

Abstract
Facioscapulohumeral muscular dystrophy (FSHD) is associated with aberrant epigenetic regulation of the chromosome 4q35 D4Z4 macrosatellite repeat. The resulting DNA hypomethylation and relaxation of epigenetic repression leads to increased expression of the deleterious DUX4-fl mRNA encoded within the distal D4Z4 repeat. With the typical late onset of muscle weakness, prevalence of asymptomatic individuals, and an autosomal dominant mode of inheritance, FSHD is often passed on from one generation to the next and affects multiple individuals within a family. Here we have characterized unique collections of 114 lymphoblastoid cell lines (LCLs) generated from 12 multigenerational FSHD families, including 56 LCLs from large, genetically homogeneous families in Utah. We found robust expression of DUX4-fl in most FSHD LCLs and a good correlation between DNA hypomethylation and repeat length. In addition, DUX4-fl levels can be manipulated using epigenetic drugs as in myocytes, suggesting that some epigenetic pathways regulating DUX4-fl in myocytes are maintained in LCLs. Overall, these FSHD LCLs provide an alternative cellular model in which to study many aspects of D4Z4, DUX4, and FSHD gene regulation in a background of low genetic variation. Significantly, these non-adherent immortal LCLs are amenable for high-throughput screening of potential therapeutics targeting DUX4-fl mRNA or protein expression.
Keywords: FSHD, DUX4, D4Z4, disease model, epigenetic, DNA methylation
1. Introduction
Facioscapulohumeral muscular dystrophy (FSHD), affecting ~ 1:7,500–15,000 individuals, is the most prevalent muscular dystrophy that indiscriminately afflicts children and adults of all ages and both genders [1–5]. All forms of FSHD are genetically and epigenetically linked to the chromosome 4q35 D4Z4 macrosatellite array, with the interplay between these factors accounting for much of the high variability in disease penetrance and severity characteristic of the disease [1, 6–15]. The predominant form of the disease, FSHD1 (OMIM 158900), represents >95% of reported cases and results from large DNA deletions within the 4q35 D4Z4 repeat array [16, 17]. Healthy, genetically unaffected individuals are typically defined as having more than 10 D4Z4 repeat units (RUs) on both 4q chromosome arms (generally 25–35 RUs and as high as 120 RUs per array [18, 19]), whereas individuals with genetic FSHD1 have a contracted D4Z4 array in the range of 1 to 10 D4Z4 RUs on one 4q chromosome arm, consistent with an autosomal dominant mode of inheritance (Fig. 1) [20]. These polymorphic FSHD1-sized D4Z4 contractions are not sufficient for pathogenesis; development of FSHD also requires a disease permissive allele of the chromosome 4q subtelomere (4A) in cis with the contracted array [19, 21–23]. FSHD2 (OMIM 158901), the far less common form of FSHD, presents with similar clinical features as FSHD1, but is caused by unlinked mutations in genes encoding chromatin regulatory proteins [7, 8, 24, 25]. However, FSHD2 is also genetically linked to the 4q35 array by the requirement of at least one permissive 4A-type subtelomere and a specific range of D4Z4 RUs (~11–28 RUs) in order to develop disease [7, 23]. Thus, with these genetic requirements, FSHD2 is considered a digenic disease [8, 25].
Fig. 1.
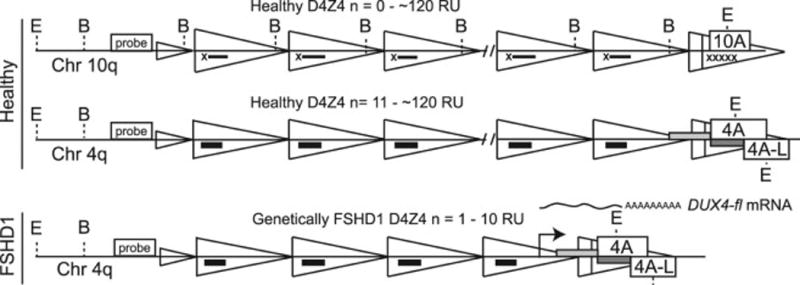
Positions of restriction enzyme sites, probe, and PCR primers used to distinguish D4Z4 sequence. Scheme depicting the chromosome 4q35 D4Z4 macrosatellite array, which is contracted in FSHD1, leading to epigenetic changes specific to the contracted chromosome. De-repression of the pathogenic allele leads to aberrant expression of the DUX4-fl mRNA from the distal-most D4Z4 repeat unit (RU). The 4q35 and 10q26 D4Z4 arrays are distinguished from each other by combined EcoRI (E) + AvrII (B) digestion followed by Southern blotting and probing with p13E-11 (probe). DNA methylation changes were assayed by BSS specific for the distal D4Z4 RU of 4qA and its allelic variant 4qA-L [12] (gray bars) or all D4Z4 RUs (black bars).
Despite the clear genetic distinctions between these two forms of FSHD, the effects on D4Z4 chromatin are quite similar. In FSHD1, the 4q35 deletions result in the loss of regulatory heterochromatin that significantly alters the local epigenetic landscape of the contracted allele and is characterized by allele-specific DNA hypomethylation [12, 26–30]. FSHD2 mutations reside in the genes encoding the epigenetic machinery responsible for establishing and maintaining repression of the D4Z4 arrays; therefore, FSHD2 is characterized by DNA hypomethylation of both 4q35 D4Z4 arrays and often both 10q26 arrays as well [7, 8, 25]. Thus, although epigenetic dysregulation is characteristic of FSHD in general, FSHD1 and FSHD2 subjects can be distinguished from each other and even individually diagnosed based on the DNA methylation profiles of the 4q35 D4Z4 arrays and disease permissive alleles [11, 31, 32]. In addition, some FSHD1 subjects may also have FSHD2-type mutations, with the combined effect presenting as a very severe form of FSHD, thus characterizing these genes as modifiers of disease severity [9]. To date, there have been two FSHD1 modifier genes identified, SMCHD1 and DNMT3B [8, 25], both of which encode epigenetic regulator proteins, further highlighting the importance of the 4q35 epigenetic status with respect to disease presentation.
In both forms of FSHD, chromatin de-repression at the 4q35 D4Z4 macrosatellite has similar downstream molecular consequences, resulting in the aberrant expression of a pathogenic isoform of the DUX4 (double homeobox 4) gene, DUX4-fl, in skeletal muscle [33] [34, 35] [15, 23, 26, 35–38]. Each D4Z4 RU encodes a copy of DUX4 [33]; however, only DUX4-fl produced from the distal-most 4q35 D4Z4 RU is stably expressed in FSHD. This is due to an array-distal polyadenylation signal (PAS) in the noncoding exon 3 of DUX4, which is only present in permissive 4q alleles (termed 4qA) [23, 35]. This PAS is required to stably express DUX4 mRNAs in somatic cells and is essential for developing both forms of FSHD. However, the DUX4 PAS is absent from about half of the 4q chromosomes (4qB alleles) in the human population. These non-permissive 4qB chromosomes lack exon 3 of the DUX4 gene and are therefore unable to generate polyadenylated DUX4 mRNA in somatic cells, supporting the requirement for stable DUX4 expression in the development of FSHD [21, 23, 35, 39, 40]. Interestingly, the highly homologous chromosome 10q26 D4Z4 arrays have the A-type exon 3 sequence distal to the array; however, the putative DUX4 PAS is a sequence variant that does not signal polyadenylation, thus explaining why D4Z4 contractions on chromosome 10 are not associated with FSHD [21, 23, 39].
With the identification of DUX4 as the key pathogenic gene in FSHD, the field is now engaged in designing DUX4-targeted therapies [23, 33, 35, 41, 42]. Therapies can also be targeted upstream of DUX4 expression, as the epigenetic disruption at the FSHD locus is a major determinant in disease presentation and progression. In addition, there are multiple genetic and epigenetic modifiers of FSHD severity, which provide additional therapeutic targets [7, 9, 12, 25, 36, 43]. However, the fundamental nature of FSHD presents several obstacles to traditional drug development and screening (e.g., DUX4 is a primate-specific gene expressed at very low levels primarily in differentiated FSHD skeletal myocytes, which are not amenable to high-throughput screening [35–37]). In addition, the high variability of genetic and epigenetic features found among FSHD patients can confound studies that are under-powered. Thus, there is a great need in the field for large numbers of well-characterized and freely available cohorts of cells, both FSHD and related healthy controls, as well as the development of readily screenable cellular models of FSHD. We and others have previously shown that primary blood cells recapitulate the epigenetic signature of the FSHD locus seen in skeletal myocytes [26, 31]. Here we perform an initial characterization of 114 lymphoblastoid cell lines (LCLs) derived from healthy and FSHD1 affected individuals from 12 multigenerational families to provide valuable new tools for the FSHD field. Previous studies using the genetically homogeneous families in the Utah region have been informative in early clinical descriptions and linkage studies for FSHD [20, 44–48]. The present study includes an updated genetic and molecular characterization of 56 LCLs from six families in the Utah cohort; thus, the availability of these LCLs, along with 58 LCLs not known to be linked to the Utah cohort (although distant linkage has not been excluded), may provide a wealth of new information with respect to the role of familial genetic background and the identification of factors that act as disease modifiers [48]. Importantly, despite being immortalized and non-myogenic, these cells represent useful models of FSHD-dependent DUX4 expression that can be therapeutically targeted and manipulated in a format suitable for high-throughput drug screening.
2. Patients and methods
2.1. Subjects and cells
All cells used in this study are Epstein-Barr virus-transformed B-LCLs generated in the laboratory of Stephen J. Jacobsen at the University of California School of Medicine, La Jolla, CA, USA and subjected to the same process for immortalization. LCLs were transferred to the NIGMS Human Genetic Cell Repository at the Coriell Institute for Medical Research (CIMR) repository and are now available by catalog using the Repository ID numbers (Table 1). Informed consent was initially obtained from >600 participants from at least 36 families prior to the initial sample collection. Consent was obtained a second time from each of the participants in this study, allowing only the cells from these 114 reconsented subjects to be transferred to and distributed by CIMR for analysis. Subjects were examined by experienced neuromuscular physicians for clinical weakness characteristic of FSHD and diagnosed as either FSHD or healthy [44]. All exams were performed and samples were collected between 1987 and 1992, prior to the discovery of the FSHD1 or FSHD2 genetic defects [8, 16, 17]; therefore, none of the subjects had undergone genetic testing for FSHD at the time of sample collection. The original clinical notes, other than the final diagnosis of FSHD-affected or unaffected, were lost in the process of salvaging and moving cell lines to CIMR. Therefore, all cell lines have been kept in cryostorage and out of circulation since their original derivation until they could be properly validated and characterized for distribution.
Table 1.
