

Abstract
The interferon (IFN)‐γ‐activated inhibitor of translation (GAIT) system directs transcript‐selective translational control of functionally related genes. In myeloid cells, IFN‐γ induces formation of a multiprotein GAIT complex that binds structural GAIT elements in the 3′‐untranslated regions (UTRs) of multiple inflammation‐related mRNAs, including ceruloplasmin and VEGF‐A, and represses their translation. The human GAIT complex is a heterotetramer containing glutamyl‐prolyl tRNA synthetase (EPRS), NS1‐associated protein 1 (NSAP1), ribosomal protein L13a (L13a), and glyceraldehyde‐3‐phosphate dehydrogenase (GAPDH). A network of IFN‐γ‐stimulated kinases regulates recruitment and assembly of GAIT complex constituents. Activation of cyclin‐dependent kinase 5 (Cdk5), mammalian target of rapamycin complex 1 (mTORC1), and S6K1 kinases induces EPRS release from its parental multiaminoacyl tRNA synthetase complex to join NSAP1 in a ‘pre‐GAIT’ complex. Subsequently, the DAPK‐ZIPK kinase axis phosphorylates L13a, inducing release from the 60S ribosomal subunit and binding to GAPDH. The subcomplexes join to form the functional GAIT complex. Each constituent has a distinct role in the GAIT system. EPRS binds the GAIT element in target mRNAs, NSAP1 negatively regulates mRNA binding, L13a binds eIF4G to block ribosome recruitment, and GAPDH shields L13a from proteasomal degradation. The GAIT system is susceptible to genetic and condition‐specific regulation. An N‐terminus EPRS truncate is a dominant‐negative inhibitor ensuring a ‘translational trickle’ of target transcripts. Also, hypoxia and oxidatively modified lipoproteins regulate GAIT activity. Mouse models exhibiting absent or genetically modified GAIT complex constituents are beginning to elucidate the physiological role of the GAIT system, particularly in the resolution of chronic inflammation. Finally, GAIT‐like systems in proto‐chordates suggests an evolutionarily conserved role of the pathway in innate immunity. WIREs RNA 2018, 9:e1441. doi: 10.1002/wrna.1441
This article is categorized under:
-
1
Translation > Translation Regulation
-
2
RNA Interactions with Proteins and Other Molecules > RNA–Protein Complexes
-
3
Regulatory RNAs/RNAi/Riboswitches > Riboswitches
IFN‐γ‐stimulated activation of the GAIT complex in human myeloid cells. A network of phosphorylation events recruits EPRS and ribosomal protein L13a to join GAPDH and NSAP1 to form the functional, heterotetrameric GAIT complex that binds GAIT element‐bearing mRNAs for translation‐inhibition.
INTRODUCTION
Regulation of gene expression profoundly influences nearly all aspects of cellular life, including proliferation, metabolism, host defense, and death.1 Gene expression depends on both transcriptional and posttranscriptional control mechanisms.2 The latter is particularly important for fine or localized control of protein accumulation. Translational control can be global, regulating expression of most transcripts, or transcript‐selective, modulating a defined subset of mRNAs.3, 4, 5 Transcript‐selective regulation is generally mediated by proteins or complexes binding to specific elements in the 5′‐ or 3′‐untranslated region (UTR) of target mRNAs.6, 7, 8 Such coordinate, posttranscriptional control of expression of functionally related genes has been termed as ‘posttranscriptional regulon.’9, 10 We have described a 3′‐UTR‐specific, transcript‐selective translational control mechanism that regulates a group of functionally related genes.11 In human myeloid cells, the pro‐inflammatory cytokine interferon‐γ (IFN‐γ) induces recruitment of several abundant ‘housekeeping’ proteins for assembly of a heterotetrameric IFN‐γ‐activated inhibitor of translation (GAIT) complex (Figure 1). The human GAIT complex consists of glutamyl‐prolyl tRNA synthetase (EPRS), NS1‐associated protein 1 (NSAP1), ribosomal protein L13a, and glyceraldehyde‐3‐phosphate dehydrogenase (GAPDH).12 Interestingly, the heterotrimeric murine GAIT complex lacks NSAP1.13 The GAIT complex binds a defined bipartite GAIT element consisting of a 29‐ to 33‐nucleotide (nt) stem‐loop in the 3′‐UTR of multiple inflammation‐responsive mRNAs and inhibits their translation without affecting total protein synthesis.11, 14
Figure 1.
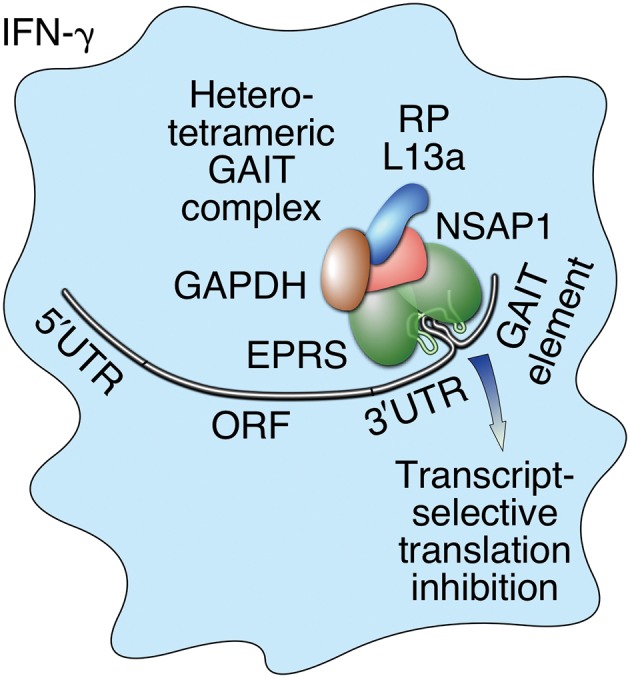
Schematic diagram depicting human interferon γ‐activated inhibitor of translation (GAIT) system. Interferon‐γ (IFN‐γ) induces formation of heterotetrameric GAIT complex in myeloid cells. The human GAIT complex contains glutamyl‐prolyl tRNA synthetase (EPRS), NS1‐associated protein 1 (NSAP1), glyceraldehyde‐3‐phosphate dehydrogenase (GAPDH), and ribosomal protein (RP) L13a. Murine GAIT complex is essentially identical but lacks NSAP1.
