

Abstract
Rationale:
Hemophagocytic lymphohistiocytosis (HLH) is a life-threatening disease entity primarily described in children, but not less relevant in adults. It is characterized by a misdirected activation of the immune system, resulting in an uncontrolled cytokine release from macrophages and cytotoxic T-cells (CTLs). Primary HLH relies on a genetic predisposition, whereas secondary HLH develops in the context of infections, malignancies or autoimmune diseases. However, the awareness and therapeutic knowledge for HLH in adulthood is limited. Most therapy protocols are almost exclusively validated in pediatric cohorts and for primary HLH. Their transferability to adult individuals with mostly secondary HLH is doubtful. Especially the high liver and bone marrow toxicity of applied etoposide-based protocols is discussed controversially and connected to overwhelming infections and death.
Patient concern:
A 51-year old, male, kidney transplant recipient was admitted to our center suffering from diarrhea, fever, nausea, hyponatremia, kidney graft failure, disorientation, progressive hemodynamic instability, and multiorgan failure.
Diagnoses:
Clinical and laboratory findings resembled those of a septic shock. Ferritin and soluble interleukin-2 receptor (sCD25) levels were disproportionally elevated. Only a mild hepatosplenomegaly was diagnosed in a CT scan. A T2-weighted, fluid-attenuated inversion recovery MRI showed marked, bilateral and periventricular white matter hyperintensities. The cerebrospinal fluid (CSF) analysis showed a moderately elevated protein content and cell count. There was no evidence of any bacterial, viral, or parasitic infection. The diagnosis of HLH was made.
Interventions & Outcomes:
The patient was successfully treated by a combined approach consisting of plasma exchange (PE), corticosteroids, anakinra, and cyclosporine (CsA).
Lessons:
HLH is an important differential diagnosis in critically ill patients. Its unspecific clinical picture complicates an early diagnosis and may be misclassified as sepsis. A combination of plasma exchange (PE), corticosteroids, anakinra, and cyclosporine (CsA) may be a promising and less toxic approach for HLH therapy in adults.
Keywords: cyclosporine, hemophagocytic lymphohistiocytosis, interleukin-1-directed therapy, kidney transplant recipient, plasma exchange
1. Introduction
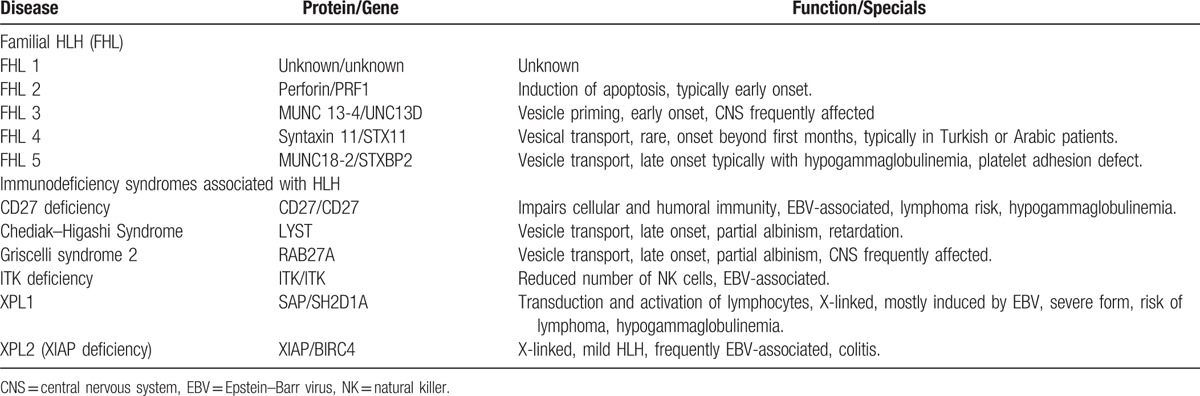
Hemophagocytic lymphohistiocytosis (HLH) is a life-threatening disease characterized by massive cytokine production from activated blood monocytes, macrophages (histiocytes), and cytotoxic T-lymphocytes (CTLs).[1] The ubiquitous cellular organ infiltration and cytokine release evoke an unspecific and often sepsis-like clinical picture.[2] Refractory and prolonged fever, hepatosplenomegaly, hemophagocytosis in the bone marrow, and several laboratory findings such as cytopenia, very high ferritin levels, low or absent natural killer (NK) cell activity, elevated soluble interleukin-2 receptor (sIL-2r = sCD25), hypertriglyceridemia and/or low fibrinogen are considered as typical HLH features. These parameters form the widely applied HLH-2004 diagnostic criteria.[3] In general, one has to distinguish between primary and secondary HLH. Primary HLH is either of genetic origin, also called familial HLH (FHL), or associated with genetic immunodeficiency syndromes (Table 1). Secondary or acquired HLH occurs mostly in the context of infections, malignancies, and autoimmune diseases.[1] In addition, cases of acquired HLH are described in patients receiving immunosuppressive therapy after solid organ transplantation.[4] The term “macrophage activation syndrome” (MAS) is particularly used for autoimmune-related secondary HLH.[5] Secondary HLH can occur at any age, whereas FHL manifests mainly during infancy or early childhood.[6] The epidemiological data for HLH are limited, especially in adulthood. Thus, its true incidence is probably unknown. The best data for primary HLH or HLH in childhood comes from three studies, indicating a yearly incidence of 1.2 per million children in Sweden[7] and of 7.5 and 3.3 per 10000 hospitalized children in Turkey and the United States of America, respectively.[8,9] Only one epidemiological study exists for HLH in adults, reporting an incidence of 3.6 per million for malignancy associated HLH.[10] The overall mortality is high and ranges between 45 and 60% for FHL[11–13] and 5 and 30% for autoimmune-related MAS in children.[14–17] The situation in adults is even worse. Recent data suggest an overall mortality of 41%.[18] Thus, early diagnosis and initiation of appropriate measures are essential to improve outcomes and quality of life.
Table 1.
Unfortunately, its nonspecific clinical presentation and sepsis-like appearance makes the diagnosis challenging and suggests a large number of undetected cases with potentially fatal outcomes in adult critically ill patients.[2] A major problem is thereby the limited awareness for HLH, leading at least in part to the high mortality in adults. In addition, most clinical guidelines, diagnostic criteria, and treatment protocols are developed and validated in pediatric patients. It is unclear to what extent these approaches are transferable into an adult patient population. Concerns exist especially with regard to the use of the cytotoxic topoisomerase II inhibitor etoposide that is widely applied during pediatric disease manifestations. Herein, we report for the first time on a HLH case in an adult kidney transplant recipient, who was successfully treated by a less toxic approach consisting of plasma exchange (PE), cyclosporine (CsA), anakinra, and corticosteroids.
2. Case report
In April 2017, a 51-year old, male, kidney transplant recipient was admitted to our center in poor general condition due to excessive diarrhea and dehydration along with fever, nausea, hyponatremia (126 mEq/L), and kidney graft failure. There were no signs for rush or polyarthralgia. The patient had a living donor kidney transplantation in 2002 due to unknown primary renal disease. On admission, the patient was on an immunosuppressive therapy with tacrolimus, mycophenolic acid, and low dose methylprednisolone. An empirical, antibiotic therapy with ciprofloxacin and metronidazole was started and the fluid losses were replaced. The patient's condition improved over the next 2 days. On the fourth day after admission, diarrhea worsened again and the patient developed progressive tachypnea, hypotonia, and disorientation. An arterial blood gas analysis revealed a metabolic acidosis with partial, respiratory compensation (pH 7.35, pCO2 15 mm Hg, pO2 161 mm Hg, sHCO3− 8 mmol/L, base excess −15.6, sodium 135 mEq/L, chloride 114 mEq/L). Under the assumption of hyperchloremic metabolic acidosis (normal anion gap) due to severe gastrointestinal bicarbonate and fluid losses and a septic clinical picture, the patient was transferred to our intensive care unit. The antibiotic regime was changed to intravenous meropenem and immunosuppressive therapy was reduced accordingly to a methylprednisolone (20 mg) monotherapy. Typical viral (norovirus, rotavirus, adenovirus, astrovirus cytomegalovirus [CMV]) and bacterial (clostridium difficile, shigella, yersenia, and campylobacter species) pathogens of gastrointestinal infection were excluded and multiple blood cultures remained negative. The neurological condition worsened and C-reactive protein (CRP) levels increased further to 120.9 mg/L. T2-weighted, fluid-attenuated inversion recovery (FLAIR) MRI images showed a marked, heterogeneous white matter hyperintensity of the bilateral periventricular regions with occipital, parieto-frontal, and especially dextral accentuation (Fig. 1).
