

Abstract
Research on acetaminophen (APAP) toxicity over the last several decades has focused on the pathophysiology of liver injury, but increasingly attention is paid to other known and possible adverse effects. It has been known for decades that APAP causes acute kidney injury, but confusion exists regarding prevalence, and the mechanisms have not been well investigated. More recently, evidence for pulmonary, endocrine, neurological, and neurodevelopmental toxicity has been reported in a number of published experimental, clinical, and epidemiological studies, but the quality of those studies has varied. It is important to view those data critically due to implications for regulation and clinical practice. Here, we review evidence and proposed mechanisms for extrahepatic adverse effects of APAP and weigh weaknesses and strengths in the available data.
Relevance for patients:
APAP is one of the most commonly used drugs in the West. Although it is generally considered safe when used according to manufacturer recommendations, it has been known for decades that overdose can cause liver injury. Recent studies have suggested that APAP can damage cells in other organs as well, leading to calls for more and stricter regulations, which would limit use of this otherwise effective drug. It is especially important to view claims of developmental effects of antenatal APAP exposure with a critical eye because APAP is currently the only over-the-counter medication recommended for pregnant women to self-treat pain and fever.
Keywords: liver injury, kidney injury, endocrine disruptors, neurotoxicity, ototoxicity
1. Introduction
Acetaminophen (APAP; a.k.a. paracetamol) is one of the most commonly used drugs in the US [1] and throughout the West, but has a relatively low therapeutic index. The major target organ of APAP toxicity is the liver. In fact, APAP is the principal cause of acute liver failure (ALF) and related deaths in several countries [2]. The hepatotoxicity of APAP was first reported in the 1960s [3-5]. In the five decades since those initial reports, studies of APAP toxicity have focused almost exclusively on the prevalence and mechanisms of liver injury. Recently, however, attention has shifted toward other adverse effects. A large number of studies have reported neurological [6-14], pulmonary [15-21] and developmental toxicity [6,7,11,14,22] in both preclinical models and humans.
It is important to critically evaluate the evidence for toxic effects of any drug or other xenobiotic. Claims of toxicity can lead to changes in clinical practice or regulation that can affect patient care. Recently, concerns regarding liver injury caused by APAP have led the US FDA to reduce the maximum amount of APAP allowed in prescription formulations to 325 mg, and to recommend lower daily doses for over-the-counter use [23]. It is especially important to view claims of developmental and congenital effects of intrauterine APAP exposure with a critical eye because APAP is currently the most commonly used drug among pregnant women and for many years was the only analgesic considered safe for use during pregnancy [24,25], a perception that still exists among many clinicians and patients. An association between APAP use in pregnancy and disease in offspring could easily lead to changes in clinical practice, just as associations between NSAIDs and various adverse outcomes such as low birth weight, birth defects, and child mortality led the FDA to classify aspirin and others as category D for pregnancy, meaning that there is positive evidence for maternal fetal risk, and caused clinicians to recommend against their use [24].
The purpose of this review is to summarize studies of adverse extrahepatic effects of APAP and to evaluate the evidence for those effects. Animal studies, human studies and epidemiological reports are discussed. Special attention is given to the pathophysiological mechanisms that have been proposed to explain the phenotypic findings from those data. The review begins with what is known about the mechanisms of toxicity in the liver, and findings from other organs are discussed with reference to those well-known mechanisms. Overall, it is clear that APAP is toxic in other organs, but the quality of the evidence and mechanisms varies. In many cases, there is a paucity of mechanistic data, or the available mechanistic studies suffer from poor design. However, that does not necessarily invalidate observations of adverse effects. We strongly recommend that future investigations use only reliable in vivo models and doses that are relevant for the human context.
Table 1.
Proposed extra-hepatic adverse effects of APAP
| Toxicity | Evidence | Proposed mechanisms | Comments |
|---|---|---|---|
| Renal | Clinical and rodent studies | Protein binding, ɤ-glutamyl cycling | Strong human and rodent data |
| Pulmonary | Epidemiology, limited preclinical studies | GSH depletion, oxidative stress, neurogenic inflammation | Better study designs needed |
| Endocrine | Epidemiology, limited preclinical studies | Altered sex steroid metabolism, inhibition of prostaglandin synthesis | Conflicting human and experimental data |
| Ototoxicity | Case reports, limited preclinical studies | Oxidative stress, ER stress | Strong human data, conflicting experimental data |
| Neurobehavioral | Epidemiology, limited preclinical studies | Endocrine disruption, endocannabinoid signaling, direct neurotoxicity | Better study designs needed |
2. Overview of APAP metabolism and hepatotoxicity
Although several critical details are still missing, the metabolism and toxicity of APAP in the liver have been thoroughly investigated [26] (Figure 1). After therapeutic doses, approximately one-third is glucuronidated while another third is sulfated [26, 27]. Any remaining parent compound is converted by cytochrome P450 enzymes to an electrophilic intermediate, believed to be N-acetyl-p-benzoquinone imine (NAPQI) [28]. Binding of the reactive metabolite to proteins is known to be the initiating event in liver injury [29-32]. Binding to mitochondrial proteins appears to be particularly important. Changes in mitochondrial function and integrity are known to occur in the liver after APAP overdose in both mice and humans [15, 33 - 36]. Interestingly, the reactive metabolite of N-acetyl-p-aminophenol (AMAP), an isomer of APAP, binds much less to mitochondrial proteins in primary mouse hepatocytes (PMH) than the metabolite of APAP, and PMH are much less susceptible to the toxicity of AMAP than of APAP [37]. Furthermore, unlike PMH, AMAP treatment does result in mitochondrial protein adducts in primary human hepatocytes (PHH) [37], which are damaged by AMAP [37,38]. Finally, rats are less susceptible to APAP hepatotoxicity than mice and also have less mitochondrial protein binding after APAP overdose [39]. Together, those data strongly suggest that mitochondrial protein binding is critical.
Figure 1. Pathophysiology of APAP-induced liver and kidney injury. Most of a dose of acetaminophen (APAP) is glucuronidated or sulfated in the liver and then excreted. A small percentage in both the liver and kidney is converted to the electrophilic intermediate N-acetyl-p-benzoquinone imine (NAPQI). NAPQI can be detoxified by reaction with glutathione (GSH), which depletes GSH stores. NAPQI can also bind to proteins, which leads to cell death. The mechanisms of cell death in the liver include mitochondrial oxidative stress, c-Jun N-terminal kinase (JNK) activation and nuclear DNA fragmentation (inset). In the kidney, GSH depletion is exacerbated by the GGT cycle, which enhances the nephrotoxicity.
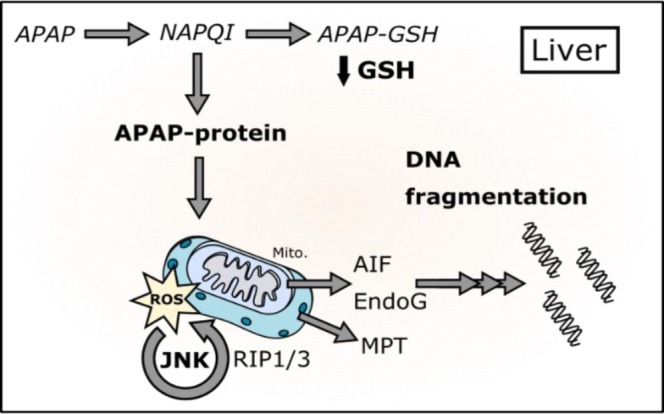
Although it is not known exactly how it occurs, the mitochondrial protein binding seems to cause oxidative stress. The major reactive oxygen species (ROS) in APAP hepatotoxicity are superoxide (O2-) and peroxynitrite (ONOO-) [40], which form primarily within mitochondria and drive the injury [40-46]. Replenishment of glutathione by treatment with the precursor N-acetylcysteine (NAC) protects against APAP hepatotoxicity not only by scavenging the reactive metabolite of APAP, but also by reducing oxidative stress [47,48].
The initial oxidative stress after APAP overdose activates mitogen - activated protein kinases (MAPKs), including the cJun N-terminal kinases (Jnk) 1/2 [49,50] (Figure 1). The role of Jnk 1/2 is controversial. The Jnk 1/2 inhibitor SP600125 protects against APAP toxicity in mice in vivo and in both PMH and PHH [51,52]. Although some groups have also shown protection with knockdown or knockout of Jnk isoforms, particularly Jnk2 [51], others have failed to reproduce those results [52-55]. The discrepancy between different studies that utilized Jnk2 deficient mice may be due to use of control animals from different substrains [56]. Interestingly, one recent study demonstrated that neither Jnk 1 nor combined Jnk 1/2 deficiency in the liver is protective against APAP hepatotoxicity [55]. In fact, Jnk1/2 knockout appeared to worsen injury [55]. Furthermore, SP600125 protected in the double knockout mice [55]. The authors concluded that Jnk 1/2 is not part of the mechanism of toxicity and that SP600125 protects through off-target effects [55]. However, those results do not explain why other Jnk 1/2 inhibitors also protect against APAP [53,57]. Overall, the weight of the evidence favors a role for Jnk [58]. Once activated, Jnk 1/2 translocates to mitochondria [44,59], and it is thought that it enhances the mitochondrial oxidative stress [59,60]. Other kinases that have been shown to play a role in mice include the mixed lineage kinase 3 (Mlk3) [61] and the receptor interacting protein kinases (Ripk) 1 and 3 [62-64]; however, their exact mechanisms are unclear.
The mitochondrial permeability transition (MPT) is also a critical step in the mechanism of APAP-induced liver injury (Figure 1). MPT inhibitors and genetic deletion of MPT pore components protect against APAP hepatotoxicity both in vitro and in vivo [34,65-67]. The resulting mitochondrial swelling leads to lysis of mitochondria and release of mitochondrial contents [35,68,69]. Mitochondrial endonucleases, in particular, are liberated and translocate to nuclei where they cleave genomic DNA [69]. Although nuclear DNA fragmentation is widely considered a hallmark of apoptosis, oncotic necrosis is actually the major mode of cell death in the liver after APAP overdose. Studies in both humans and mice demonstrate that apoptosis has, at most, a minor role [70-73].
In addition to the intracellular mechanisms of toxicity described above, results from numerous studies have demonstrated that inflammation may enhance APAP-induced liver injury [74,75]. The earliest evidence for a contribution of inflame-mation to APAP hepatotoxicity was the finding that resident macrophages in the liver (Kupffer cells) are activated after APAP overdose in rats [76] and that inhibition of macrophages with gadolinium chloride was protective in that model [77]. The latter finding was later repeated in mice [78]. Similarly, it was also reported that antibodies against neutrophils can protect against APAP hepatotoxicity in rats and mice [79,80]. Finally, damage-associated molecular patterns (DAMPs) are released during APAP hepatotoxicity in both mice and humans [35,36] and several studies revealed that inhibition of Nalp3 inflammasome-mediated DAMP signaling in myeloid cells can reduce the injury [81-84]. However, the conclusions from those studies are controversial. Gadolinium chloride has numerous effects other than macrophage inactivation that could also explain protection against hepatotoxicity, and it was reported that targeting macrophages with liposomal clodrinate actually exacerbated the APAP-induced liver injury [85]. Furthermore, deficiency of Nalp3 signaling components does not protect against APAP toxicity, and modulation of IL-1β signaling also has no effect [86,87]. For more detailed information about sterile inflammation in APAP hepatotoxicity, the reader is directed to two excellent reviews that have recently been published [74,75].
Importantly, it appears that the mechanisms of APAP hepatotoxicity are the same in both humans and mice. Both GSH depletion [88,89] and APAP-protein binding are known to occur in humans [27,90] and oxidative stress, Jnk 1/2 activation and the MPT have been demonstrated in human hepatocytes treated with APAP [50,73]. Finally, there is evidence that mitochondrial damage is important in human APAP hepatotoxicity too [35,36,91].