ACTIVATION, ASSEMBLY, AND FUNCTION OF THE GAIT COMPLEX
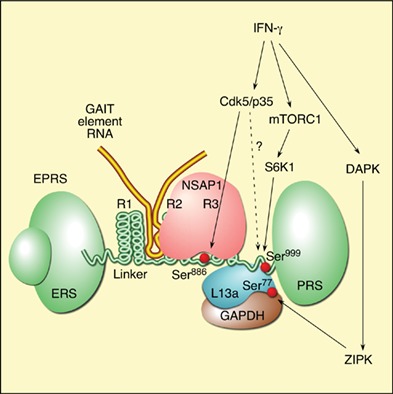
IFN‐γ‐inducible recruitment of the four human GAIT constituent proteins, and their assembly in the GAIT complex, occurs in two distinct, tightly regulated stages (Figure 2). In the first stage, completed after about 2–4 h, human EPRS is phosphorylated at Ser886 and Ser999 by two kinase systems.15 IFN‐γ induces cyclin‐dependent kinase 5 (Cdk5) in conjunction with ERK2 and its activator, p35 (Cdk5R1); activated Cdk5/p35 directly phosphorylates Ser886 in EPRS.16 A downstream event, phosphorylation of EPRS, Ser999, is mediated by the kinase axis of mammalian target of rapamycin complex 1 (mTORC1) and p70 ribosomal protein S6 kinase 1 (S6K1).17 Interestingly, Ser999 phosphorylation also requires Cdk5 activity, but the specific role of Cdk5 in this event has not been elucidated. Phosphorylated EPRS is released from the multiaminoacyl tRNA synthetase complex (MSC) to join NSAP1 to generate an inactive ‘pre‐GAIT complex’ that does not bind GAIT element RNA or silence translation. NSAP1 binding specifically requires EPRS Ser886 phosphorylation.13 About 12–16 h later, L13a is phosphorylated at Ser77 by DAPK‐activated ZIPK, and is released from the 60S ribosomal subunit.18 Released L13a binds GAPDH and the pre‐GAIT complex to form the functional heterotetrameric GAIT complex competent for translation‐repression of GAIT element‐bearing transcripts.
Figure 2.
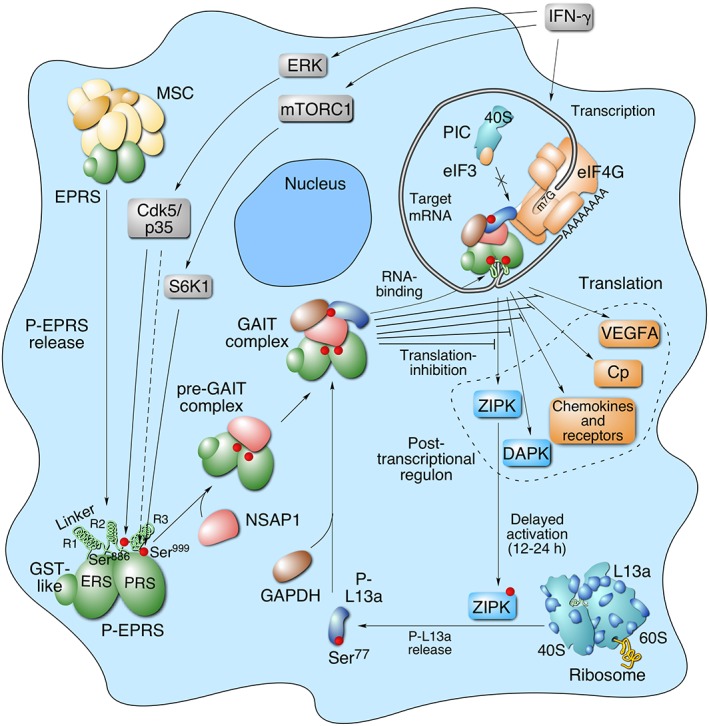
Schematic diagram of interferon γ‐activated inhibitor of translation (GAIT) system activation for transcript‐selective translation inhibition of inflammation‐related genes in myeloid cells. Interferon‐γ (IFN‐γ) induces early activation of human EPRS by two‐step phosphorylation at Ser886 and Ser999 in the linker region by Cdk5/p35, mTORC1, and S6K1 kinases. Cdk5/p35 directly phosphorylates Ser886, and is also required, in conjunction with mTORC1 and S6K1 for phosphorylation of Ser999. Phosphorylated‐EPRS (P‐EPRS) is released from the multiaminoacyl tRNA synthetase complex (MSC). Phospho‐Ser886 EPRS interacts with NSAP1 to form an inactive pre‐GAIT complex (in humans only). After ~12–16 h, L13a is phosphorylated at Ser77 (P‐L13a) by DAPK‐activated ZIPK, and released from the 60S ribosomal subunit. Free P‐L13a interacts with glyceraldehyde‐3‐phosphate dehydrogenase (GAPDH) and joins the pre‐GAIT complex to form the functional GAIT complex. The complex binds GAIT elements in the 3′‐UTR of functionally related transcripts induced by IFN‐γ, and inhibits their translation by a mechanism that requires a circularized mRNA. L13a in the GAIT complex interferes with the eukaryotic translation‐initiation factor, eIF4G, near the eIF3‐binding site, and blocks translation‐initiation. DAPK and ZIPK translations are repressed by GAIT system constituting a negative‐feedback module to limit L13a phosphorylation and GAIT system activity.
