

Abstract
Rationale:
We present a case of hemophagocytic lymphohistiocytosis (HLH) with severe pulmonary complication and acute respiratory distress syndrome (ARDS) hospitalized in our intensive care unit (ICU) in 2014; distinctive trait of this case has been the challenging diagnosis, with a bone marrow biopsy always negative, the severe pulmonary complication with ARDS and severe pulmonary hypertension, and the ferritin temporal kinetics that precisely followed the clinical course of disease.
Patient concerns:
A 32-year-old woman from the Philippines first diagnosed with upper airway infection, was subsequently hospitalized in infectious disease department and treated for community acquired pneumonia.
Diagnoses:
After clinical picture worsened with a profound respiratory insufficiency, the patient was intubated and transferred to our ICU. During this hospitalization, the clinical picture of fever, cutaneous rashes, lymphadenitis, hepatitis, leukopenia, anemia, hyperferritinemia, hypertriglyceridemia, high level of auto-antibodies, and low NK activity suggested an hemophagocytic lymphohistiocytosis syndrome, even if bone marrow biopsy was negative for hemophagocytosis.
Interventions:
Immunosuppressive therapy with dexamethasone and etoposide was started, and the patient was discharged from ICU 4 months after admission.
Lessons:
HLH is a rare disorder of the mononuclear phagocytic system, characterized by systemic proliferation of non- neoplastic histiocytes. The diagnosis is often challenging and not all of the diagnostic criteria may be present at the same time; this case shows how complex the diagnosis could be, how hematic ferritin levels could help in following the course of the disease, and the possibility of severe pulmonary complication either due to the disease itself and to possible sovra infections.
Keywords: hemophagocytic lymphohistiocytosis, immunosuppressive therapy, multiorgan failure, pulmonary fibrosis
1. Introduction
Hemophagocytic lymphohistiocytosis (HLH) is a rare disorder of the mononuclear phagocytic system, characterized by systemic proliferation of non-neoplastic histiocytes.[1,2] It comprises 2different conditions: a primary familiar one, familial hemophagocytic lymphohistiocytosis (FHL), and the secondary HLH form (sHLH).[3–6] sHLH has an incidence of 1 case per every 50,000 births and it may develop as a result of strong immunological activation of the immune system.[1] It has been described in association with viral infection (Epstein–Barr virus, cytomegalovirus, human immunodeficiency virus, influenza virus, and rubella), during other kinds of infections (Mycobacterium tuberculosis, Mycoplasma pneumoniae, fungi, and parasites), during autoimmune disease (systemic rheumatoid arthritis and macrophage activation syndrome)[7] and during malignancies (lymphoma–lymphoma-associated hemophagocytic syndrome, LAHS- and other malignancies-malignancy associated hemophagocytic syndrome, MAHS).[4]
HLH pathophysiology relies on the deficiency of cytotoxic functioning of natural killer lymphocytes (NK cells) and cytotoxic T lymphocytes (CTLs), their hyperproliferation and the consequent cytokine storm; in affected individuals, the activation of immune response to antigens is preserved, but immune response modulation and termination are defective. The deficit is primarily attributed to a defect of perforin-mediated cytotoxic activity of the cluster of differentiation 8 transmembrane glycoprotein (CD8) T cells or NK cells: without perforin activity, the immune system cannot get rid of the antigen, creating a short circuit that maintains an active immune system stimulation.