Figure 1.
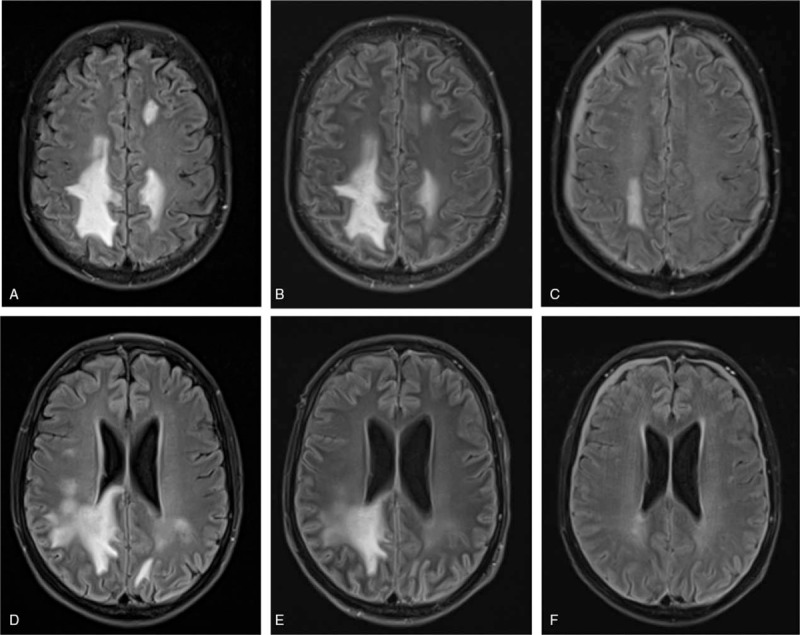
HLH-associated T2-/FLAIR-weighted white matter hyperintensities parietal (A–C) and around periventricular regions (D–F) in axial plane. (A+D) ICU admission (day 5 after admission), (B+E) after 4 plasma exchange procedures (day 13 after admission), (C+F) under maintenance therapy (day 68 after admission). FLAIR = fluid-attenuated inversion recovery, HLH = hemophagocytic lymphohistiocytosis
In the context of immunosuppressive therapy in this patient, a diagnosis of progressive multifocal leukoencephalopathy (PML) due to JC-virus (JCV) reactivation, any other virus-associated encephalitis or posterior reversible encephalopathy syndrome (PRES) was suspected. The cerebrospinal fluid (CSF) analysis showed a moderately elevated spinal fluid protein content (480 mg/L), reduced lactate levels (1.6 mmol/L) and an elevated leucocyte count (12×106/L), consistent with inflammatory CSF alterations. Antiviral therapy with acyclovir and cidofovir was initiated, but the neurological condition worsened. The patient developed hemodynamic instability together with liver and renal failure and needed mechanical ventilation and vasopressor therapy (norepinephrine 23 μg/kg/min). A CT scan of the thorax and abdomen showed only mild hepatosplenomegaly without further pathologies. The most striking laboratory findings at this time were as follows: ferritin 23623 μg/L, serum creatinine 6.6 mg/dL, platelet count 66×109/L, leucocyte count 10.79×106/L, hemoglobin 91 g/L, CRP 160 mg/L, total bilirubin 4.6 mg/dL, aspartate aminotransferase (AST) 6106 U/L, alanine aminotransferase (ALT) 1421 U/L, and lactate dehydrogenase (LDH) 2032 U/L. On the basis of therapy-refractory fever, hepatosplenomegaly, massive hyperferritinemia, progressive thrombocytopenia and anemia (though not fulfilling HLH-2004 criteria), central nervous system (CNS) involvement and a sepsis-like clinical picture without any evidence of a microbiological pathogen, the suspected diagnosis of secondary HLH was established.
At this time, only 3 of the HLH-2004 criteria (fever, splenomegaly, and hyperferritinemia, Table 2) were fulfilled, but the clinical picture was highly suspicious for HLH. Thus, a therapy with 250 mg prednisolone and PE were initiated on day 9 after admission. The laboratory findings prior to initiation of PE are displayed in Table 3. The decision for PE was based on data retrieved from children whit autoimmune related MAS that was successfully treated by PE.[23,24] A therapy with etoposide and CsA according to the HLH-2004 protocol (Table 4) was avoided due to high liver toxicity of etoposide and known association of CsA with suspected PRES and PML development.
Table 2.
HLH-2004 diagnostic criteria.[3]
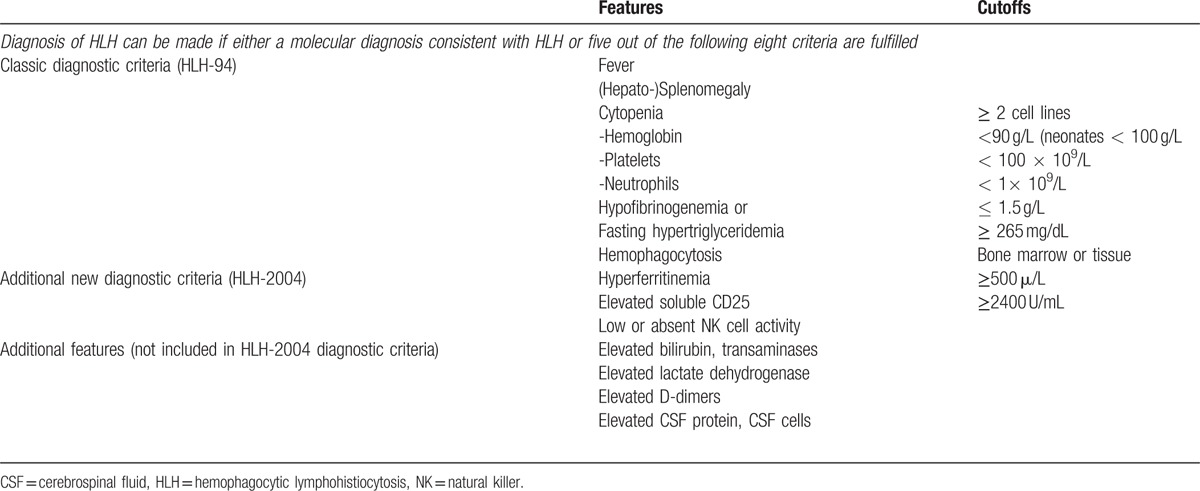
Table 3.
Overview of laboratory findings on admission, prior plasma exchange, Peak values for HLH-classification and ambulant follow-up after 2 months.