Several unique features of the GAIT system distinguish it from other stimulus‐induced pathways that regulate gene expression. For example, the requirement for two independent, temporally distinct signaling pathways, namely, early induction of EPRS phosphorylation and delayed induction of L13a phosphorylation.15, 18 Also, the complex is formed from proteins with canonical activities distinct from their function in the GAIT system. For example, EPRS and L13a are ubiquitous components of translation machinery required for mRNA translation, but when assembled into the GAIT complex they adapt to perform ‘moonlighting’ functions in translation repression.11 Likewise, GAPDH activity is essential for glycolysis and energy production, but it displays a noncanonical chaperone‐like activity in the GAIT system.19 Also remarkable is the stimulus‐dependent release of EPRS and L13a from their macromolecular complexes: the MSC and ribosome, respectively.12, 20 These findings introduced the concept that otherwise stable macromolecular complexes, particularly the MSC, can act as depots of stimulus‐dependent releasable regulatory proteins that perform specialized auxiliary functions unrelated to their primary function within the parental complex.21 Here, we review the function of the individual constituents in GAIT complex assembly and the 3′‐UTR GAIT element‐bearing targets subject to translation‐repression, as well as the auxiliary activities of the GAIT complex proteins. Also addressed are the genetic and environmental stresses that influence the GAIT system, and their potential pathophysiological consequences.
AUXILIARY, NONCANONICAL ACTIVITIES OF GAIT COMPLEX PROTEINS
Individual proteins can exhibit more than one function, thereby expanding the classical ‘one gene–one enzyme’ theory proposed by George Beadle and Edward Tatum in 1941.22 In many cases, such multifunctional (or ‘moonlighting’) proteins exhibit a primary, constitutive function and a secondary (or noncanonical), context‐dependent role.23 Frequently, the new cellular activity is directed by posttranslational modification(s) that can cause conformational changes and recruit new binding partners, or fewer partners in the case of release from a parental complex.24, 25 Alternatively, a secondary function is driven by an isoform with a distinct primary sequence generated, e.g., by alternative RNA splicing or translation‐initiation, or proteolytic cleavage.26, 27 Remarkably, each of the GAIT complex constituents are ubiquitously expressed, essential housekeeping protein with a primary function unrelated to its function in the GAIT system (Figure 3). In this section, we briefly summarize canonical and noncanonical activities of the GAIT complex proteins.
Figure 3.
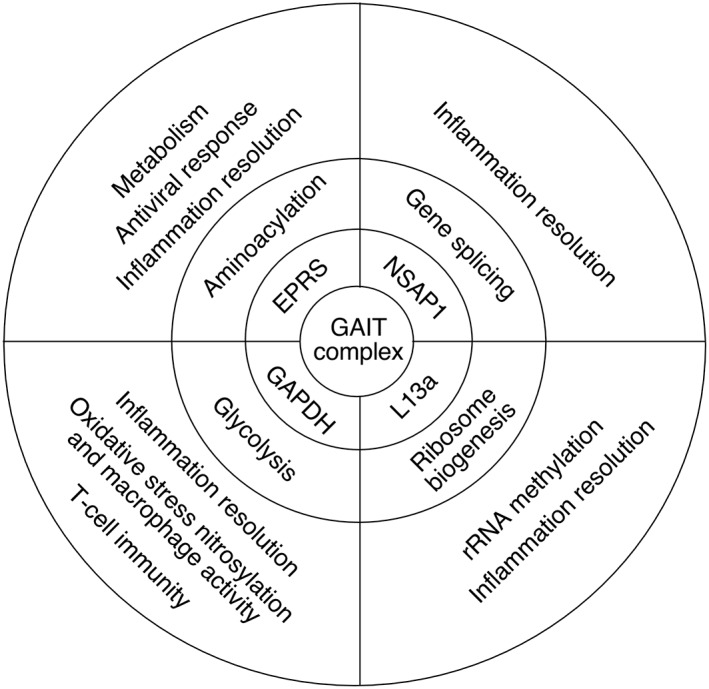
Summary of canonical and noncanonical activities of interferon γ‐activated inhibitor of translation (GAIT) complex constituent proteins.
