

Abstract
Rationale:
Carney complex (CNC) is a multiple neoplasia syndrome with autosomal dominant inheritance. CNC is characterized by the presence of myxomas, spotty skin pigmentation, and endocrine overactivity. No direct correlation has been established between disease-causing mutations and phenotype.
Patient concerns:
A 16-year-old boy was admitted because of excessive weight gain over 3 years and purple striae for 1 year. Physical examination revealed Cushingoid features and spotty skin pigmentation on his face, lip, and sclera.
Diagnoses:
The patient was diagnosed as Carney complex.
Interventions:
the patient underwent right adrenalectomy and partial adrenalectomy of the left adrenal gland.
Outcome:
Results of imaging showed bilateral adrenal nodular hyperplasia, multiple microcalcifications of the bilateral testes, and compression fracture of the thoracolumbar spine. Histopathological results confirmed multiple pigmented nodules in the adrenal glands. DNA sequencing revealed a nonsense mutation in the gene encoding regulatory subunit type 1-alpha of protein kinase A (PRKAR1A; c.205C > T). After the second adrenalectomy, the Cushingoid features disappeared, and cortisol levels returned to normal.
Lessons:
Carney complex is a rare disease that lacks consistent genotype–phenotype correlations. Our patient, who carried a germline PRKAR1A nonsense mutation (c.205C > T), clinical features included spotty skin pigmentation, osteoporosis, and primary pigmented nodular adrenal disease. Adrenalectomy is the preferred treatment for Cushing syndrome due to primary pigmented nodular adrenal disease.
Keywords: adrenalectomy, Carney complex, Cushing syndrome, PRKAR1A, rare diseases
1. Introduction
Carney complex (CNC) is a multiple neoplasia syndrome with an autosomal dominant inheritance.[1] The clinical manifestations of CNC vary widely but may include myxomas of the heart, skin, and other tissues and multiple other endocrine and nonendocrine neoplasms including pituitary tumors, adrenocortical tumors, thyroid neoplasms, psammomatous melanotic schwannomas, testicular tumors, breast tumors, ovarian lesions, and bone lesions.[2] More than 80% of CNC patients develop spotty skin pigmentation or skin growths, which typically appear early in life and may be located anywhere on the body, typically on the face, lips, genital area, and mucosa.[3] The most common noncutaneous lesions found in CNC are cardiac myxomas (affecting 20–40% of patients), which are responsible for >50% of CNC-related mortality.[4–6]
CNC is a genetically heterogeneous disease, with >70% patients with CNC carrying mutations of the PRKAR1A gene,[5] which encodes the 1-α regulatory subunit (RIα) of the cAMP-dependent protein kinase A (PKA) and functions as a tumor suppressor gene.[7] Pathogenic PRKAR1A mutations include single base substitutions, small (≤15 bp) deletions/insertions, combined rearrangements, and large deletions.[8–10] More than 125 PRKAR1A gene mutations have been identified; however, linking genotype to phenotype has been challenging.[11] Although previous studies [5,9,10,12–15] have demonstrated associations between specific mutations and CNC manifestations, only 3 pathogenic variants (c.82C>T, c.491_492delTG, and c.709–2_709–7 delATTTTT) have been identified in >3 unrelated pedigrees.[5,7,13] The other mutations are unique (present in a single kindred). To better understand genotype–phenotype relationships in this rare heterogeneous disease, it is essential to describe clinical features associated with specific mutations.
Here we present a case of CNC with a known germline PRKAR1A nonsense mutation (c.205C>T) [5]; however, the phenotype associated with this mutation has not yet been established. Our results indicate that this PRKAR1A mutation may be associated with increased pigmentation on the face, lip, and sclera; osteoporosis resulting in compression fracture; and primary pigmented nodular adrenal disease (PPNAD)-associated Cushing syndrome.