3. Nephrotoxicity
Evidence. Numerous studies have shown that large doses of APAP can cause kidney injury in rodent models [4,15,92-96] and many reports of kidney injury in humans after APAP overdose have been published [3,97-102]. An often-cited figure for the overall incidence of renal dysfunction in patients diagnosed with APAP overdose is approximately 1%. However, this was derived from a single early review of unselected patients diagnosed with “APAP poisoning” at a single center in the UK [103]. Multiple reports suggest that the prevalence of renal injury among APAP overdose patients who develop liver injury is much greater; values from 10% to 79% have been reported [98,99,102-105]. One study found that circulating creatinine levels were ≥ 2 mg/dL (177 µmol/l) (reference interval: 0.7-1.2 mg/dL or 60–115 µmol/l) in approximately 50% of APAP-induced ALF patients, and the levels were higher in non-survivors compared to survivors [105]. Those data were supported by later studies that showed plasma creatinine level at admission and serum kidney injury molecule 1 (KIM-1) are predictive of poor patient outcome after APAP overdose [98,106]. Interestingly, some evidence suggests that chronic use of low doses of APAP can increase risk for kidney disease and cause analgesic nephropathy [107,108], although that has been questioned by findings from very large studies of “healthy” individuals who regularly use over-the-counter analgesics including APAP [108].
Proposed mechanisms. Although the nephrotoxicity of APAP has been known about for decades, surprisingly few studies have explored the mechanisms. Early on, it was thought that endotoxemia as a result of failure of the damaged liver to eliminate endotoxins from the normal GI flora was responsible for the renal damage [104], but results from later studies suggested a more direct effect involving reactive metabolites of APAP and APAP-protein binding [109]. There are significant species differences, and even within-species strain differences, in renal metabolism of APAP [110]. In Fischer F344 rats, APAP and NAPQI appear to be converted to p-aminophenol (PAP) by deacetylation in the kidney, and PAP can be further metabolized to a reactive quinone imine other than NAPQI, possibly by a prostaglandin endoperoxide synthase (PGES; aka cyclooxygenase, COX) [110-115]. Based on those data, it was initially thought that APAP nephrotoxicity was mediated by PAP. However, it was later demonstrated that inhibition of deacetylation had no effect on covalent protein binding in renal microsomes from Sprague-Dawley (SD) rats [116], and an antibody against the N-acetyl moiety of APAP-cysteine could bind to APAP-protein adducts in the kidneys of mice after APAP treatment but not after treatment with p-aminophenol [117]. Furthermore, covalent binding in renal microsomes from SD rats can be prevented by the P450 inhibitor 1-aminobenzotriazole [116], and the nephrotoxicity of APAP in mice is reduced by the P450 inhibitor piperonylbutoxide [117]. It is also apparent that sex differences in APAP nephrotoxicity in mice are due to differences in renal P450s. Female mice are resistant to renal injury even at doses of APAP that cause hepatotoxicity, and that is likely due to hormone-induced differences in P450 expression. Castration of male mice reduces APAP metabolism and protects against APAP-induced kidney injury [118], while testosterone injections induce Cyp2e1 and render female mice susceptible to APAP nephrotoxicity [119]. Together, those data strongly suggest that APAP nephrotoxicity in mice is mediated at least in part by P450s and the same reactive metabolite of APAP that causes liver injury. Which species (mouse or rat) and which strain (F344 or SD rats) is more relevant for human APAP nephrotoxicity is not yet known. PAP and PAP metabolites have been detected in urine from humans after APAP ingestion [120,121], which may suggest that deacetylation of APAP to PAP occurs in humans. However, PAP and APAP metabolism are difficult to disentangle. Furthermore, we know that the mouse is a better model for the liver injury caused by APAP [39]. Aside from cytochrome P450s, results from studies using isolated rabbit and human kidney microsomes have indicated that a PGES/COX can also convert APAP to NAPQI (via a phenoxy radical intermediate) [122]. Interestingly, more recent studies showed that renal injury after APAP overdose in mice is exacerbated by free APAP-cysteine from APAP-GSH [95,96]. APAP-cysteine generated from the breakdown of APAP-GSH in the GI tract and kidneys can act as an acceptor of the γ-glutamyl moiety of GSH in the GSH cycle, and thereby exacerbate GSH depletion in the kidneys [96].
Overall, it appears that NAPQI formation and protein binding are critical, similar to the liver. There is also some evidence that APAP can inhibit mitochondrial respiration in kidney cells from rodents [123,124]. However, little is currently known about APAP nephrotoxicity beyond those results. Although it is tempting to assume that the mechanisms are the same as in the liver due to the involvement of protein binding and mitochondria, there is currently no direct evidence for oxidative stress, kinase activation, or the MPT in APAP nephrotoxicity.
Biological relevance of proposed mechanisms. Nephrotoxicity is clearly a risk after APAP overdose. Available data suggest that protein binding and mitochondrial dysfunction occur in the liver after APAP overdose and that the injury is exacerbated by glutathione cycling, but much more work is needed to prove the importance of those phenomena in APAP nephrotoxicity. This is especially important because acute kidney injury is a predictor of poor patient outcome after APAP overdose [99,106], possibly because it contributes to death after APAP overdose through multi-organ failure. The high affinity of the PGES for APAP has prompted some to speculate that it is responsible for the increased risk of kidney disease after chronic low-dose exposure to the drug [122,110,115], but again, the occurrence of APAP nephrotoxicity among therapeutic users is controversial. We recommend that future research on APAP nephrotoxicity be focused on the importance of mitochondrial dysfunction and kinase signaling and treatments that could address those, as well as mechanisms of renal cell recovery that have been demonstrated to be important in other models of acute kidney injury [125].
4. Pulmonary toxicity
Evidence. There is evidence for a link between chronic APAP exposure at therapeutic doses and respiratory disease. A survey of general practice clinic patients in the UK found a positive association between frequency of APAP use and signs of asthma [20]. The same group also found that regional sales of acetaminophen in Europe correlated with incidence of respiratory illnesses [126] and that prenatal exposure to APAP may be associated with asthma, wheezing and other respiratory problems later in life [127]. Since then, other groups have obtained similar findings [128-130]. APAP exposure has also been associated with development of chronic obstructive pulmonary disease [131]. However, the conclusions from these studies are controversial. Several possible confounding factors have been suggested [132-134]. Among these, indication bias (“reverse causation”) is probably of greatest concern. For example, children with respiratory infections are more likely to be exposed to APAP as a part of normal treatment [135], which may lead to a false association between APAP exposure early in life and later asthma when in fact the later respiratory problems may be a result of the infection or related issues. There is some evidence of pulmonary toxicity in rodent models. Bronchiolar epithelium necrosis has been observed in mice treated with very large doses of APAP [15,16,136], but those data are clearly not relevant for the chronic low-dose exposures that are thought by some to cause asthma and other lung diseases. There is some evidence that low doses of APAP are proinflammatory in the lungs [17]. Furthermore, adult mice that were exposed to APAP in utero were found to have a greater response to an allergic challenge later in life [18]. However, additional work is needed to understand the pathophysiological significance of the latter phenomena. Overall, there is currently a tentative link between APAP and pulmonary disease that requires further investigation.
Proposed mechanisms. It has been suggested that chronic exposure to APAP can deplete GSH in the lungs and that this could explain a connection between APAP and respiratory diseases if it enhances susceptibility to oxidants, such as reactive-oxygen species produced by inflammatory cells or even environmental oxidants [20]. GSH depletion and increased expression of oxidative stress response genes have been detected in lungs from mice treated with large, acutely toxic doses of APAP and that could suggest oxidative stress [137-139]. APAP-protein binding in the lung has also been demonstrated in mice [137,140-142]. In fact, one study found that a polymorphism in glutathione-s-transferase (GST) P1 that reduces its activity was associated with wheeze in children exposed to APAP prenatally [129], although a conflicting study reported that wheezing and asthma in children of mothers who used APAP during pregnancy is greater when the mother possesses multiple copies of GSTP1 and/or GSTM1 compared with null genotypes [139].
A more specific mechanism of APAP-induced lung disease that has been proposed is neurogenic inflammation. Nassini et al. [17] suggested that inflammation develops in the lungs after APAP treatment due to activation of the transient receptor potential ankyrin 1 (TRPA1) channel in peptidergic neurons by NAPQI. They demonstrated that direct treatment with NAPQI can enhance Ca2+ uptake in cells expressing TRPA1. Importantly, there was also evidence for increased TRPA1 signaling and evidence of inflammation in lungs from rodents treated intratracheally with NAPQI or either intragastrically or intraperitoneally with relatively low doses of APAP (15-300 mg/kg). The authors were even able to detect sulfhydryl adducts after the 15 mg/kg dose, though it’s not clear what effect this had on total GSH levels or if protein binding actually occurred.
Biological relevance and future studies. Although GSH depletion has been demonstrated in lungs from mice overdosed with APAP, it is not clear if that occurs after repeated exposure to APAP at therapeutic doses, which would be more relevant for the reported epidemiological connections between APAP and chronic lung disease. Moreover, the GSH depletion that has been observed in lung is unimpressive: only about 30% of total lung GSH is lost even after treatment with a dose as large as 500 mg/kg [137]. It is possible that the GSH depletion selectively occurs in certain cell types in the lungs (e.g. Clara cells), in which case the total GSH would not be expected to dramatically change; however, covalent protein binding also has not been observed except at very high doses [137,140-142]. The TRPA1 hypothesis has more data to support its biological relevance. Unfortunately, the authors of that study used multiple models, including cultured cells, rat liver slices, isolated guinea pig trachea and mice to perform different experiments in the same study [17], and it’s not clear how each model is related. Furthermore, there was no assessment of pulmonary function in an in vivo model treated with APAP, so the physiological consequences of the inflammation are unknown. The authors did, however, test the effect of APAP on pulmonary insufflation pressure in vivo in guinea pigs and reported no change [17]. Thus, the evidence for TRPA1-mediated lung damage in animals is preliminary and should be further explored. Overall, it is not yet clear if or how APAP causes lung disease. We recommend that experiments measuring GSH and protein binding in the lungs be repeated in mice using low, therapeutic doses to determine if those mechanisms are actually relevant for humans. Presently, the most compelling data suggest that NAPQI can activate TRPA1 on neurons and lead to neurogenic airway inflammation, but a more detailed study using only the mouse model, and that includes assessment of pulmonary function, is needed to test that.
5. Endocrine disruption and sexual development
Evidence. It is critical to evaluate claims regarding long-term effects of intrauterine APAP exposure because APAP is currently the only drug recommended for pregnant women to reduce pain and fever. Modestly increased risk of cryptorchidism after prenatal exposure to APAP has been reported in humans in a few studies [143-145], which suggests some estrogenic or anti-androgen activity of APAP. However, the results are inconsistent and difficult to interpret together. For example, one study examined two patient cohorts and discovered an effect in only one of them [145]. Another study found that the risk of cryptorchidism was increased in offspring of mothers who used APAP for ≥ 4 weeks during pregnancy, but the likelihood of the child undergoing orchiopexy (surgical treatment, and therefore a surrogate marker of long-term cryptorchidism) was not [144]. Another study failed to find an association between APAP alone and other measures of androgen exposure, such as penis width and anogenital distance (AGD), commonly associated with reproductive disorders, despite an association with APAP and NSAIDs together [146]. Overall, there does not seem to be a clear relationship between APAP exposure during development and reproductive effects in humans. Nevertheless, several studies using rodent models have indicated a connection. One group has reported that intrauterine APAP exposure modestly affects AGD in male and female rodents [145,147,148] and may affect germ cell proliferation in female mice [148]. However, although they claimed to use subtoxic doses, the authors treated the animals with 50-350 mg/kg of APAP every morning for 7 days. While the maximum recommended dose of APAP in humans is approximately 50-60 mg/kg/day, that amount is typically divided into multiple smaller doses over a 24 h period. In fact, it is well known that a single treatment with ≥150 mg/kg is hepatotoxic in mice, resulting in significantly elevated plasma ALT values and evidence of hepatocyte necrosis by histology [149]. It is not surprising that there may be developmental abnormalities in offspring of animals that suffer liver injury during pregnancy. In fact, the most surprising finding from these studies may be that the effects were not more pronounced. Adding confusion to the debate, the same group recently found that 50 mg/kg/day has no effect on masculine behaviors or morphology in a region of the brain associated with those behaviors in male offspring [150], though the 150 mg/kg/day dose did have an effect. Overall, there is currently no clear association between APAP and reproductive effects in offspring.
