/ja311795d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gadd M. S.; Testa A.; Lucas X.; Chan K.; Chen W.; Lamont D. J.; Zengerle M.; Ciulli A. Structural basis of PROTAC cooperative recognition for selective protein degradation. Nat. Chem. Biol. 2017, 13, 514–521. 10.1038/nchembio.2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petzold G.; Fischer E. S.; Thomä N. H. Structural basis of lenalidomide-induced CK1α degradation by the CRL4(CRBN) ubiquitin ligase. Nature 2016, 532, 127–130. 10.1038/nature16979. [DOI] [PubMed] [Google Scholar]
- Angers S.; Li T.; Yi X.; MacCoss M. J.; Moon R. T.; Zheng N. Molecular architecture and assembly of the DDB1–CUL4A ubiquitin ligase machinery. Nature 2006, 443, 590–593. 10.1038/nature05175. [DOI] [PubMed] [Google Scholar]
- A recent publication demonstrated for a VHL-PROTAC targeting a kinase that a wide range of linker lengths were tolerated but with an apparent lower limit cutoff of 12 linear non-H linker atoms, see:; a Crew A. P.; Raina K.; Dong H.; Qian Y.; Wang J.; Vigil D.; Serebrenik Y. V.; Hamman B. D.; Morgan A.; Ferraro C.; Siu K.; Neklesa T. K.; Winkler J. D.; Coleman K. G.; Crews C. M. Identification and Characterization of Von Hippel-Lindau-Recruiting Proteolysis Targeting Chimeras (PROTACs) of TANK-Binding Kinase 1. J. Med. Chem. 2017, 10.1021/acs.jmedchem.7b00635. [DOI] [PubMed] [Google Scholar]; b While shorter linker lengths were not investigated during the course of this work, shorter linker lengths in degradation probes have been successfully previously employed against other protein targets, see:Zhou B.; Hu J.; Xu F.; Chen Z.; Bai L.; Fernandez-Salas E.; Lin M.; Liu L.; Yang C.-Y.; Zhao Y.; McEachern D.; Przybranowski S.; Wen B.; Sun D.; Wang S. Discovery of a small-molecule degrader of bromodomain and extra-terminal (BET) proteins with picomolar cellular potencies and capable of achieving tumor regression. J. Med. Chem. 2017, 10.1021/acs.jmedchem.6b01816. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Robb C. M.; Contreras J. I.; Kour S.; Taylor M. A.; Abid M.; Sonawane Y. A.; Zahid M.; Murry D. J.; Natarajan A.; Rana S. Chemically induced degradation of CDK9 by a proteolysis targeting chimera (PROTAC). Chem. Commun. 2017, 53, 7577–7580. 10.1039/C7CC03879H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruchelman A. L.; Man H.; Zhang W.; Chen R.; Capone L.; Kang J.; Parton A.; Corral L.; Schafer P. H.; Babusis D.; Moghaddam M. F.; Tang Y.; Shirley M. A.; Muller G. W. Isosteric analogs of lenalidomide and pomalidomide: Synthesis and biological activity. Bioorg. Med. Chem. Lett. 2013, 23, 360–365. 10.1016/j.bmcl.2012.10.071. [DOI] [PubMed] [Google Scholar]
- Lohbeck J.; Miller A. K. Practical synthesis of a phthalimide-based cereblon ligand to enable PROTAC development. Bioorg. Med. Chem. Lett. 2016, 26, 5260–5262. 10.1016/j.bmcl.2016.09.048. [DOI] [PubMed] [Google Scholar]
- a Georgsson J.; Hallberg A.; Larhed M. Rapid palladium-catalyzed synthesis of esters from aryl halides utilizing Mo(CO)6 as a solid carbon monoxide source. J. Comb. Chem. 2003, 5, 350–352. 10.1021/cc0201086. [DOI] [PubMed] [Google Scholar]; b Georgsson J.; Sköld C.; Plouffe B.; Lindeberg G.; Botros M.; Larhed M.; Nyberg F.; Gallo-Payet N.; Gogoll A.; Karlén A.; Hallberg A. Angiotensin II pseudopeptides containing 1,3,5-trisubstituted benzene scaffolds with high AT2 receptor affinity. J. Med. Chem. 2005, 48, 6620–6631. 10.1021/jm050280z. [DOI] [PubMed] [Google Scholar]
- Chan K.; Zengerle M.; Testa A.; Ciulli A. Impact of target warhead and linkage vector on inducing protein degradation: Comparison of bromodomain and extra-terminal (BET) degraders derived from triazolodiazepine (JQ1) and tetrahydroquinoline (I-BET726) BET inhibitor scaffolds. J. Med. Chem. 2017, 10.1021/acs.jmedchem.6b01912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Our FP-probe was generated using a Sulfo-Cy5 fluorophore rather than Cy5 but displayed similar Ki values for thalidomide and lenalidomide as:Fischer E. S.; Böhm K.; Lydeard J. R.; Yang H.; Stadler M. B.; Cavadini S.; Nagel J.; Serluca F.; Acker V.; Lingaraju G. M.; Tichkule R. B.; Schebesta M.; Forrester W. C.; Schirle M.; Hassiepen U.; Ottl J.; Hild M.; Beckwith R. E. J.; Harper J. W.; Jenkins J. L.; Thomä N. H. Structure of the DDB1-CRBN E3 ubiquitin ligase in complex with thalidomide. Nature 2014, 512, 49–53. 10.1038/nature13527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ki values were calculated from the geometric mean IC50 and the FP-assay probe affinity using the methods described by:; a Huang X. Fluorescence polarization competition assay: The range of resolvable inhibitor potency is limited by the affinity of the fluorescent ligand. J. Biomol. Screening 2003, 8, 34–38. 10.1177/1087057102239666. [DOI] [PubMed] [Google Scholar]; b Munson P. J.; Rodbard D. An exact correction to the ″Cheng-Prusoff″ correction. J. Recept. Res. 1988, 8, 533–456. 10.3109/10799898809049010. [DOI] [PubMed] [Google Scholar]
- Wes: Simple Westerns Just Got Even Simpler; Proteinsimple: San Jose, CA, 2017; https://www.proteinsimple.com/wes.html (accessed August 28, 2017).