LCL characteristics
| Family | Catalog No. | Gender | Genotype |
EcoRI/ AvrII |
Clinical FSHD |
Genetic FSHD |
Epigenetic FSHD |
DUX4-fl Expression |
|---|---|---|---|---|---|---|---|---|
| 2 | GM16250 | M | 4A161/4B163 | 22kb (6RU) | Yes | Yes | Yes | 220.1 |
| 2 | GM16251 | F | 4A161/4A161 | NC | No | No | No | 0.2 |
| 2 | GM16277 | F | 4A161/4C166H | 22kb (6RU) | Yes | Yes | Yes | 5.4 |
| 2 | GM16275 | F | 4C166H/4B163 | NC | No | No | No | 0.1 |
| 2 | GM16276 | F | 4A161/4B163 | NC | No | No | No | 1.6 |
| 2 | GM16278 | M | 4A161/4C166H | 22kb (6RU) | Yes | Yes | Yes | 67.7 |
| 2 | GM16279 | M | 4A161/4A161 | 22kb (6RU) | Yes | Yes | Yes | 71.1 |
| 2 | GM16280 | F | 4C166H/4B163 | NC | No | No | No | 10.1 |
| 2 | GM16281 | F | 4A161/4B163 | NC | No | No | No | 0.4 |
| 2 | GM16282 | F | 4A161/4A161 | NC | No | No | No | 0.8 |
| 2 | GM16283 | F | 4A161/4A161 | 22kb (6RU) | Yes | Yes | Yes | 476.9 |
| 2 | GM16284 | F | 4A161/4A161 | NC | No | No | No | 0.2 |
| 2 | GM16361 | M | 4A161/4A161 | 22kb (6RU) | Yes | Yes | Yes | 158.7 |
| 2 | GM16412 | M | 4A161/4C166H | NC | No | No | No | 0.5 |
| 2 | GM16427 | M | 4A161/4A161 | 22kb (6RU) | Yes | Yes | No | 13.3 |
|
| ||||||||
| 4 | GM16285 | F | 4A161/4A161 | 22kb (6RU) | Yes | Yes | Yes | 38.3 |
| 4 | GM16286 | F | 4A161/4A-L161 | 22kb (6RU) | Yes | Yes | Yes | 78.5 |
| 4 | GM16287 | M | 4A161/4A161 | 22kb (6RU) | Yes | Yes | Yes | 19.8 |
| 4 | GM16288 | M | 4A161/4A-L161 | 22kb (6RU) | Yes | Yes | No | 39.9 |
| 4 | GM16289 | F | 4A161/4A-L161 | NC | No | No | No | 0.1 |
| 4 | GM16290b | F | 4A161/4A161 | NC | Yes | No | No | 1.6 |
| 4 | GM16291 | F | 4A161/4A161 | 22kb (6RU) | Yes | Yes | No | 416.1 |
| 4 | GM16292 | F | 4A-L161/4A-L161 | NC | No | No | No | ND |
| 4 | GM16293 | F | 4A-L161/4A-L161 | NC | No | No | No | ND |
| 4 | GM16294 | F | 4A161/4A161 | 22kb (6RU) | Yes | Yes | No | 24.7 |
| 4 | GM16295 | M | 4A161/4A161 | NC | No | No | No | 0.7 |
|
| ||||||||
| 6 | GM16253 | F | 4A161/4A-L161 | 22kb (6RU) | Yes | Yes | Yes | 69.3 |
| 6 | GM16257 | M | 4A161/4B163 | 22kb (6RU) | Yes | Yes | Yes | 10.5 |
| 6 | GM16296 | F | 4A161/4B163 | 22kb (6RU) | Yes | Yes | Yes | 76.7 |
| 6 | GM16297 | F | 4A161/4A166 | 22kb (6RU) | Yes | Yes | Yes | 1277.3 |
| 6 | GM16298 | F | 4A161/4A-L161 | 22kb (6RU) | Yes | Yes | Yes | 11.3 |
| 6 | GM16299b | F | 4A-L161/4*168 | NC | Yes | No | No | 0.0 |
| 6 | GM16300 | F | 4A161/4B163 | 22kb (6RU) | Yes | Yes | Yes | 12.4 |
| 6 | GM16301 | M | 4B163/4B163 | NC | No | No | No | 0.0 |
| 6 | GM16302 | F | 4A161/4*168 | 22kb (6RU) | Yes | Yes | Yes | 28.7 |
| 6 | GM16303 | M | 4A-L161/4B163 | NC | No | No | No | 0.0 |
| 6 | GM16304 | F | 4A161/4A166 | 22kb (6RU) | Yes | Yes | Yes | 208.3 |
| 6 | GM16305 | M | 4C166H/4*168 | NC | No | No | No | 0.0 |
| 6 | GM16306 | F | 4A161/4A-L161 | 22kb (6RU) | Yes | Yes | Yes | 24.5 |
| 6 | GM16307 | F | 4A161/4*168 | 22kb (6RU) | Yes | Yes | Yes | 1393.0 |
| 6 | GM16308a | M | 4A161/4B163 | 22kb (6RU) | No | Yes | Yes | 169.4 |
| 6 | GM16309 | F | 4A161/4*168 | 22kb (6RU) | Yes | Yes | Yes | 33.3 |
|
| ||||||||
| 7 | GM16310 | M | 4A161/4B163 | NC | No | No | No | 0.2 |
| 7 | GM16311 | F | 4A161/4B163 | 22kb (6RU) | Yes | Yes | No | 24.7 |
| 7 | GM16312 | M | 4A-L161/4*168 | NC | No | No | No | ND |
| 7 | GM16313 | M | 4A-L161/4*168 | NC | No | No | No | ND |
| 7 | GM16314 | M | 4A-L161/4A161 | 22kb (6RU) | Yes | Yes | Yes | 4.1 |
| 7 | GM16315 | F | 4*168/4*168 | NC | No | No | No | 0.1 |
| 7 | GM16316 | M | 4A-L161/4A161 | NC | No | No | No | 0.2 |
| 7 | GM16317 | F | 4A-L161/4A161 | 22kb (6RU) | Yes | Yes | Yes | 240.7 |
| 7 | GM16318 | F | 4A161/4*168 | 22kb (6RU) | Yes | Yes | Yes | 208.9 |
| 7 | GM16319 | F | 4A-L161/4*168 | NC | No | No | No | 0.1 |
|
| ||||||||
| 11 | GM16320 | F | 4C166H/4*162 | NC | No | No | No | 0.1 |
| 11 | GM16321 | F | 4A161/4C166H | 22kb (6RU) | Yes | Yes | No | 111.5 |
| 11 | GM16322b | M | 4A161/4C166H | NC | Yes | No | No | 0.1 |
| 11 | GM16324b | F | 4A-L161/4C166H | NC | Yes | No | No | 0.4 |
| 11 | GM16325 | F | 4A-L161/4A166 | NC | No | No | No | 0.0 |
| 11 | GM16413 | M | 4A161/4A-L161 | 22kb (6RU) | Yes | Yes | Yes | 46.7 |
| 11 | GM16414 | F | 4A161/4C166H | 22kb (6RU) | Yes | Yes | Yes | 2928.5 |
| 11 | GM16415 | M | 4A-L161/4C166H | NC | No | No | No | 0.1 |
| 11 | GM16416a | M | 4A161/4*162 | 22kb (6RU) | No | Yes | Yes | 72.8 |
| 11 | GM16418 | F | 4A-L161/4C166H | NC | No | No | No | 0.1 |
| 11 | GM16428 | M | 4A-L161/4A161 | NC | No | No | No | 0.2 |
| 11 | GM16429a | M | 4A-L161/4A161 | 22kb (6RU) | No | Yes | Yes | 199.1 |
| 11 | GM16430a | M | 4A161/4*162 | 22kb (6RU) | No | Yes | Yes | 19.5 |
| 11 | GM16417 | M | 4A161/4C166H | 22kb (6RU) | Yes | Yes | No | 28.8 |
|
| ||||||||
| 12 | GM16326 | M | 4A161/4B165 | 22kb (6RU) | Yes | Yes | No | 7.7 |
| 12 | GM16327 | F | 4A161/4A161 | 22kb (6RU) | Yes | Yes | Yes | 312.3 |
| 12 | GM16328 | M | 4A161/4B165 | NC | No | No | No | 0.1 |
| 12 | GM16419 | M | 4A161del/4*165 | NC | No | No | ND | 0.1 |
| 12 | GM16420 | M | 4A161/4A161 | 22kb (6RU) | Yes | Yes | Yes | 26.7 |
| 12 | GM16421 | F | 4A161/4A161 | NC | No | No | No | 0.2 |
| 12 | GM16422 | M | 4A161/4A161 | 22kb (6RU) | Yes | Yes | No | 382.9 |
|
| ||||||||
| 15 | GM16329 | M | 4A161/4A166 | 22kb (6RU) | Yes | Yes | No | 203.1 |
| 15 | GM16130 | F | 4A161/4A161 | 22kb (6RU) | Yes | Yes | Yes | 147.2 |
| 15 | GM16331 | M | 4A161/4C166H | NC | No | No | No | 0.2 |
| 15 | GM16332 | F | 4C166H/4B163 | NC | No | No | No | 0.2 |
| 15 | GM16333 | F | 4A161/4B163 | 22kb (6RU) | Yes | Yes | Yes | 65.5 |
|
| ||||||||
| 18 | GM16334 | F | 4A161/4A166 | 18kb (5RU) | Yes | Yes | Yes | 1668.3 |
| 18 | GM16335 | M | 4A161/4A-L161 | NC | No | No | No | 0.3 |
| 18 | GM16336 | F | 4A161/4A-L161 | 18kb (5RU) | Yes | Yes | Yes | 1468.1 |
| 18 | GM16337 | F | 4A161/4A-L161 | 18kb (5RU) | Yes | Yes | Yes | 535.3 |
|
| ||||||||
| 25 | GM16338 | F | 4A161/4A161 | 23kb (6RU) | Yes | Yes | No | 56.9 |
| 25 | GM16339a | F | 4A-L161/4A161 | 23kb (6RU) | No | Yes | Yes | 163.1 |
| 25 | GM16340 | M | 4A161/4B163 | NC | No | No | No | 0.1 |
| 25 | GM16341 | M | 4A-L161/4B163 | NC | No | No | No | 0.2 |
|
| ||||||||
| 32 | GM16100 | F | 4C166H/4B163 | NC | No | No | No | 3.0 |
| 32 | GM16342 | M | 4A161/4A166 | 22kb (6RU) | Yes | Yes | No | 39.5 |
| 32 | GM16258 | M | 4A161/4B163 | 22kb (6RU) | Yes | Yes | No | 4.9 |
| 32 | GM16343 | M | 4A161/4C166H | 22kb (6RU) | Yes | Yes | Yes | 613.9 |
| 32 | GM16431b | M | 4C166H/4A166 | NC | Yes | No | No | 12.5 |
|
| ||||||||
| 33 | GM16101 | F | 4A-L161/4*162 | NC | No | No | No | 0.3 |
| 33 | GM16126 | F | 4A-L161/4A166 | 33kb (9RU) | Yes | Yes | Yes | 19.7 |
| 33 | GM16131 | M | 4A161/4A161 | 22kb (6RU) | Yes | Yes | Yes | 59.2 |
| 33 | GM16154 | M | 4A161/4*162 | 22kb (6RU) | Yes | Yes | No | 1.5 |
| 33 | GM16254a | F | 4A-L161/4A166 | 33kb (9RU) | No | Yes | No | 318.3 |
| 33 | GM16259 | M | 4A-L161/4A161 | 22kb (6RU) | Yes | Yes | Yes | 30.3 |
| 33 | GM16354 | F | 4A-L161/4A166 | 33kb (9RU) | Yes | Yes | Yes | 248.5 |
| 33 | GM16344 | F | 4A161/4*162 | NC | No | No | No | 0.3 |
| 33 | GM16345 | F | 4A-L161/4A161 | 22kb (6RU) | Yes | Yes | No | 85.4 |
| 33 | GM16355 | F | 4A-L161/4A166 | NC | No | No | No | 0.2 |
| 33 | GM16425a | F | 4A166/4*168 | 33kb (9RU) | No | Yes | No | 1.0 |
| 33 | GM16426 | M | 4A-L161/4A161 | NC | No | No | No | 0.2 |
| 33 | GM16357 | M | 4A166/4*168 | NC | No | No | No | 0.2 |
| 33 | GM16423 | F | 4A161/4*162 | 22kb (6RU) | Yes | Yes | No | 41.5 |
| 33 | GM16424a | F | 4A166/4A166 | 33kb (9RU) | No | Yes | No | 8.4 |
| 33 | GM16353 | F | 4A-L161/4*162 | NC | No | No | No | 0.3 |
| 33 | GM16356 | M | 4A-L161/4A166 | NC | No | No | No | 0.1 |
|
| ||||||||
| 36 | GM16347 | M | 4B163/4B168 | NC | No | No | No | 0.0 |
| 36 | GM16348 | F | 4A-L161/4B168 | 16kb (4RU) | Yes | Yes | Yes | 1.1 |
| 36 | GM16349 | F | 4B163/4B168 | NC | No | No | No | 0.0 |
| 36 | GM16350 | F | 4A-L161/4B163 | NC | No | No | No | 0.0 |
| 36 | GM16351 | M | 4A-L161/4B168 | 16kb (4RU) | Yes | Yes | No | 1.0 |
| 36 | GM16352 | F | 4A166/4B163 | NC | No | No | No | 0.3 |
|
| ||||||||
| NA | GM16330 | ND | 4A161/4B163 | NC | ND | No | No | 0.2 |
EcoRI/AvrII (equivalent to EcoRI/BlnI) below 37kb considered contracted allele and genetic FSHD1; DNA methylation of the distal D4Z4 gene body <35% considered epigenetic FSHD; Clinical notes from physician listing FSHD considered clinical FSHD (if FSHD was not noted, considered healthy). DUX4-fl mRNA expression shown as fold expression compared to the lowest expressing FSHD1 LCL (GM16154), which was set to 1X. NC (Non-Contracted 4qA), ND (Not Determined), NA (information is Not Available),
cannot determine 4A or 4B for this haplotype.