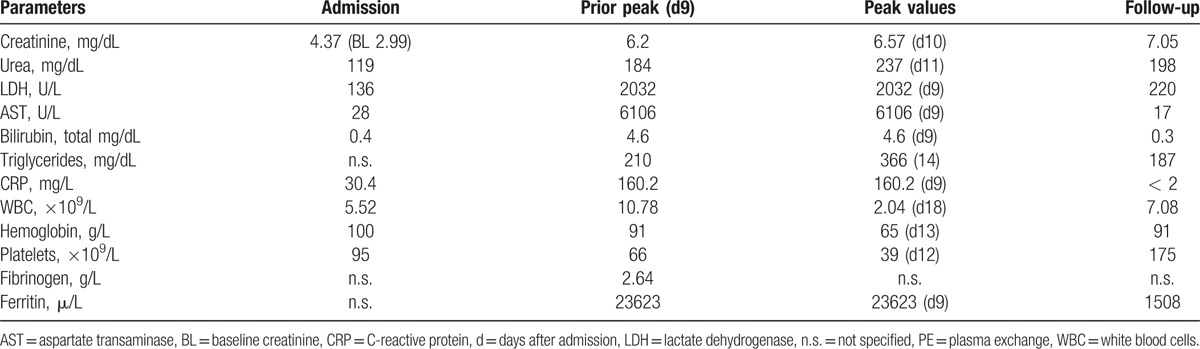
Table 4.
HLH-2004 treatment protocol.[3]
Before steroid therapy was started, an active viral infection of the CSF (JCV, varizella zoster (VZV), herpes simplex virus (HSV)) was ruled out by polymerase chain reaction (PCR). During the following 5 days, the patient developed relevant hypertriglyceridemia (366 mg/dL), progressive thrombocytopenia (39×109/L), anemia (nadir of 6.5 g/dL) and leukocytopenia (3.37×109/L), before all cell lines recovered under PE therapy. The diagnosis of hemophagocytosis was confirmed in a bone marrow biopsy, but admittedly of minor severity. The diagnosis of HLH could be made according to the HLH-2004 criteria 5 days after initiation of therapeutic measures. In total, 18 plasmaphereses were performed and with initiation of plasma exchange the patient's condition improved impressively, vasopressor therapy was terminated after the first procedure and the neurological impairment as well as the CNS lesions were regressive in a control MRI only 4 days after initiation of therapy (Fig. 1). The laboratory parameters in relation to PE procedures are shown in Figure 2. Anakinra, a recombinant interleukin-1 (IL-1) receptor antagonist, was started as maintenance therapy on day 19 with 200 mg every other day based on 3 recent publications, which showed a beneficial effect of IL-1 blockade via anakinra in adults suffering from HLH and MAS-like syndrome.[25–27] After improvement of CNS lesions during PE and establishment of a definite diagnosis of HLH, PRES, and PML were considered as unlikely and a CsA therapy at 3 mg/kg per day was added to the maintenance therapy on day 27. Target CsA blood concentration was 150 μg/L. All cell lines recovered further and the PE therapy was terminated on day 31. The prednisolone dose was tapered from 250 mg for 2 days to 125 mg for 5 days to a maintenance dose of 20 mg/day. During the further clinical course a ventilator-associated sepsis with pseudomonas aeruginosa in blood cultures and tracheal secretion was successfully treated with pipieracilline/tazobactam and ciprofloxacin that was changed to meropenem according to the antibiotic resistance pattern.
Figure 2.
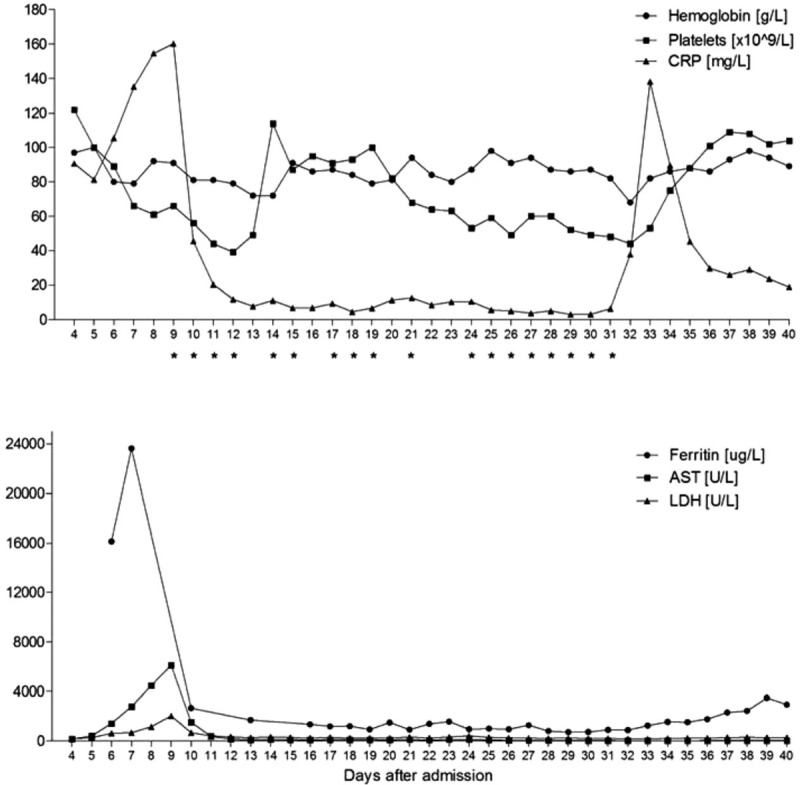
Laboratory findings in relation to plasma exchange. ∗Plasma exchange (one plasma volume). AST = aspartate aminotransferase, CRP = C-reactive protein, LDH = lactate dehydrogenase.
After termination of plasma exchange the ferritin levels remained stable at 2000 to 2500 μg/L during maintenance therapy with CsA, anakinra and low-dose prednisolone. After 48 days in the ICU, the patient was transferred to the general ward and improved further. A potential infectious trigger for HLH and the reason for the initial gastrointestinal symptoms could not be identified. A virologic screening for BK-virus (BKV), CMV, Ebstein–Barr virus (EBV), enterovirus, hepatitis virus A/B/C/E (HAV, HBV, HCV, HEV), HSV, human herpes virus 6 (HHV6), human immune-deficiency virus (HIV), JC-virus (JCV), parainfluenza virus, parvovirus B19, and VZV showed no evidence of an active viral infection. The patient was discharged 75 days after the initial admission.
2.1. Concept of secondary HLH
Secondary or acquired HLH can occur at any age,[18] but generally manifests in older ages, is more frequent than primary HLH, and appears mostly in the context of infectious, malignant or autoimmune diseases.[1] One of the most common factors that trigger the disease are viruses of the herpes family (CMV, HSV, HHV8), especially EBV.[22,28] In addition, other viral (HAV, HBV, HCV, measles, dengue, enterovirus, parvovirus B19, etc.), bacterial (campylobacter, chlamydia, staphylococcus, salmonella, tuberculosis), fungal (aspergillus, candida) and parasitic (leishmanial, malaria, toxoplasma) pathogens can serve as a trigger.[28] Malignant disease, most notably leukemia, lymphoma and especially t-cell driven disease entities, are also able to trigger HLH.[1]
The term “Macrophage activation syndrome” (MAS) is particularly used for autoimmune-related, secondary HLH[5] and by some authors also for HLH in adults.[29] The highest incidence of MAS in pediatric rheumatology is found in patients with systemic juvenile idiopathic arthritis (SJIA), systemic lupus erythematosus (SLE), and Kawasaki syndrome,[30] whereas adult-onset Still's disease (AOSD) and SLE play a major role in adulthood.[18] About 7 to 11% of children with SJIA seem to develop MAS[17,31] and according to newer reports up to 30 to 40% may suffer from subclinical MAS.[32] Epidemiological data on secondary HLH/MAS in adults are almost absent. Studies are needed to generate a better understanding of the epidemiology of secondary HLH, especially in adults and nonautoimmune diseases.
2.2. Diagnostic criteria
The first guidelines for the diagnosis of HLH were published by the Histiocyte Society in 1991[6] and in revised version in 2004 (HLH-2004).[3] Today, at least 6 relevant diagnostic guidelines exist[3,32–36] (Table 5), but only 2 are proposed for use in adult patient population.[35,36] All other guidelines either focus on primary HLH or HLH secondary to autoimmune diseases in children.[3,32–34] Thus, validated scores for a reliable and early HLH/MAS diagnosis in adults are still missing apart from autoimmune or malignant diseases (Table 5). Furthermore, the differentiation between sepsis and HLH/MAS remains a major problem in clinical practice, especially when liver, kidney, and multiorgan failure occur.[37]
Table 5.