Glutamyl‐prolyl tRNA Synthetase (EPRS)
EPRS belongs to an ancient group of 20 aminoacyl‐tRNA synthetases (AARSs), one for each amino acid, that catalyze ligation of amino acids to their cognate tRNAs to ensure accurate decryption of the genetic code during protein synthesis.28, 29 Present in nearly all animals, EPRS is the only bifunctional AARS containing two synthetase catalytic cores in a single polypeptide chain (Figure 4).30 Specifically, the ERS and PRS domains ligate Glu and Pro, respectively. In human EPRS, the two enzymatic domains are joined by a noncatalytic linker containing three helix‐turn‐helix WHEP domains (named after the AARSs first shown to contain these domains, i.e., Trp(W)RS, His(H)RS, and EPRS).31 The N‐terminus of EPRS also contains an appended glutathione‐S‐transferase (GST)‐like domain that has an important role in MSC assembly and structure.32 EPRS along with seven other AARSs and three non‐AARS proteins reside in the MSC, a cytoplasmic 1.5‐mDa macromolecular complex.33 The MSC has been shown to bind ribosomes and is thought to improve translation efficiency by direct channeling of charged tRNAs to the ribosome A‐site.34, 35 Activation of EPRS during GAIT complex assembly in myeloid cells requires IFN‐γ‐induced phosphorylation of Ser886 and Ser999 in the linker region, followed by release of EPRS from the MSC.15 Free, phosphorylated EPRS binds NSAP1 and the other GAIT components to form the active translational silencing complex. Recently, EPRS has been observed to exhibit several GAIT‐independent, noncanonical activities related to adiposity and antiviral defense, and possibly in breast cancer progression.17, 36, 37, 38 Interestingly, EPRS is not unique among AARSs in the expression of a noncanonical function as recent studies have shown many, if not most, AARS also express secondary activities unrelated to tRNA charging.39, 40 Beyond the noncanonical function, elucidation of EPRS's mode of activation revealed an unexpected finding, namely, stimulus‐dependent release of EPRS from its parental complex, the MSC.12, 15 This observation, coupled with stimulus‐dependent release of L13a from the 60S ribosome (see below), led us to propose the ‘depot hypothesis’ in which macromolecular complexes serve as depots or reservoirs of proteins that can be released under specific conditions or stimuli to perform new functions unrelated to the primary function within the complex.20, 21
Figure 4.

Schematic diagram of glutamyl‐prolyl tRNA synthetase (EPRS) domains, phospho‐sites and their interactors. An N‐terminal glutathione‐S‐transferase (GST)‐like domain is upstream of the ERS domain, which is joined to the PRS domain by a linker containing three WHEP repeats: R1, R2, and R3. In humans, NSAP1 binding to EPRS in the R2–R3 region requires Ser886 phosphorylation. Phospho‐Ser77‐L13a and glyceraldehyde‐3‐phosphate dehydrogenase (GAPDH) interaction with EPRS requires Ser999 phosphorylation. The interferon γ‐activated inhibitor of translation (GAIT) element RNA binds in the upstream R1–R2 repeat region.
EPRS is the sole GAIT complex constituent that recognizes and binds the 3′‐UTR GAIT element in target mRNAs.12 Deletion analysis showed that GAIT element RNA binds the upstream pair of EPRS WHEP repeats (R1 and R2) in the noncatalytic linker41 (Figure 4). Surprisingly, although EPRS is released from the MSC within a few hours following IFN‐γ stimulation, the interaction with the GAIT RNA element is delayed for at least 10 h.12 Interaction of NSAP1 with the downstream, overlapping pair of WHEP repeats, R2 and R3, prevents EPRS binding to target mRNAs.41 The inhibition is reversed by delayed interaction of EPRS with Ser77‐phosphorylated L13a and GAPDH that induces a conformational shift in the EPRS/NSAP1 complex thereby exposing the RNA binding site (Figures 2 and 4).18, 42 A proteomic screen using RBDmap to identify RNA‐binding proteins (RBPs) and domains identified the EPRS linker as an atypical RNA binding site.43 Each of the phospho‐sites in the human EPRS linker has its own protein‐binding function. Phosphorylation of Ser886, located in the spacer between R2 and R3, is essential for NSAP1 binding, whereas phosphorylation of Ser999, located just upstream of the PRS domain, facilitates the binding of L13a and GAPDH, completing the formation of the GAIT complex15, 41 (Figure 4).
Through accretive evolutionary processes, many metazoan AARSs bear appended domains not present in bacterial, archaeal, and yeast orthologs.31 These acquired domains, most often at the N‐ or C‐termini, are thought to be nonessential for aminoacylation, but rather participate in executing noncanonical activities.39 Importantly, the N‐terminal GST‐like domain and the WHEP domain‐containing linker connecting the synthetase cores are essential for the known noncanonical activities of EPRS.15, 36 The ERS and PRS catalytic domains became joined to form a single polypeptide by a gene fusion event that occurred about a billion years ago in a unicellular animal ancestor; the earliest detection of the linked synthetases in the filasterean, Capsaspora owczarzaki that contains a linker with two WHEP domains.44 ERS and PRS have remained joined by linkers containing a variable number up to six WHEP domains in nearly all known animals with the exception of the nematode Caenorhabditis elegans and closely related species. In the cnidarian, Nematostella vectensis (stinging sea anemone), EPRS mRNA is alternatively spliced to generate three isoforms of variable number and sequence of WHEP domains with differential RNA binding capabilities (described below).45
NS1‐Associated Protein 1 (NSAP1)
NSAP1 (also known as heterogeneous nuclear ribonucleoprotein (hnRNP) Q; glycine‐ and tyrosine‐rich RBP, i.e., GRY‐RBP; or synaptotagmin‐binding cytoplasmic RNA‐interacting protein, i.e., SYNCRIP) is a member of the hnRNP family of RBPs.46 NSAP1 contains three RNA‐recognition motifs (RRMs) and participates in packaging of nuclear transcripts, RNA editing, splicing, stabilization, and transport, nonsense‐mediated mRNA decay, transcriptional control, and internal ribosome entry site (IRES)‐dependent translation control.47, 48, 49, 50, 51 In the human GAIT system, NSAP1 binds EPRS released from the MSC, in a Ser886 phosphorylation‐dependent manner, to form a transient binary complex (Figures 2 and 4). NSAP1 prevents EPRS binding of GAIT element‐bearing mRNAs until the negative regulation is overcome by a conformational shift induced by interaction with phospho‐L13a and GAPDH.15 The presence of the pre‐GAIT complex for about 10 h in human myeloid cells suggests the possibility of an as‐yet unidentified function. Interestingly, the negative regulation by NSAP1 is primate‐specific, and is absent in the GAIT system activated in mouse macrophages, likely due to the absence of the Ser886 phospho‐site in murine EPRS.13 Species‐specific differences between the human and mouse GAIT systems are not too surprising as inflammatory responses differ substantially between these organisms.52 Future studies in which the mice are generated bearing a knock‐in of the human NSAP1‐binding site in EPRS might shed light on the consequences of NSAP1‐mediated negative regulation, as well as the function of the human‐specific pre‐GAIT complex.