Moreover, perforin-mediated T lymphocytes apoptosis that usually modulates immune response is impaired, resulting in T lymphocytes hyperproliferation and consequent persistent immune system activation. This abnormous immune response stimulation leads to an excessive production of cytokines, macrophage activation and, finally, tissue damage, because of lymphocytes invasions.[8–13]
High levels of cytokines and organ infiltration are associated with the signs and symptoms of HLH: prolonged fever is caused by high levels of interleukin 1 (IL1), IL6, and tumor necrosis factor alpha (TNFα); cytopenia is attributed to high concentration of TNFα and bone marrow infiltration with hemophagocytes, macrophages phagocytizing hematopoietic components (erythrocytes, leukocytes, platelets, their precursors, and the cellular fragments).[2,8] Hypofibrinogenemia, resulting from increased plasminogen activator expressed by activated macrophages will lead to increased serum levels of plasmin that, together with liver dysfunction, coming from tissue infiltration by macrophages, explain HLH coagulopathy. A high level of ferritin is secreted by activated macrophages and high concentration of the soluble form of the IL-2 receptor (sCD25) is produced by activated lymphocytes. Hepatosplenomegaly, liver dysfunction with hyperbilirubinemia, hypoalbuminemia, elevated levels of liver lysis tests (aspartate aminotransferase, alanine aminotransferase) and jaundice, skin manifestations (nonspecific rash, maculopapular rash, erythroderma, purpuric macules, and morbilliform papules) and neurological symptoms (seizures, ataxia, hemiplegia, mental status changes, or simply irritability that may be associated with a spinal fluid hyperproteinemia and moderate pleocytosis) are explained either by organ infiltration with lymphocytes and histiocytes and by high levels of cytokines and their direct cytotoxicity to the tissues and organs involved with lymphocytes and histiocytes infiltration.[14]
Diagnostic guidelines for HLH have recently been reviewed: the 5 diagnostic criteria from Henter et al have been published in 1991: (1) fever, (2) splenomegaly, (3) cytopenia, (4) hypertriglyceridemia, and/or hypofibrinogenemia, (5) hemophagocytosis in bone marrow, spleen, or lymph node.[14] In addition, in 2004 3 additional criteria have been introduced: (6) low or absent NK cells activity, (7) hyperferritinemia, (8) high levels of s CD25.[2] Five of the 8 criteria must be fulfilled in order to make the diagnosis of HLH, but the diagnosis is often challenging because not all of the diagnostic criteria may be present at the same time.[2]
HLH therapy is based on immunosuppression with the early use of etoposide (in 10 doses, cumulatively 1.5 g/m2)[15,16] and its early combination with cyclosporin A (continuous infusion of 1.5–3 mg/kg/h for 1 or 2 weeks until absolute neutrophil counts recover, then wean off or switch to oral administration).[15,17] Dexamethasone (0.25 mg/kg given 3–5 times in a week for a minimum of 2 weeks) can be added to HLH therapy,[15,18] together with supportive care (gastroprotection, appropriate antibiotic or antiviral therapy in patients with ongoing infection, oral antimycotics, antithrombin III, granulocyte colony-stimulating factor, and intravenous immunoglobulin).[2,15] Alternative treatment protocols include antithymocite globulin (ATG) (dose 10 mg/kg/day for 5 days),[2,19,20] and other immunosuppressants that can be classified as salvage therapy: Tacrolimus (FK 506) and methotrexate (MTX).[8,21] Finally, nonsteroidal anti-inflammatory (FANS) drugs such as indomethacin, naproxen, and ibuprofen may be useful, but therapeutic experience is limited in HLH patients.[22] In refractory HLH cases, there is a rationale for the use of salvage chemotherapy (adriamycin/cyclophosphamide/vincristine/procarbazine/prednisolone (ACOPP) and/or adriamycin/bleomycin/vinblastine/prednisolone (ABVD));[23] hematopoietic cell transplantation, stem cell transplantation (HSCT) or bone marrow transplantation (BMT), is the mandatory therapy for FHL patients.[2,15]
2. Case report
A 32-year-old previously healthy woman from the Philippines arrived in our emergency department (ED) on January 2014 with fever, sore throat, and a productive cough; objective examination was otherwise negative. Remote pathologic and familial anamnesis were negative, except for a travel to the Philippines 3 months before.
She was discharged home with a diagnosis of upper airway infection and antibiotic therapy, first with amoxicillin and then with levofloxacin.
One week after the first ED consultation, the patient came back with an ongoing fever, asthenia, and vomiting. Physical examination revealed bilateral scleral hemorrhage, a rash to face, thorax and arms bilaterally and a sovraclavear, axillar, and inguinal lymphadenopathy.