2.3. Pathogenesis
Central to the pathogenesis of HLH is a massive production of pro-inflammatory cytokines (“cytokine storm”) through an uncontrolled activation of macrophages and CTLs.[30] An impairment of cytotoxic activity of NK cells and CTLs (mainly cytotoxic CD 8+ T-cells) is considered a hallmark of most HLH entities.[22,38] The general understanding is that on the basis of impaired cytotoxic cell function, NK cells, and CTLs fail to sufficiently eliminate antigen-presenting cells, especially macrophages, leading to an inability to terminate immune responses.[30] As a consequence, uncontrolled expansion of macrophages and CTLs with production of large amounts of cytokines occurs with IL-1, IL-6, IL-18, tumor necrosis factor alpha (TNFα), interferon gamma (IFNγ) being the driving forces.[30] The result is an ubiquitous inflammation and cellular organ infiltration, causing the diverse, syndrome-like disease pattern.[28] This hypothesis applies to primary HLH, where genetic mutations cause a dysfunction of perforin-mediated cytotoxicity in 97% of FHL patients.[38] However, in patients suffering from secondary HLH the postulated NK cell deficiency is only seen in 22%.[38] Thus, the precise pathogenesis for secondary HLH where normal NK cell function is found remains speculative. In this setting, an acquired immunodeficiency on the basis of a malignancy, a HIV infection,[39] an iatrogenic immunodeficiency due to immunosuppressive drug therapy (solid organ- and stem cell transplantation)[4,40,41] or any other reason is still discussed as key role in adult HLH pathogenesis.[28,42]
However, in otherwise healthy and immune competent individuals, it remains unclear how viral infections, auto-inflammatory diseases and malignancies induce secondary HLH. Single nucleotide polymorphisms in cytokine genes, which lead to an aggravated and prolonged immune response are just as considered[43,44] as repeated stimulation of toll-like receptors due to genetic predisposition.[45–47] In summary, the general pathophysiology in acquired HLH remains incompletely understood, especially when NK cell deficiency is absent.
2.4. Clinical picture & classic laboratory findings
HLH has a syndrome-like appearance with characteristic clinical and laboratory findings. Particularly during the disease onset, symptoms are rather unspecific and patients develop a clinical picture with fever, hemodynamic instability, hepatosplenomegaly, CNS affection, and an increase in leukocytes and CRP. Unfortunately, early diagnosis is often hampered by delayed appearance of the characteristic laboratory findings.[2] Fever is induced by a misdirected cytokine release, unresponsive to antibiotic treatment and often of subacute, prolonged and recurrent character.[48] It is one of the most consistent symptoms, but certainly not specific for HLH diagnosis.[2,48] CNS symptoms were initially described in FHL,[49–51] but manifest as well in adults suffering from secondary HLH.[52] They range from minor alterations of consciousness, headache, memory loss and disorientation to Guillain–Barré-like syndromes, nerve palsy, meningismus, and seizures. The CSF in HLH patients usually shows an elevated spinal fluid leukocyte count and a mild to moderate elevation of protein content.[49,52] MRI scans typically display polymorphic, bilateral and especially periventricular white matter hyperintensities in T2-weighted, FLAIR images.[51,53] Therefore, HLH patients with CNS affection may often be misdiagnosed with viral or other inflammatory CNS diseases.
2.4.1. Hyperferritinemia
Hyperferritinemia is generally considered as one of the most characteristic findings in HLH. According to a recent study, median and maximal ferritin levels in adults range between 2367 and 25,720 ng/mL, and 6067–684,000 ng/mL, respectively. However, there are also patients, who are diagnosed with HLH with significantly lower ferritin levels (181–2424 ng/mL).[2] Several studies investigated the significance of ferritin levels for HLH diagnosis in children. One study showed a 84% sensitivity for FHL diagnosis for ferritin levels above 500 ng/mL,[3] whereas 2 other studies exhibited a 70% sensitivity and 68% specificity and a 90% sensitivity and 96% specificity for concentrations over 2000 ng/mL and 10,000 ng/mL, respectively.[8,54] The diagnostic value of ferritin concentrations in adult HLH is unknown. A study by Schram et al[55] suggested that ferritin levels beyond 50,000 ng/mL are associated with a variety of disorders (hemochromatosis, hepatocellular injury, hematologic malignancy, solid malignancy, inflammatory conditions, and hemolytic anemia) and are nonspecific.
2.4.2. Cytopenia
Cytopenia is caused by cytokines, especially IFNγ and TNFα and usually affects two or more cell lines. Hemophagocytosis, however, seems to play a minor role.[48] Median thrombocyte counts appear to be lower in adults than in children (6–52×109/L vs. 44–69×109/L) and are reduced below 100×109/L in 86 to 93% of all HLH cases. Median hemoglobin (Hb) levels range from 6.7 to 11.4 g/dL in adults.[2] Hemolysis, however, has to be ruled out in this context. Haptoglobin measurement and exclusion of auto-antibody-mediated hemolysis via Coombs-test are obligate. The degree of neutrocytopenia differs in the literature. While severe agranulocytosis is described in most but not all pediatric cases,[48] neutrocytopenia does not represent a characteristic finding in adults.[56]
2.4.3. Hypertriglyceridemia and hypofibrinogenemia
Hypertriglyceridemia is caused by inhibition of the lipoprotein lipase through TNFα. It occurs in 30 to 89% of HLH patients. Fibrinogen concentrations differ substantially in literature and have to be analyzed together with standard coagulation parameters and platelet counts, especially to exclude other coagulopathies of critically ill patients like disseminated intravascular coagulation (DIC).[57]
2.5. Special features
The characteristic clinical picture and the classic laboratory findings are supplemented by special diagnostics, which play a major role in diagnostic criteria of HLH and MAS: hemophagocytosis and sCD25.
2.5.1. Hemophagocytosis
Despite the fact that the disease owes its name to this feature, hemophagocytosis is rather of minor relevance as diagnostic criterion. Hemophagocytosis occurs in many inflammatory conditions such as sepsis, bacterial, and viral infections (influenza, malaria, leishmaniosis), rheumatologic diseases and after blood transfusions.[58,59] It manifests late in the disease course and was found in only 32% of children on admission, but in 85% at the time of diagnosis.[60] In other reports, hemophagocytosis was present in 59 to 100% of children and in 52 to 100% of adults. Although one study claims a high sensitivity of hemophagocytosis for HLH diagnosis of 83%, the specificity is rather low and additional criteria need to be fulfilled to establish a definite diagnosis.
2.5.2. Soluble interleukin-2 receptor (sCD25)
The soluble interleukin-2 receptor (sIL-2r = sCD25) is a surrogate parameter for T-cell activation[61] and suggested as a sensitive marker for HLH detection. However, the primary data on its sensitivity is derived from only 3 pediatric patient cohorts.[62] On the basis of one report, which showed an 93% sensitivity and 100% specificity for values ≥ 2400 U/mL, this threshold was incorporated in the 2004 HLH diagnostic criteria.[3,62] sCD25 levels are supposed to correlate with higher disease activity, worse clinical response to therapy and worse clinical outcomes, but fail to reliably distinguish between subtypes of HLH/MAS.[62]
In general, median sCD25 values lie between 2963 an d21,500 U/mL, but data in adults are limited. Two studies in adults showed absolute values between 1891 an d206,567 U/mL[63,64] and in a recent review 79% of 775 HLH cases had sCD25 concentrations > 2400 U/mL and 38% > 10,000 U/mL.[18] Prospective clinical trials are needed to clarify the diagnostic relevance of sCD25 in adults and nonautoimmune disease entities.