- For the origin of our SK-OV-3 ovarian carcinoma cell line, see the Supporting Information.
- Erales J.; Coffino P. Ubiquitin-independent proteasomal degradation. Biochim. Biophys. Acta, Mol. Cell Res. 2014, 1843, 216–221. 10.1016/j.bbamcr.2013.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai A. C.; Toure M.; Hellerschmied D.; Salami J.; Jaime-Figueroa S.; Ko E.; Hines J.; Crews C. M. Modular PROTAC design for the degradation of oncogenic BCR-ABL. Angew. Chem., Int. Ed. 2016, 55, 807–810. 10.1002/anie.201507634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mateus A.; Gordon L. J.; Wayne G. J.; Almqvist H.; Axelsson H.; Seashore-Ludlow B.; Treyer A.; Matsson P.; Lundbäck T.; West A.; Hann M. M.; Artursson P. Prediction of intracellular exposure bridges the gap between target- and cell-based drug discovery. Proc. Natl. Acad. Sci. U. S. A. 2017, 114, E6231–E6239. 10.1073/pnas.1701848114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Matsson P.; Kihlberg J. How big is too big for cell permeability?. J. Med. Chem. 2017, 60, 1662–1664. 10.1021/acs.jmedchem.7b00237. [DOI] [PubMed] [Google Scholar]; b Pye C. R.; Hewitt W. M.; Schwochert J.; Haddad T. D.; Townsend C. E.; Etienne L.; Lao Y.; Limberakis C.; Furukawa A.; Mathiowetz A. M.; Price D. A.; Liras S.; Lokey R. S. Nonclassical size dependence of permeation defines bounds for passive adsorption of large drug molecules. J. Med. Chem. 2017, 60, 1665–1672. 10.1021/acs.jmedchem.6b01483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hann M. M.; Keserü G. M. Finding the sweet spot: The role of nature and nurture in medicinal chemistry. Nat. Rev. Drug Discovery 2012, 11, 355–365. 10.1038/nrd3701. [DOI] [PubMed] [Google Scholar]
- Palm K.; Luthman K.; Ungell A. L.; Strandlund G.; Beigi F.; Lundahl P.; Artursson P. Evaluation of dynamic polar molecular surface area as predictor of drug absorption: Comparison with other computational and experimental predictors. J. Med. Chem. 1998, 41, 5382–5392. 10.1021/jm980313t. [DOI] [PubMed] [Google Scholar]
- Gillis E. P.; Eastman K. J.; Hill M. D.; Donnelly D. J.; Meanwell N. A. Applications of fluorine in medicinal chemistry. J. Med. Chem. 2015, 58, 8315–8359. 10.1021/acs.jmedchem.5b00258. [DOI] [PubMed] [Google Scholar]
- George I. R.; Lewis W.; Moody C. J. Synthesis of iodopyridone. Tetrahedron 2013, 69, 8209–8215. 10.1016/j.tet.2013.07.033. [DOI] [Google Scholar]
- In mouse fibroblasts, pirin has a measured inherent protein half-life of 64 h:Schwanhäusser B.; Busse D.; Li N.; Dittmar G.; Schuchhardt J.; Wolf J.; Chen W.; Selbach M. Global quantification of mammalian gene expression control. Nature 2011, 473, 337–342. 10.1038/nature10098. [DOI] [PubMed] [Google Scholar]
- The stability of the thalidomide derived probes in DMSO stock appears dependent on the water content of the DMSO. We recommend fresh DMSO stocks are prepared from solid prior to each use. For a discussion of thalidomide stability, see:; a Lepper E. R.; Smith N. F.; Cox M. C.; Scripture C. D.; Figg W. D. Thalidomide metabolism and hydrolysis: mechanisms and implications. Curr. Drug Metab. 2006, 7, 677–685. 10.2174/138920006778017777. [DOI] [PubMed] [Google Scholar]; b Reist M.; Carrupt P.; Francotte E.; Testa B. Chiral inversion and hydrolysis of thalidomide: mechanisms and catalysis by bases and serum albumin, and chiral stability of teratogenic metabolites. Chem. Res. Toxicol. 1998, 11, 1521–1528. 10.1021/tx9801817. [DOI] [PubMed] [Google Scholar]