Chest x-ray (CXR) and thorax computed tomography scan (CT scan) (Fig. 1) revealed pulmonary infiltrates to the middle and inferior right lobe with ground-glass-appearing opacities and a pre-scissural right nodule.
Figure 1.
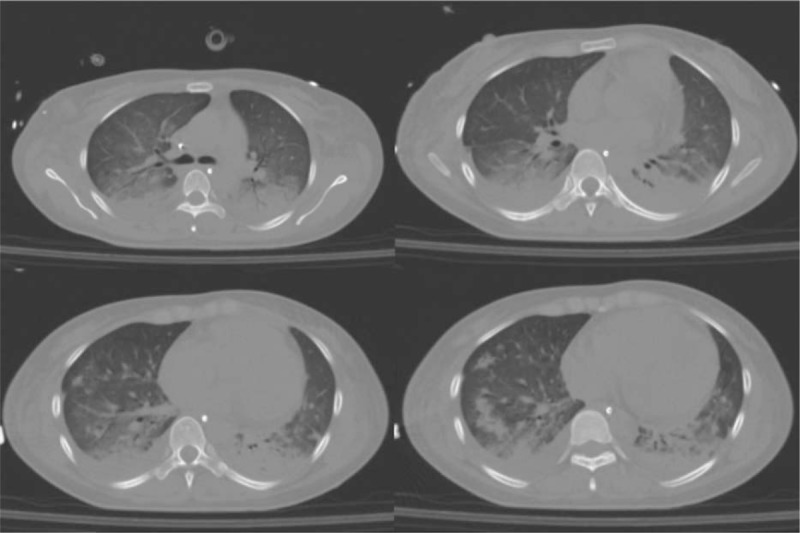
First CT scan revealed pulmonary infiltrates to the middle and inferior right lobe with ground-glass-appearing opacities and a pre-scissural right nodule. CT = computed tomography.
The patient was then admitted to Infectious Disease Department where a large spectrum antibiotic therapy with ceftriaxone and clarithromicyn was started; research for an opportunistic infection resulted negative. When after 5 days the clinical picture did not ameliorate, antibiotic therapy was changed to meropenem, rifampicin, and doxycycline to cover a possible rickettsia infection, considering her recent travel to the Philippines.
A biopsy of an axillar lymph node revealed necrotizing lymphadenitis, suspected for Kikuchi lymphadenitis. After 3 more days, 10 days after hospitalization, the patient developed a severe hypoxic respiratory insufficiency and was admitted to ICU, intubated and mechanically ventilated.
CXR and CT scan (Fig. 2) revealed a picture of ARDS with multiple bilateral pulmonary infiltrates with an hypoxic normocapnic respiratory insufficiency with a ratio of arterial oxygen partial pressure (PaO2) to fractional inspired oxygen (FiO2) (PaO2/FiO2) of 230 with FiO2 0.5, arterial carbon dioxide partial pressure (PaCO2) 37 mm Hg and pH 7.34.
Figure 2.
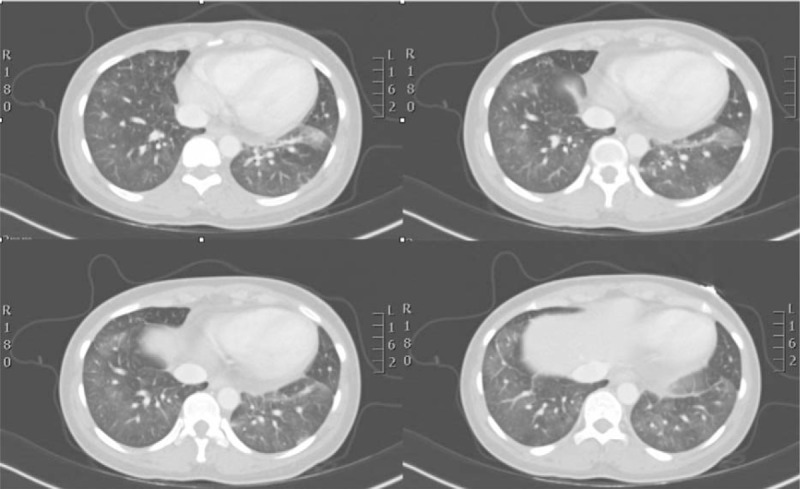
CT scan revealed a picture of ARDS with multiple bilateral pulmonary infiltrates. CT = computed tomography, ARDS = acute respiratory distress syndrome.