2.6. Therapy/treatment
The first prospective treatment protocol was established in 1994 by the Histiocyte Society,[33] but revised in 2004.[3] The HLH-2004 protocol now recommends a combined, early chemo-immunotherapy approach consisting of CsA and pulse therapy with dexamethasone and etoposide for at least 8 weeks.[3] Nevertheless, the severe side-effects of etoposide and especially the high liver toxicity and bone marrow suppression remain a general concern.[30] In this context, deaths due to overwhelming infections are reported in primary and secondary HLH in childhood.[11,12,65] Therefore, most clinicians start with an intravenous dexamethasone pulse therapy and added CsA (2–7 mg/kg per day) if there is no rapid response to steroid monotherapy. In most of these patients, CsA induces quick disease control and allows for rapid steroid reduction,[16] but one has to be aware of neurological and renal toxicity. In the end, the HLH-2004 protocol is reserved for more severe and therapy-refractory cases and efforts exist to establish less toxic therapy alternatives. Previous attempts on the basis of PE, intravenous immunoglobulins (IVIG), and methylprednisolone are under investigation.[23,24,66,67]
However, treatment studies in adults, mostly include <20 patients and are of retrospective character.
Furthermore, the studies are extremely heterogenic and drugs were applied in various doses and combinations.[18] Thus, treatment decisions are rather based on clinical expertise, than on reliable data. The situation in patients who develop HLH under immunosuppressive therapy (see also our case report) is even more complex when HLH may be triggered by malignancies or overwhelming infectious diseases. In these patients, the underlying triggering event must be treated but HLH specific therapy may also be needed.
2.6.1. Plasma exchange
In the 1980s, PE was shown to induce transient clinical remission in FHL patients.[68] However, PE was later replaced by chemo-immunotherapy. Lately, PE is regaining attention for acquired HLH as a less toxic therapeutic alternative to the HLH-2004 protocol.[24,67] Especially in autoimmune-related MAS in children, PE combined with corticosteroids has proven therapeutic efficacy by showing an effective disease suppression and improved survival compared to etoposide-based regimens.[23,24,69] Furthermore, in case reports PE seems to be beneficial as salvage therapy, when a CsA and steroid regime fail to control autoimmune-related MAS on the basis of SJIA or AOSD.[23,66,70] In adults and apart from autoimmune diseases, there exist only a few case reports about PE as HLH therapy.[71–73] Most of them showed fatal outcomes, although PE was initially capable of stabilizing the patient's condition. Taken together, PE may be a promising less toxic therapeutic option, especially to control disease onsets of secondary HLH. However, the data are mostly derived from case reports and after termination of PE; the choice of maintenance therapy has to be discussed. A congeneric therapeutic option may be a highly effective cytokine clearance via high cutoff membranes (cytosorb),[74] but up to now, there is no report of a use in the context of HLH therapy.
2.6.2. Interleukine-1-directed therapy
Independent from the underlying pathogenesis, a common feature of HLH entities is a massive cytokine release. Thus, cytokine-directed therapy, targeting cytokines that are typically involved in HLH pathogenesis, is a rising therapeutic alternative. According to the current knowledge, especially IL-1 may be one promising target.[30]
IL-1 is a pro-inflammatory cytokine produced by peripheral mononuclear cells.[75] It leads to leukocyte activation and production of other cytokines like IL-6 and is considered as a relevant molecule in the pathogenesis of SJIA.[47,76] This is supported by data showing an IL-1 related gene expression profile in the blood of patients with newly diagnosed SJIA.[46,76] In addition, serum of SJIA patients is capable of activating IL-1 related genes in monocytes of healthy individuals.[47] Anakinra, a recombinant IL-1 receptor antagonist, is an established treatment agent in SJIA, inducing a long-lasting remission in more than half of SJIA patients[77,78] and represents a major therapeutic option in AOSD.[79] The contribution of IL-1 to HLH development is, however, unclear. Several studies showed a beneficial effect of anakinra in SJIA-associated MAS after inadequate response to steroids and cyclosporine,[80,81] as well as in AOSD complicated by MAS.[26,82] The data of anakinra in autoimmune independent HLH entities is rather limited. To our knowledge three studies reported a beneficial effect of anakinra in a heterogeneous adult HLH cohort (after transplantation, autoimmune disease, acute lymphoblastic leukemia, EBV infection),[27] AOSD related MAS[26] and a sepsis-related MAS-like syndrome, defined by hepatobiliary dysfunction and DIC.[25]
3. Conclusions
To our knowledge this is the first report of an adult kidney transplant recipient suffering from severe, acquired HLH years after transplantation, who was successfully treated by an initial combination of PE and steroids, followed by a maintenance therapy with anakinra, CsA and low dose steroids. HLH has been reported in kidney transplant recipients, mostly weeks after renal transplantation. In only a few patients HLH occurred years after surgery.[4] Late occurrence of HLH was seen in patients with neoplasia (T-cell lymphoma, angiosarcoma, karposi sarcoma), parasitic infection (leishmaniosis, toxoplasmosis, babeiosis, histoplasmosis), parvovirus B19 and histoplasmosis.[4] In our patient, HLH occurred 15 years after living donor kidney transplantation and both solid and hematological malignancies were excluded by means such as multiple CT scans, bone marrow aspiration and flow cytometry. Although, there was no evidence of a viral, bacterial or parasitic infection on admission, the patient presented with severe gastroenteritis, diarrhea, dehydration, and nausea suggesting an infectious HLH trigger. This in line with previous reports where HLH in kidney transplant patients was associated with viral infections such as CMV, EBV, HHV6, HHV8, BKV or with bacterial infections like tuberculosis.[4]
In our patient, the sepsis-like appearance delayed the diagnosis of HLH and the initiation of adequate therapeutic measures. The patient suffered from hepatic and renal transplant failure, severe neurologic dysfunction and relevant hemodynamic instability. Until the time of initiation of PE therapy, none of the 6 known HLH diagnostic criteria were fulfilled and characteristic features like hypertriglyceridemia and relevant pancytopenia according to the HLH 2004 criteria occurred late.[3] Additionally, hemophagocytosis was of only minor severity in an early bone marrow aspiration and could rather be attributable to any other inflammatory condition like sepsis. This observation is consistent with reports from other authors, who found that hemophagocytosis lacked specificity for HLH, when not supported by other criteria.[83] sCD25 is considered as a parameter with higher sensitivity.[62] A retrospective analysis of a sample that was drawn in our patient 3 days before initiation of PE, confirmed a significantly increased sCD25 level of 5450 U/mL. Thus, although we did not measure NK cell activity, our case report suggests that the HLH diagnostic criteria (HLH-2004) may not be sufficient to diagnose the disease in adults at early stages.
High ferritin together with persistent fever, progressive thrombocytopenia or anemia (even when not fulfilling HLH 2004 cutoffs) and CNS impairment pointed to the suspected diagnosis of HLH in our case. But once the diagnosis was considered the question came to therapeutic measures. In our patient, several contraindications existed for the HLH-2004 protocol (Table 4). Other causes of the CNS symptoms such as PRES, PML or viral infection have not been ruled out completely during MRI scan and CSF analysis and the patient suffered from progressive pancytopenia and severe hepatic failure, so that the application of immunosuppressive therapy or etoposide were not reasonable at that time. Thus, we considered cytokine clearance via a PE and corticosteroid-based regime as less toxic therapy alternative, especially to account for the mentioned contraindications. It has already been shown in children that secondary HLH can be successfully treated with a combination of PE, IVIG and methylprednisolone. This approach led to improved survival compared to etoposide- or CsA- based regimens.[24] In adults, however, treatment with PE had only been reported in the form of case reports[71–73] with mostly fatal outcomes. The PE frequency was low, ranging from one procedure at all[71,73] to twice per week[72] and maintenance therapies were diverse including etoposide- and CsA-based regimes,[71] or corticosteroid monotherapy,[73] suggesting an insufficient concept for disease control after PE termination.