Proposed mechanisms. APAP does not seem to be directly estrogenic [151], so other mechanisms have been proposed. One possible mechanism for the suggested endocrine-disrupting effects of APAP is altered sex steroid metabolism. Interestingly, one research group obtained moderately elevated values for total estrogen metabolites in urine from premenopausal women who reported high APAP use [152]. The only rodent in vivo study to address this issue revealed that plasma testosterone decreased after APAP treatment in castrated mice with human testis xenografts, which suggests that APAP decreases testosterone production in human testes [153]. Finally, a few in vitro studies have demonstrated that cytochrome P450-mediated steroid metabolism can be altered by APAP [154,155], though other studies have provided partially conflicting results [156]. Treatment of an adrenocortical carcinoma cell line resulted in increased pregnenolone and decreased androstenedione and testosterone in two studies [144,157]. Estrone and β-estradiol were also increased by APAP [147]. However, another study found no effect of APAP on testosterone production in human fetal testis [156]. Another mechanism that has been proposed for the possible endocrine-disrupting effects of APAP is reduced prostaglandin synthesis due to cyclooxygenase inhibition. Certain prostaglandin levels have been shown to decrease in cultured human fetal testis after APAP treatment [156].
Biological relevance and future studies. Altogether, there are limited and conflicting results regarding the endocrine effects of APAP. There is some epidemiological evidence for modestly increased risk of indirect markers of abnormal sexual development after intrauterine exposure to APAP in humans, but those data are by no means conclusive. Although one human study reported increased urine estrogen in humans after APAP use [152], it is unlikely that the modest effect that was observed would have a major impact on development. Even the evidence for developmental effects of prenatal use of potent, direct estrogens like oral contraceptives on sexual development in offspring is weak at best [157]. While results from some studies using cell culture models do support an effect of APAP on hormone metabolism, others have revealed conflicting results. Moreover, most of those studies involved prolonged treatment (24-72 h) with µM to mM concentrations of APAP, which is not consistent with the pharmacokinetics of APAP in vivo. Finally, the data from the human testis xenograft model are compelling, but the human relevance of that model is unclear. Overall, there is currently no strong evidence that intrauterine exposure to APAP can significantly alter sexual development or reproductive health later in life. Before any further research on the endocrine and reproductive effects of APAP or the mechanisms involved, we recommend that a simple study be performed in which pregnant mice receive a low dose of APAP (15 mg/kg) one to four times per day for several days and multiple developmental parameters of offspring health, including AGD and other measurements of reproductive health, is assessed. That will also require an evidence-based consensus on what are the most important or relevant reproductive health parameters to measure.
6. Ototoxicity
Evidence. At least 19 reports of rapidly progressive sensorineural hearing loss caused by abuse of APAP/opioid combinations have been published [158-160]. In most cases, the hearing loss is bilateral, suggesting a systemic cause consistent with drug exposure. In vitro studies have demonstrated that long-term (≥24 h) exposure to high concentrations (mM) of APAP can reduce the number of viable cells in isolated cochlea (particularly in the outer hair cells) and cause evidence of apoptotic cell death in an auditory cell line (HEI-OC1) that was derived from the organ of Corti in the ImmortomouseTM model [161] and is generally thought to represent cochlear hair cells [3,8]. Interestingly, co-treatment with hydromorphone enhanced APAP ototoxicity in these models, though hydromor-phone or hydrocodone alone did not cause cell death [8]. NAPQI was shown to have similar effects [13]. Those data suggested that APAP is the primary cause of hearing loss due to APAP/opioid abuse. However, no clinical reports of hearing loss after overdose of APAP alone have been published. Furthermore, the same group published a more recent study indicating that APAP does not actually cause cell death in HEIOC1 cells, despite evidence of reduced energy metabolism and even increased caspase activity [162]. Finally, a recent in vivo study in mice found no evidence for hearing loss based on auditory brainstem response (ABR) in a clinically relevant model of acute APAP overdose [163]. Thus, it seems unlikely that APAP by itself causes ototoxicity in humans or mice. Nevertheless, a practical clinical problem clearly exists in patients treated with opioid/APAP combinations and further investigation may be warranted.
Proposed mechanisms. Kalinec et al. [13] found that APAP can cause evidence of oxidative stress in HEI-OC1 cells 12-48 h after initiation of treatment, but that NAPQI does not have this effect. Furthermore, increased endoplasmic reticulum (ER) fragmentation was observed in these cells after treatment with NAPQI but not APAP [13]. Despite the latter, both treatments altered levels of ER stress markers. Based on these findings, the authors concluded that APAP and NAPQI exert toxic effects through different mechanisms in cochlear cells: APAP ototoxicity involves oxidative stress and ER stress, while NAPQI causes ER stress without oxidative stress [13]. The only in vivo study of APAP ototoxicity to date also revealed that there is oxidative stress in cochleae after acute APAP overdose [163]; however, no ototoxicity was observed in that study based on auditory brainstem thresholds (ABR) [163].
Biological relevance and future studies. While interesting, the results from cell culture studies thus far are questionable. First, APAP has a very short half-life in circulation [26]. Thus, it is unlikely to persist at the cochlea for ≥ 24 h, as in the in vitro experiments described above. Although some drugs (e.g. aminoglycosides) may become trapped within the cochlear fluid, this is unlikely to occur with APAP because it is neutral at physiological pH and readily crosses membranes [26]. Next, it is not known if HEI-CO1 cells, or cochlear cells in general, express P450s at concentrations sufficient to convert APAP to NAPQI. The only study to address that issue revealed that mice treated with a hepatotoxic dose of APAP had no evidence of GSH depletion or protein binding in cochlea [163]. Finally, it is clear that APAP toxicity in vitro does not necessarily translate to toxicity in vivo. Many cell lines succumb to APAP toxicity through mechanisms that are not physiologically relevant. For example, both Hepa 1-6 and SK-Hep1 liver cells will die after prolonged exposure to mM concentrations of APAP, despite the fact that these cells do not form the reactive metabolite of APAP [164 , 165]. Importantly, the primary mode of cell death in these cells was found to be apoptosis, which is not a major contributor to APAP-induced hepatocyte death in vivo [35,50,70, 166]. Furthermore, APAP is also toxic to human lymphocytes in culture [165], but there is little or no evidence that that is true in vivo. Clearly, it is important to realize that cell culture studies do not necessarily mimic the in vivo situation. Overall, it is clear that APAP/opioid combinations are ototoxic in humans, but there is no strong evidence that APAP is ototoxic by itself. Future research in this area is encouraged, and should focus on hearing loss caused by the combination drugs, and should use only in vivo models with clear human relevance.
7. Neurodevelopmental and neurobehavioral disorders
Evidence. Several groups have claimed that APAP may be a cause of autism spectrum disorder (ASD) [7,11,14]. Two major pieces of evidence led to that hypothesis. First, it was observed that at least some patients with ASD exhibit defective xenobiotic sulfation [167]. In fact, when APAP was used as a probe drug to assess sulfation capacity, the ratio of APAP-sulfate to APAP-glucuronide was lower in severely autistic subjects compared to healthy controls [176]. Initially, it was suggested that this could lead to poor clearance of, and therefore increased exposure to, certain chemicals present in food or in the environment that may have neurological effects, but it was later proposed that APAP itself might be a problem. Schultz et al. [7] suggested that reduced sulfation may lead to increased NAPQI formation with neurotoxic effects. Second, it was found that diagnoses of ASD began to increase in the 1980s, after the CDC issued a warning regarding the risk of Reye’s syndrome and birth defects when treating children or pregnant women with aspirin, and sales of children’s APAP rose [168]. However, it is unlikely that reduced sulfation would lead to a significant increase in NAPQI formation at therapeutic doses of APAP. Sulfation is a low capacity route of elimination and is already saturated in healthy subjects at pharmacologic doses of APAP [169]. Glucuronidation, on the other hand, is a high capacity process and does not appear to be saturable [27]. In fact, the hepatotoxicity of APAP is probably not due to saturation of Phase II metabolism resulting in greater NAPQI formation; the percentage of APAP converted to the reactive metabolite is likely the same regardless of dose. Rather, it is probably the greater absolute amount of NAPQI that is produced that initiates liver injury after overdose [27]. Furthermore, the observed correlation between children’s APAP sales and ASD diagnoses does not prove causation.
Nevertheless, several groups have reported results from epidemiological studies that seem to show an association between APAP exposure early in life and development of ASD [7,11,170]. One of the earliest such studies revealed that parents of children with autism were more likely to report use of APAP after receiving the measles-mumps-rubella vaccine [7]. However, it has been pointed out by others that the parents were solicited from autism websites and thus were likely to be biased [171]. In addition, there is the possibility of recall bias in parents of children with autism who are in search of a cause [171]. More recent studies have employed more rigorous methods [170]. Unfortunately, even those that have marginalized the risk of indication bias may still be affected by genetic factors or other residual bias [172]. Overall, the only human data available to support the idea that APAP causes ASD are from epidemiological studies that may be subject to significant bias.
In addition to ASD, it has recently been suggested that antenatal exposure to APAP may cause hyperactivity or ADD / ADHD-like behavior in offspring. Liew et al. [7] found an association between APAP and these disorders in a large prospective cohort study, and their results are supported by data from a few other groups [173-176]. However, significant sources of bias have been pointed out in three of these studies as well [177], and earlier work by Streissguth et al. [178] provided conflicting results. Interestingly, one group has even tested the association between prenatal APAP exposure and ADD/ADHD-like behavior in mice and found no evidence to support it [179], although it should be noted that there were clear experimental deficiencies such as a lack of well-validated endpoints for ADD/ADHD in mice and the fact that a positive control is not available for comparison. Overall, there is currently no strong evidence that APAP causes ADD/ADHD.
Although the evidence for neurobehavioral effects of APAP in humans is poor, multiple studies have demonstrated that exposure to relatively low doses of APAP during early development can affect behavioral measures in adult mice [12,180]. While it is not possible to make a direct connection between non-specific behavioral studies in mice and ASD or ADD/ADHD in humans, these observations are intriguing and may warrant further investigation. Typically, pregnant women are advised not to use NSAIDs due to the increased risk of birth defects and miscarriage that has been reported in a few studies. As a result, most pregnant women rely on APAP to control fever and pain. If it can be shown that APAP also poses a significant risk of congenital abnormalities, then that may result in removal of the only remaining treatment option for those patients.
Proposed mechanisms. The proposed mechanisms by which APAP could cause ASD and ADD/ADHD are similar. Endocrine disruption, activation of endocannabinoid receptors during development [181], oxidative stress and inflammation [182] have all been suggested. However, no studies have been done to directly test those possibilities. A more straightforward hypothesis is that APAP is directly toxic to neurons. Posadas et al. [9] tested that by treating rat cortical neurons with APAP in vitro and by injecting rats with APAP in vivo and measuring neuron death. They demonstrated that APAP overdose was moderately toxic to cortical neurons. However, the purpose of their study was to determine if large doses of APAP (250-500 mg/kg) are neurotoxic, and it is not known if typical human doses for therapeutic use (approximately 10-20 mg/kg) have similar effects. Cell death in APAP-treated cultured neurons has also been reported [9], but again most cell culture models do not accurately reflect APAP toxicity in vivo. Finally, it is not clear exactly how neuron death would lead to ASD and ADD/ADHD.
Biological relevance and future studies. Currently, the association between APAP and ASD or ADD/ADHD is based on conflicting results from epidemiological studies. No mechanistic studies have been performed, and the few mechanisms that have been proposed have not been directly tested. In fact, there is strong evidence that ASD, in particular, is driven by genetics [183], so exposure to APAP or other xenobiotics may not be important. Males are far more likely to develop ASD, and siblings of children with ASD are at greater risk [183]. There is also striking evidence for a genetic component of social behaviors associated with ASD, such as viewing of social scenes [184]. Nevertheless, the importance of APAP as a treatment option during pregnancy, together with the seriousness of ASD and ADD/ADHD, warrants future research in this area to enable more definitive conclusions. Even a simple study could be performed in which pregnant mice receive 15 mg/kg APAP one to four times per day for several days and behaviors associated with ASD and ADD/ADHD are measured in offspring over time.
8. APAP toxicity in other tissues or systems
APAP toxicity has been reported in other tissues, but the evidence is limited. For example, APAP is also known to cause ocular opacity or cataracts in mice, but only after direct induction of P450 enzymes in ocular tissue [185,186]. It has also been suggested that APAP can be cardiotoxic, but this is based on case reports with no direct evidence [187]. Currently, there is no compelling evidence for clinically-relevant APAP toxicity in tissues other than those discussed above.