WBC-PaO2/FiO2 patient's laboratory tests revealed anemia (hemoglobin [Hb] 10 g/dL) and leucopenia (white blood cell count [WBC] 3000/L), acute hepatitis with cytolysis (total bilirubin 3.02 mg/dL, alanine aminotransferase 649 U/L, aspartate aminotransferase 649 U/L, alkaline phosphatase 176 U/L), hypertriglyceridemia (325 mg/dL), and high ferritin level (20.826 g/mL). Antinuclear antibodies (ANA) level titration was 5120 and anti-ribonucleoprotein antibodies (ENArnp) was 249 U/mL with a complement component partially consumed (C3 = 60 mg/dL, C4 = 17 mg/dL); dosage of IL2 receptor was also elevated (21.130) while NK cells’ activity was low.
Broncho-alveolar lavage (BAL) was compatible with a macrophagic and hemolymphophagocytic activation, while all the cultures for microbic infections were negative.
The clinical picture of fever, cutaneous rashes, lymphadenitis, hepatitis, leukopenia, anemia, hyperferritinemia, hypertriglyceridemia, high level of auto-antibodies, and low NK activity suggested an hemophagocytic lymphohistiocytosis syndrome, even if bone marrow biopsy was negative for hemophagocytosis.
Immunosuppressive therapy with dexamethasone, etoposide, and cyclophosphamide was started; the patient was ventilated with FiO2 of 1 and positive end expiratory pressure (PEEP) of 12 cm H2O with pronation cycles and nitrous oxide 10 parts per million because of refractory hypoxia in ARDS.
After 11 days of ICU, the patient was extubated and undergone noninvasive ventilation.
Ferritin levels were monitored as illness activity indicator: after immunosuppressive therapy, ferritin level fell to 1853 g/mL (first sample 20.826 g/mL).
Three days after extubation, the patient developed a severe respiratory insufficiency with CT scan showing increased bilateral infiltrates (Fig. 3); BAL was diagnostic for herpes virus type 1 (HSV1) infection with a plasmatic genome level of 50.176 copies/mL. The patient was then started on antiviral therapy with acyclovir that resulted in a negativization of BAL after 21 days.
Figure 3.
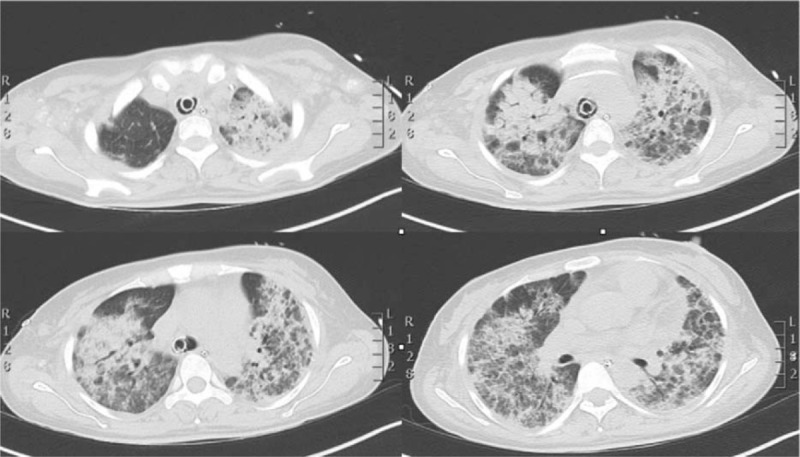
HR-CT scan showed increased bilateral infiltrates and a worsening of ARDS picture. ARDS = acute respiratory distress syndrome, HR-CT = high resolution computer tomogrphy.