2. Case report
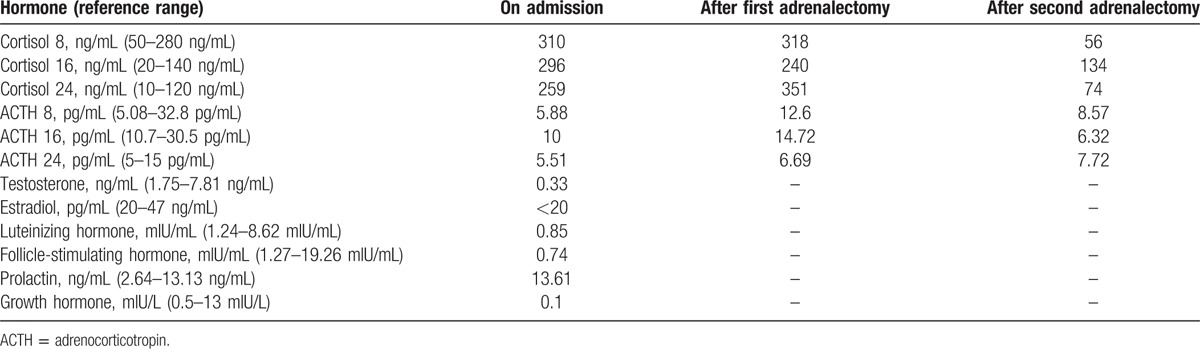
A 16-year-old boy was admitted to our department because of excessive weight gain for 3 years and purple striate for 1 year. His weight was 60 kg, and height was 1.2 m. Physical examination revealed typical Cushingoid features (moon face, buffalo hump, central obesity, and purple striae of the bilateral axilla and lower abdomen) (Fig. 1A), and spotty skin pigmentation on the face, lip, and sclera (Fig. 1C–E). Laboratory findings revealed hypercortisolism (Table 1). After low- and high-dose dexamethasone suppression tests, cortisol levels were still high (462.9 and 828.3 nmol/L, respectively), indicating adrenocorticotropic hormone (ACTH)-independent Cushing syndrome. His grandparents have no relevant disease, and his parents died early with unknown cause.
Figure 1.
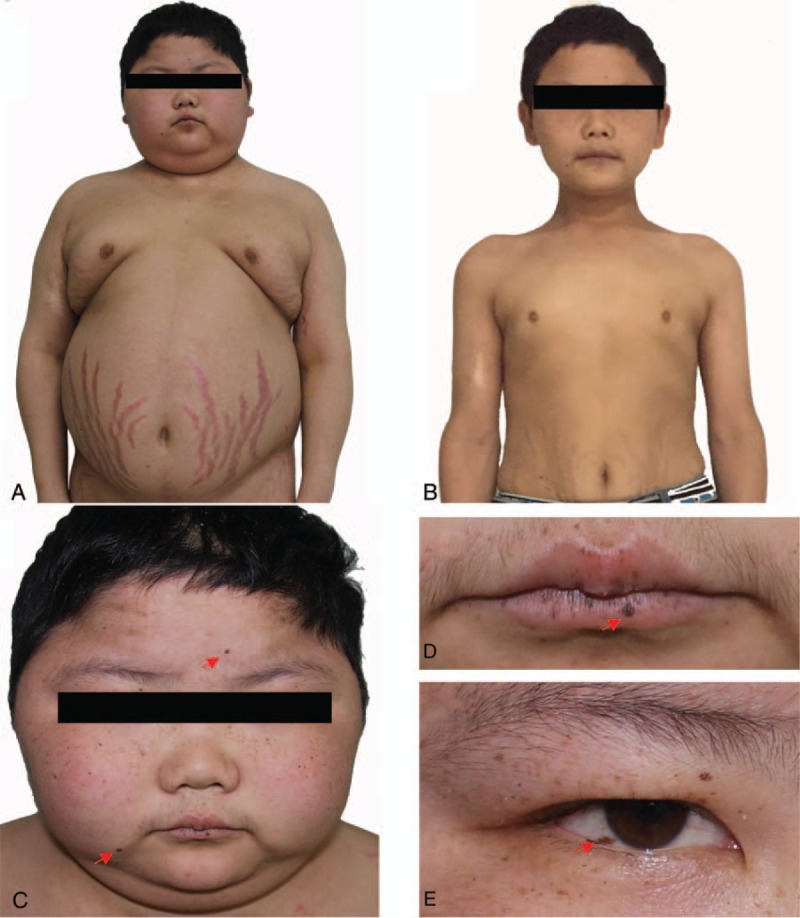
Patient characteristics before and after adrenalectomies. (A) Cushingoid features include moon face, central obesity, and purple striae of the bilateral axillary and lower abdomen before the operations. (B) Cushingoid features are decreased after left and right adrenalectomies. (C–E) Spotty skin pigmentation (indicated by red arrows) on the face (C), lip (D), and sclera (E).