Glyceraldehyde‐3‐Phosphate Dehydrogenase (GAPDH)
GAPDH is an essential and abundant ‘housekeeping’ enzyme required for the breakdown of glucose for energy production in the sixth step of glycolysis, i.e., conversion of glyceraldehyde‐3‐phosphate to 1.3‐bisphosphoglycerate.19, 53 Despite the essential glycolytic activity, GAPDH is an archetypal moonlighting protein exhibiting diverse nonglycolytic activities in transcriptional as well as posttranscriptional regulation, cell motility, cytokine production, and apoptosis among others.54, 55, 56, 57 Several GAPDH functions require incorporation into multiprotein complexes, e.g., it acts as a redox‐sensitive sensor that regulates transcription following integration into the OCA‐S coactivator complex.58 GAPDH is also a RBP, targeting both ribosomal RNA (rRNA) and the UTRs of several eukaryotic and viral mRNAs.59, 60, 61 Studies on the pathophysiological regulation of the GAIT system revealed that GAPDH exhibits chaperone‐like activity and binding to L13a prevents its proteasomal degradation.62 The requirement for protection of a free ribosomal protein is not unexpected as the 1:1 stoichiometry of ribosomal proteins (and rRNAs) is maintained, in part, by rapid degradation of newly synthesized ribosomal proteins not assembled into ribosome subcomplexes.63 During ribosome biogenesis, heat shock proteins are the predominant protectors.64 However, a specialized mechanism (and protein) might be required for protection of a ribosomal protein released from a mature ribosome, as in the case of L13a. Possibly, GAPDH might bind and protect other ribosomal proteins with stimulus‐inducible extraribosomal functions.65 The pathological dysregulation of the protective activity of GAPDH is described below.
Ribosomal Protein L13a
The eukaryotic 80S ribosome is a complex consisting of four rRNAs and about 80 proteins split between the large 60S and small 40S subunits.66, 67 The rRNA component of the ribosome drives the central peptidyltransferase activity necessary for interpretation of the triplet genetic code. Although the precise role of most ribosomal proteins in protein synthesis is not well understood, ribosomal proteins are essential for biogenesis and structural integrity of the ribosome.68 Similar to the AARSs, several ribosomal proteins exhibit ribosome‐independent moonlighting activities influencing various pathophysiological processes.65, 69 Ribosome‐free ribosomal proteins can arise from de novo synthesis or from dissociation from the ribosome.20, 21
Mammalian ribosomal protein L13a is an integral constituent of the ribosome 60S subunit.70, 71 RPL13A mRNA is co‐transcribed with four box C/D small nucleolar RNA (snoRNA) genes, U32a, U33, U34, and U35a, located within four RPL13A gene introns.72 During ribosome biogenesis, L13a is first incorporated into the 90S pre‐ribosome in the nucleolus and is essential for rRNA methylation within that complex.73 Delayed release of L13a from the mature 60S ribosomal subunit, about 16 h after IFN‐γ stimulation, represents the rate‐determining step of GAIT complex formation.20 Ribosomal release requires L13a phosphorylation at Ser77 by IFN‐γ‐mediated activation of the DAPK–ZIPK kinase axis, with the latter serving as the proximal kinase.18 The activity of both kinases is maximal at about 16 h following stimulation, nearly coinciding with assembly of active GAIT complex and translational arrest of target transcripts. L13a release from the 60S subunit does not perturb overall ribosome structural integrity or global protein synthesis, consistent with the location of L13a far from the tRNA binding sites and exit tunnel of the ribosome.20, 71 Stimulus‐dependent release is consistent with structural data indicating that many ribosomal proteins essentially ‘float’ on the rRNA core, without extensive protein–protein interactions and with limited RNA‐penetrating extensions.20, 21 However, the specific molecular details of L13a release from the ribosome have not been elucidated beyond identification of Arg68 as a residue site near the phosphorylation site and is required for L13a binding to rRNA and incorporation into the ribosome during hierarchical assembly.73
Following its release, Ser77 phosphorylated L13a binds GAPDH and the pre‐GAIT complex to form the functional heterotetrameric (or heterotrimeric in mice) GAIT complex18 (Figure 2). As described above, EPRS is the GAIT RNA element‐binding protein in the mature GAIT complex.13, 15 Ser77 phosphorylated L13a has an equally important function in the GAIT system. Phospho‐L13a in the RNA‐bound GAIT complex interacts with eukaryotic translation‐initiation factor 4G (eIF4G) of the translation‐initiation complex at or near the eIF3‐binding site.15, 18, 42 This interaction blocks recruitment of the eIF3‐containing 43S pre‐initiation complex, thereby inhibiting the initiation of translation of GAIT element‐bearing target mRNAs. A ‘closed loop’ model of mRNA, in which the 5′ and 3′ termini are connected by protein–protein interactions involving both ends, has been proposed to increase translation by enhancing the efficiency of ribosome subunit recycling.74 Importantly, the GAIT system also requires mRNA circularization, presumably to bring the 3′‐UTR‐bound GAIT complex into the vicinity of the 5′ terminus where it can effectively block 43S joining and translation‐initiation11, 75 (Figure 2).