9. Conclusions
It has been 50 years since the first reports of APAP-induced liver injury, and we are only beginning to investigate the extrahepatic toxicity of the drug in earnest. Renal toxicity after APAP overdose is known to occur, but the mechanisms have not been fully elucidated. It is also not known if common comorbidities like alcoholism or obesity affect that outcome. The pulmonary and neuro- toxicity of APAP are more controversial. Most data regarding the non-hepatic and non-renal effects of APAP are from epidemiological studies that do not prove causation and frequently suffer from bias and/or conflicting results. Published experimental data provide support for many of these adverse effects, but too often the data come from flawed models. However, we believe that some additional research may be appropriate in at least two areas. The sheer volume of epidemiological studies that have revealed increased risk of lung disease after exposure to APAP early in life and the fact that at least one group has reported a plausible mechanism based on data from animal models using low doses of APAP may warrant further investigation of the pulmonary toxicity of chronic APAP use. Also, the fact that APAP is a very important drug for pregnant women combined with the several rodent studies suggesting adverse neurodevelopmental effects in offspring may warrant further investigation of neurodevelopmental toxicity to fully evaluate that possibility. Overall, however, the data for extrahepatic toxicity of APAP are weak and significant changes in clinical or consumer use would be not advisable at this time.
Acknowledgements
This work was supported in part by start-up funds from the University of Arkansas for Medical Sciences, including the Arkansas Translational Research Institute (NIH U54TR001629).
Disclosure
The authors have no financial conflicts to disclose.
References
- [1].Kaufman DW, Kelly JP, Rosenberg L, Anderson TE, Mitchell AA. Recent patterns of medication use in the ambulatory adult population of the United States: the Slone survey. JAMA. 2002;287:337–344. doi: 10.1001/jama.287.3.337. [DOI] [PubMed] [Google Scholar]
- [2].Lee WM. Etiologies of acute liver failure. Semin Liver Dis. 2008;28:142–52. doi: 10.1055/s-2008-1073114. [DOI] [PubMed] [Google Scholar]
- [3].Davidson DG, Eastham WN. Acute liver necrosis following overdose of paracetamol. Br Med J. 1966;2:497–499. doi: 10.1136/bmj.2.5512.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Boyd EM, Bereczky GM. Liver necrosis from paracetamol. Br J Pharmacol Chemother. 1966;26:606–614. doi: 10.1111/j.1476-5381.1966.tb01841.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Proudfoot AT, Wright N. Acute paracetamol poisoning. Br Med J. 1970;3:557–558. doi: 10.1136/bmj.3.5722.557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Torres AR. Is fever suppression involved in the etiology of autism and neurodevelopmental disorders? BMC Pediatr. 2003;3:9. doi: 10.1186/1471-2431-3-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Schultz ST, Klonoff-Cohen HS, Wingard DL, Akshoomoff NA, Macera CA, Ji M. Acetaminophen (paracetamol) use, measles-mumps-rubella vaccination, and autistic disorder: the results of a parent survey. Autism. 2008;12:293–307. doi: 10.1177/1362361307089518. [DOI] [PubMed] [Google Scholar]
- [8].Yorgason JG, Kalinec GM, Luxford WM, Warren FM, Kalinec F. Acetaminophen ototoxicity after acetaminophen / hydrocodone abuse: evidence from two parallel in vitro mouse models. Otolaryngol Head Neck Surg. 2010;142:814–9. 819.e1–2. doi: 10.1016/j.otohns.2010.01.010. [DOI] [PubMed] [Google Scholar]
- [9].Posadas I, Santos P, Blanco A, Munoz-Fernández M, Cena V. Acetaminophen induces apoptosis in rat cortical neurons. PLoS One. 2010;5:e15360. doi: 10.1371/journal.pone.0015360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].da Silva MH, da Rosa EJ, de Carvalho NR, Dobrachinski F, da Rocha JB, Mauriz JL, González-Gallego J, Soares FA. Acute brain damage induced by acetaminophen in mice: effect of diphenyl diselenide on oxidative stress and mitochondrial dysfunction. Neurotox Res. 2012;21:334–344. doi: 10.1007/s12640-011-9288-1. [DOI] [PubMed] [Google Scholar]
- [11].Bauer AZ, Kriebel D. Prenatal and perinatal analgesic exposure and autism: an ecological link. Environ Health. 2013;12:41. doi: 10.1186/1476-069X-12-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Viberg H, Eriksson P, Gordh T, Fredriksson A. Paracetamol (acetaminophen) administration during neonatal brain development affects cognitive function and alters its analgesic and anxiolytic response in adult male mice. Toxicol Sci. 2014;138:139–47. doi: 10.1093/toxsci/kft329. [DOI] [PubMed] [Google Scholar]
- [13].Kalinec GM, Thein P, Parsa A, Yorgason J, Luxford W, Urrutia R, Kalinec F. Acetaminophen and NAPQI are toxic to auditory cells via oxidative and endoplasmic reticulum stress-dependent pathways. Hear Res. 2014;313:26–37. doi: 10.1016/j.heares.2014.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Liew Z, Ritz B, Rebordosa C, Lee PC, Olsen J. Acetaminophen use during pregnancy, behavioral problems, and hyperkinetic disorders. JAMA Pediatr. 2014;168:313–20. doi: 10.1001/jamapediatrics.2013.4914. [DOI] [PubMed] [Google Scholar]
- [15].Placke ME, Wyand DS, Cohen SD. Extrahepatic lesions induced by acetaminophen in the mouse. Toxicol Pathol. 1987;15:381–387. doi: 10.1177/019262338701500401. [DOI] [PubMed] [Google Scholar]
- [16].Bartolone JB, Beierschmitt WP, Birge RB, Hart SG, Wyand S, Cohen SD, Khairallah EA. Selective acetaminophen metabolite binding to hepatic and extrahepatic proteins: an in vivo and in vitro analysis. Toxicol Appl Pharmacol. 1989;99:240–9. doi: 10.1016/0041-008x(89)90006-9. [DOI] [PubMed] [Google Scholar]
- [17].Nassini R, Materazzi S, André E, Sartiani L, Aldini G, Trevisani M, Carnini C, Massi D, Pedretti P, Carini M, Cerbai E, Preti D, Villetti G, Civelli M, Trevisan G, Azzari C, Stokesberry S, Sadofsky L, McGarvey L, Patacchini R, Geppetti P. Acetaminophen, via its reactive metabolite Nacetyl-p-benzo-quinoneimine and transient receptor potential ankyrin-1 stimulation, causes neurogenic inflammation in the airways and other tissues in rodents. FASEB J. 2010;24:4904–4916. doi: 10.1096/fj.10-162438. [DOI] [PubMed] [Google Scholar]
- [18].Karimi K, Kebler T, Thiele K, Ramisch K, Erhardt A, Huebener P, Barikbin R, Arck P, Tiegs G. Prenatal acetaminophen induces liver toxicity in dams, reduces fetal liver stem cells, and increases airway inflammation in adult offspring. J Hepatol. 2015;62:1085–1091. doi: 10.1016/j.jhep.2014.12.020. [DOI] [PubMed] [Google Scholar]
- [19].Shaheen SO, Newson RB, Sherriff A, Henderson AJ, Heron JE, Burney PG. Golding J; ALSPAC Study Team. Paracetamol use in pregnancy and wheezing in early childhood. Thorax. 2002;57:958–963. doi: 10.1136/thorax.57.11.958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Shaheen SO, Sterne JA, Songhurst CE, Burney PG. Frequent paracetamol use and asthma in adults. Thorax. 2000;55:266–270. doi: 10.1136/thorax.55.4.266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Shaheen S, Potts J, Gnatiuc L, Makowska J, Kowalski ML, Joos G, van Zele T, van Durme Y, De Rudder I, Wӧhrl S, Godnic-Cvar J, Skadhauge L, Thomsen G, Zuberbier T, Bergmann KC, Heinzerling L, Gjomarkaj M, Bruno A, Pace E, Bonini S, Fokkens W, Weersink EJ. The relation between paracetamol use and asthma: a GA2LEN European case-control study. EurRespir J. 2008;32:1231–1236. doi: 10.1183/09031936.00039208. [DOI] [PubMed] [Google Scholar]
- [22].Thiele K, Solano ME, Huber S, Flavell RA, Kessler T, Barikbin R, Jung R, Karimi K, Tiegs G, Arck PC. Prenatal acetaminophen affects maternal immune and endocrine adaptation to pregnancy, induces placental damage, and impairs fetal development in mice. Am J Pathol. 2015;185:2805–2818. doi: 10.1016/j.ajpath.2015.06.019. [DOI] [PubMed] [Google Scholar]
- [23].Lee WM. Acetaminophen (APAP) hepatotoxicity – isn’t it time for APAP to go away? J Hepatol. 2017 doi: 10.1016/j.jhep.2017.07.005. [Epub ahead of print] doi: 10.1016/j.jhep.2017.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Black RA, Hill DA. Over-the-counter medications in pregnancy. Am Fam Physician. 2003;67:2517–2524. [PubMed] [Google Scholar]
- [25].Thiele K, Kessler T, Arck P, Erhardt A, Tiegs G. Acetaminophen and pregnancy: short-and long-term consequences for mother and child. J Reprod Immunol. 2013;97:128–139. doi: 10.1016/j.jri.2012.10.014. [DOI] [PubMed] [Google Scholar]
- [26].McGill MR, Jaeschke H. Metabolism and disposition of acetaminophen: recent advances in relation to hepatotoxicity and diagnosis. Pharm Res. 2013;30:2174–2187. doi: 10.1007/s11095-013-1007-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Xie Y, McGill MR, Cook SF, Sharpe MR, Winefield RD, Wilkins DG, Rollins DE, Jaeschke H. Time course of acetaminophen-protein adducts and acetaminophen metabolites in circulation of overdose patients and in HepaRG cells. Xenobiotica. 2015;45:921–929. doi: 10.3109/00498254.2015.1026426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Dahlin DC, Miwa GT, Lu AY, Nelson SD. N-acetyl-pbenzoquinone imine: a cytochrome P-450-mediated oxidation product of acetaminophen. Proc Natl Acad Sci U S A. 1984;81:132713–31. doi: 10.1073/pnas.81.5.1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Potter WZ, Davis DC, Mitchell JR, Jollow DJ, Gillette JR, Brodie BB. Acetaminophen-induced hepatic necrosis. 3. Cytochrome P-450-mediated covalent binding in vitro. J Pharmacol Exp Ther. 1973 Oct;187:203–210. [PubMed] [Google Scholar]
- [30].Jollow DJ, Mitchell JR, Potter WZ, Davis DC, Gillette JR, Brodie BB. Acetaminophen-induced hepatic necrosis. II. Role of covalent binding in vivo. J Pharmacol Exp Ther. 1973;187:195–202. [PubMed] [Google Scholar]
- [31].Mitchell JR, Jollow DJ, Potter WZ, Davis DC, Gillette JR, Brodie BB. Acetaminophen-induced hepatic necrosis. I. Role of drug metabolism. J Pharmacol Exp Ther. 1973;187:185–194. [PubMed] [Google Scholar]
- [32].Mitchell JR, Jollow DJ, Potter WZ, Gillette JR, Brodie BB. Acetaminophen-induced hepatic necrosis. IV. Protective role of glutathione. J Pharmacol Exp Ther. 1973;187:211–217. [PubMed] [Google Scholar]
- [33].Meyers LL, Beierschmitt WP, Khairallah EA, Cohen SD. Acetaminophen-induced inhibition of hepatic mitochondrial respiration in mice. Toxicol Appl Pharmacol. 1988;93:378–387. doi: 10.1016/0041-008x(88)90040-3. [DOI] [PubMed] [Google Scholar]
- [34].Kon K, Kim JS, Jaeschke H, Lemasters JJ. Mitochondrial permeability transition in acetaminophen-induced necrosis and apoptosis of cultured mouse hepatocytes. Hepatology. 2004;40:1170–1179. doi: 10.1002/hep.20437. [DOI] [PubMed] [Google Scholar]