indicates asymptomatic FSHD1,
indicates clinical diagnosis of FSHD is not supported by genetic or epigenetic testing.
2.2 Cell culture and drug treatment
LCL cultures were grown in suspension at 37°C with 5% CO2 in RPMI 1640 supplemented with 15% fetal bovine serum, glutamine, and antimycotics. For drug treatments, 5-aza-2’-deoxycytidine (ADC) (Sigma-Aldrich, #A3656) was added daily to the LCL cultures at 5 µM final concentration for a total of 3 days. Trichostatin A (TSA) (Sigma-Aldrich, #T1952) was added to the cultures at 200 nM final concentration for the last 24 h prior to sampling.
2.3. D4Z4 deletion analysis
Genomic Southern blotting to identify subjects possessing 4q35 deletions was performed as described [49], and following personal communication with Dr. Yukiko Hayashi. Briefly, 7.5 µg of LCL genomic DNA was digested overnight with either EcoRI or both EcoRI and AvrII (New England Bioloabs), electrophoresed through a 0.3% agarose TAE gel for 36 h at 12 mAmps, denatured, and transferred to a nylon membrane. Blots were probed with radiolabeled p13E-11 fragment [16] and assessed for EcoRI restriction fragments that decreased in size by 3.2 kb when combined with AvrII digestions, indicating the particular fragment was from chromosome 4q [50, 51]. AvrII is an isoschizomer of BlnI, the enzyme traditionally used for FSHD1 diagnosis to distinguish 4q35 D4Z4 arrays from the BlnI-sensitive chromosome 10q26 D4Z4 arrays.
2.4. DNA methylation analysis
The DNA methylation status of the 4q35 D4Z4 array was determined by bisulfite sequencing (BSS) analysis. BSS analysis of the 4qA distal D4Z4 repeat unit was performed on genomic DNAs isolated from LCLs, as described [12, 31]. BSS analysis for the distal D4Z4 repeat unit containing a B-type subtelomeric region was performed using the MetB-For and MetB-Rev PCR primers [30]. PCR products were cloned into the pGEM-T Easy vector (Promega) for sequencing and analyzed using web-based analysis software BISMA (http://biochem.jacobs-university.de/BDPC/BISMA/) [52] with the default parameters.
2.5 Chromosome 4q trace by Simple Tandem Repeat analysis (4qSTR analysis)
In order to confirm the position of each cell line in its family pedigree and to trace inheritance of 4q D4Z4 repeats, PCR-based 4qSTR analysis was performed using three STR markers: D4S2930 and D4S1523 that have high heterozygosity and most proximity to D4Z4 (900kb and 500kb, respectively), and D4F104S1/SSLP (4q/10q STR marker) [53] (https://www.urmc.rochester.edu/fields-center.aspx). For Family36, D4S1652 was also analyzed to further identify parental origin of 4q. Gender of each sample was also confirmed by STR analysis of amelogenin [54].
2.6 Haplotyping and polyadenylation signal (PAS) analysis
Standard genomic PCR was performed on non-converted LCL DNA and about 30% of each PCR reaction was run on 2% agarose gels to identify the 4qA, 4qA-L, and 4qB chromosomes, as described [23]. The rest of the 4A and 4AL PCR fragments were purified with a PCR purification kit (Qiagen) and used for direct DNA sequencing to confirm PAS and D4F104S1/SSLP haplotypes. Specific 4q and 10q haplotypes were identified and assigned as described [22, 40].
2.7 Gene expression analysis
Total RNA was prepared from each cell line using TRIzol (ThermoFisher), as per manufacturer’s instructions, followed by clean-up using the RNeasy mini kit (Qiagen). First strand cDNA was generated from 2 µg total RNA using SuperScript III reverse transcriptase and Oligo (dT)16 primer at 55°C for 1 hr. DUX4 expression was analyzed using nested PCR. For the first reaction, 200 ng cDNA was amplified with primers DUX4 For and DUX4 Rev2 in 1X GC Buffer supplied with Phusion Hot Start II DNA Polymerase (Thermo Fisher) and cycled 10 times at 98°C for 15 sec, 64°C for 20 sec, and 72°C for 15 sec. For the quantitative reaction, 5% of the first reaction was amplified by qPCR using 1X IQ SYBR mix (BioRad) and primers DUX4 For and DUX4 Rev with cycling parameters 95°C for 10 sec, 64°C for 15 sec, and 72°C for 20 sec, with readings taken at 86°C for 10 sec. Expression of DUX4 target genes was analyzed by qPCR using 5 to 10 ng cDNA, as described [55]. Expression of all genes was normalized to 18S rRNA, analyzed by qPCR using 5 ng of cDNA. All oligonucleotide primer sequences for DUX4, downstream targets, and 18S rRNA are previously reported [37]. Some DUX4-fl qPCR products were purified with a PCR purification kit (Qiagen) and used for direct DNA sequencing to confirm haplotype.
3. Results
3.1 Genetic characterization of FSHD1 in cohorts of LCLs
We have characterized and validated 114 LCLs originally obtained from 12 multigenerational FSHD-affected families and validated them as genetically FSHD1 or healthy (Figs. 2 – 7, A. 1). Six of the families (Family 2, 7, 11, 12, 25, and 33) originate from a historically informative region of Utah [20, 44, 48], and six families have no known relationship to the Utah cohort. Family pedigrees and diagnosis with respect to the presence of clinical FSHD were available for each cell line. However, samples were collected and LCLs were generated more than 25 years ago by a third party; therefore, we first evaluated the position of each LCL on the accompanying pedigrees by identifying the gender and parental origin of 4q chromosomes using short tandem repeat (STR) markers (D4S2930, D4S1523, and D4F104S1). Overall, the identities of the cell lines with respect to their placement within families and throughout the pedigrees was >99% accurate and only one of the 114 cell lines, GM16330, could not be placed in a pedigree. We conclude that despite the minimal records available, these 113 cell lines are accurately represented by the accompanying pedigrees. We have no information on the accuracy of the clinical diagnosis for additional subjects whose cell lines were not available (NA on the pedigrees), nor do we know if the pedigrees are complete representations of all members of each generation in a particular family.
Fig. 2.

Pedigree for Family 2 from southern Utah. Families 7 and 11 (Fig. 3) are related as indicated. Those subjects determined by Southern blotting to be FSHD1 are indicated in black, with the contracted chromosome in red. The clinical diagnosis of FSHD is labeled. Relations were confirmed by 4qSTR analysis and 4q subtelomere haplotyping (4A, 4L, 4B, 4C) as indicated for each chromosome. When samples were not available (NA), the predicted 4qSTR based on the offspring is within an open oval. DNA methylation levels for each chromosome are indicated as % methylation above the number of D4Z4 RUs, when known.
Fig. 7.
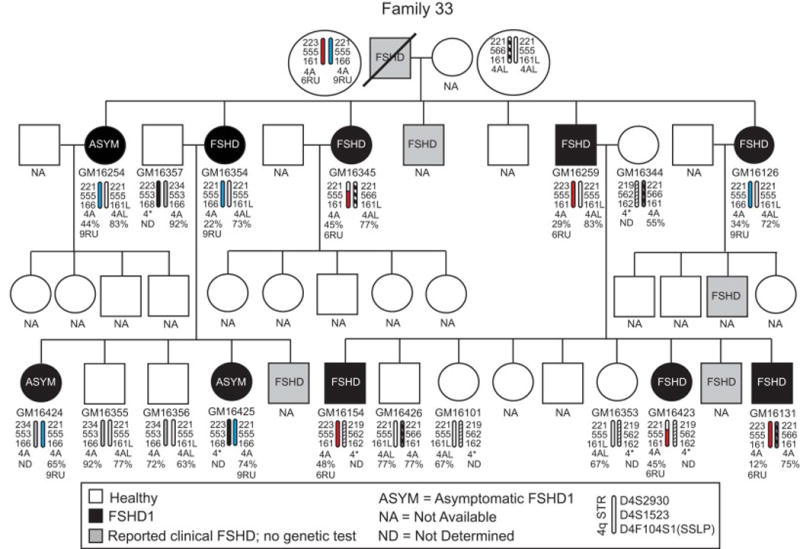
Pedigree for Family 33 from northern Utah. See legend for Fig 3. This family contains two different FSHD1 contracted chromosomes, one with 6RUs (red) and one with 9RUs (blue).