In our case, a high frequency PE led to a tremendous improvement of the patient's general condition as well as the neurological status. After the occurrence of further characteristic HLH symptoms (hypertriglyceridemia, relevant pancytopenia) over the next days, PRES, viral disease and PML were considered as unlikely and a maintenance therapy with anakinra and CsA was started. PE was terminated after 18 procedures. All laboratory findings turned to normal, the disease activity was sufficiently suppressed under a maintenance therapy with CsA, anakinra and low dose corticosteroids and the patient could be discharged 75 days after admission. Several follow-ups showed a sufficient disease control. The kidney graft, however, did not recover and we started with intermittent hemodialysis two months after discharge.
In summary, this case report suggests high frequency PE in combination with steroids as an effective and less-toxic alternative to the HLH-2004 protocol for severe HLH onsets in adults or as a bridging approach as long as contraindications to more aggressive therapies exist. Furthermore, the maintenance therapy with CsA, anakinra and low dose corticosteroids showed excellent disease control after termination of PE. The relevance of PE as first line treatment option for severe HLH in adulthood as well as the here applied combination of CsA and IL-1-directed maintenance therapy by anakinra has to be assessed in prospective future studies.
Footnotes
Abbreviations: ALT = alanine aminotransferase, AOSD = adult onset Still's disease, APC = antigen presenting cell, AST = aspartate aminotransferase, BKV = BK-virus, CMV = cytomegalovirus, CNS = central nervous system, CRP = C-reactive protein, CsA = cyclosporine A, CSF = cerebrospinal fluid, CTLs = cytotoxic T-lymphocytes, DIC = disseminated intravascular coagulation, EBV = Ebstein–Barr virus, FHL = familial hemophagocytic lymphohistiocytosis, HAV = hepatitis A virus, HBV = hepatitis B virus, HCV = hepatitis C virus, HEV = hepatitis E virus, HHV6 = human herpes virus 6, HIV = human immune-deficiency virus, HLH = hemophagocytic lymphohistiocytosis, HSV = herpes simplex virus, IFNγ = interferon gamma, IL = interleukin, IVIG = intravenous immunoglobulins, JCV = JC-virus, LDH = lactate dehydrogenase, MAS = macrophage activation syndrome, NK cells = natural killer cells, PCR = polymerase chain reaction, PE = plasma exchange, PML = progressive multifocal leukoencephalopathy, PRES = posterior reversible encephalopathy syndrome, sCD25 or sIL-2r = soluble interleukin-2 receptor, SJIA = systemic juvenile idiopathic arthritis, SLE = systemic lupus erythematosus, SNP = single nucleotide polymorphisms, TLR = toll-like receptors, TNFα = tumor necrosis factor alpha, VZV = varicella zoster virus.
TB and UM both contributed equally to this work.
Consent for publication: Written informed consent was obtained from the participant.
We acknowledge financial support by Deutsche Forschungsgemeinschaft within the funding programme Open Access Publishing, by the Baden-Württemberg Ministry of Science, Research and the Arts and by Ruprecht-Karls-Universität Heidelberg.
The authors have no conflicts of interest to disclose.
References
- [1].Janka GE, Lehmberg K. Hemophagocytic syndromes—an update. Blood Rev 2014;28:135–42. [DOI] [PubMed] [Google Scholar]
- [2].Machowicz R, Janka G, Wiktor-Jedrzejczak W. Similar but not the same: differential diagnosis of HLH and sepsis. Crit Rev Oncol Hematol 2017;114:1–2. [DOI] [PubMed] [Google Scholar]
- [3].Henter JI, Horne A, Aricó M, et al. HLH-2004: diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer 2007;48:124–31. [DOI] [PubMed] [Google Scholar]
- [4].Ponticelli C, Alberighi ODC. Haemophagocytic syndrome—a life-threatening complication of renal transplantation. Nephrol Dial Transplant 2009;24:2623–7. [DOI] [PubMed] [Google Scholar]
- [5].Atteritano M, David A, Bagnato G, et al. Haemophagocytic syndrome in rheumatic patients. A systematic review. Eur Rev Med Pharmacol Sci 2012;16:1414–24. [PubMed] [Google Scholar]
- [6].Henter JI, Elinder G, Ost A. Diagnostic guidelines for hemophagocytic lymphohistiocytosis. The FHL Study Group of the Histiocyte Society. Semin Oncol 1991;18:29–33. [PubMed] [Google Scholar]
- [7].Henter JI, Elinder GR, Söder O, et al. Incidence in Sweden and clinical features of familial hemophagocytic lymphohistiocytosis. Acta Paediatrica 1991;80:428–35. [DOI] [PubMed] [Google Scholar]
- [8].Allen CE, Yu X, Kozinetz CA, et al. Highly elevated ferritin levels and the diagnosis of hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer 2008;50:1227–35. [DOI] [PubMed] [Google Scholar]
- [9].Gürgey A, Göğüş S, Ozyürek E, et al. Primary hemophagocytic lymphohistiocytosis in Turkish children. Pediatric Hematology and Oncology 2003;20:367–71. [PubMed] [Google Scholar]
- [10].Machaczka M, Vaktnäs J, Klimkowska M, et al. Malignancy-associated hemophagocytic lymphohistiocytosis in adults: a retrospective population-based analysis from a single center. Leuk Lymphoma 2011;52:613–9. [DOI] [PubMed] [Google Scholar]
- [11].Henter JI, Samuelsson-Horne A, Aricó M, et al. Treatment of hemophagocytic lymphohistiocytosis with HLH-94 immunochemotherapy and bone marrow transplantation. Blood 2002;100:2367–73. [DOI] [PubMed] [Google Scholar]
- [12].Sung L, King SM, Carcao M, et al. Adverse outcomes in primary hemophagocytic lymphohistiocytosis. J Pediatr Hematol Oncol 2002;24:550–4. [DOI] [PubMed] [Google Scholar]
- [13].Karapinar B, Yilmaz D, Balkan C, et al. An unusual cause of multiple organ dysfunction syndrome in the pediatric intensive care unit: hemophagocytic lymphohistiocytosis. PediatrCritic Care Med 2009;10:285–90. [DOI] [PubMed] [Google Scholar]
- [14].Minoia F, Davì S, Horne A, et al. Clinical features, treatment, and outcome of macrophage activation syndrome complicating systemic juvenile idiopathic arthritis: a multinational, multicenter study of 362 patients. Arthritis Rheumatol 2014;66:3160–9. [DOI] [PubMed] [Google Scholar]
- [15].Bennett TD, Fluchel M, Hersh AO, et al. Macrophage activation syndrome in children with systemic lupus erythematosus and children with juvenile idiopathic arthritis. Arthritis Rheum 2012;64:4135–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Stéphan JL, Koné-Paut I, Galambrun C, et al. Reactive haemophagocytic syndrome in children with inflammatory disorders. A retrospective study of 24 patients. Rheumatology (Oxford) 2001;40:1285–92. [DOI] [PubMed] [Google Scholar]
- [17].Sawhney S, Woo P, Murray KJ. Macrophage activation syndrome: a potentially fatal complication of rheumatic disorders. Arch Dis Child 2001;85:421–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Ramos-Casals M, Brito-Zerón P, López-Guillermo A, et al. Adult haemophagocytic syndrome. Lancet 2014;383:1503–16. [DOI] [PubMed] [Google Scholar]
- [19].Seidel MG. CD27: a new player in the field of common variable immunodeficiency and EBV-associated lymphoproliferative disorder? J Allergy Clin Immunol 2012;129:1175–6. 1175–authorreply1175–6. [DOI] [PubMed] [Google Scholar]
- [20].Sandrock K, Nakamura L, Vraetz T, et al. Platelet secretion defect in patients with familial hemophagocytic lymphohistiocytosis type 5 (FHL-5). Blood 2010;116:6148–50. [DOI] [PubMed] [Google Scholar]