- [35].McGill MR, Sharpe MR, Williams CD, Taha M, Curry SC, Jaeschke H. The mechanism underlying acetaminophen-induced hepatotoxicity in humans and mice involves mitochondrial damage and nuclear DNA fragmentation. J Clin Invest. 2012;122:1574–1583. doi: 10.1172/JCI59755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].McGill MR, Staggs VS, Sharpe MR, Lee WM, Jaeschke H. Acute Liver Failure Study Group. Serum mitochondrial biomarkers and damage-associated molecular patterns are higher in acetaminophen overdose patients with poor outcome. Hepatology. 2014;60:1336–1345. doi: 10.1002/hep.27265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Xie Y, McGill MR, Du K, Dorko K, Kumer SC, Schmitt TM, Ding WX, Jaeschke H. Mitochondrial protein adducts formation and mitochondrial dysfunction during N-acetyl-maminophenol (AMAP)-induced hepatotoxicity in primary human hepatocytes. Toxicol Appl Pharmacol. 2015;289:213–222. doi: 10.1016/j.taap.2015.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Hadi M, Dragovic S, van Swelm R, Herpers B, van de Water B, Russel FG, Commandeur JN, Groothuis GM. AMAP, the alleged non-toxic isomer of acetaminophen, is toxic in rat and human liver. Arch Toxicol. 2013;87:155–165. doi: 10.1007/s00204-012-0924-1. [DOI] [PubMed] [Google Scholar]
- [39].McGill MR, Williams CD, Xie Y, Ramachandran A, Jaeschke H. Acetaminophen-induced liver injury in rats and mice: comparison of protein adducts, mitochondrial dysfunction, and oxidative stress in the mechanism of toxicity. Toxicol Appl Pharmacol. 2012;264:387–394. doi: 10.1016/j.taap.2012.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Cover C, Mansouri A, Knight TR, Bajt ML, Lemasters JJ, Pessayre D, Jaeschke H. Peroxynitrite - induced mitochondrial and endonuclease - mediated nuclear DNA damage in acetaminophen hepatotoxicity. J Pharmacol Exp Ther. 2005;315:879–887. doi: 10.1124/jpet.105.088898. [DOI] [PubMed] [Google Scholar]
- [41].Jaeschke H. Glutathione disulfide formation and oxidant stress during acetaminophen-induced hepatotoxicity in mice in vivo: the protective effect of allopurinol. J Pharmacol Exp Ther. 1990;255:935–941. [PubMed] [Google Scholar]
- [42].Fujimoto K, Kumagai K, Ito K, Arakawa S, Ando Y, Oda S, Yamoto T, Manabe S. Sensitivity of liver injury in heterozygous Sod2 knockout mice treated with troglitazone or acetaminophen. ToxicolPathol. 2009;37:193–200. doi: 10.1177/0192623308329282. [DOI] [PubMed] [Google Scholar]
- [43].Yoshikawa Y, Morita M, Hosomi H, Tsuneyama K, Fukami T, Nakajima M, Yokoi T. Knockdown of superoxide dismutase 2 enhances acetaminophen-induced hepatotoxicity in rat. Toxicology. 2009;264:89–95. doi: 10.1016/j.tox.2009.07.017. [DOI] [PubMed] [Google Scholar]
- [44].Ramachandran A, Lebofsky M, Weinman SA, Jaeschke H. The impact of partial manganese superoxide dismutase (SOD2)-deficiency on mitochondrial oxidant stress, DNA fragmentation and liver injury during acetaminophen hepatotoxicity. Toxicol Appl Pharmacol. 2011;251:226–233. doi: 10.1016/j.taap.2011.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Du K, McGill MR, Xie Y, Bajt ML, Jaeschke H. Resveratrol prevents protein nitration and release of endonucleases from mitochondria during acetaminophen hepatotoxicity. Food Chem Toxicol. 2015;81:62–70. doi: 10.1016/j.fct.2015.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].McGill MR, Du K, Weemhoff JL, Jaeschke H. Critical review of resveratrol in xenobiotic-induced hepatotoxicity. Food ChemToxicol. 2015;86:309–318. doi: 10.1016/j.fct.2015.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Knight TR, Ho YS, Farhood A, Jaeschke H. Peroxynitrite is a critical mediator of acetaminophen hepatotoxicity in murine livers: protection by glutathione. J Pharmacol Exp Ther. 2002;303:468–475. doi: 10.1124/jpet.102.038968. [DOI] [PubMed] [Google Scholar]
- [48].Bajt ML, Knight TR, Lemasters JJ, Jaeschke H. Acetaminophen - induced oxidant stress and cell injury in cultured mouse hepatocytes: protection by N-acetyl cysteine. Toxicol Sci. 2004;80:343–349. doi: 10.1093/toxsci/kfh151. [DOI] [PubMed] [Google Scholar]
- [49].Nakagawa H, Maeda S, Hikiba Y, Ohmae T, Shibata W, Yanai A, Sakamoto K, Ogura K, Noguchi T, Karin M, Ichijo H, Omata M. Deletion of apoptosis signal-regulating kinase 1 attenuates acetaminophen-induced liver injury by inhibiting c-Jun N-terminal kinase activation. Gastroenterology. 2008;135:1311–1321. doi: 10.1053/j.gastro.2008.07.006. [DOI] [PubMed] [Google Scholar]
- [50].Xie Y, McGill MR, Dorko K, Kumer SC, Schmitt TM, Forster J, Jaeschke H. Mechanisms of acetaminophen-induced cell death in primary human hepatocytes. Toxicol ApplPharmacol. 2014;279:266–274. doi: 10.1016/j.taap.2014.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Gunawan BK, Liu ZX, Han D, Hanawa N, Gaarde WA, Kaplowitz N. c-Jun N-terminal kinase plays a major role in murine acetaminophen hepatotoxicity. Gastroenterology. 2006;131:165–178. doi: 10.1053/j.gastro.2006.03.045. [DOI] [PubMed] [Google Scholar]
- [52].Saito C, Lemasters JJ, Jaeschke H. c-Jun N-terminal kinase modulates oxidant stress and peroxynitrite formation independent of inducible nitric oxide synthase in acetaminophen hepatotoxicity. Toxicol Appl Pharmacol. 2010;246:8–17. doi: 10.1016/j.taap.2010.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Henderson NC, Pollock KJ, Frew J, Mackinnon AC, Flavell RA, Davis RJ, Sethi T, Simpson KJ. Critical role of c-jun (NH2) terminal kinase in paracetamol- induced acute liver failure. Gut. 2007;56:982–990. doi: 10.1136/gut.2006.104372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Bourdi M, Korrapati MC, Chakraborty M, Yee SB, Pohl LR. Protective role of c-Jun N-terminal kinase 2 in acetaminophen-induced liver injury. Biochem Biophys Res Commun. 2008;374:6–10. doi: 10.1016/j.bbrc.2008.06.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Cubero FJ, Zoubek ME, Hu W, Peng J, Zhao G, Nevzorova YA, Al Masaoudi M, Bechmann LP, Boekschoten MV, Muller M, Preisinger C, Gassler N, Canbay AE, Luedde T, Davis RJ, Liedtke C, Trautwein C. Combined activities of JNK1 and JNK2 in hepatocytes protect against toxic liver injury. Gastroenterol. 2016 Apr;;150:968–981. doi: 10.1053/j.gastro.2015.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Bourdi M, Davies JS, Pohl LR. Mispairing C57BL/6 substrains of genetically engineered mice and wild-type controls can lead to confounding results as it did in studies of JNK2 in acetaminophen and concanavalin A liver injury. Chem Res Toxicol. 2011;24:794–796. doi: 10.1021/tx200143x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Latchoumycandane C, Goh CW, Ong MM, Boelsterli UA. Mitochondrial protection by the JNK inhibitor leflunomide rescues mice from acetaminophen-induced liver injury. Hepatology. 2007;45:412–421. doi: 10.1002/hep.21475. [DOI] [PubMed] [Google Scholar]
- [58].Du K, Xie Y, McGill MR, Jaeschke H. Pathophysiological significance of c-jun N-terminal kinase in acetaminophen hepatotoxicity. Expert Opin Drug Metab Toxicol. 2015;11:1769–1779. doi: 10.1517/17425255.2015.1071353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Hanawa N, Shinohara M, Saberi B, Gaarde WA, Han D, Kaplowitz N. Role of JNK translocation to mitochondria leading to inhibition of mitochondria bioenergetics in acetaminophen-induced liver injury. J Biol Chem. 2008;283:13565–13577. doi: 10.1074/jbc.M708916200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Jaeschke H, McGill MR, Ramachandran A. Oxidant stress, mitochondria, and cell death mechanisms in drug-induced liver injury: lessons learned from acetaminophen hepatotoxicity. Drug Metab Rev. 2012;44:88–106. doi: 10.3109/03602532.2011.602688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Sharma M, Gadang V, Jaeschke A. Critical role for mixed-lineage kinase 3 in acetaminophen-induced hepatotoxicity. MolPharmacol. 2012;82:1001–1007. doi: 10.1124/mol.112.079863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Ramachandran A, McGill MR, Xie Y, Ni HM, Ding WX, Jaeschke H. Receptor interacting protein kinase 3 is a critical early mediator of acetaminophen-induced hepatocyte necrosis in mice. Hepatology. 2013;58:2099–2108. doi: 10.1002/hep.26547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Zhang YF, He W, Zhang C, Liu XJ, Lu Y, Wang H, Zhang ZH, Chen X, Xu DX. Role of receptor interacting protein (RIP)1 on apoptosis-inducing factor-mediated necroptosis during acetaminophen-evoked acute liver failure in mice. Toxicol Lett. 2014;225:445–453. doi: 10.1016/j.toxlet.2014.01.005. [DOI] [PubMed] [Google Scholar]
- [64].Deutsch M, Graffeo CS, Rokosh R, Pansari M, Ochi A, Levie EM, van Heerden E, Tippens DM, Greco S, Barilla R, Tomkӧtter L, Zambirinis CP, Avanzi N, Gulati R, Pachter HL, Torres-Hernandez A, Eisenthal A, Daley D, Miller G. Divergent effects of RIP1 or RIP3 blockade in murine models of acute liver injury. Cell Death Dis. 2015;6:e1759. doi: 10.1038/cddis.2015.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Reid AB, Kurten RC, McCullough SS, Brock RW, Hinson JA. Mechanisms of acetaminophen-induced hepatotoxicity: role of oxidative stress and mitochondrial permeability transition in freshly isolated mouse hepatocytes. J Pharmacol Exp Ther. 2005;312:509–516. doi: 10.1124/jpet.104.075945. [DOI] [PubMed] [Google Scholar]
- [66].Ramachandran A, Lebofsky M, Baines CP, Lemasters JJ, Jaeschke H. Cyclophilin D deficiency protects against acetaminophen-induced oxidant stress and liver injury. Free Radic Res. 2011;45:156–164. doi: 10.3109/10715762.2010.520319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Hu J, Ramshesh VK, McGill MR, Jaeschke H, Lemasters JJ. Low dose acetaminophen induces reversible mitochondrial dysfunction associated with transient c-Jun N-terminal kinase activation in mouse liver. Toxiol Sci. 2016;150:204–215. doi: 10.1093/toxsci/kfv319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Placke ME, Ginsberg GL, Wyand DS, Cohen SD. Ultrastructural changes during acute acetaminophen-induced hepatotoxicity in the mouse: a time and dose study. Toxicol Pathol. 1987;15:431–438. doi: 10.1177/019262338701500407. [DOI] [PubMed] [Google Scholar]
- [69].Bajt ML, Cover C, Lemasters JJ, Jaeschke H. Nuclear translocation of endonuclease G and apoptosis-inducing factor during acetaminophen-induced liver cell injury. Toxicol Sci. 2006;94:217–225. doi: 10.1093/toxsci/kfl077. [DOI] [PubMed] [Google Scholar]
- [70].Gujral JS, Knight TR, Farhood A, Bajt ML, Jaeschke H. Mode of cell death after acetaminophen overdose in mice: apoptosis or oncotic necrosis? Toxicol Sci. 2002;67:322–328. doi: 10.1093/toxsci/67.2.322. [DOI] [PubMed] [Google Scholar]