Nonetheless, the CT scan showed worsened lung infiltrates with initial inter- and intralobular fibrosis; the clinical picture was complicated by an hypercapnic respiratory insufficiency with a lowest dynamic compliance of 10 mL/cmH2O for a patient weight of 40 kg.
For severe hypercapnia and respiratory acidosis with a PaCO2 of 83 and a pH of 7.27, the patient was reintubated and extracorporeal CO2 removal with Decap was necessary; after 25 days from ICU admission, the patient underwent percutaneous tracheostomy.
A new CT scan (Fig. 4) confirmed pulmonary fibrosis, and methylprednisolone therapy was reaugmented to high doses.
Figure 4.
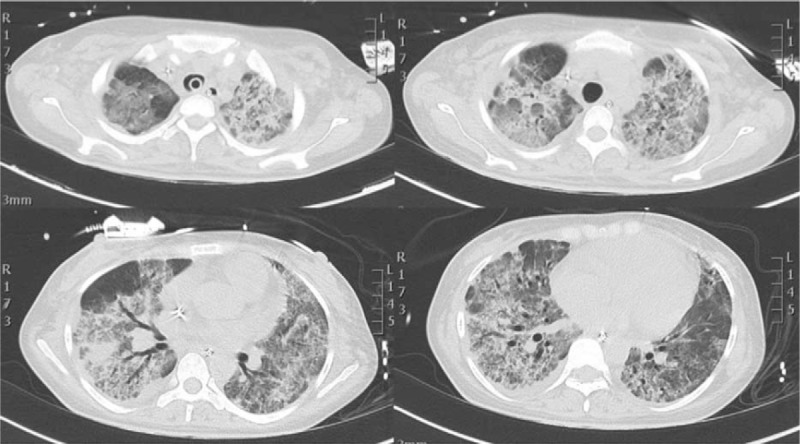
HR-CT scan showed worsened lung infiltrates with initial inter and intralobular fibrosis. HR-CT = high resolution computer tomogrphy.
The patient was autonomous from mechanical ventilation 3 months after the first event; she developed a severe pulmonary hypertension (pulmonary arterial pressure, PAP, of 80 mm Hg) possibly either due to the hypoxic vasoconstriction that followed the 3 episodes of respiratory insufficiency and to tricuspid insufficiency. The responsiveness of PAP to nitrous oxide induced the introduction of Sildenafil in patient's treatment.
The patient received enteral nutrition via nasogastric tube and undergone daily physiotherapy during ICU stay; nonetheless, she developed muscular weakness. The electromyography revealed a distal motor sufferance typical of ICU patients that resolved with time.
At ICU discharge, 4 months after the patient was admitted, she was in a good mood, capable of walking with help, spontaneously breathing with a PaO2/FiO2 of 250 with FiO2 0.4, tachypnoeic but not dyspnoeic while walking (Fig. 5).
Figure 5.
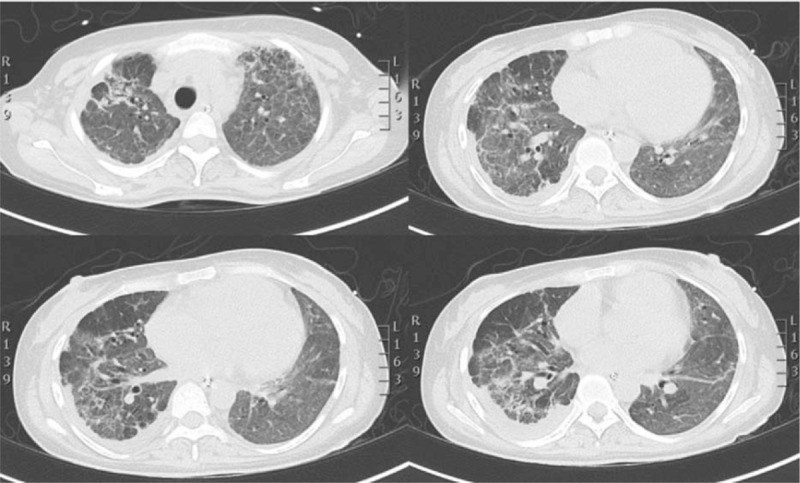
Last HR-CT scan showed fibrosis evolution. HR-CT = high resolution computer tomogrphy.