- [21].Huck K, Feyen O, Niehues T, et al. Girls homozygous for an IL-2-inducible T cell kinase mutation that leads to protein deficiency develop fatal EBV-associated lymphoproliferation. J Clin Invest 2009;119:1350–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Ramachandran S, Zaidi F, Aggarwal A, et al. Recent advances in diagnostic and therapeutic guidelines for primary and secondary hemophagocytic lymphohistiocytosis. Blood Cells Mol Dis 2016;64:53–7. [DOI] [PubMed] [Google Scholar]
- [23].Shi L, Hu F, Xu C, et al. Plasma exchange successfully treated macrophage activation syndrome in rheumatoid factor-positive polyarticular juvenile idiopathic arthritis with combined pneumonia. Int J Rheum Dis March 2017;DOI: 10.1111/1756-185X.13064. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- [24].Demirkol D, Yildizdas D, Bayrakci B, et al. Hyperferritinemia in the critically ill child with secondary hemophagocytic lymphohistiocytosis/sepsis/multiple organ dysfunction syndrome/macrophage activation syndrome: what is the treatment? Crit Care 2012;16:R52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Shakoory B, Carcillo JA, Chatham WW, et al. Interleukin-1 receptor blockade is associated with reduced mortality in sepsis patients with features of macrophage activation syndrome: reanalysis of a prior phase III trial. Crit Care Med 2016;44:275–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Parisi F, Paglionico A, Varriano V, et al. Refractory adult-onset Still disease complicated by macrophage activation syndrome and acute myocarditis: a case report treated with high doses (8 mg/kg/d) of anakinra. Medicine 2017;96:e6656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Wohlfarth P, Agis H, Gualdoni GA, et al. Interleukin 1 receptor antagonist anakinra, intravenous immunoglobulin, and corticosteroids in the management of critically Ill adult patients with hemophagocytic lymphohistiocytosis. J Int Care Med 2017;15: 885066617711386. [DOI] [PubMed] [Google Scholar]
- [28].Janka G, Imashuku S, Elinder G, et al. Infection- and malignancy-associated hemophagocytic syndromes. Secondary hemophagocytic lymphohistiocytosis. Hematol Oncol Clin North Am 1998;12:435–44. [DOI] [PubMed] [Google Scholar]
- [29].Emmenegger U, Reimers A, Frey U, et al. Reactive macrophage activation syndrome: a simple screening strategy and its potential in early treatment initiation. Swiss Med Wkly 2002;132:230–6. [DOI] [PubMed] [Google Scholar]
- [30].Schulert GS, Grom AA. Pathogenesis of macrophage activation syndrome and potential for cytokine- directed therapies. Annu Rev Med 2015;66:145–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Moradinejad MH, Ziaee V. The incidence of macrophage activation syndrome in children with rheumatic disorders. Minerva Pediatr 2011;63:459–66. [PubMed] [Google Scholar]
- [32].Ravelli A, Minoia F, Davì S, et al. 2016 classification criteria for macrophage activation syndrome complicating systemic juvenile idiopathic arthritis: A European League Against Rheumatism/American College of Rheumatology/Paediatric Rheumatology International Trials Organisation Collaborat. Arthritis Rheumatol 2016;68:566–76. [DOI] [PubMed] [Google Scholar]
- [33].Henter JI, Aricó M, Egeler RM, et al. HLH-94: a treatment protocol for hemophagocytic lymphohistiocytosis. Med Pediatr Oncol 1997;28:342–7. [DOI] [PubMed] [Google Scholar]
- [34].Ravelli A, Magni-Manzoni S, Pistorio A, et al. Preliminary diagnostic guidelines for macrophage activation syndrome complicating systemic juvenile idiopathic arthritis. J Pediatr 2005;146:598–604. [DOI] [PubMed] [Google Scholar]
- [35].Fardet L, Galicier L, Lambotte O, et al. Development and validation of the HScore, a score for the diagnosis of reactive hemophagocytic syndrome. Arthritis Rheumatol 2014;66:2613–20. [DOI] [PubMed] [Google Scholar]
- [36].Tamamyan GN, Kantarjian HM, Ning J, et al. Malignancy-associated hemophagocytic lymphohistiocytosis in adults: relation to hemophagocytosis, characteristics, and outcomes. Cancer 2016;122:2857–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Nusshag C, Weigand MA, Zeier M, et al. Issues of acute kidney injury staging and management in sepsis and critical illness: a narrative review. Int J Mol Sci 2017;18:1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Bryceson YT, Pende D, Maul-Pavicic A, et al. A prospective evaluation of degranulation assays in the rapid diagnosis of familial hemophagocytic syndromes. Blood 2012;119:2754–63. [DOI] [PubMed] [Google Scholar]
- [39].Fardet L, Lambotte O, Meynard J-L, et al. Reactive haemophagocytic syndrome in 58 HIV-1-infected patients: clinical features, underlying diseases and prognosis. AIDS 2010;24:1299–306. [DOI] [PubMed] [Google Scholar]
- [40].Karras A, Thervet E, Legendre C. Hemophagocytic syndrome in renal transplant recipients: report of 17 cases and review of literature. Transplantation 2004;77:238–43. [DOI] [PubMed] [Google Scholar]
- [41].Abe Y, Choi I, Hara K, et al. Hemophagocytic syndrome: a rare complication of allogeneic nonmyeloablative hematopoietic stem cell transplantation. Bone Marrow Transplant 2002;29:799–801. [DOI] [PubMed] [Google Scholar]
- [42].Risdall RJ, McKenna RW, Nesbit ME, et al. Virus-associated hemophagocytic syndrome: a benign histiocytic proliferation distinct from malignant histiocytosis. Cancer 1979;44:993–1002. [DOI] [PubMed] [Google Scholar]
- [43].Sinha S, Mishra SK, Sharma S, et al. Polymorphisms of TNF-enhancer and gene for FcgammaRIIa correlate with the severity of falciparum malaria in the ethnically diverse Indian population. Malaria J 2008;7:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Poggi A, Costa P, Tomasello E, et al. IL-12-induced up-regulation of NKRP1A expression in human NK cells and consequent NKRP1A-mediated down-regulation of NK cell activation. Eur J Immunol 1998;28:1611–6. [DOI] [PubMed] [Google Scholar]
- [45].Behrens EM, Canna SW, Slade K, et al. Repeated TLR9 stimulation results in macrophage activation syndrome-like disease in mice. J Clin Invest 2011;121:2264–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Fall N, Barnes M, Thornton S, et al. Gene expression profiling of peripheral blood from patients with untreated new-onset systemic juvenile idiopathic arthritis reveals molecular heterogeneity that may predict macrophage activation syndrome. Arthritis Rheum 2007;56:3793–804. [DOI] [PubMed] [Google Scholar]
- [47].Pascual V, Allantaz F, Arce E, et al. Role of interleukin-1 (IL-1) in the pathogenesis of systemic onset juvenile idiopathic arthritis and clinical response to IL-1 blockade. J Exp Med 2005;201:1479–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Janka GE. Familial and acquired hemophagocytic lymphohistiocytosis. Eur J Pediatr 2007. [DOI] [PubMed] [Google Scholar]
- [49].Horne A, Trottestam H, Aricó M, et al. Frequency and spectrum of central nervous system involvement in 193 children with haemophagocytic lymphohistiocytosis. Br J Haematol 2008;140:327–35. [DOI] [PubMed] [Google Scholar]
- [50].Yang S, Zhang L, Jia C, et al. Frequency and development of CNS involvement in Chinese children with hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer 2010;54:408–15. [DOI] [PubMed] [Google Scholar]