- [71].Williams CD, Koerner MR, Lampe JN, Farhood A, Jaeschke H. Mouse strain-dependent caspase activation during acetaminophen hepatotoxicity does not result in apoptosis or modulation of inflammation. Toxicol Appl Pharmacol. 2011;257:449–458. doi: 10.1016/j.taap.2011.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Jaeschke H, Williams CD, Farhood A. No evidence for caspase-dependent apoptosis in acetaminophen hepatotoxicity. Hepatology. 2011;53:718–719. doi: 10.1002/hep.23940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].McGill MR, Yan HM, Ramachandran A, Murray GJ, Rollins DE, Jaeschke H. HepaRG cells: a human model to study mechanisms of acetaminophen hepatotoxicity. Hepatology. 2011;53:974–82. doi: 10.1002/hep.24132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Woolbright BL, Jaeschke H. The impact of sterile inflammation in acute liver injury. J Clin Transl Res. 2017;3:170–188. doi: 10.18053/jctres.03.2017S1.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Woolbright BL, Jaeschke H. Role of the inflammasome in acetaminophen-induced liver injury and acute liver failure. J Hepatol. 2017;66((4)):836–848. doi: 10.1016/j.jhep.2016.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Laskin DL, Pilaro AM. Potential role of activated macrophages in acetaminophen hepatotoxicity. I. Isolation and characterization of activated macrophages from rat liver. Toxicol Appl Pharmacol. 1986;86:204–215. doi: 10.1016/0041-008x(86)90051-7. [DOI] [PubMed] [Google Scholar]
- [77].Laskin DL, Gardner CR, Price VF, Jollow DJ. Modulation of macrophage functioning abrogates the acute hepatotoxicity of acetaminophen. Hepatology. 1995;21:1045–1050. [PubMed] [Google Scholar]
- [78].Michael SL, Pumford NR, Mayeux PR, Niesman MR, Hinson JA. Pretreatment of mice with macrophage inactivators decreases acetaminophen hepatotoxicity and the formation of reactive oxygen and nitrogen species. 1999;30((1)):186–195. doi: 10.1002/hep.510300104. [DOI] [PubMed] [Google Scholar]
- [79].Smith GS, Nadiw DE, Kokoska ER, Solomon H, Tiniakos DG, Miller TA. Role of neutrophils in hepatotoxicity induced by oral acetaminophen administration in rats. J Surg Res. 1998;80((2)):252–258. doi: 10.1006/jsre.1998.5441. [DOI] [PubMed] [Google Scholar]
- [80].Liu ZX, Han D, Gunawan B, Kaplowitz N. Neutrophil depletion protects against murine acetaminophen hepatotoxicity. Hepatology. 2006;43((6)):1220–1230. doi: 10.1002/hep.21175. [DOI] [PubMed] [Google Scholar]
- [81].Imaeda AB, Watanabe A, Sohail MA, Mahmood S, Mohamadnejad M, Sutterwala FS, Flavell RA, Mehal WZ. Acetaminophen-induced hepatotoxicity in mice is dependent on Tlr9 and the Nalp3 inflammasome. J Clin Invest. 2009;119((2)):305–314. doi: 10.1172/JCI35958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Kono H, Chen CJ, Ontiveros F, Rock KL. Uric acid promotes an acute inflammatory response to sterile cell death in mice. J Clin Invest. 2010;120:1939–1949. doi: 10.1172/JCI40124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Dear JW, Simpson KJ, Nicolai MP, Catterson JH, Street J, Huizinga T, Craig DG, Dhaliwal K, Webb S, Bateman DN, Webb DJ. Cycophilin A is a damage-associated molecular pattern molecule that mediates acetaminophen-induced liver injury. J Immunol. 2011;187:3347–3352. doi: 10.4049/jimmunol.1100165. [DOI] [PubMed] [Google Scholar]
- [84].Marques PE, Amaral SS, Pires DA, Nogueira LL, Soriani FM, Lima BH, Lopes GA, Russo RC, Avila TV, Megaco JG, Oliveira AG, Pinto MA, Lima CX, De Paula AM, Cara DC, Leite MF, Teixeira MM, Menezes GB. Chemokines and mitochondrial products activate neutrophils to amplify organ injury during mouse acute liver failure. Hepatology. 2012;56:1971–1982. doi: 10.1002/hep.25801. [DOI] [PubMed] [Google Scholar]
- [85].Ju C, Reilly TP, Bourdi M, Radonovich MF, Brady JN, George JW, Pohl LR. Protective role of Kupffer cells in acetaminophen-induced hepatic injury in mice. Chem Res Toxicol. 2002;15:1504–1513. doi: 10.1021/tx0255976. [DOI] [PubMed] [Google Scholar]
- [86].Williams CD, Antoine DJ, Shaw PJ, Benson C, Farhood A, Williams DP, Kanneganti TD, Park BK, Jaeschke H. Role of the Nalp3 inflammasome in acetaminophen-induced sterile inflammation and liver injury. Toxicol Appl Pharmacol. 2011;252((3)):289–297. doi: 10.1016/j.taap.2011.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Williams CD, Farhood A, Jaeschke H. Role of caspase-1 and interleukin-1beta in acetaminophen-induced hepatic inflammation and liver injury. Toxicol Appl Pharmacol. 2010;247:169–178. doi: 10.1016/j.taap.2010.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Davis M, Ideo G, Harrison NG, Williams R. Hepatic glutathione depletion and impaired bromosulphthalein clearance early after paracetamol overdose in man and the rat. Clin Sci Mol Med. 1975;49:495–502. doi: 10.1042/cs0490495. [DOI] [PubMed] [Google Scholar]
- [89].Lauterburg BH, Mitchell JR. Therapeutic doses of acetaminophen stimulate the turnover of cysteine and glutathione in man. J Hepatol. 1987;4:206–211. doi: 10.1016/s0168-8278(87)80081-8. [DOI] [PubMed] [Google Scholar]
- [90].Muldrew KL, James LP, Coop L, McCullough SS, Hendrickson HP, Hinson JA, Mayeux PR. Determination of acetaminophen-protein adducts in mouse liver and serum and human serum after hepatotoxic doses of acetaminophen using high-performance liquid chromatography with electrochemical detection. Drug Metab Dispos. 2002;30:446–451. doi: 10.1124/dmd.30.4.446. [DOI] [PubMed] [Google Scholar]
- [91].Bhattacharyya S, Yan K, Pence L, Simpson PM, Gill P, Letzig LG, Beger RD, Sullivan JE, Kearns GL, Reed MD, Marshall JD, Van Den Anker JN, James LP. Targeted liquid chromatography-mass spectrometry analysis of serum acylcarnitines in acetaminophen toxicity in children. Biomark Med. 2014;8:147–159. doi: 10.2217/bmm.13.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Mitchell JR, McMurtry RJ, Statham CN, Nelson SD. Molecular basis for several drug-induced nephropathies. Am J Med. 1977;62:518–526. doi: 10.1016/0002-9343(77)90407-7. [DOI] [PubMed] [Google Scholar]
- [93].Trumper L, Girardi G. Elías MM. Acetaminophen nephrotoxicity in male Wistar rats. Arch Toxicol. 1992;66:107–111. doi: 10.1007/BF02342503. [DOI] [PubMed] [Google Scholar]
- [94].Lucas AM, Hennig G, Dominick PK, Whiteley HE, Roberts JC, Cohen SD. Ribose cysteine protects against acetaminophen- induced hepatic and renal toxicity. Toxicol Pathol. 2000;28:697–704. doi: 10.1177/019262330002800510. [DOI] [PubMed] [Google Scholar]
- [95].Stern ST, Bruno MK, Hennig GE, Horton RA, Roberts JC, Cohen SD. Contribution of acetaminophen-cysteine to acetaminophen nephrotoxicity in CD-1 mice: I. Enhancement of acetaminophen nephrotoxicity by acetaminophen-cysteine. Toxicol Appl Pharmacol. 2005;202:151–159. doi: 10.1016/j.taap.2004.06.030. [DOI] [PubMed] [Google Scholar]
- [96].Stern ST, Bruno MK, Horton RA, Hill DW, Roberts JC, Cohen SD. Contribution of acetaminophen-cysteine to acetaminophen nephrotoxicity II. Possible involvement of the gamma-glutamyl cycle. Toxicol Appl Pharmacol. 2005;202:160–171. doi: 10.1016/j.taap.2004.06.029. [DOI] [PubMed] [Google Scholar]
- [97].Boyer TD, Rouff SL. Acetaminophen-induced hepatic necrosis and renal failure. JAMA. 1971;218((3)):440–1. [PubMed] [Google Scholar]
- [98].Mour G, Feinfeld DA, Caraccio T, McGuigan M. Acute renal dysfunction in acetaminophen poisoning. Ren Fail. 2005;27:381–383. [PubMed] [Google Scholar]
- [99].Pakravan N, Simpson KJ, Waring WS, Bates CM, Bateman DN. Renal injury at first presentation as a predictor for poor outcome in severe paracetamol poisoning referred to a liver transplant unit. Eur J Clin Pharmacol. 2009;65:163–168. doi: 10.1007/s00228-008-0580-9. [DOI] [PubMed] [Google Scholar]
- [100].O'Riordan A, Brummell Z, Sizer E, Auzinger G, Heaton N, O'Grady JG, Bernal W, Hendry BM, Wendon JA. Acute kidney injury in patients admitted to a liver intensive therapy unit with paracetamol-induced hepatotoxicity. Nephrol Dial Transplant. 2011;26:3501–3508. doi: 10.1093/ndt/gfr050. [DOI] [PubMed] [Google Scholar]
- [101].Chen YG, Lin CL, Dai MS, Chang PY, Chen JH, Huang TC, Wu YY, Kao CH. Risk of acute kidney Injury and Long-Term Outcome in Patients With Acetaminophen Intoxication: A Nationwide Population-Based Retrospective Cohort Study. Medicine (Baltimore). 2015;94:e2040. doi: 10.1097/MD.0000000000002040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Curry SC, Padilla-Jones A, O'Connor AD, Ruha AM, Bikin DS, Wilkins DG, Rollins DE, Slawson MH, Gerkin RD. Acetaminophen adduct study group. Prolonged acetaminophen-protein adduct elimination during renal failure, lack of adduct removal by hemodiafiltration, and urinary adduct concentrations after acetaminophen overdose. J Med Toxicol. 2015;11:169–178. doi: 10.1007/s13181-014-0431-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Prescott LF, Critchley JA. The treatment of acetaminophen poisoning. Annu Rev Pharmacol Toxicol. 1983;23:87–101. doi: 10.1146/annurev.pa.23.040183.000511. [DOI] [PubMed] [Google Scholar]
- [104].Wilkinson SP, Moodie H, Arroyo VA, Williams R. Frequency of renal impairment in paracetamol overdose compared with other causes of acute liver damage. J Clin Pathol. 1977;30:141–143. doi: 10.1136/jcp.30.2.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Larson AM, Polson J, Fontana RJ, Davern TJ, Lalani E, Hynan LS, Reisch JS, Schiødt FV, Ostapowicz G, Shakil AO. Lee WM; Acute Liver Failure Study Group. Acetaminophen-induced acute liver failure: results of a United States multicenter, prospective study. Hepatology. 2005;42:1364–1372. doi: 10.1002/hep.20948. [DOI] [PubMed] [Google Scholar]
- [106].Antoine DJ, Sabbisetti VS, Francis B, Jorgenson AL, Craig DG, Simpson KJ, Bonventre JV, Park BK, Dear JW. Circulating kidney injury molecule 1 predicts prognosis and poor outcome in patients with acetaminophen-induced liver injury. Hepatology. 2015;62:591–599. doi: 10.1002/hep.27857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Perneger TV, Whelton PK, Klag MJ. Risk of kidney failure associated with the use of acetaminophen, aspirin, and nonsteroidal antiinflammatory drugs. N Eng J Med. 1994;331:1675–1679. doi: 10.1056/NEJM199412223312502. [DOI] [PubMed] [Google Scholar]
- [108].Blantz RC. Acetaminophen: acute and chronic effects on renal function. Am J Kidney Dis. 1996;28:S3–6. doi: 10.1016/s0272-6386(96)90561-2. [DOI] [PubMed] [Google Scholar]
- [109].Mudge GH, Gemborys MW, Duggin GG. Covalent binding of metabolites of acetaminophen to kidney protein and depletion of renal glutathione. J Pharmacol Exp Ther. 1978;206:218–226. [PubMed] [Google Scholar]