She was transferred to the subintensive department where she remained for a month and was finally discharged to a rehabilitation clinic.
3. Discussion
We have reported a case of HLH with a presentation in combination to an infectious disease.
Kumakura et al[24] in 1997 proposed a disease entity called autoimmune-associated hemophagocytic syndrome (AAHS). The pathogenesis of AAHS could be explained by autoantibody-mediated, immune-complex-mediated, or cytokine-mediated mechanism: at first, autoantibodies may react to blood cells, resulting in hemophagocytosis by stimulated histiocytes through the Fc receptor, primarily in the bone marrow. In a second time, blood cells sensitized by immune complexes may be phagocytosed by histiocytes via complement–receptor interactions; finally, uncontrolled production of inflammatory cytokines may activate histiocytes.[25–27]
The correlation between HLH, systemic inflammatory response syndrome (SIRS), and multiorgan dysfunction syndrome (MODS) has been proposed by Castillo and Carcillo[28]: according to these authors, the conditions of SIRS/sepsis/MODS/HLH form a continuum of immune dysregulation in the presence of a trigger.
We can hypothesize a combined pathophysiological mechanism in our patient, mediated either by autoantibody and cytokines, even if macrophage activation and hemophagocytosis have never been demonstrated by biopsy. In our patient, the trigger for immune response initiation and for the elevated cytokines’ production seems to be the upper airway infection.
In conclusion, in a pre-existing genetic mutations context, the trigger can induce an extensive and lethal cytotoxic dysfunction of NK cells and CTLs, resulting in HLH; fewer alterations of CTLs-NKs function (genetic polymorphism) can, on the other hand, result in partial cytotoxicity and under conditions of the right immune or infectious stimulus sepsis and MODs could occur.
Our case is characterized by a highly severe respiratory insufficiency with the overlap of different clinical pictures: ARDS in suspected lymphohistiocytosis, pulmonary infection from HSV 1, pulmonary fibrotic evolution and finally initial cardiac decompensation in cor pulmonale.
We can hypothesize that the good outcome of this patient with a very bad prognosis is due to different factors: first the promptness of ICU admission, before the diagnosis of lymphohistiocytosis, the early beginning of immunosuppressive therapy and finally the multidisciplinary approach to the patient.
Our clinical case might suggest to think of the diagnosis of lymphohistiocytosis in a patient with respiratory insufficiency without a clear infective cause, fever and rash with adenopathies, hyperferritinemia and high level of ANA and ENArnp and low NK cells activity.
Footnotes
Abbreviations: AAHS = autoimmune-associated hemophagocytic syndrome, ABVD = adriamycin/bleomycin/vinblastine/prednisolone, ACOPP = adriamycin/cyclophosphamide/vincristine/procarbazine/prednisolone, ANA = antinuclear antibodies, ARDS = acute respiratory distress syndrome, ATG = antithymocite globulin, BAL = broncho-alveolar lavage, BMT = bone marrow transplantation, C3, C4 = complement component 3, 4, CD8 = cluster of differentiation 8 transmembrane glycoprotein, cmH2O = centimeters of water, CT scan = computed tomography scan PaO2/FiO2—ratio of arterial oxygen partial pressure (PaO2) to fractional inspired oxygen (FiO2), CTLs = cytotoxic T lymphocytes, CXR = chest x-ray, dL = deciliter, ED = emergency department, ENArnp = anti-ribonucleoprotein antibodies, FANS = nonsteroidal anti-inflammatory drugs, FHL = familial hemophagocytic lymphohistiocytosis, FK 506 = tacrolimus, gr = grams, Hb = hemoglobin, HLH = hemophagocytic lymphohistiocytosis, HSCT = hematopoietic stem cell transplantation, HSV1 = herpes virus type 1, ICU = intensive care unit, IL = interleukin, kg = kilograms, L = liter, LAHS = lymphoma-associated hemophagocytic syndrome, m2 = square meters, MAHS = malignancy-associated hemophagocytic syndrome, mg = milligrams, mm Hg = millimeters of mercury, MODS = multiorgan dysfunction syndrome, MTX = methotrexate, NK = natural killer lymphocytes cells, PaCO2 = arterial carbon dioxide partial pressure, PAP = pulmonary arterial pressure, PEEP = positive end expiratory pressure (PEEP), sCD25 = soluble form of the IL-2 receptor, sHLH = secondary HLH form, SIRS = systemic inflammatory response syndrome, TNFα = tumor necrosis factor alpha, U = units, WBC = white blood cell count.