All subjects were evaluated for signs of FSHD [44] and clinical samples from which LCLs were derived were collected between 1987 and 1992, prior to the availability of genetic or epigenetic testing for FSHD. Thus, at the time of collection, none of the subjects was genetically tested to confirm a diagnosis of FSHD1 or FSHD2. Therefore, we analyzed each LCL from these family cohorts for FSHD-permissive 4q haplotypes, the presence of a 4qA PAS, and the presence of FSHD1-sized 4q35 D4Z4 arrays (Tables 1 and A. 1) [16, 17, 23, 30, 40, 49]. Linear Southern blot analysis of EcoRI and EcoRI/AvrII digested genomic DNA performed with the p13E-11 probe [56] identified 66 LCLs that contain contracted 4q35 D4Z4 arrays with EcoRI/AvrII fragments below the 37kb cutoff for FSHD1 (10 RUs) and ranging in size from 16 kb (4 RU) to 33 kb (9 RU), as shown in Tables 1 and A.1. Chromosome 4q haplotyping determined that 63 of these LCLs with contracted alleles have a predicted FSHD-permissive 4qA subtelomere and the exon 3 PAS. Overall, the genetic characterizations indicated that 63 cell lines met the standard genetic criteria for FSHD1. For the remaining 3 cell lines (GM16349, GM16350, and GM16352) with FSHD1-sized D4Z4 contractions (33 kb), all from Family 36, the contracted arrays were found on non-permissive 4qB chromosomes, with no PAS detected, and were therefore genetically characterized as healthy controls [39]. Interestingly, none of these three subjects was clinically classified as FSHD; however, two other members of Family 36, GM16348 and GM16351, who were clinically diagnosed with FSHD, were confirmed to have FSHD1-sized D4Z4 contractions (16kb) on FSHD-permissive 4qA chromosomes. Thus, this one family illustrates the requirement for both a contracted 4q allele and the permissive 4qA exon 3 PAS for developing FSHD. The remaining 48 LCLs did not meet these FSHD1 genetic criteria and were therefore classified as healthy controls, bringing the total number of control LCLs to 51.
FSHD1 displays an autosomal dominant mode of inheritance with incomplete and variable penetrance [13, 20]. Including the FSHD1 genetic diagnosis for each subject on the pedigrees showing multiple affected generations in all families (e.g., 6 affected generations in Family 18, Fig. A.1) clearly reveals this autosomal dominant mode of inheritance (Figs. 2–7, A.1). Interestingly, the pedigrees accompanying the LCLs provided a limited clinical diagnosis for each subject with respect to FSHD presentation (either healthy or affected). Although the specific criteria for each individual clinical diagnosis in this study are not known, previous published work by the coordinating physician, Dr. S. J. Jacobsen, described the FSHD diagnostic criteria his group used at the time [44]. We compared the recorded clinical diagnosis of FSHD with our genetic diagnosis of FSHD1 and found a strong correlation (55/63). However, 8/63 LCLs from four families (Family 6: GM16308; Family 11: GM16416, GM16429, GM16430; Family 25: GM16339; Family 33: GM16254, GM16424, GM16425) that fit the genetic FSHD1 criteria were derived from subjects that did not show any clinical signs of FSHD at the time of their neuromuscular examination and blood donation, suggesting that these subjects are FSHD1 carriers. In addition, the FSHD1 EcoRI/AvrII deletion sizes (22–33kb or 6–9RU) for each of these subjects is within the range reported for asymptomatic FSHD1 [11, 12, 14, 36]; therefore, these 8 subjects are considered in this study to be asymptomatic. It is possible that these subjects were examined prior to developing clinical FSHD, which is typically diagnosed in males by age 20 and females by age 30. Fortunately, the identifying information provided with these samples allowed us to determine the age, within ~4 years, at which each designated asymptomatic subject was analyzed. Six of the subjects were >24 years old, with the oldest being >65 years, and two of the subjects from Family 11 (GM16416 and GM16430), both brothers, were <20 years old. The designation for these two youngest asymptomatic subjects raises the concern that they had yet to manifest noticeable symptoms. However, these brothers have two FSHD-affected siblings who were also <20 years old, indicating that FSHD can present at a young age in this family. Interestingly, among these four genetically FSHD1 siblings, the two affected subjects inherited the same nonpathogenic chromosome from their mother while the two asymptomatic subjects inherited the other nonpathogenic maternal chromosome (Figure 3), suggesting the nonpathogenic chromosome 4q allele may be contributing to disease presentation. Overall, the 8 asymptomatic LCLs in this study may prove to be informative in the future identification of disease modifiers, although caution should always be used in the interpretation of data from asymptomatic lines, particularly from younger subjects.
Fig. 3.
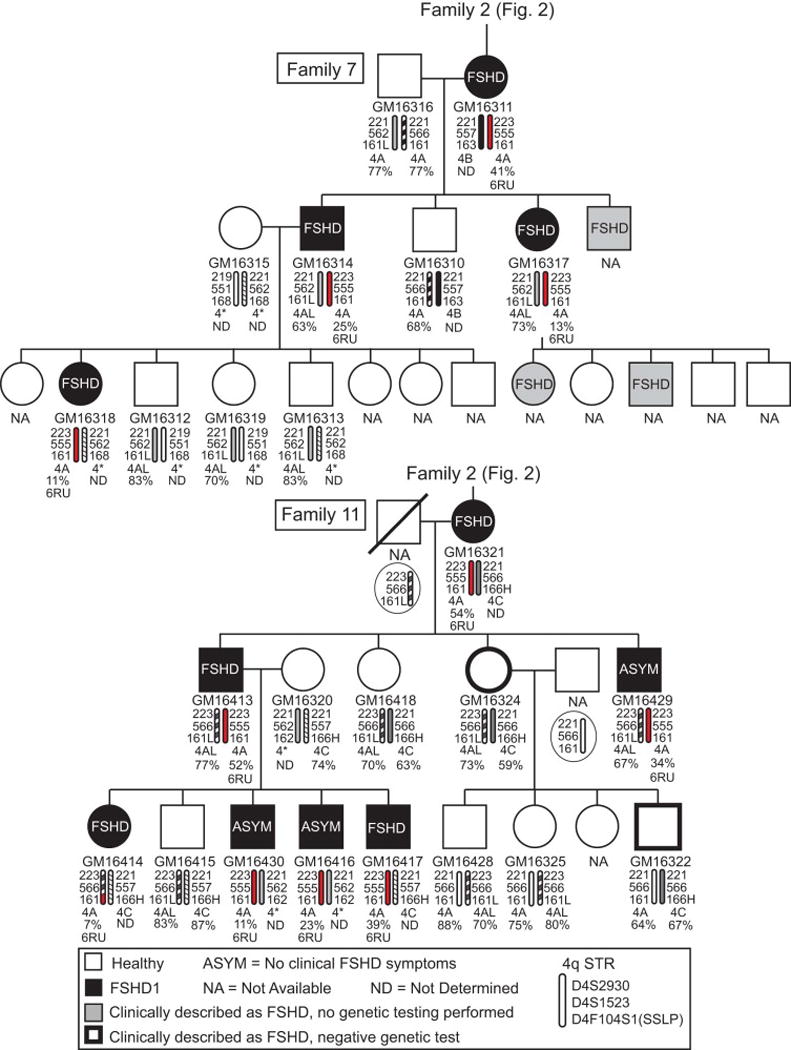
Pedigrees for Families 7 and 11, direct relatives of Family 2 where indicated, from southern Utah. See legend for Fig 2. Those subjects determined to be genetically permissive for FSHD1 but reported as clinically healthy at the time of evaluation are designated as asymptomatic (ASYM).
3.2 Correlation between DNA methylation levels and 4q35 D4Z4 repeat length in FSHD1 LCLs
FSHD is ultimately caused by the disruption of epigenetic regulation governing the chromosome 4q35 D4Z4 macrosatellite array, combined with an FSHD-permissive 4q35 PAS, resulting in the pathogenic expression of the DUX4 gene [15, 26, 35, 57]. In FSHD1, the epigenetic disruption is caused by the loss of large regions of regulatory heterochromatin. However, it is well established that certain FSHD1-sized deletions (6–10 D4Z4 RUs) can exist in different epigenetic states that correlate with clinical presentation [11, 12, 32]. In addition, genetic mutations in epigenetic modifiers of the D4Z4 array, typical of FSHD2, can also affect the epigenetic status and clinical manifestation of disease in FSHD1 subjects [9, 25]. Although FSHD is a myopathy, the epigenetic status of the disease locus is similar between myogenic cells and peripheral blood leukocytes isolated from the same individual [26, 31]; it is not known if immortalized LCLs similarly maintain the epigenetic status of this locus in the cells from which they were derived. Studies assessing global DNA methylation levels in LCLs compared with primary blood cells have reported both similarities and differences, thus necessitating caution in the interpretation of epigenetic profiles from LCLs [58–60]. Therefore, in addition to screening for FSHD1-sized deletions, we characterized the epigenetic status of this cohort of cells by assaying the DNA methylation levels of the region using FSHD diagnostic bisulfite sequencing (BSS) [12, 31] (Figs. A.2 and A.3).
Genomic DNA isolated from primary cells, saliva, or peripheral blood leukocytes of FSHD1 subjects displays <35% DNA methylation on the distal D4Z4 RU of the contracted 4qA allele when assayed using our chromosome 4qA-specific BSS assay [12, 31]. BSS analysis of FSHD1 LCLs revealed that 42/63 had levels of DNA hypomethylation considered diagnostic for FSHD1 (<35% methylation) (Table A. 1). All eleven families produced at least one LCL with FSHD1 levels of DNA methylation, while four families (Family 6, 12/12; Family 18, 3/3; Family 2, 6/7; Family 7, 3/4) showed a consistently strong correlation between having an FSHD1 deletion and DNA hypomethylation. Interestingly, the remaining 21 LCLs from FSHD1 subjects across seven families showed a lack of concordance between DNA methylation and disease manifestation among cells derived from individuals containing the same deletion. For example, in Family 25, GM16338 was derived from a subject that exhibited clinical signs of FSHD, yet had >57% methylation, while GM16339 was derived from their daughter who had yet to show any symptoms of FSHD despite ~5% DNA methylation in her LCLs. Since the D4Z4 deletion is the same within each family, this lack of concordance between DNA methylation levels and disease manifestation suggests the presence of a mutation in a modifier gene or differences in other underlying genetic conditions that could account for this, such as overall D4Z4 content.
In contrast to the FSHD1 LCLs, none of the cell lines derived from healthy subjects displayed FSHD1 levels of DNA hypomethylation (0/51; n=44 hypermethylated >35% and n=7 no 4qA BSS product). However, 3 LCLs from healthy members of Family 36 (GM16349, GM16350, GM16352) contained contracted 4q35 D4Z4 arrays with non-permissive 4qB alleles (Fig. 6, Table A.1). Since all six members of this family had at least 1 4qB allele, this family provided an opportunity to investigate the epigenetics of 4qB D4Z4 arrays. We performed 4qB-specific BSS on the 6 LCLs from Family 36 (Fig. A.2) and found that the 3 healthy LCLs with contracted 4qB alleles displayed FSHD1-like levels of DNA hypomethylation (GM16349, Q1=7.1%; GM16350, Q1=0.0%; GM16352, Q1=0.0%) as compared with the non-contracted 4qB alleles with healthy levels of DNA methylation. This data supports the hypothesis that the physical removal of heterochromatin at D4Z4 arrays causes the epigenetic disruption in FSHD1 [15, 26, 28, 57]. In addition, this data provides further support for the unifying model for FSHD [23], again confirming that a 4q35 D4Z4 contraction alone is not causal for FSHD, but requires a permissive 4qA subtelomere in cis to cause disease [39].
Fig. 6.