- [110].Bessems JG, Vermeulen NP. Paracetamol (acetaminophen)-induced toxicity: molecular and biochemical mechanisms, analogues and protective approaches. Crit Rev Toxicol. 2001;31:55–138. doi: 10.1080/20014091111677. [DOI] [PubMed] [Google Scholar]
- [111].Carpenter HM, Mudge GH. Acetaminophen nephrotoxicity: studies on renal acetylation and deacetylation. J Pharmacol Exp Ther. 1981;218:161–167. [PubMed] [Google Scholar]
- [112].Newton JF, Braselton WE Jr, Kuo CH, Kluwe WM, Gemborys MW, Mudge GH, Mudge GH, Hook JB. Metabolism of acetaminophen by the isolated perfused kidney. J Pharmacol Exp Ther. 1982;221:76–79. [PubMed] [Google Scholar]
- [113].Newton JF, Kuo CH, De Shone GM, Hoefle D, Bernstein J, Hook JB. The role of p-aminophenol in acetaminophen-induced nephrotoxicity: effect of bis(p-nitrophenyl) phosphate on acetaminophen and p-aminophenol nephrotoxicity and metabolism in Fischer 344 rats. Toxicol Appl Pharmacol. 1985;81:416–430. doi: 10.1016/0041-008x(85)90413-2. [DOI] [PubMed] [Google Scholar]
- [114].Newton JF, Pasino DA, Hook JB. Acetaminophen nephrotoxicity in the rat: quantitation of renal metabolic activation in vivo. Toxicol Appl Pharmacol. 1985;78:39–46. doi: 10.1016/0041-008x(85)90302-3. [DOI] [PubMed] [Google Scholar]
- [115].Ściskalska M, Śliwinska-Mosson M, Podawacz M, Sajewicz W, Milnerowicz H. Mechanisms of interaction of the N-acetylp-aminophenol metabolites in terms of nephrotoxicity. Drug Chem Toxicol. 2015;38:121–125. doi: 10.3109/01480545.2014.928722. [DOI] [PubMed] [Google Scholar]
- [116].Mugford CA, Tarloff JB. Contribution of oxidation and deacetylation to the bioactivation of acetaminophen in vitro in liver and kidney from male and female Sprague-Dawley rats. Drug Metab Dispos. 1995;23:290–294. [PubMed] [Google Scholar]
- [117].Emeigh Hart SG, Beierschmitt WP, Bartolone JB, Wyand DS, Khairallah EA, Cohen SD. Evidence against deacetylation and for cytochrome P450-mediated activation in acetaminophen-induced nephrotoxicity in the CD-1 mouse. Toxicol Appl Pharmacol. 1991;107:1–15. doi: 10.1016/0041-008x(91)90325-9. [DOI] [PubMed] [Google Scholar]
- [118].Hart SG, Beierschmitt WP, Wyand DS, Khairallah EA, Cohen SD. Acetaminophen nephrotoxicity in CD-1 mice. I. Evidence of a role for in situ activation in selective covalent binding and toxicity. Toxicol Appl Pharmacol. 1994;126:267–275. doi: 10.1006/taap.1994.1116. [DOI] [PubMed] [Google Scholar]
- [119].Hoivik DJ, Manatou JE, Tviet A, Hart SG, Khairallah EA, Cohen SD. Gender-related differences in susceptibility to acetaminophen-induced protein arylation and nephrotoxicity in the CD-1 mouse. Toxicol Appl Pharmacol. 1995;130:257–271. doi: 10.1006/taap.1995.1031. [DOI] [PubMed] [Google Scholar]
- [120].Clark PM, Clark JD, Wheatley T. Urine discoloration after acetaminophen overdose. Clin Chem. 1986;32:1777–1778. [PubMed] [Google Scholar]
- [121].Chen CF, Tseng TY, Tseng HK, Liu TZ. Automated spectrophotometric assay for urine p-aminophenol by oxidative coupling reaction. Ann Clin Lab Sci. 2004;34:336–340. [PubMed] [Google Scholar]
- [122].Larsson R, Ross D, Berlin T, Olsson LI, Moldeus P. Prostaglandin synthase catalyzed metabolic activation of pphenetidine and acetaminophen by microsomesisoalted from rabbit and human kidney. J Pharmacol Exp Ther. 1985;235:475–480. [PubMed] [Google Scholar]
- [123].Porter KE, Dawson AG. Inhibition of respiration and gluconeogenesis by paracetamol in rat kidney preparations. Biochem Pharmacol. 1979;28:3057–3062. doi: 10.1016/0006-2952(79)90613-0. [DOI] [PubMed] [Google Scholar]
- [124].Satav JG, Bhattacharya RK. Respiratory functions in kidney mitochondria following paracetamol administration to young-adult & old rats. Indian J Med Res. 1997;105:131–135. [PubMed] [Google Scholar]
- [125].Stallons LJ, Funk JA, Schnellmann RG. Mitochondrial homeostasis in acute organ failure. Curr Pathobiol Rep. 2013;1 doi: 10.1007/s40139-013-0023-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Newson RB, Shaheen SO, Chinn S, Burney PG. Paracetamol sales and atopic disease in children and adults: an ecological analysis. Eur Respir J. 2000;16:817–823. doi: 10.1183/09031936.00.16581700. [DOI] [PubMed] [Google Scholar]
- [127].Shaheen SO, Newson RB, Henderson AJ, Headley JE, Stratton FD, Jones RW, Strachan DP. ALSPAC Study Team. Prenatal paracetamol exposure and risk of asthma and elevated immunoglobulin E in childhood. Clin Exp Allergy. 2005;35:18–25. doi: 10.1111/j.1365-2222.2005.02151.x. [DOI] [PubMed] [Google Scholar]
- [128].Beasley R, Clayton T, Crane J, von Mutius E, Lai CK, Montefort S. Stewart A; ISAAC Phase Three Study Group. Association between paracetamol use in infancy and childhood, and risk of asthma, rhinoconjunctivitis, and eczema in children aged 6-7 years: analysis from Phase Three of the ISAAC programme. Lancet. 2008;372:1039–1048. doi: 10.1016/S0140-6736(08)61445-2. [DOI] [PubMed] [Google Scholar]
- [129].Perzanowski MS, Miller RL, Tang D, Ali D, Garfinkel RS, Chew GL, Goldstein IF, Perera FP, Barr RG. Prenatal acetaminophen exposure and risk of wheeze at age 5 years in an urban low-income cohort. Thorax. 2010;65:118–123. doi: 10.1136/thx.2009.121459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Kelkar M, Cleves MA, Foster HR, Hogan WR, James LP, Martin BC. Prescription-acquired acetaminophen use and the risk of asthma in adults: a case-control study. Ann Pharmacother. 2012;46:1598–1608. doi: 10.1345/aph.1R430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].McKeever TM, Lewis SA, Smit HA, Burney P, Britton JR, Cassano PA. The association of acetaminophen, aspirin, and ibuprofen with respiratory disease and lung function. Am J Respir Crit Care Med. 2005;171:966–971. doi: 10.1164/rccm.200409-1269OC. [DOI] [PubMed] [Google Scholar]
- [132].Singh A. Acetaminophen and asthma. CMAJ. 2009;181:616. doi: 10.1503/cmaj.109-2026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Henderson AJ, Shaheen SO. Acetaminophen and asthma. aediatr Respir Rev. 2013;14:9–15. doi: 10.1016/j.prrv.2012.04.004. [DOI] [PubMed] [Google Scholar]
- [134].Weatherall M, Ioannides S, Braithwaite I, Beasley R. The association between paracetamol use and asthma: causation or coincidence? Clin Exp Allergy. 2015;45:108–113. doi: 10.1111/cea.12410. [DOI] [PubMed] [Google Scholar]
- [135].Rusconi F, Gagliardi L, Galassi C, Forastiere F, Brunetti L, La Grutta S, Piffer S, Talassi F. SIDRIA-2 Collaborative Group. Paracetamol and antibiotics in childhood and subsequent development of wheezing/asthma: association or causation? Int J Epidemiol. 2011;40:662–667. doi: 10.1093/ije/dyq263. [DOI] [PubMed] [Google Scholar]
- [136].Gu J, Cui H, Behr M, Zhang L, Zhang QY, Yang W, Hinson JA, Ding X. In vivo mechanisms of tissue-selective drug toxicity: effects of liver-specific knockout of the NADPHcytochrome P450 reductase gene on acetaminophen toxicity in kidney, lung, and nasal mucosa. Mol Pharmacol. 2005;67:623–630. doi: 10.1124/mol.104.007898. [DOI] [PubMed] [Google Scholar]
- [137].Chen TS, Richie JP Jr, Lang CA. Life span profiles of glutathione and acetaminophen detoxification. Drug Metab Dispos. 1990;18:882–887. [PubMed] [Google Scholar]
- [138].Smith GJ, Cichocki JA, Doughty BJ, Manautou JE, Jordt SE, Morris JB. Effects of acetaminophen on oxidant and irritant respiratory tract responses to environmental tobacco smoke in female mice. Environ Health Perspect. 2016;124:642–650. doi: 10.1289/ehp.1509851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Shaheen SO, Newson RB, Ring SM, Rose-Zerilli MJ, Holloway JW, Henderson AJ. Prenatal and infant acetaminophen exposure, antioxidant gene polymorphisms, and childhood asthma. J Allergy Clin Immunol. 2010;126:1141–8e7. doi: 10.1016/j.jaci.2010.08.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Breen K, Wandscheer JC, Peignoux M, Pessayre D. In situ formation of the acetaminophen metabolite covalently bound in kidney and lung. Supportive evidence provided by total hepatectomy. BiochemPharmacol. 1982;31:115–116. doi: 10.1016/0006-2952(82)90247-7. [DOI] [PubMed] [Google Scholar]
- [141].Bartolone JB, Beierschmitt WP, Birge RB, Hart SG, Wyand S, Cohen SD, Khairallah EA. Selective acetaminophen metabolite binding to hepatic and extrahepatic proteins: an in vivo and in vitro analysis. Toxicol Appl Pharmacol. 1989;99:240–249. doi: 10.1016/0041-008x(89)90006-9. [DOI] [PubMed] [Google Scholar]
- [142].Bulera SJ, Cohen SD, Khairallah EA. Acetaminophenarylated proteins are detected in hepatic subcellular fractions and numerous extra-hepatic tissues in CD-1 and C57B1/6J mice. Toxicology. 1996;109:85–99. doi: 10.1016/0300-483x(96)03309-4. [DOI] [PubMed] [Google Scholar]
- [143].Berkowitz GS, Lapinski RH. Risk factors for cryptorchidism: a nested case-control study. Paediatr Perinat Epidemiol. 1996;10:39–51. doi: 10.1111/j.1365-3016.1996.tb00024.x. [DOI] [PubMed] [Google Scholar]
- [144].Jensen MS, Rebordosa C, Thulstrup AM, Toft G, Sørensen HT, Bonde JP, Henriksen TB, Olsen J. Maternal use of acetaminophen, ibuprofen, and acetylsalicylic acid during pregnancy and risk of cryptorchidism. Epidemiology. 2010;21:779–785. doi: 10.1097/EDE.0b013e3181f20bed. [DOI] [PubMed] [Google Scholar]
- [145].Kristensen DM, Hass U, Lesné L, Lottrup G, Jacobsen PR, Desdoits-Lethimonier C, Boberg J, Petersen JH, Toppari J, Jensen TK, Brunak S, Skakkebaek NE, Nellemann C, Main KM, Jacgou B, Leffers H. Intrauterine exposure to mild analgesics is a risk factor for development of male reproductive disorders in human and rat. Hum Reprod. 2011;26:235–244. doi: 10.1093/humrep/deq323. [DOI] [PubMed] [Google Scholar]
- [146].Lind DV, Main KM, Kyhl HB, Kristensen DM, Toppari J, Andersen HR, Andersen MS, Skakkebæk NE, Jensen TK. Maternal use of mild analgesics during pregnancy associated with reduced anogenital distance in sons: a cohort study of 1027 mother-child pairs. Hum Reprod. 2017;32:223–231. doi: 10.1093/humrep/dew285. [DOI] [PubMed] [Google Scholar]
- [147].Holm JB, Chalmey C, Modick H, Jensen LS, Dierkes G, Weiss T, Jensen BA, Nørregård MM, Borkowski K, Styrishave B, Martin Koch H, Mazaud-Guittot S, Jegou B, Kristiansen K, Kristensen DM. Aniline is rapidly converted into paracetamol impairing male reproductive development. Toxicol Sci. 2015;148:288–298. doi: 10.1093/toxsci/kfv179. [DOI] [PubMed] [Google Scholar]
- [148].Holm JB, Mazaud-Guittot S, Danneskiold-Samsøe NB, Chalmey C, Jensen B, Nørregård MM, Hansen CH, Styrishave B, Svingen T, Vinggaard AM, Koch HM, Bowles J, Koopman P, Jegou B, Kristiansen K, Kristensen DM. Intrauterine exposure to paracetamol and aniline impairs female reproductive development by reducing follicle reserves and fertility. Toxicol Sci. 2016;150:178–189. doi: 10.1093/toxsci/kfv332. [DOI] [PubMed] [Google Scholar]