- [51].Deiva K, Mahlaoui N, Beaudonnet F, et al. CNS involvement at the onset of primary hemophagocytic lymphohistiocytosis. Neurology 2012;78:1150–6. [DOI] [PubMed] [Google Scholar]
- [52].Cai G, Wang Y, Liu X, et al. Central nervous system involvement in adults with haemophagocytic lymphohistiocytosis: a single-center study. Ann Hematol 2017;96:1279–85. [DOI] [PubMed] [Google Scholar]
- [53].Rego I, Severino M, Micalizzi C, et al. Neuroradiologic findings and follow-up with magnetic resonance imaging of the genetic forms of haemophagocytic lymphohistiocytosis with CNS involvement. Pediatr Blood Cancer 2011;58:810–4. [DOI] [PubMed] [Google Scholar]
- [54].Lehmberg K, McClain KL, Janka GE, et al. Determination of an appropriate cut-off value for ferritin in the diagnosis of hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer 2014;61:2101–3. [DOI] [PubMed] [Google Scholar]
- [55].Schram AM, Campigotto F, Mullally A, et al. Marked hyperferritinemia does not predict for HLH in the adult population. Blood 2015;125:1548–52. [DOI] [PubMed] [Google Scholar]
- [56].Aulagnon F, Lapidus N, Canet E, et al. Acute kidney injury in adults with hemophagocytic lymphohistiocytosis. Am J Kidney Dis 2015;65:851–9. [DOI] [PubMed] [Google Scholar]
- [57].Asakura H, Takahashi H, Uchiyama T, et al. Proposal for new diagnostic criteria for DIC from the Japanese Society on Thrombosis and Hemostasis. Thromb J 2016;14:42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Jordan MB, Allen CE, Weitzman S, et al. How I treat hemophagocytic lymphohistiocytosis. Blood 2011;118:4041–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Zoller EE, Lykens JE, Terrell CE, et al. Hemophagocytosis causes a consumptive anemia of inflammation. J Exp Med 2011;208:1203–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Janka GE, Schneider EM. Modern management of children with haemophagocytic lymphohistiocytosis. Br J Haematol 2004;124:4–14. [DOI] [PubMed] [Google Scholar]
- [61].Coca A, Bundy KW, Marston B, et al. Macrophage activation syndrome: serological markers and treatment with anti-thymocyte globulin. Clin Immunol 2009;132:10–8. [DOI] [PubMed] [Google Scholar]
- [62].Lin M, Park S, Hayden A, et al. Clinical utility of soluble interleukin-2 receptor in hemophagocytic syndromes: a systematic scoping review. Ann Hematol 2017;15:92–111. [DOI] [PubMed] [Google Scholar]
- [63].Beutel K, Gross-Wieltsch U, Wiesel T, et al. Infection of T lymphocytes in Epstein–Barr virus-associated hemophagocytic lymphohistiocytosis in children of non-Asian origin. Pediatr Blood Cancer 2009;53:184–90. [DOI] [PubMed] [Google Scholar]
- [64].Chellapandian D, Das R, Zelley K, et al. Treatment of Epstein Barr virus-induced haemophagocytic lymphohistiocytosis with rituximab-containing chemo-immunotherapeutic regimens. Br J Haematol 2013;162:376–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Gupta AA, Tyrrell P, Valani R, et al. Experience with hemophagocytic lymphohistiocytosis/macrophage activation syndrome at a single institution. J Pediatr Hematol Oncol 2009;31:81–4. [DOI] [PubMed] [Google Scholar]
- [66].Nakakura H, Ashida A, Matsumura H, et al. A case report of successful treatment with plasma exchange for hemophagocytic syndrome associated with severe systemic juvenile idiopathic arthritis in an infant girl. Ther Apher Dial 2009;13:71–6. [DOI] [PubMed] [Google Scholar]
- [67].Song KS, Sung HJ. Effect of plasma exchange on the circulating IL-6 levels in a patient with fatal hemophagocytic syndrome associated with bile ductopenia. Ther Apher Dial 2006;10:87–9. [DOI] [PubMed] [Google Scholar]
- [68].Ladisch S, Ho W, Matheson D, et al. Immunologic and clinical effects of repeated blood exchange in familial erythrophagocytic lymphohistiocytosis. Blood 1982;60:814–21. [PubMed] [Google Scholar]
- [69].Bosnak M, Erdogan S, Aktekin EH, et al. Therapeutic plasma exchange in primary hemophagocytic lymphohistiocytosis: reports of two cases and a review of the literature. Transfus Apher Sci 2016;55:353–6. [DOI] [PubMed] [Google Scholar]
- [70].Komiya Y, Takenaka K, Nagasaka K. Successful treatment of glucocorticoid and cyclosporine refractory adult-onset Still's disease complicated with hemophagocytic syndrome with plasma exchange therapy and tocilizumab: a case report. Nihon Rinsho Meneki Gakkai Kaishi 2013;36:478–83. [DOI] [PubMed] [Google Scholar]
- [71].Arai A, Nogami A, Imadome K-I, et al. Sequential monitoring of serum IL-6, TNF-α, and IFN-γ levels in a CAEBV patient treated by plasma exchange and immunochemotherapy. Int J Hematol 2012;96:669–73. [DOI] [PubMed] [Google Scholar]
- [72].Lin S, Li Y, Long J, et al. Acute liver failure caused by hemophagocytic lymphohistiocytosis in adults: a case report and review of the literature. Medicine 2016;95:e5431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Pastula DM, Burish M, Reis GF, et al. Adult-onset central nervous system hemophagocytic lymphohistiocytosis: a case report. BMC Neurol 2015;15:203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Kogelmann K, Jarczak D, Scheller M, et al. Hemoadsorption by CytoSorb in septic patients: a case series. Crit Care 2017;21:74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Dinarello CA, Renfer L, Wolff SM. Human leukocytic pyrogen: purification and development of a radioimmunoassay. Proc Natl Acad Sci USA 1977;74:4624–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Gattorno M, Piccini A, Lasigliè D, et al. The pattern of response to anti-interleukin-1 treatment distinguishes two subsets of patients with systemic-onset juvenile idiopathic arthritis. Arthritis Rheum 2008;58:1505–15. [DOI] [PubMed] [Google Scholar]
- [77].Nigrovic PA, Mannion M, Prince FHM, et al. Anakinra as first-line disease-modifying therapy in systemic juvenile idiopathic arthritis: Report of forty-six patients from an international multicenter series. Arthritis Rheum 2011;63:545–55. [DOI] [PubMed] [Google Scholar]
- [78].Quartier P, Allantaz F, Cimaz R, et al. A multicentre, randomised, double-blind, placebo-controlled trial with the interleukin-1 receptor antagonist anakinra in patients with systemic-onset juvenile idiopathic arthritis (ANAJIS trial). Ann Rheum Dis 2011;70:747–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Fitzgerald AA, LeClercq SA, Yan A, et al. Rapid responses to anakinra in patients with refractory adult-onset Still's disease. Arthritis Rheum 2005;52:1794–803. [DOI] [PubMed] [Google Scholar]
- [80].Kelly A, Ramanan AV. A case of macrophage activation syndrome successfully treated with anakinra. Nat Clin Pract Rheumatol 2008;4:615–20. [DOI] [PubMed] [Google Scholar]
- [81].Miettunen PM, Narendran A, Jayanthan A, et al. Successful treatment of severe paediatric rheumatic disease-associated macrophage activation syndrome with interleukin-1 inhibition following conventional immunosuppressive therapy: case series with 12 patients. Rheumatology (Oxford) 2011;50:417–9. [DOI] [PubMed] [Google Scholar]
- [82].Durand M, Troyanov Y, Laflamme P, et al. Macrophage activation syndrome treated with anakinra. J Rheumatol 2010;37:879–80. [DOI] [PubMed] [Google Scholar]
- [83].Ho C, Yao X, Tian L, et al. Marrow assessment for hemophagocytic lymphohistiocytosis demonstrates poor correlation with disease probability. Am J Clin Pathol 2014;141:62–71. [DOI] [PubMed] [Google Scholar]