Pedigrees for unrelated Families 32 and 36. See legend for Fig 2.
We next sought to investigate the potential for these FSHD1 cell lines to possess FSHD2-type modifier mutations. Since these mutations result in hypomethylation of both alleles at 4q and 10q [9, 25], we analyzed all FSHD1 LCLs using our D4Z4 5’ BSS assay that analyzes the methylation status of all 4q and 10q D4Z4 RUs [31]. For each LCL, we found large differences in DNA methylation among all 4q and 10q D4Z4 RUs (Table A.1), strongly suggesting that these subjects do not have known FSHD2-type modifier mutations, although we cannot rule out that the immortalization procedure and/or culturing conditions and selection may play a role in these epigenetic differences. While a comprehensive search for modifier mutations in SMCHD1, DNMT3B, and elsewhere in the genome is beyond the scope of this study, this cohort of cell lines with appropriate family controls will be useful for addressing these types of questions. Overall, we conclude that many, but not all, LCLs exhibit the appropriate healthy or FSHD1 DNA methylation profiles, and that this large cohort displays genetic and epigenetic characteristics that may be useful for investigating certain lines of inquiry pertinent to FSHD.
3.3 FSHD1 LCLs display robust and variable expression of DUX4-fl, which correlates with expression of DUX4-FL target genes
To further assess the utility of these cells, we performed targeted gene expression analysis. The loss of transcriptional repression at the disease locus in FSHD results in the aberrant stable expression of the pathogenic DUX4-fl mRNA in skeletal muscle, due in part to the presence of two DUX4 myogenic enhancers [37]. Although LCLs are not myogenic, DUX4-fl expression has also been reported in non-myogenic cells [35]. Therefore, we analyzed DUX4-fl mRNA expression by qRT-PCR in all cell lines (Fig. 9). We readily detected robust levels of DUX4-fl mRNA in all 61 of the 4A161 contracted FSHD1 LCLs; however, the relative expression levels were highly variable within families and across the entire cohort, displaying a nearly 3000-fold range among all FSHD1 samples (Table 1). In contrast, DUX4-fl expression, when reliably detected at all, was generally very low in the healthy LCLs (Table 1), only five of which displayed DUX4-fl levels above the lowest FSHD1 level. Three of these lines (GM16276, GM16290, and GM16100) expressed 3-fold or less DUX4-fl expression compared to the lowest-expressing FSHD1 line, which is lower expression than 59 of the 61 FSHD1 LCLs, while DUX4-fl levels in GM16280 (10.1-fold) and GM16431 (12.5-fold) were still in the bottom 20% compared to the FSHD1 cells. Overall, FSHD1 LCLs expressed on average >300-fold more DUX4-fl mRNA than the healthy control LCLs. Variability among FSHD1 cell lines and slight overlap in expression levels between a few healthy LCLs and the lowest DUX4-fl expressing FSHD1 LCLs likely reflect individual differences between donor subjects and are consistent with the DUX4-fl mRNA expression profiles previously reported for family cohorts of FSHD1-derived and healthy myocytes [12, 36]. We conclude that this cohort of cell lines maintains the disease specificity and individual variability of DUX4-fl mRNA expression profiles typical of FSHD families.
Fig. 9.
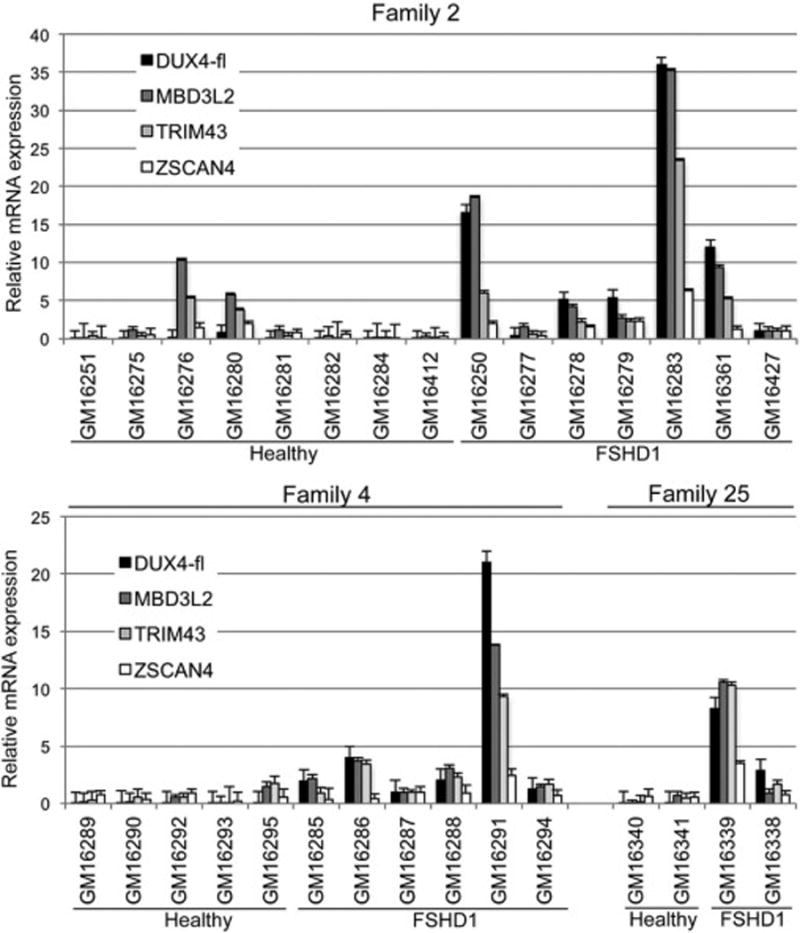
Expression of DUX4-fl in FSHD1 lymphoblastoid cell lines correlates with increased expression of DUX4-FL target genes. mRNA levels of DUX4-fl and three of its downstream target genes, MBD3L2, TRIM43, and ZSCAN4, were determined by qRT-PCR and normalized to levels of 18S RNA.
In myocytes, DNA methylation at the 4q35 D4Z4 arrays typically correlates with DUX4-fl expression levels; healthy myocytes display D4Z4 hypermethylation and express very low or undetectable levels of DUX4-fl mRNA, while FSHD1 myocytes are hypomethylated on the contracted allele and typically express significantly higher levels of DUX4-fl mRNA [12, 31, 36]. As expected, the healthy LCLs in this cohort showed a similar correlation, with DNA hypermethylation at the 4q35 D4Z4 array and correspondingly low or undetectable levels of DUX4-fl (Table A.1). Intriguingly, the five DUX4-fl expressing healthy LCLs, described above, all have healthy levels of DNA methylation on both 4q D4Z4 arrays (>35% methylated). It is interesting that these five lines represent only three families (GM16276 and GM16280 in Family 2; GM16431 and GM16100 in Family 32; GM16290 in Family 4), suggesting that these families may have interesting modifier mutations that affect DUX4-fl expression levels independent of DNA methylation levels.
Surprisingly, levels of DUX4-fl mRNA in FSHD1 LCLs did not necessarily correlate with levels of DNA methylation at the contracted 4qA allele (Table A.1). The top five DUX4-fl expressing lines exhibited extreme DNA hypomethylation as expected (GM16414, 6.0%; GM16334, 1.8%; GM16336, 5.54%; GM16307, 0.0%; GM16267, 7.1%); however, there are several striking inconsistencies throughout the cohort. For example, in Family 6, GM16297 and GM16298 both exhibit 7.1% DNA methylation on the contracted allele, yet there is a >100-fold difference in DUX4-fl expression between the two cell lines, again suggesting the presence of genetic modifiers of DUX4-fl expression in this family. In contrast, Family 18 behaves exactly as expected, with the three FSHD1 lines (18kb D4Z4 contraction) exhibiting DNA hypomethylation of the contracted allele coupled with very high DUX4-fl expression, while the healthy cell line was hypermethylated with nearly undetectable levels of DUX4-fl mRNA (Tables 1 and A.1, Figs. 8, A.3 and A.4.). This data suggests that there are no FSHD modifying mutations in this family. Overall, the FSHD1 LCLs showed more variability in DNA methylation at the 4q35 D4Z4 array, often extending above the diagnostic threshold of 35% methylation, than we have seen in primary cells (myocytes, white blood cells, or saliva). Since we do not have a source of primary cells from the same individuals to compare with, it is not clear if these methylation levels are true representations of each donor individual or are a result of immortalization, cell culturing, modifier mutations, or some combination of these factors. Regardless, it is important for the design of future experiments to know the relationship between DNA methylation and DUX4-fl expression as we have documented it for each of these cell lines (Tables 1 and A.1 and Fig. A.4).
Fig. 8.
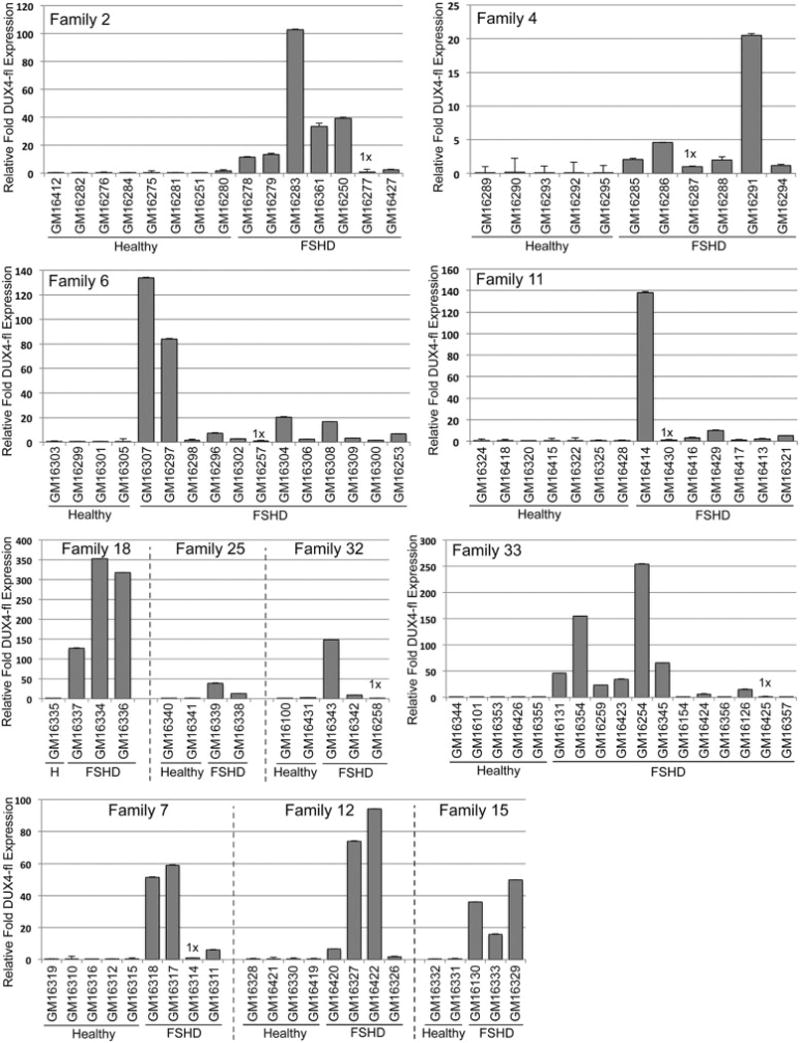
FSHD1 lymphoblastoid cell lines express variable levels of DUX4-fl. DUX4-fl mRNA levels were determined by qRT-PCR and normalized to levels of 18S RNA. The lowest expressing FSHD sample in each family or group was set to 1 and relative expression levels are shown in each graph. See Table 1 for a relative comparison of DUX4-fl expression across all LCLs.