Table 1.
Laboratory parameters of the patient.
Adrenal computed tomography showed bilateral adrenal nodular hyperplasia (Fig. 2A and B). Ultrasonography showed a normal thyroid but multiple microcalcifications of the bilateral testes (Fig. 2C and D), suggesting large-cell calcifying Sertoli cell tumor (LCCST). Considering the young age and the will of the patient, the needle biopsy of the testes was not conducted to confirm the LCCST. However, testicular ultrasound is conducted every 6 month to monitor the lesions. Results of magnetic resonance imaging showed a pituitary normal but a compression fracture of the thoracolumbar spine (T5–T12 and L1). Echocardiography did not reveal any cardiac myxomas.
Figure 2.
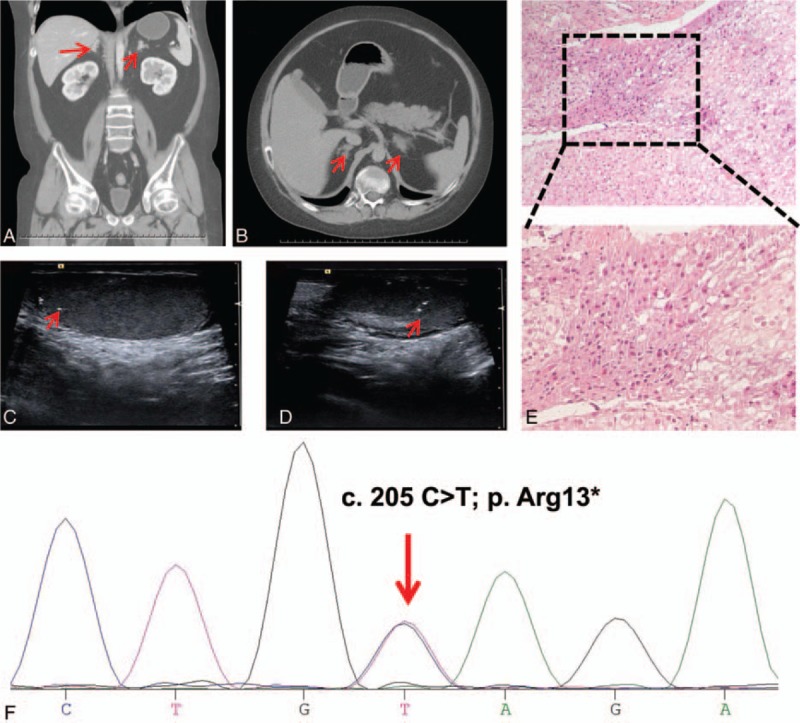
Results of imaging, histopathology, and DNA sequencing. Adrenal computed tomography scan showed bilateral adrenal nodular hyperplasia (A and B). Ultrasonography demonstrated multiple microcalcifications of the bilateral testes (C and D). (E) Hematoxylin and eosin staining of the adrenal tissue revealed multiple pigmented nodules. (F) Sequencing of DNA extracted from peripheral leukocytes identified a heterozygous C>T substitution in PRKAR1A exon 3 (indicated by the red arrow).
On February 19, 2017, the patient underwent right adrenalectomy via retroperitoneal laparoscopy; however, cortisol levels did not normalize (Table 1). On March 1, 2017, the patient underwent partial adrenalectomy of the left adrenal gland. Histopathology findings of multiple pigmented nodules substantiated the diagnosis of PPNAD (Fig. 2E). After the second adrenalectomy, the Cushingoid features disappeared (Fig. 1B), and cortisol levels returned to normal (Table 1).