- [149].McGill MR, Lebofsky M, Norris HR, Slawson MH, Bajt ML, Xie Y, Williams CD, Wilkins DG, Rollins DE, Jaeschke H. Plasma and liver acetaminophen-protein adduct levels in mice after acetaminophen treatment: dose-response, mechanisms, and clinical implications. Toxicol Appl Pharmacol. 2013;269:240–249. doi: 10.1016/j.taap.2013.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [150].Hay-Schmidt A, Finkielman OTE, Jensen BAH, Hogsbro CF, Bak Holm J, Johansen KH, Jensen TK, Andrade AM, Swan SH, Bornehag CG, Brunak S, Jeqou B, Kristiansen K, Kristensen DM. Prenatal exposure to paracetamol/ acetaminophen and precursor aniline impairs masculinisation of male brain and behavior. Reproduction. 2017;154:145–152. doi: 10.1530/REP-17-0165. [DOI] [PubMed] [Google Scholar]
- [151].Patel R, Rosengren RJ. Acetaminophen elicits anti-estrogenic but not estrogenic responses in the immature mouse. Toxicol Lett. 2001;122:89–96. doi: 10.1016/s0378-4274(01)00352-6. [DOI] [PubMed] [Google Scholar]
- [152].Fortner RT, Oh H, Daugherty SE, Xu X, Hankinson SE, Ziegler RG, Eliassen AH. Analgesic use and patterns of estrogen metabolism in premenopausal women. Horm Cancer. 2014;5:104–112. doi: 10.1007/s12672-013-0167-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [153].van den Driesche S, Macdonald J, Anderson RA, Johnston ZC, Chetty T, Smith LB, McKinnell C, Dean A, Homer NZ, Jorgensen A, Camacho-Moll ME, Sharpe RM, Mitchell RT. Prolonged exposure to acetaminophen reduces testosterone production by the human fetal testis in a xenograft model. SciTransl Med. 2015;7:288ra80. doi: 10.1126/scitranslmed.aaa4097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].Kristensen DM, Lesné L, Le Fol V, Desdoits-Lethimonier C, Dejucq-Rainsford N, Leffers H. Jégou B. Paracetamol (acetaminophen), aspirin (acetylsalicylic acid) and indomethacin are anti-androgenic in the rat foetal testis. Int J Androl. 2012;35:377–384. doi: 10.1111/j.1365-2605.2012.01282.x. [DOI] [PubMed] [Google Scholar]
- [155].Albert O, Desdoits-Lethimonier C, Lesné L, Legrand A, Guillé F, Bensalah K, Dejucq-Rainsford N. Jégou B. Paracetamol, aspirin and indomethacin display endocrine disrupting properties in the adult human testis in vitro. Hum Reprod. 2013;28:1890–1898. doi: 10.1093/humrep/det112. [DOI] [PubMed] [Google Scholar]
- [156].Mazaud-Guittot S, Nicolas Nicolaz C, Desdoits-Lethimonier C, Coiffec I, Ben Maamar M, Balaguer P, Kristensen DM, Chevrier C, Lavoué V, Poulain P, Dejucq-Rainsford N, Jégou B. Paracetamol, aspirin, and indomethacin induce endocrine disturbances in the human fetal testis capable of interfering with testicular descent. J Clin Endocrinol Metab. 2013;98:1757–1767. doi: 10.1210/jc.2013-2531. [DOI] [PubMed] [Google Scholar]
- [157].Nohynek GJ, Borgert CJ, Dietrich D, Rozman KK. Endocrine disruption: fact or urban legend? Toxicol Lett. 2013;223:295–305. doi: 10.1016/j.toxlet.2013.10.022. [DOI] [PubMed] [Google Scholar]
- [158].Friedman RA, House JW, Luxford WM, Gherini S, Mills D. Profound hearing loss associated with hydrocodone/ acetaminophen abuse. Am J Otol. 2000;21:188–191. doi: 10.1016/s0196-0709(00)80007-1. [DOI] [PubMed] [Google Scholar]
- [159].Oh AK, Ishiyama A, Baloh RW. Deafness associated with abuse of hydrocodone/acetaminophen. Neurology. 2000;54:2345. doi: 10.1212/wnl.54.12.2345. [DOI] [PubMed] [Google Scholar]
- [160].Ho T, Vrabec JT, Burton AW. Hydrocodone use and sensorineural hearing loss. Pain Physician. 2007;10:467–472. [PubMed] [Google Scholar]
- [161].Kalinec GM, Webster P, Lim DJ, Kalinec F. A cochlear cell line as an in vitro system for drug ototoxicity screening. Audiol Neurootol. 2003;8:177–189. doi: 10.1159/000071059. [DOI] [PubMed] [Google Scholar]
- [162].Kalinec G, Thein P, Park C, Kalinec F. HEI-OC1 cells as a model for investigating drug cytotoxicity. Hear Res. 2016;335:105–17. doi: 10.1016/j.heares.2016.02.019. [DOI] [PubMed] [Google Scholar]
- [163].McGill MR, Kennon-McGill S, Durham D, Jaeschke H. Hearing, reactive metabolite formation, and oxidative stress in cochleae after a single acute overdose of acetaminophen: an in vivo study. Toxicol Mech Methods. 2016;26:104–111. doi: 10.3109/15376516.2015.1122136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [164].Boulares HA, Giardina C, Navarro CL, Khairallah EA, Cohen SD. Modulation of serum growth factor signal transduction in Hepa 1-6 cells by acetaminophen: an inhibition of c-myc expression, NF-kappaB activation, and Raf-1 kinase activity. Toxicol Sci. 1999;48:264–274. doi: 10.1093/toxsci/48.2.264. [DOI] [PubMed] [Google Scholar]
- [165].Boulares AH, Zoltoski AJ, Stoica BA, Cuvillier O, Smulson ME. Acetaminophen induces a caspase-dependent and BclXL sensitive apoptosis in human hepatoma cells and lymphocytes. Pharmacol Toxicol. 2002;90:38–50. doi: 10.1034/j.1600-0773.2002.900108.x. [DOI] [PubMed] [Google Scholar]
- [166].Antoine DJ, Jenkins RE, Dear JW, Williams DP, McGill MR, Sharpe MR, Craig DG, Simpson KJ, Jaeschke H, Park BK. Molecular forms of HMGB1 and keratin-18 as mechanistic biomarkers for mode of cell death and prognosis during clinical acetaminophen hepatotoxicity. J Hepatol. 2012;56:1070–1079. doi: 10.1016/j.jhep.2011.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [167].Alberti A, Pirrone P, Elia M, Waring RH, Romano C. Sulphation deficit in "low-functioning" autistic children: a pilot study. Biol Psychiatry. 1999;46:420–424. doi: 10.1016/s0006-3223(98)00337-0. [DOI] [PubMed] [Google Scholar]
- [168].Good P. Did acetaminophen provoke the autism epidemic? Altern Med Rev. 2009;14:364–372. [PubMed] [Google Scholar]
- [169].Prescott LF. Kinetics and metabolism of paracetamol and phenacetin. Br J Clin Pharmacol. 1980;10:291S–298S. doi: 10.1111/j.1365-2125.1980.tb01812.x. Suppl 2: [DOI] [PMC free article] [PubMed] [Google Scholar]
- [170].Liew Z, Ritz B, Virk J, Olsen J. Maternal use of acetaminophen during pregnancy and risk of autism spectrum disorders in childhood: A Danish national birth cohort study. Autism Res. 2016;9:951–958. doi: 10.1002/aur.1591. [DOI] [PubMed] [Google Scholar]
- [171].Cox AR, McDowell S. A response to the article on the association between paracetamol/acetaminophen: use and autism by Stephen T. Schultz. Autism. 2009;13:123–4. doi: 10.1177/1362361308101816. [DOI] [PubMed] [Google Scholar]
- [172].Olson J, Liew Z. Commentary: acetaminophen use in pregnancy and neurodevelopment: attention function and autism spectrum symptoms. Int J Epidemiol. 2016;45:1996–1997. doi: 10.1093/ije/dyw169. [DOI] [PubMed] [Google Scholar]
- [173].Brandlistuen RE, Ystrom E, Nulman I, Koren G, Nordeng H. Prenatal paracetamol exposure and child neurodevelopment: a sibling-controlled cohort study. Int J Epidemiol. 2013;42:1702–1713. doi: 10.1093/ije/dyt183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [174].Thompson JM, Waldie KE, Wall CR, Murphy R, Mitchell EA. ABC study group. Associations between acetaminophen use during pregnancy and ADD/ADHD symptoms measured at ages 7 and 11 years. PLoS One. 2014;9:e108210. doi: 10.1371/journal.pone.0108210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [175].Avella-Garcia CB, Julvez J, Fortuny J, Rebordosa C, Garcia-Esteban R, Galan IR, Tardon A, Rodriguez-Bernal CL, Iniguez C, Andiarena A, Santa-Marina L, Sunyer J. Acetaminophen use in pregnancy and neurodevelopment: attention function and autism spectrum symptoms. Int J Epidemiol. 2016;45:1987–1996. doi: 10.1093/ije/dyw115. [DOI] [PubMed] [Google Scholar]
- [176].Stergiakouli E, Thapar A, Davey Smith G. Association of acetaminophen use during pregnancy with behavioral problems in childhood: evidence against confounding. JAMA Pediatr. 2016;170:964–970. doi: 10.1001/jamapediatrics.2016.1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [177].Hoover RM, Hayes VA, Erramouspe J. Association between prenatal acetaminophen exposure and future risk of attention deficit/hyperactivity disorder in children. Ann Pharmacother. 2015;49:1357–1361. doi: 10.1177/1060028015606469. [DOI] [PubMed] [Google Scholar]
- [178].Streissguth AP, Treder RP, Barr HM, Shepard TH, Bleyer WA, Sampson PD, Martin DC. Aspirin and acetaminophen use by pregnant women and subsequent child IQ and attention decrements. Teratology. 1987;35:211–219. doi: 10.1002/tera.1420350207. [DOI] [PubMed] [Google Scholar]
- [179].Saad A, Hegde S, Kechichian T, Gamble P, Rahman M, Stutz SJ, Anastasio NC, Alshehri W, Lei J, Mori S, Kajs B, Cunningham KA, Saade G, Burd I, Costantine M. Is There a Causal Relation between Maternal Acetaminophen Administration and ADD/ADHD? PLoS One. 2016;11:e0157380. doi: 10.1371/journal.pone.0157380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [180].Philippot G, Gordh T, Fredriksson A, Viberg H. Adult neurobehavioral alterations in male and female mice following developmental exposure to paracetamol (acetaminophen): characterization of a critical period. J Appl Toxicol. 2017 doi: 10.1002/jat.3473. doi: 10.1002/jat.3473.[Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- [181].Schultz ST. Can autism be triggered by acetaminophen activation of the endocannabinoid system? Acta Neurobiol Exp (Wars). 2010;70:227–231. doi: 10.55782/ane-2010-1793. [DOI] [PubMed] [Google Scholar]
- [182].Parker W, Hornik CD, Bilbo S, Holzknecht ZE, Gentry L, Rao R, Lin SS, Herbert MR, Nevison CD. The role of oxidative stress, inflammation and acetaminophen exposure from birth to early childhood in the induction of autism. J Int Med Res. 2017;45:407–438. doi: 10.1177/0300060517693423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [183].Constantino JN, Marrus N. The early origins of autism. Child Adolesc Psychiatr Clin N Am. 2017;26:555–570. doi: 10.1016/j.chc.2017.02.008. [DOI] [PubMed] [Google Scholar]
- [184].Constantino JN, Kennon-McGill S, Weichselbaum C, Marrus N, Haider A, Glowinski AL, Gillespie S, Klaiman C, Klin A, Jones W. Infant viewing of social scenes is under genetic control and is atypical in autism. Nature. 2017;547:340–344. doi: 10.1038/nature22999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [185].Zhao C, Shichi H. Histocytological study on the possible mechanism of acetaminophen cataractogenesis in mouse eye. Exp Mol Pathol. 1995;63:118–128. doi: 10.1006/exmp.1995.1036. [DOI] [PubMed] [Google Scholar]
- [186].Qian W, Amin RH, Shichi H. Cytotoxic metabolite of acetaminophen, N-acetyl-p-benzoquinone imine, produces cataract in DBA2 mice. J Ocul Pharmacol Ther. 1999;15:537–545. doi: 10.1089/jop.1999.15.537. [DOI] [PubMed] [Google Scholar]
- [187].Armour A, Slater SD. Paracetamol cardiotoxicity. Postgrad Med J. 1993;69:52–54. doi: 10.1136/pgmj.69.807.52. [DOI] [PMC free article] [PubMed] [Google Scholar]