The DUX4-FL protein is a transcription factor and its aberrant expression in FSHD leads to aberrant expression of its downstream target genes, with consequent pathology [55, 61–67]. To determine if DUX4-mediated aberrant gene regulation occurs in our cohorts of FSHD1 LCLs, we analyzed the expression levels of three robustly upregulated DUX4-FL target genes, ZSCAN4, TRIM43, and MBD3L2 [55], in 30 cell lines from Families 2, 4, and 25 (Fig. 9). Similar to DUX4-fl expression, the expression levels of these target genes were highly variable between the 15 FSHD1 cell lines analyzed. However, for each cell line, the relative expression level of each target gene precisely correlated with the relative level of DUX4-fl, supporting the likelihood that DUX4-FL induces expression of its target genes in these cells. Similarly, in the healthy cell lines, the levels of these target genes correlated with the levels of DUX4-fl and were very low in 13/15 lines. Only two of the healthy cell lines, GM16276 and GM16295, displayed appreciable expression of the target genes without detectable DUX4-fl expression. We conclude that, as with FSHD myocytes, these FSHD LCLs aberrantly express DUX4-FL protein, which functions as a transcriptional activator of its direct target genes.
3.4 Treatment with inhibitors of epigenetic repression induces DUX4-fl expression in FSHD1 LCLs
In myogenic cultures, treatment with the DNA methyltransferase inhibitor 5-aza-2’-deoxycytidine (ADC) and the histone deacetylase inhibitor Trichostatin A (TSA) induces DUX4-fl expression in FSHD cells, but not in healthy controls. To determine if these LCLs respond similarly to epigenetic drug treatment, we tested 4 FSHD1 and 2 healthy LCLs from Family 33 (Fig. 10). Combined treatment with ADC and TSA resulted in very strong induction (up to >3000 fold) of DUX4-fl expression in all four FSHD1 LCLs, while failing to induce DUX4-fl to appreciable levels in the two healthy lines. This responsiveness to ADC and TSA is consistent with what is found in FSHD skeletal myocytes; however, the level of induction is significantly higher in the LCLs.
Fig. 10.

DUX4-fl mRNA is induced in FSHD1 LCLs, but not in healthy controls, by treatment with epigenetic drugs. LCLs from Family 33 were treated (+) or not treated (NT) with 5-aza-2’-deoxycytidine (ADC) and Trichostatin A (TSA). DUX4-fl mRNA levels were determined by qRT-PCR and normalized to levels of 18S RNA. The lowest expressing NT FSHD1 sample (GM16126) is set to 1.
Interestingly, as with FSHD myocytes [12], cells from asymptomatic subjects (GM16254 and GM16425) are readily inducible and appear epigenetically poised to express DUX4-fl. Since ADC can induce MyoD expression in non-myogenic cell types [68, 69] and DUX4-fl is regulated in part by myogenic factors, we assayed levels of MyoD and Myogenin in these LCLs; however, these genes were not induced by the drug treatment (data not shown). We conclude that the epigenetic regulation of DUX4-fl expression and repression is maintained in the LCLs.
3.5 Family 33
The analysis of Family 33 (Fig. 7, Table 1) revealed several unusual characteristics that present interesting opportunities for further investigation. The FSHD1 deletion analysis indicated that this family has two different FSHD1 chromosomes, 6RUs and 9RUs, originating from one parent. Cell lines were available for 5 of the 7 children and, as expected, all 5 children contained one of the FSHD1 chromosomes with 4/5 being clinically diagnosed with FSHD. Overall, the complement of Family 33 cell lines contains 5 with 6RUs (all from affected subjects), 5 with 9RUs (2 from affected and 3 from asymptomatic subjects), and 7 healthy (2 from unaffected spouses). Thus, both 6RU and 9RU are independently associated with clinically described FSHD; however, only the 9RU chromosome was found in cell lines from asymptomatic subjects, consistent with the reported asymptomatic range of D4Z4 RUs [11, 12]. In addition, the three asymptomatic subjects were all >35 years of age at the time of their clinical evaluation, with one subject being >65 years old, supporting the classification of these subjects as asymptomatic. The 9RU chromosome in this family is particularly interesting because it has the 4qA166 haplotype, which is typically described in the literature as nonpermissive despite having an exon 3 PAS. We confirmed the presence of the PAS and, importantly, the expression of 4A166-type polyadenylated DUX4-fl mRNA in all 9RU cell lines by sequencing the RT-PCR product (Fig A.5) [35]. Interestingly, two of the 4A166 9RU LCLs (GM16254 and GM16354) expressed very high levels of DUX4-fl mRNA while another 4A166 9RU LCL (GM16425) expressed the lowest levels of DUX4-fl mRNA among all 63 FSHD1 lines in the cohort. Overall, these cell lines from first and second degree relatives of Family 33 present the opportunity to compare cell lines from FSHD1 affected, asymptomatic, and healthy relatives, the differences between 6 and 9RUs in first degree relatives, and to analyze FSHD1 affected and asymptomatic cells with the 4A166 haplotype.
4. Discussion
The goal of this study was to perform an initial characterization of 114 LCLs long held out of circulation due to a lack of characterization, that would allow these valuable resources to be made freely available to the FSHD community through the NIGMS Human Genetic Cell Repository at the CIMR repository, where they have been held in cryostorage. This study does not represent a complete characterization of the cell lines; for example, determining the sizes of both 4q and 10q D4Z4 arrays for each cell line or the genetic status of SMCHD1, mutations in which can be causal for FSHD2 and modify severity of FSHD1, were beyond our capabilities and outside the scope of this study [8, 9, 11]. Because the original clinical samples were collected prior to genetic testing for FSHD, we decided, in consultation with officials at CIMR, to confirm the identity of each cell line with respect to the pedigree, identify those with FSHD1-sized deletions or FSHD2-type DNA hypomethylation, determine 4q and 10q SSLPs and subtelomere haplotypes, and assess the presence or absence of an exon 3 PAS. We have successfully completed this analysis for all LCLs and provided the information to CIMR, thus making these cells immediately available to the research community through their online catalog.
One notable omission for these LCLs is information on the clinical characteristics, specific methods for evaluation, and specific diagnostic criteria used for each of the individual subjects from which they were derived. Multiple clinicians across the United States examined these subjects and their family members, and ultimately >600 clinical samples were obtained from at least 36 families, with numerous subjects across generations clinically diagnosed with FSHD; however, many samples were lost over time, some subjects were not available for reconsent, and most of the specific clinician notes were lost or destroyed. For the samples that remained, the only clinical information available was the final clinical assessment of “FSHD”; the specific measurements and criteria for each individual (e.g., which muscles were affected, clinical severity, or level of weakness) are not known. However, an FSHD study performed by Dr. Jacobsen and colleagues, who obtained the Utah samples described in our current study and published soon thereafter, describes the clinical assessment used for FSHD subjects [44]. The criteria for FSHD were: 1) inheritance consistent with autosomal dominant genetics, 2) muscle weakness of at least one of several specific muscles of the face and shoulder girdle, and 3) muscle atrophy by the age of 20 years in at least one affected family member [44]. When matched with our blinded genetic testing, the original clinical diagnosis for FSHD was 12/12 with respect to families (all are, in fact, now known to be genetically FSHD families) and five families (2, 7, 12, 18, and 36) had a 100% match between the clinical and our genetic diagnosis for healthy and FSHD. Out of a total of 63 genetically defined FSHD1 subjects, 55 showed clinical FSHD and 8 are designated as asymptomatic (lacking clinical symptoms at the time of their assessment). We feel that the designation of asymptomatic is accurate, since these 8 subjects were examined by neurologists specifically looking for signs of clinical FSHD in families affected by the disease. This information, as with all FSHD asymptomatic or non-manifesting diagnoses, comes with the caveat that these subjects may have developed FSHD at a later time in their lives. Interestingly, 5 subjects (GM16290, GM16299, GM16322, GM16324, and GM16431) clinically characterized as FSHD were found not to contain an FSHD1-sized deletion. Unfortunately, in the absence of more detailed notes we can only speculate on the reasons for this discrepancy. However, since these subjects represent 4 different families (Family 4, 6, 11, and 32) not known to be related and all exhibit DNA methylation levels well within the range for healthy subjects, these cases likely represent over-interpretation of clinical signs. Alternatively, one or more of these could represent clinical mis-classification of a neuromuscular disease phenocopy of FSHD, but a second rare disorder spontaneously arising in even one of the families is highly unlikely. Now that nearly 30 years has passed, a potential follow-up study of all of these subjects would be extremely interesting and informative.
In addition to the minimal information required to make these LCLs available, we also collected data that would be important and useful for making decisions on which cell lines to use for different lines of investigation relevant to FSHD. As DUX4-fl expression in skeletal muscle is the key pathogenic consequence of the epigenetic disruption in FSHD, it stands to reason that DUX4-fl expression is typically and best studied in myogenic cells. Thus, the relevance of LCLs for assaying DUX4 expression has been in question. Here we show that FSHD1 lymphoblastoid lines are clearly capable of expressing DUX4-fl mRNA and protein, with consequent expression of several DUX4-FL target genes that are also activated in myocytes. Additionally, as with FSHD myocytes, epigenetic drugs readily induce high levels of DUX4-fl in these FSHD1 LCLs, suggesting that these cells may be useful for studies investigating or manipulating the epigenetic status of the 4q35 D4Z4 array. Importantly, we confirmed that only genetically FSHD1 LCLs express significant levels of DUX4-fl mRNA; however, the variable levels of DUX4-fl expression across 63 FSHD1 lines, many with the same deletion, might help to explain the variable success labs have reported with lymphoblastoid cell lines. Surprisingly, we typically found a range of >100-fold differences in DUX4-fl expression levels in FSHD1 LCLs within families. We do not know the underlying cause of this variability, which would represent an interesting follow-up study, but a knowledge of these differences is important for the planning of future experiments.
5. Conclusions
We have provided a preliminary genetic and molecular characterization of 114 LCLs from 12 FSHD1 affected families, enabling these cell lines to become publicly available for use by the FSHD field. We have also provided additional evidence that this cohort of cells will be a valuable resource for investigating many aspects of gene expression and regulation in FSHD. We recognize that FSHD is a muscle disease and investigating FSHD in myogenic cells is important; however, there are limitations imposed by working with primary differentiated myogenic cells, and alternative models are needed. We propose that these LCLs may fill this need, serving as a tool enabling high-throughput screening of therapeutics targeting DUX4 expression.
Supplementary Material
Fig. 4.

Pedigrees for unrelated Families 6, 12, and 15. See Figure legends for Figs 2 and 3.
Fig. 5.