2.1. Genetic analysis
Peripheral blood was collected after obtaining informed consent from the patient's grandparents. DNA was extracted from leukocytes and amplified by the polymerase chain reaction. Bidirectional DNA sequence analysis of the PRKAR1A gene identified a known CNC-causing germline mutation (c.205C>T) in exon 3 (Fig. 2F) and a second mutation (c.34 G>T) in the intron preceding exon 9.
3. Discussion
In this study, we describe a patient with patient typical features of CNC such as spotty skin pigmentation on the face, lip, and sclera; osteoporosis resulting in compression fracture; and PPNAD-associated Cushing syndrome. DNA sequencing identified a known pathogenic mutation of PRKAR1A. Taken together, our patient met the criteria for the diagnosis of CNC [2]: (1) spotty pigmentation with the typical distribution (face, lip, and sclera); (2) osteoporotic bone changes due primarily to glucocorticoid excess, which accelerates bone resorption and decreases intestinal calcium absorption and bone formation [16] (3); probable LCCST, as demonstrated by ultrasonography of the bilateral testes, which revealed multiple microcalcifications; and (4) Cushing syndrome, which may be ACTH-dependent or ACTH-independent (e.g., caused by PPNAD, as in our patient).
CNC has diverse clinical manifestations, which typically develop over a period of years.[2] Mutations in PRKAR1A appear to be the most common cause of CNC. Patients carrying PRKAR1A mutations have more severe disease, with earlier presentation and higher frequency of myxomas, thyroid, and gonadal tumors, schwannomas, and lentigines compared withPRKAR1A-negative patients.[5,17] In a small number of PRKAR1A missense mutations, the mRNA escapes nonsense-mediated decay, and the R I-α mutant proteins are associated with a more severe phenotype.[14,15] Two PRKAR1A mutations (c.709–7del6 and M1V c.1A>G/p.M1V substitution) are associated with low penetrance, early-life isolated PPNAD, and Cushing syndrome.[18,19] However, in most cases, genotype cannot predict phenotype or penetrance. Therefore, it is necessary to describe clinical features associated with specific mutations. According to our case presentation, the mutation (c. 205 C>T) in our case is inclined to cause pigmented spots, osteoporosis, as well as PPNAD.
In conclusion, we have identified a reported PRKAR1A nonsense mutation (c.205C>T) in a sporadic case of CNC characterized by the spotty skin pigmentation, osteoporosis resulting in compression fracture, and PPNAD-associated Cushing syndrome. The description of clinical features of this patient adds to our knowledge of CNC and PRKAR1A mutations. The ability to recognize CNC is important for early diagnosis and prevention of severe complications. PRKAR1A mutation analysis should be conducted as soon as possible in patients with suspected CNC.
3.1. Compliance with ethical standards
The institutional review board (IRB) of Daping Hospital of Third Military Medical University waived IRB approval for the study. Written informed consent was obtained from the patient for the use of medical records and related images.
Footnotes
Abbreviations: ACTH = adrenocorticotropic hormone, CNC = Carney complex, IRB = institutional review board, LCCST = large-cell calcifying Sertoli cell tumor, PKA = protein kinase A, PPNAD = primary pigmented nodular adrenal disease, RIα = 1-α regulatory subunit.
The authors have no funding and conflicts of interest to disclose.