Ethical approval was waived because of the nature of the case report; no informed consent was asked for reporting this case report because any sign of recognition was revealed in this paper.
The authors have no conflicts of interest to disclose.
References
- [1].Dhote R, Simon J, Papo T, et al. Reactive hemophagocytic syndrome in adult system disease: report of twenty-six cases and literature review. Arthritis Rheum 2003;49:633–9. [DOI] [PubMed] [Google Scholar]
- [2].Henter JI, Horne A, Aricó M, et al. HLH-2004: diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatric Blood Cancer 2007;48:124–31. [DOI] [PubMed] [Google Scholar]
- [3].Henter JI, Aricò M, Elinder G, et al. Familial hemophagocytic lymphohistiocytosis (primary HLH). Hematol Oncol Clin North Am 1998;12:417–33. [DOI] [PubMed] [Google Scholar]
- [4].Janka G, Imashuku S, Elinder G, et al. Infection and malignancy-associated hemophagocytic syndrome: secondary hemophagocytic lymphohistiocytosis. Hematol Oncol Clin North Am 1998;12:435–44. [DOI] [PubMed] [Google Scholar]
- [5].De Saint Basile G, Ménasché G, Fischer A. Molecular mechanisms of biogenesis and exocytosis of cytotoxic granules. Nat Rev Immunol 2010;10:568–79. [DOI] [PubMed] [Google Scholar]
- [6].Voskoboinik I, Smyth MJ, Trapani JA. Perforin-mediated target-cell death and immune homeostasis. Nat Rev Immunol 2006;6:940–52. [DOI] [PubMed] [Google Scholar]
- [7].Ramanan AV, Baildam EM. Macrophage activation syndrome is hemophagocytic lymphohistiocytosis: need for the right terminology. J Rheumatol 2002;29:1105. [PubMed] [Google Scholar]
- [8].Tang YM, Xu XJ. Advances in hemophagocytic lymphohistiocytosis: pathogenesis, early diagnosis/differential diagnosis, and treatment. Scientif Word J 2011;11:697–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Jordan MB, Hildeman D, Kappler J, et al. An animal model of hemophagocytic lymphohistiocytosis (HLH): CD8 T cells and interferon gamma are essential for the disorder. Blood 2004;104:735–43. [DOI] [PubMed] [Google Scholar]
- [10].Spaner D, Raju K, Radvanyi L, et al. A role for perforin in activation-induced cell death. J Immunol 1998;160:2655–64. [PubMed] [Google Scholar]
- [11].Fadeel B, Orrenius S, Henter JI. Induction of apoptosis and caspase activation in cells obtained from familial haemophagocytic lymphohistiocytosis patients. Br J Haematol 1999;106:406–15. [DOI] [PubMed] [Google Scholar]
- [12].Osugi Y, Hara J, Tagawa S, et al. Cytokine production regulating Th1 and Th2 cytokines in hemophagocytic lymphohistiocytosis. Blood 1997;89:4100–3. [PubMed] [Google Scholar]
- [13].Takada H, Ohga S, Mizuno Y, et al. Oversecretion of IL18 in hemophagocytic lymphohistiocytosis: a novel marker of disease activity. Br J Haematol 1999;106:182–9. [DOI] [PubMed] [Google Scholar]
- [14].Henter J-I, Elinder G, Soder O, et al. Hypercytokinemia in familial hemophagocytic lymphohistiocytosis. Blood 1991;78:2918–22. [PubMed] [Google Scholar]
- [15].Teramura T, Morimoto A, Imashuku S, et al. Recent developments in the management of hemophagocytic lymphohistiocytosis. Expert Opin Pharmacother 2001;2:1437–48. [DOI] [PubMed] [Google Scholar]