shRNA‐mediated knockdown of L13a in myeloid cells inactivates the GAIT system and rescues target mRNA translation.76 Most protein synthetic functions are not disturbed by L13a knockdown as shown by the lack of effect on rRNA processing, polysome formation, or global translation. However, knockdown significantly reduces rRNA methylation as well as cap‐independent translation of several mRNAs containing IRES elements. These alternative activities of L13a potentially confound interpretation of genetic knockdown experiments.
L13a Phosphorylation by DAPK–ZIPK is Autoregulated by Negative Feedback
Approximately 12–16‐h delay before DAPK and ZIPK activation is likely due to a complex transcriptional upregulation of DAPK possibly involving CCAAT/enhancer‐binding protein (C/EBP)‐β transcription factor.77, 78 Despite continued elevated mRNA levels, DAPK and ZIPK protein levels decline markedly soon after L13a phosphorylation, consistent with translational silencing. Remarkably, both DAPK and ZIPK bear stem‐loop structures in their 3′‐UTR similar to that of Cp and VEGFA GAIT elements, and translational silencing by both elements was confirmed experimentally.18 The attenuation of the expression of both kinases by the GAIT complex establishes an auto‐regulatory feedback loop, in which the kinases that activate the pathway are themselves regulated by the same pathway. Thus, DAPK–ZIPK–L13a axis constitutes an endogenous negative feedback module that limits the negative regulation of inflammatory gene expression by the GAIT system. DAPK and ZIPK are established regulators of programmed cell death pathways.79 However, the possibility that GAIT‐mediated translational repression of the kinases contributes to regulation of apoptosis has not been explored. A similar regulatory mechanism has been reported for the cytosolic tyrosine kinase, c‐Src.80 Upon phosphorylation by c‐Src, phospho‐hnRNP K binds the c‐Src mRNA 3′‐UTR and inhibits the expression during erythroid maturation. These examples of repression of kinase mRNA translation by their substrate represent a unique ‘network motif’ in regulation of gene expression.
THE 3′‐UTR GAIT RNA ELEMENT
The mature GAIT complex binds a cis‐acting element featuring unique structural characteristics in target mRNA.81 A GAIT element was first identified in the 3′‐UTR of human Cp mRNA by deletion and site‐directed mutagenesis‐based mapping.14 The minimal 29‐nt GAIT element was sufficient to confer robust translational silencing to a heterologous reporter transcript. The Cp GAIT element has a bipartite stem‐loop structure consisting of a 5‐nt terminal loop, a 3‐bp helix, an asymmetric internal bulge, and a proximal 6‐bp helical stem (Figure 5). Recognition of the GAIT element by the GAIT complex largely depends on the structure, not the primary sequence, with the exception of two relatively invariant nucleotides in the internal bulge. There appears to be substantial positional tolerance as the distance between the GAIT element and termination codon or poly(A) tail has little effect on activity. To identify additional GAIT element‐bearing mRNAs, the PatSearch pattern‐matching algorithm was used to mine UTRdb, a nonredundant 3′‐UTR database, based on the required sequence and secondary structure features of the Cp GAIT element.82 The query revealed GAIT element‐like patterns in 52 human 3′‐UTRs, and several have been functionally validated, e.g., VEGFA and DAPK1, as well as an atypical GAIT element in ZIPK (DAPK3) that contains a symmetric bulge in the proximal stem and a 4–6‐bp distal stem.18, 82. Posttranscriptional base modifications, such as N6‐methyladenosine or pseudouridinylation, in the GAIT element certainly could influence element structure and function, likewise, single nucleotide polymorphisms might prevent (or permit) GAIT element formation.83 These intriguing possibilities could influence condition‐dependent GAIT‐mediated translational repression, but they have not yet been investigated.
Figure 5.
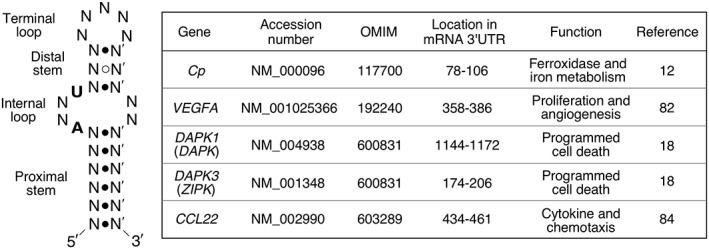
Sequence and structure characteristics of a typical bipartite stem‐loop interferon γ‐activated inhibitor of translation (GAIT) element with conserved nucleotides in bold (left), and examples of validated functional GAIT elements (right). The structure is derived from the biochemical and mutagenesis experiments on human 3′‐UTR Cp GAIT element.