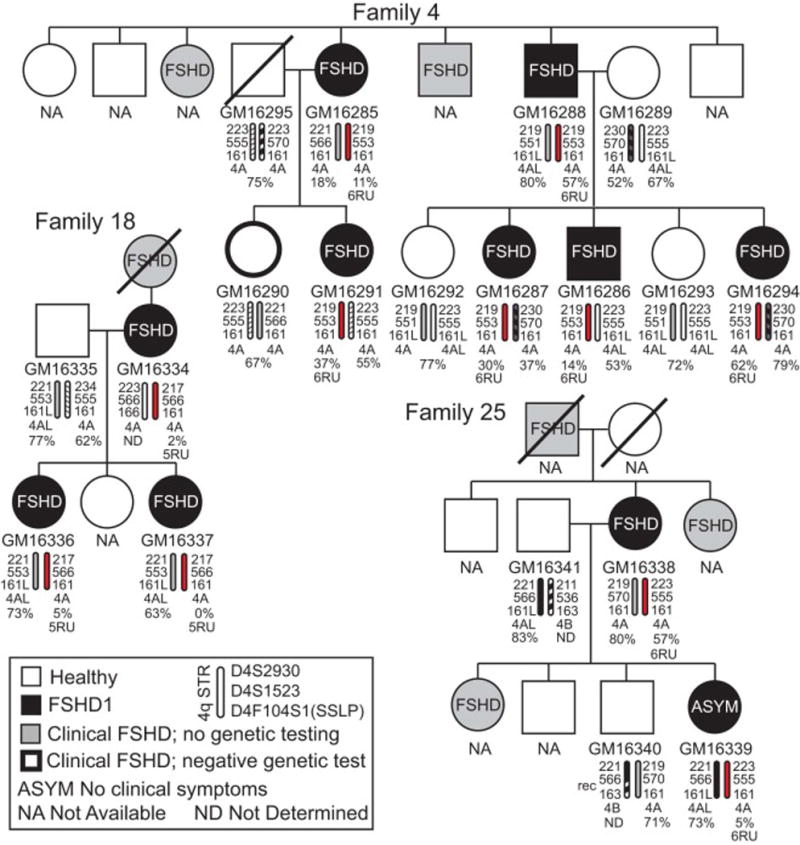
Pedigrees for unrelated Families 4, 18, and 25. See legend for Fig. 2.
Highlights.
Genetic characterization of lymphoblastoid cell lines from 12 large FSHD1 families
Relevant FSHD gene expression and DNA methylation in lymphoblastoid lines
Cellular model for FSHD gene expression suitable for therapeutic screening
Acknowledgments
We are grateful to these families for participating in the study. We thank Dr. Stephen Jacobsen for clinically evaluating many of the subjects and collecting the clinical samples, and the NIGMS Human Genetic Cell Repository at the Coriell Institute for Medical Research repository for culturing, maintaining, and distributing the cell lines. We thank Dr. Yukiko K. Hayashi, Department of Neurophysiology, Tokyo Medical University, for sharing the protocol for linear southern blotting analysis.
Funding
This work was funded by the National Institute of Arthritis and Musculoskeletal and Skin Diseases [grant number 1R01AR062587], the FSH Society [grant number FSHS-2015-SG02], and the OCTV Technology Development Fund at the University of Massachusetts. The FSH Society initiated the project and facilitated the work through its completion. The funders played no role in the data analysis or interpretation.
Appendix
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author contributions
Takako Jones: Drafting/revising the manuscript; study concept and design; acquisition and analysis of data
Charis Himeda: Drafting/revising the manuscript; study design; acquisition and analysis of data
Daniel Perez: Drafting/revising the manuscript; study concept, coordination and design; interfacing with clinicians, patients, families, and Coriell Biorepositories; obtaining bioresources
Peter Jones: Drafting/revising the manuscript; study concept and design; acquisition and analysis of data
All authors approved the final manuscript.
References
- 1.Padberg GW. Facioscapulohumeral Disease [thesis] Leiden University; Leiden, the Netherlands: 1982. p. 243. [Google Scholar]
- 2.Flanigan KM. The muscular dystrophies. Semin Neurol. 2012;32:255–63. doi: 10.1055/s-0032-1329199. [DOI] [PubMed] [Google Scholar]
- 3.Orphanet. Prevalence and incidence of rare diseases: Bibliographic data. Orphanet Report Series: Rare Diseases collection. 2016 [cited; Available from: http://www.orpha.net/orphacom/cahiers/docs/GB/Prevalence_of_rare_diseases_by_alphabetical_list.pdf.
- 4.Deenen JC, Arnts H, van der Maarel SM, et al. Population-based incidence and prevalence of facioscapulohumeral dystrophy. Neurology. 2014;83:1056–9. doi: 10.1212/WNL.0000000000000797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang LH, Tawil R. Facioscapulohumeral Dystrophy. Curr Neurol Neurosci Rep. 2016;16:66. doi: 10.1007/s11910-016-0667-0. [DOI] [PubMed] [Google Scholar]
- 6.Padberg GW, van Engelen BG. Facioscapulohumeral muscular dystrophy. Curr Opin Neurol. 2009;22:539–42. doi: 10.1097/WCO.0b013e328330a572. [DOI] [PubMed] [Google Scholar]
- 7.de Greef JC, Lemmers RJ, van Engelen BG, et al. Common epigenetic changes of D4Z4 in contraction-dependent and contraction-independent FSHD. Hum Mutat. 2009;30:1449–59. doi: 10.1002/humu.21091. [DOI] [PubMed] [Google Scholar]
- 8.Lemmers RJ, Tawil R, Petek LM, et al. Digenic inheritance of an SMCHD1 mutation and an FSHD-permissive D4Z4 allele causes facioscapulohumeral muscular dystrophy type 2. Nat Genet. 2012;44:1370–4. doi: 10.1038/ng.2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sacconi S, Lemmers RJ, Balog J, et al. The FSHD2 gene SMCHD1 is a modifier of disease severity in families affected by FSHD1. Am J Hum Genet. 2013;93:744–751. doi: 10.1016/j.ajhg.2013.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Larsen M, Rost S, El Hajj N, et al. Diagnostic approach for FSHD revisited: SMCHD1 mutations cause FSHD2 and act as modifiers of disease severity in FSHD1. Eur J Hum Genet. 2014;23:808–16. doi: 10.1038/ejhg.2014.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lemmers RJ, Goeman JJ, Van Der Vliet PJ, et al. Inter-individual differences in CpG methylation at D4Z4 correlate with clinical variability in FSHD1 and FSHD2. Hum Mol Genet. 2015;24:659–69. doi: 10.1093/hmg/ddu486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jones TI, King OD, Himeda CL, et al. Individual epigenetic status of the pathogenic D4Z4 macrosatellite correlates with disease in facioscapulohumeral muscular dystrophy. Clinical epigenetics. 2015;7:37. doi: 10.1186/s13148-015-0072-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zatz M, Marie SK, Cerqueira A, Vainzof M, Pavanello RC, Passos-Bueno MR. The facioscapulohumeral muscular dystrophy (FSHD1) gene affects males more severely and more frequently than females. Am J Med Genet. 1998;77:155–61. [PubMed] [Google Scholar]
- 14.Tonini MM, Passos-Bueno MR, Cerqueira A, Matioli SR, Pavanello R, Zatz M. Asymptomatic carriers and gender differences in facioscapulohumeral muscular dystrophy (FSHD) Neuromuscul Disord. 2004;14:33–8. doi: 10.1016/j.nmd.2003.07.001. [DOI] [PubMed] [Google Scholar]
- 15.Himeda CL, Jones TI, Jones PL. Facioscapulohumeral muscular dystrophy as a model for epigenetic regulation and disease. Antioxidants & redox signaling. 2015;22:1463–82. doi: 10.1089/ars.2014.6090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wijmenga C, Hewitt JE, Sandkuijl LA, et al. Chromosome 4q DNA rearrangements associated with facioscapulohumeral muscular dystrophy. Nat Genet. 1992;2:26–30. doi: 10.1038/ng0992-26. [DOI] [PubMed] [Google Scholar]
- 17.van Deutekom JC, Wijmenga C, van Tienhoven EA, et al. FSHD associated DNA rearrangements are due to deletions of integral copies of a 3.2 kb tandemly repeated unit. Hum Mol Genet. 1993;2:2037–42. doi: 10.1093/hmg/2.12.2037. [DOI] [PubMed] [Google Scholar]
- 18.Schaap M, Lemmers RJ, Maassen R, et al. Genome-wide analysis of macrosatellite repeat copy number variation in worldwide populations: evidence for differences and commonalities in size distributions and size restrictions. BMC Genomics. 2013;14:143. doi: 10.1186/1471-2164-14-143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rossi M, Ricci E, Colantoni L, et al. The Facioscapulohumeral muscular dystrophy region on 4qter and the homologous locus on 10qter evolved independently under different evolutionary pressure. BMC medical genetics. 2007;8:8. doi: 10.1186/1471-2350-8-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tyler FH, Stephens FE. Studies in disorders of muscle. II Clinical manifestations and inheritance of facioscapulohumeral dystrophy in a large family. Ann Intern Med. 1950;32:640–60. doi: 10.7326/0003-4819-32-4-640. [DOI] [PubMed] [Google Scholar]
- 21.Lemmers RJ, de Kievit P, Sandkuijl L, et al. Facioscapulohumeral muscular dystrophy is uniquely associated with one of the two variants of the 4q subtelomere. Nat Genet. 2002;32:235–6. doi: 10.1038/ng999. [DOI] [PubMed] [Google Scholar]
- 22.Lemmers RJ, Wohlgemuth M, van der Gaag KJ, et al. Specific sequence variations within the 4q35 region are associated with facioscapulohumeral muscular dystrophy. Am J Hum Genet. 2007;81:884–94. doi: 10.1086/521986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lemmers RJ, van der Vliet PJ, Klooster R, et al. A unifying genetic model for facioscapulohumeral muscular dystrophy. Science. 2010;329:1650–3. doi: 10.1126/science.1189044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.de Greef JC, Lemmers RJ, Camano P, et al. Clinical features of facioscapulohumeral muscular dystrophy 2. Neurology. 2010;75:1548–54. doi: 10.1212/WNL.0b013e3181f96175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.van den Boogaard ML, Lemmers RJ, Balog J, et al. Mutations in DNMT3B Modify Epigenetic Repression of the D4Z4 Repeat and the Penetrance of Facioscapulohumeral Dystrophy. Am J Hum Genet. 2016;98:1020–9. doi: 10.1016/j.ajhg.2016.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.van Overveld PG, Lemmers RJ, Sandkuijl LA, et al. Hypomethylation of D4Z4 in 4q–linked and non-4q-linked facioscapulohumeral muscular dystrophy. Nat Genet. 2003;35:315–7. doi: 10.1038/ng1262. [DOI] [PubMed] [Google Scholar]
- 27.van Overveld PG, Enthoven L, Ricci E, et al. Variable hypomethylation of D4Z4 in facioscapulohumeral muscular dystrophy. Ann Neurol. 2005;58:569–76. doi: 10.1002/ana.20625. [DOI] [PubMed] [Google Scholar]
- 28.Zeng W, de Greef JC, Chen YY, et al. Specific loss of histone H3 lysine 9 trimethylation and HP1gamma/cohesin binding at D4Z4 repeats is associated with facioscapulohumeral dystrophy (FSHD) PLoS Genet. 2009;5:e1000559. doi: 10.1371/journal.pgen.1000559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Balog J, Thijssen PE, de Greef JC, et al. Correlation analysis of clinical parameters with epigenetic modifications in the DUX4 promoter in FSHD. Epigenetics. 2012;7:1–6. doi: 10.4161/epi.20001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Calandra P, Cascino I, Lemmers RJ, et al. Allele-specific DNA hypomethylation characterises FSHD1 and FSHD2. J Med Genet. 2016;53:348–55. doi: 10.1136/jmedgenet-2015-103436. [DOI] [PubMed] [Google Scholar]
- 31.Jones TI, Yan C, Sapp PC, et al. Identifying diagnostic DNA methylation profiles for facioscapulohumeral muscular dystrophy in blood and saliva using bisulfite sequencing. Clinical epigenetics. 2014;6:23. doi: 10.1186/1868-7083-6-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gaillard MC, Roche S, Dion C, et al. Differential DNA methylation of the D4Z4 repeat in patients with FSHD and asymptomatic carriers. Neurology. 2014;83:733–42. doi: 10.1212/WNL.0000000000000708. [DOI] [PubMed] [Google Scholar]