- Goodwin J. T.; Conradi R. A.; Ho N. F. H.; Burton P. S. Physicochemical determinants of passive membrane permeability: Role of solute hydrogen-bonding potential and volume. J. Med. Chem. 2001, 44, 3721–3729. 10.1021/jm010253i. [DOI] [PubMed] [Google Scholar]
- Evans L. E.; Jones K.; Cheeseman M. D. Targeting secondary protein complexes in drug discovery: studying the druggability and chemical biology of the HSP70/BAG1 complex. Chem. Commun. 2017, 53, 5167–5170. 10.1039/C7CC01376K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Youdim K. A.; Avdeef A.; Abbott N. J. In vitro trans-monolayer permeability calculations: Often forgotten assumptions. Drug Discovery Today 2003, 8, 997–1003. 10.1016/S1359-6446(03)02873-3. [DOI] [PubMed] [Google Scholar]; b Filipe H. A. L.; Salvador A.; Silvestre J. M.; Vaz W. L. C.; Moreno M. J. Beyond Overton’s rule: Quantitative modeling of passive permeation through tight cell monolayers. Mol. Pharmaceutics 2014, 11, 3696–3706. 10.1021/mp500437e. [DOI] [PubMed] [Google Scholar]; c Owen S. C.; Doak A. K.; Ganesh A. N.; Nedyalkova L.; McLaughlin C. K.; Shoichet B. K.; Shoichet M. S. Colloidal drug formulations can explain “bell-shaped” concentration-response curves. ACS Chem. Biol. 2014, 9, 777–784. 10.1021/cb4007584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MoKA; Molecular Discovery: Borehamwood, Hertfordshire, UK, 2017; http://www.moldiscovery.com/software/moka (accessed August 28, 2017).
- Jones K.; Rye C.; Chessum N.; Cheeseman M.; Pasqua A. E.; Pike K. G.; Faulder P. F.. Fused 1,4-dihydrodioxin derivatives as inhibitors of heat shock transcription factor 1. WO 2015/049535 A1, April 9, 2015.
- The poor aqueous stability of the thalidomide derived PDPs made measurement of their assay free fractions by dialysis, as well as standard permeability assessments using PAMPA and CACO-2, unreliable so it was not possible to compare PDP probes at their free concentrations or to determine permeability values.
- Kisselev A. F.; Goldberg A. L. Proteasome inhibitors: From research tools to drug candidates. Chem. Biol. 2001, 8, 739–758. 10.1016/S1074-5521(01)00056-4. [DOI] [PubMed] [Google Scholar]
- a Diz A. P.; Carvajal-Rodriguez A.; Skibinski D. O. F. Multiple hypothesis testing in proteomics: A strategy for experimental work. Mol. Cell. Proteomics 2011, 10, M110.004374. 10.1074/mcp.M110.004374. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Noble W. S. How does multiple testing correction work?. Nat. Biotechnol. 2009, 27, 1135–1137. 10.1038/nbt1209-1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Thorarensen A.; Banker M. E.; Fensome A.; Telliez J.; Juba B.; Vincent F.; Czerwinski R. M.; Casimiro-Garcia A. ATP-mediated kinome selectivity: The missing link in understanding the contribution of individual JAK kinase isoforms to cellular signaling. ACS Chem. Biol. 2014, 9, 1552–1558. 10.1021/cb5002125. [DOI] [PubMed] [Google Scholar]; b Smyth L. A.; Collins I. Measuring and interpreting the selectivity of protein kinase inhibitors. J. Chem. Biol. 2009, 2, 131–151. 10.1007/s12154-009-0023-9. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Knight Z. A.; Shokat K. M. Features of selective kinase inhibitors. Chem. Biol. 2005, 12, 621–637. 10.1016/j.chembiol.2005.04.011. [DOI] [PubMed] [Google Scholar]
- a Strelow J. M. A perspective on the kinetics of covalent and irreversible inhibition. SLAS Discovery 2017, 22, 3–20. 10.1177/1087057116671509. [DOI] [PubMed] [Google Scholar]; b Competition of PDP 16 at 6 h exposure with control compounds 1 and 22 was unsuccessful at concentration > 1 μM (data not shown).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.