- [16].Imashuku S, Kuriyama K, Teramura T, et al. Requirement for etoposide in the treatment of Epstein Barr virus associated hemophagocytic lymphohistiocytosis. J Clin Oncol 2001;19:2665–7263. [DOI] [PubMed] [Google Scholar]
- [17].Imashuku S, Hibi S, Kuriyama K, et al. Management of severe neutropenia with cyclosporin during initial treatment of Epstein–Barr virus-related hemophagocytic lymphohistiocytosis. Leuk Lymphoma 2000;36:339–46. [DOI] [PubMed] [Google Scholar]
- [18].Bonanomi MH, Velvart M, Stimpel M, et al. Studies of pharmacokinetics and therapeutic effects of glucocorticoids entrapped in liposomes after intra-articular application in healthy rabbits and in rabbits with antigen-induced arthritis. Rheumatol Int 1987;7:203–12. [DOI] [PubMed] [Google Scholar]
- [19].Stephan JL, Donadieu J, Ledeist F, et al. Treatment of familial hemophagocytic lymphohistiocytosis with antithymocyte globulins, steroids, and Cyclosporin A. Blood 1993;82:2319–23. [PubMed] [Google Scholar]
- [20].Perel Y, Alos N, Ansoborlo S, et al. Dramatic efficacy of antithymocyte globulinsin childhood EBV-associated haemophagocytic syndrome. Acta Paediatr 1997;86:911. [DOI] [PubMed] [Google Scholar]
- [21].Fischer A, Virelizier JL, Arenzana-Seisdedos F, et al. Treatment of four patients with erythrophagocytic lymphohistiocytosis by a combination of VP16-213, steroids and intrathecal methotrexate and cranial irradiation. Paediatrics 1985;76:263–8. [PubMed] [Google Scholar]
- [22].Brown RE, Bowman WP, D’Cruzmd CA, et al. Endoperoxidation, hyperprostaglandinemia,;1; and hyperlipidemia in a case of erythrophagocytic lymphohistiocytosis. Reversal with VP16 and indomethacin. Cancer 1987;60:2388–93. [DOI] [PubMed] [Google Scholar]
- [23].Canellos GP, Anderson JR, Propert KJ, et al. Chemotherapy of advanced Hodgkin's disease with MOPP, ABVD or MOPP alternating with ABVD. New Engl J Med 1992;327:1478–84. [DOI] [PubMed] [Google Scholar]
- [24].Kumakura S, Ishikura H, Kondo M, et al. Autoimmune-associated hemophagocytic syndrome. Am J Med 2004;14:205–15. [DOI] [PubMed] [Google Scholar]
- [25].Yamashita H, Atsuki Y, Shimizu A, et al. Hemophagocytic lymphohistiocytosis complicated by central nervous system in a patient with dermatomyositis: a case presentation and literature review. Mod Rheumatol 2013;23:386–92. [DOI] [PubMed] [Google Scholar]
- [26].Fukaya S, Yasuda S, Hashimoto T, et al. Clinical features of haemophagocytic syndrome in patients with systemic autoimmune diseases: analysis of 30 cases. Rheumatol 2008;47:1686–91. [DOI] [PubMed] [Google Scholar]
- [27].Kumakura S, Ishikura H, Munemasa S, et al. Adult onset Still's disease associated hemophagocytosis. J Rheumatol 1997;24:1645–8. [PubMed] [Google Scholar]
- [28].Castillo L, Carcillo J. Secondary hemophagocytic lymphohistiocytosis and severe sepsis/systemic inflammatory response syndrome/multiorgan dysfunction syndrome/macrophage activation syndrome share common intermediate phenotypes on a spectrum of inflammation. Pediatr Crit Care Med 2009;10:387–92. [DOI] [PubMed] [Google Scholar]