References
- [1].Carney JA, Gordon H, Carpenter PC, et al. The complex of myxomas, spotty pigmentation, and endocrine overactivity. Medicine 1985;64:270–83. [DOI] [PubMed] [Google Scholar]
- [2].Correa R, Salpea P, Stratakis CA. Carney complex: an update. Eur J Endocrinol 2015;173:M85–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Sandrini F, Stratakis C. Clinical and molecular genetics of Carney complex. Mol Genet Metab 2003;78:83–92. [DOI] [PubMed] [Google Scholar]
- [4].Espiard S, Bertherat J. Carney complex. Front Hormone Res 2013;41:50–62. [DOI] [PubMed] [Google Scholar]
- [5].Bertherat J, Horvath A, Groussin L, et al. Mutations in regulatory subunit type 1A of cyclic adenosine 5′-monophosphate-dependent protein kinase (PRKAR1A): phenotype analysis in 353 patients and 80 different genotypes. J Clin Endocrinol Metab 2009;94:2085–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Boikos SA, Stratakis CA. Carney complex: the first 20 years. Curr Opin Oncol 2007;19:24–9. [DOI] [PubMed] [Google Scholar]
- [7].Kirschner LS, Carney JA, Pack SD, et al. Mutations of the gene encoding the protein kinase A type I-alpha regulatory subunit in patients with the Carney complex. Nat Genet 2000;26:89–92. [DOI] [PubMed] [Google Scholar]
- [8].Salpea P, Horvath A, London E, et al. Deletions of the PRKAR1A locus at 17q24.2-q24.3 in Carney complex: genotype–phenotype correlations and implications for genetic testing. J Clin Endocrinol Metab 2014;99:E183–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Horvath A, Bertherat J, Groussin L, et al. Mutations and polymorphisms in the gene encoding regulatory subunit type 1-alpha of protein kinase A (PRKAR1A): an update. Hum Mutat 2010;31:369–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Horvath A, Bossis I, Giatzakis C, et al. Large deletions of the PRKAR1A gene in Carney complex. Clin Cancer Res 2008;14:388–95. [DOI] [PubMed] [Google Scholar]
- [11].Anselmo J, Medeiros S, Carneiro V, et al. A large family with Carney complex caused by the S147G PRKAR1A mutation shows a unique spectrum of disease including adrenocortical cancer. J Clin Endocrinol Metab 2012;97:351–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Groussin L, Kirschner LS, Vincent-Dejean C, et al. Molecular analysis of the cyclic AMP-dependent protein kinase A (PKA) regulatory subunit 1A (PRKAR1A) gene in patients with Carney complex and primary pigmented nodular adrenocortical disease (PPNAD) reveals novel mutations and clues for pathophysiology: augmented PKA signaling is associated with adrenal tumorigenesis in PPNAD. Am J Hum Genet 2002;71:1433–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Groussin L, Horvath A, Jullian E, et al. A PRKAR1A mutation associated with primary pigmented nodular adrenocortical disease in 12 kindreds. J Clin Endocrinol Metab 2006;91:1943–9. [DOI] [PubMed] [Google Scholar]
- [14].Meoli E, Bossis I, Cazabat L, et al. Protein kinase A effects of an expressed PRKAR1A mutation associated with aggressive tumors. Cancer Res 2008;68:3133–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Greene EL, Horvath AD, Nesterova M, et al. In vitro functional studies of naturally occurring pathogenic PRKAR1A mutations that are not subject to nonsense mRNA decay. Hum Mut 2008;29:633–9. [DOI] [PubMed] [Google Scholar]
- [16].Papanastasiou L, Fountoulakis S, Voulgaris N, et al. Identification of a novel mutation of the PRKAR1A gene in a patient with Carney complex with significant osteoporosis and recurrent fractures. Hormones 2016;15:129–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Stratakis CA, Carney JA, Lin JP, et al. Carney complex, a familial multiple neoplasia and lentiginosis syndrome. Analysis of 11 kindreds and linkage to the short arm of chromosome 2. J Clin Invest 1996;97:699–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Groussin L, Jullian E, Perlemoine K, et al. Mutations of the PRKAR1A gene in Cushing's syndrome due to sporadic primary pigmented nodular adrenocortical disease. J Clin Endocrinol Metab 2002;87:4324–9. [DOI] [PubMed] [Google Scholar]
- [19].Pereira AM, Hes FJ, Horvath A, et al. Association of the M1V PRKAR1A mutation with primary pigmented nodular adrenocortical disease in two large families. J Clin Endocrinol Metab 2010;95:338–42. [DOI] [PMC free article] [PubMed] [Google Scholar]