- 33.Gabriels J, Beckers MC, Ding H, et al. Nucleotide sequence of the partially deleted D4Z4 locus in a patient with FSHD identifies a putative gene within each 3.3 kb element. Gene. 1999;236:25–32. doi: 10.1016/s0378-1119(99)00267-x. [DOI] [PubMed] [Google Scholar]
- 34.Dixit M, Ansseau E, Tassin A, et al. DUX4, a candidate gene of facioscapulohumeral muscular dystrophy, encodes a transcriptional activator of PITX1. Proc Natl Acad Sci U S A. 2007;104:18157–62. doi: 10.1073/pnas.0708659104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Snider L, Geng LN, Lemmers RJ, et al. Facioscapulohumeral dystrophy: incomplete suppression of a retrotransposed gene. PLoS Genet. 2010;6:e1001181. doi: 10.1371/journal.pgen.1001181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jones TI, Chen JC, Rahimov F, et al. Facioscapulohumeral muscular dystrophy family studies of DUX4 expression: evidence for disease modifiers and a quantitative model of pathogenesis. Hum Mol Genet. 2012;21:4419–30. doi: 10.1093/hmg/dds284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Himeda CL, Debarnot C, Homma S, et al. Myogenic enhancers regulate expression of the facioscapulohumeral muscular dystrophy associated DUX4 gene. Mol Cell Biol. 2014;34:1942–55. doi: 10.1128/MCB.00149-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.van der Maarel SM, Miller DG, Tawil R, Filippova GN, Tapscott SJ. Facioscapulohumeral muscular dystrophy: consequences of chromatin relaxation. Curr Opin Neurol. 2012;25:614–20. doi: 10.1097/WCO.0b013e328357f22d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lemmers RJ, Wohlgemuth M, Frants RR, Padberg GW, Morava E, van der Maarel SM. Contractions of D4Z4 on 4qB subtelomeres do not cause facioscapulohumeral muscular dystrophy. Am J Hum Genet. 2004;75:1124–30. doi: 10.1086/426035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lemmers RJ, van der Vliet PJ, van der Gaag KJ, et al. Worldwide population analysis of the 4q and 10q subtelomeres identifies only four discrete interchromosomal sequence transfers in human evolution. Am J Hum Genet. 2010;86:364–77. doi: 10.1016/j.ajhg.2010.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vanderplanck C, Ansseau E, Charron S, et al. The FSHD Atrophic Myotube Phenotype Is Caused by DUX4 Expression. PLoS One. 2011;6:e26820. doi: 10.1371/journal.pone.0026820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Marsollier AC, Ciszewski L, Mariot V, et al. Antisense targeting of 3’ end elements involved in DUX4 mRNA processing is an efficient therapeutic strategy for facioscapulohumeral dystrophy: a new gene-silencing approach. Hum Mol Genet. 2016;25:1468–78. doi: 10.1093/hmg/ddw015. [DOI] [PubMed] [Google Scholar]
- 43.Himeda CL, Jones TI, Jones PL. CRISPR/dCas9-mediated Transcriptional Inhibition Ameliorates the Epigenetic Dysregulation at D4Z4 and Represses DUX4-fl in FSH Muscular Dystrophy. Mol Ther. 2016;24:527–35. doi: 10.1038/mt.2015.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jacobsen SJ, Diala ES, Dorsey BV, et al. A clinically homogeneous group of families with facioscapulohumeral (Landouzy-Dejerine) muscular dystrophy: linkage analysis of six autosomes. Am J Hum Genet. 1990;47:376–88. [PMC free article] [PubMed] [Google Scholar]
- 45.Sarfarazi M, Wijmenga C, Upadhyaya M, et al. Regional mapping of facioscapulohumeral muscular dystrophy gene on 4q35: combined analysis of an international consortium. Am J Hum Genet. 1992;51:396–403. [PMC free article] [PubMed] [Google Scholar]
- 46.Weiffenbach B, Bagley R, Falls K, et al. Linkage analyses of five chromosome 4 markers localizes the facioscapulohumeral muscular dystrophy (FSHD) gene to distal 4q35. Am J Hum Genet. 1992;51:416–23. [PMC free article] [PubMed] [Google Scholar]
- 47.Weiffenbach B, Dubois J, Storvick D, et al. Mapping the facioscapulohumeral muscular dystrophy gene is complicated by chromsome 4q35 recombination events. Nat Genet. 1993;4:165–9. doi: 10.1038/ng0693-165. [DOI] [PubMed] [Google Scholar]
- 48.Flanigan KM, Coffeen CM, Sexton L, Stauffer D, Brunner S, Leppert MF. Genetic characterization of a large, historically significant Utah kindred with facioscapulohumeral dystrophy. Neuromuscul Disord. 2001;11:525–9. doi: 10.1016/s0960-8966(01)00201-2. [DOI] [PubMed] [Google Scholar]
- 49.Goto K, Nishino I, Hayashi YK. Very low penetrance in 85 Japanese families with facioscapulohumeral muscular dystrophy 1A. J Med Genet. 2004;41:e12. doi: 10.1136/jmg.2003.008755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Deidda G, Cacurri S, Piazzo N, Felicetti L. Direct detection of 4q35 rearrangements implicated in facioscapulohumeral muscular dystrophy (FSHD) J Med Genet. 1996;33:361–5. doi: 10.1136/jmg.33.5.361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lemmers RJL, de Kievit P, van Geel M, et al. Complete allele information in the diagnosis of facioscapulohumeral muscular dystrophy by triple DNA analysis. Ann Neurol. 2001;50:816–9. doi: 10.1002/ana.10057. [DOI] [PubMed] [Google Scholar]
- 52.Rohde C, Zhang Y, Reinhardt R, Jeltsch A. BISMA--fast and accurate bisulfite sequencing data analysis of individual clones from unique and repetitive sequences. BMC Bioinformatics. 2010;11:230. doi: 10.1186/1471-2105-11-230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Barat-Houari M, Nguyen K, Bernard R, et al. New multiplex PCR-based protocol allowing indirect diagnosis of FSHD on single cells: can PGD be offered despite high risk of recombination? Eur J Hum Genet. 2010;18:533–8. doi: 10.1038/ejhg.2009.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mannucci A, Sullivan KM, Ivanov PL, Gill P. Forensic application of a rapid and quantitative DNA sex test by amplification of the X-Y homologous gene amelogenin. Int J Legal Med. 1994;106:190–3. doi: 10.1007/BF01371335. [DOI] [PubMed] [Google Scholar]
- 55.Geng LN, Yao Z, Snider L, et al. DUX4 activates germline genes, retroelements, and immune mediators: Implications for facioscapulohumeral dystrophy. Dev Cell. 2012;22:38–51. doi: 10.1016/j.devcel.2011.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bakker E, Wijmenga C, Vossen RH, et al. The FSHD-linked locus D4F104S1 (p13E-11) on 4q35 has a homologue on 10qter. Muscle Nerve. 1995;2:S39–44. [PubMed] [Google Scholar]
- 57.Daxinger L, Tapscott SJ, van der Maarel SM. Genetic and epigenetic contributors to FSHD. Curr Opin Genet Dev. 2015;33:56–61. doi: 10.1016/j.gde.2015.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Caliskan M, Cusanovich DA, Ober C, Gilad Y. The effects of EBV transformation on gene expression levels and methylation profiles. Hum Mol Genet. 2011;20:1643–52. doi: 10.1093/hmg/ddr041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sugawara H, Iwamoto K, Bundo M, Ueda J, Ishigooka J, Kato T. Comprehensive DNA methylation analysis of human peripheral blood leukocytes and lymphoblastoid cell lines. Epigenetics. 2011;6:508–15. doi: 10.4161/epi.6.4.14876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Thompson TM, Sharfi D, Lee M, Yrigollen CM, Naumova OY, Grigorenko EL. Comparison of whole-genome DNA methylation patterns in whole blood, saliva, and lymphoblastoid cell lines. Behav Genet. 2013;43:168–76. doi: 10.1007/s10519-012-9579-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Krom YD, Thijssen PE, Young JM, et al. Intrinsic Epigenetic Regulation of the D4Z4 Macrosatellite Repeat in a Transgenic Mouse Model for FSHD. PLoS Genet. 2013;9:e1003415. doi: 10.1371/journal.pgen.1003415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yao Z, Snider L, Balog J, et al. DUX4-induced gene expression is the major molecular signature in FSHD skeletal muscle. Hum Mol Genet. 2014;23:5342–52. doi: 10.1093/hmg/ddu251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Choi SH, Gearhart MD, Cui Z, et al. DUX4 recruits p300/CBP through its C-terminus and induces global H3K27 acetylation changes. Nucleic Acids Res. 2016 doi: 10.1093/nar/gkw141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wuebbles RD, Long SW, Hanel ML, Jones PL. Testing the effects of FSHD candidate gene expression in vertebrate muscle development. Int J Clin Exp Pathol. 2010;3:386–400. [PMC free article] [PubMed] [Google Scholar]
- 65.Wallace LM, Garwick SE, Mei W, et al. DUX4, a candidate gene for facioscapulohumeral muscular dystrophy, causes p53-dependent myopathy in vivo. Ann Neurol. 2011;69:540–52. doi: 10.1002/ana.22275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Rickard AM, Petek LM, Miller DG. Endogenous DUX4 expression in FSHD myotubes is sufficient to cause cell death and disrupts RNA splicing and cell migration pathways. Hum Mol Genet. 2015;24:5901–14. doi: 10.1093/hmg/ddv315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Homma S, Beermann ML, Boyce FM, Miller JB. Expression of FSHD-related DUX4-FL alters proteostasis and induces TDP-43 aggregation. Annals of clinical and translational neurology. 2015;2:151–66. doi: 10.1002/acn3.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Taylor SM, Jones PA. Multiple new phenotypes induced in 10T1/2 and 3T3 cells treated with 5-azacytidine. Cell. 1979;17:771–9. doi: 10.1016/0092-8674(79)90317-9. [DOI] [PubMed] [Google Scholar]
- 69.Tapscott SJ. The circuitry of a master switch: Myod and the regulation of skeletal muscle gene transcription. Development. 2005;132:2685–95. doi: 10.1242/dev.01874. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.