In a separate genome‐wide approach, delayed repression of mRNAs in IFN‐γ‐treated human U937 monocytic cells was determined by polysome profiling coupled with microarray analysis.84 Transcripts encoding several chemokines and chemokine receptors were identified, including CCL22, APOL2, CCR3, CCR4, CCR6, and CXCL13. RNA secondary structure prediction algorithms identified putative GAIT elements in the 3′‐UTR of these mRNAs. Translational repression by the atypical GAIT element in CCL22 mRNA was confirmed experimentally (Figure 5). Future analysis employing state‐of‐the‐art technologies such as CLIP‐ (ultraviolet crosslinking coupled with immunoprecipitation) or RIP‐ (ribonucleoprotein‐immunoprecipitation) Seq with antibody against EPRS, the sole RNA binding constituent of the GAIT complex, will likely provide a more complete profile of the GAIT complex‐bound transcripts, and additional insights into GAIT system function.
REGULATION AND DYSREGULATION OF GAIT PATHWAY
The GAIT system is subject to endogenous checks and dysregulation by environmental conditions (Figure 6). These regulatory mechanisms are discussed in detail in the following sections.
Figure 6.
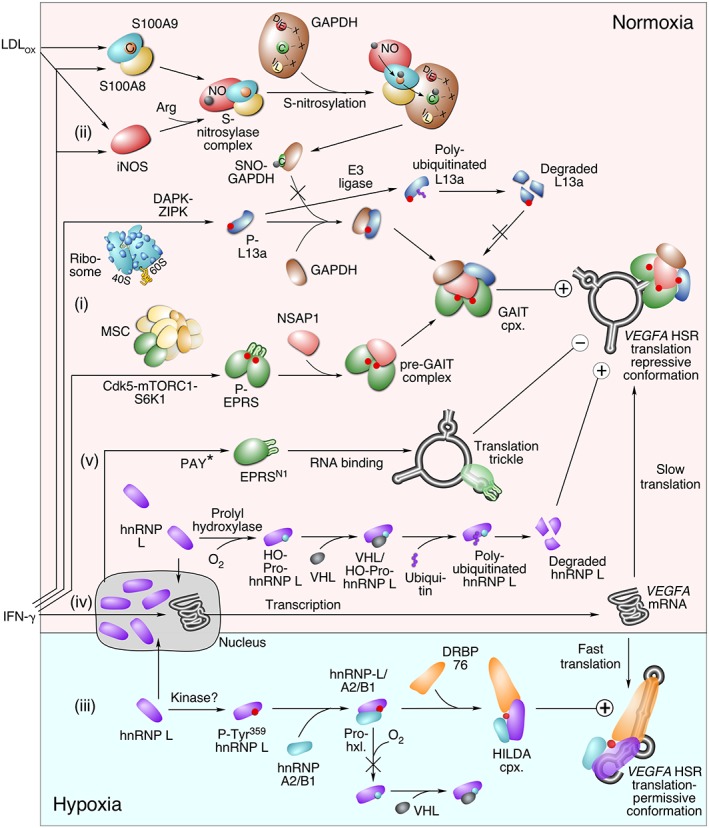
Regulation of interferon γ‐activated inhibitor of translation (GAIT) system in normoxic and hypoxic macrophages, and control of GAIT element‐bearing VEGFA mRNA. (i) The GAIT system is activated by IFN‐γ‐dependent phosphorylation of EPRS and L13a in myeloid cells. (ii) Stimulation with both IFN‐γ and oxidatively modified low density lipoprotein, LDL (LDLox) induces activation of an iNOS‐S100A8/A9 nitrosylase complex and site‐specific S‐nitrosylation of an array of targets bearing an I/L‐X‐C‐X2‐D/E motif, including Cys247 of glyceraldehyde‐3‐phosphate dehydrogenase (GAPDH). SNO‐GAPDH fails to bind L13a causing proteasomal degradation of the latter and inactivation of the GAIT system. (iii) In hypoxia, IFN‐γ‐induced hnRNP L is phosphorylated at Tyr359 and joins hnRNP A2/B1 and double‐stranded RNA‐binding protein (RBP), DRBP76, to form the active HILDA complex. The complex binds the hypoxia stability region (HSR) in VEGFA mRNA, blocks GAIT element conformation, and switches to translation‐permissive conformation. (iv) In normoxia, translational repression of IFN‐γ‐induced VEGFA mRNA by the GAIT complex is facilitated by degradation of hnRNP L following prolyl hydroxylation and von Hippel‐Lindau (VHL)‐mediated polyubiquitination. (v) Constitutive generation of an EPRS truncate, EPRSN1 by polyadenylation‐directed conversion of a Tyr to a stop codon (PAY*) binds GAIT elements in target mRNAs including VEGFA and shields it from the GAIT complex, thereby allowing a ‘translational trickle’ of expression.
Oxidatively Modified Low Density Lipoprotein Inhibits GAIT System Activity
Chronic inflammation induces expression of reactive oxygen and nitrogen species promoting pathological modifications of lipids and proteins; one example is oxidatively modified low density lipoprotein (LDLox) which accumulates in atherosclerotic lesions.85 Plasma level of LDLox is a strong predictor of acute coronary heart disease events.86 A correlation between expression of LDLox and GAIT targets, e.g., Cp and VEGF‐A, has been observed in atherosclerotic lesions.87, 88 Our studies show that in myeloid cells, IFN‐γ‐induced GAIT complex assembly (Figure 6(i)) is inhibited by LDLox (generated by in vitro treatment of human LDL with the myeloperoxidase/H2O2/NO2 − system of leukocytes) resulting in sustained production and accumulation of GAIT system target proteins, e.g., VEGF‐A, Cp, and others.62 Cell stimulation with IFN‐γ and LDLox (but not native LDL) caused rapid disappearance of nearly the entire cellular complement of L13a without diminishing the levels of other GAIT complex proteins, or other ribosomal proteins. LDLox induces degradation of L13a by a mechanism involving target‐selective, site‐specific S‐nitrosylation of GAPDH that fails to bind L13a (Figure 6(ii)). S‐nitrosylation is a ubiquitous, reversible protein modification involving covalent addition of a reactive nitric oxide (NO) moiety to Cys thiols.89 Unprotected L13a is polyubiquitinated and degraded by the proteasome system, thereby preventing assembly of a functional GAIT complex.62 A protective function of GAPDH was verified by siRNA‐mediated GAPDH depletion that also caused L13a degradation following its IFN‐γ‐induced, phosphorylation‐dependent release from the ribosome.
Human GAPDH is a homotetramer with three Cys in each monomeric subunit, each susceptible to S‐nitrosylation by chemical NO donors.90, 91 Bacterial lipopolysaccharide (LPS) induces site‐specific S‐nitrosylation at GAPDH Cys152 near the catalytic active site, suppressing glycolytic activity.92 Importantly, treatment of myeloid cells with IFN‐γ plus LDLox triggers S‐nitrosylation of GAPDH at Cys247, thereby preventing GAPDH binding to L13a.62 The site is near the enzyme surface, far from the catalytic site, and nitrosylation does not alter activity. Site‐specific Cys247 S‐nitrosylation requires expression of inducible nitric oxide synthase (iNOS), which in the presence of IFN‐γ plus LDLox, forms a novel, multiprotein S‐nitrosylase complex also containing two members of the S100 calcium‐binding family, S100A8 and S100A9.93 Mass spectrometry‐based proteomic approach identified about 95 candidate targets of the iNOS‐S100A8/A9 nitrosylase complex. In addition to GAPDH, four proteins were inducibly nitrosylated by this complex: annexin 5, ezrin, moesin, and vimentin. Alignment of the candidate sequences revealed a conserved I/L‐X‐C‐X2‐D/E motif that was validated by loss‐ and gain‐of‐function mutagenesis experiments. The discovery of a sequence motif that directs nitrosylation provides an additional parallel to posttranslational modification by phosphorylation—both are stimulus‐inducible, influence protein activity, and can be enzymatically reversed, e.g., by denitrosylases and phosphatases, respectively.
Ribosomal proteins have been implicated in several diseases, i.e., ribosomopathies, including peripheral nerve tumors, diabetes, Diamond‐Blackfan anemia, and in developmental abnormalities such as cleft palate.94, 95, 96, 97 The pathology generally is traceable to genetic alteration or deletion of a ribosomal protein. Degradation of L13a is unique in that its dysregulation is not at the genetic level, but is rather a consequence of stimulus‐dependent posttranslational modification of a distinct protein, in this case, GAPDH. Also unusual is the finding that the extraribosomal activity of L13a is subject to dysregulation. The potential role of extraribosomal functions in other ribosomopathies has been considered, e.g., in Diamond‐Blackfan anemia, but not yet established.65, 95
Regulation of VEGF‐A Expression by a Stress‐Inducible RNA Element Switch
The activity of the GAIT system can be modulated by condition‐dependent binding of other RBPs or complexes to the 3′‐UTR of GAIT element‐bearing mRNAs.7, 98 The best‐studied example is VEGFA mRNA that features a condition‐ and protein‐dependent RNA switch in the 3′‐UTR (Figure 6). Normoxia and hypoxia induce distinct conformational structures in the VEGFA 3′‐UTR driven by alternative binding of the GAIT complex and an hnRNP L‐containing complex, respectively.82, 99 When myeloid cells are stimulated with IFN‐γ under hypoxic conditions, hnRNP L is phosphorylated at Tyr359 and accumulates in the cytoplasm in a complex with hnRNP A2/B1 and DRBP76 (double‐stranded RBP) (Figure 6(iii)).100 The hnRNP L in the HILDA (hypoxia‐induced hnRNP L‐DRBP76‐hnRNP A2/B1) complex binds a CA‐rich element directly adjacent to the GAIT element, and DRBP76 binds a downstream AU‐rich stem‐loop thereby disrupting GAIT element formation, preventing GAIT complex binding, and enhancing VEGFA mRNA translation. In contrast, under normoxic condition, IFN‐γ induces prolyl hydroxylation of cytoplasmic hnRNP L, followed by pVHL (von Hippel‐Lindau)‐mediated ubiquitination and degradation by the proteasome system (Figure 6(iv)). Degradation of hnRNP L in normoxia permits GAIT complex binding to the GAIT element and represses VEGFA mRNA translation. Within the HILDA complex, hnRNP L acts as the ‘selector’ that recognizes and binds CA‐rich elements in mRNAs, thereby determining target specificity. DRBP76 acts as the ‘flipper’ that alters the conformation of the VEGFA 3′‐UTR to prevent GAIT complex binding. A comprehensive search for other target mRNAs regulated by this RNA switch is ongoing in our laboratory. A plethora of metabolite‐driven conformational alterations in RNA that regulate gene expression in bacteria, i.e., riboswitches, have been described.101 However, very limited information is available on protein‐driven RNA switches in eukaryotes, such as the one described here for VEGFA mRNA, and certainly offers much scope for discovery.
EPRSN1 Prevents Complete Translation Inhibition by GAIT Complex
Basal expression of GAIT element‐bearing transcripts is maintained despite the abundance of active GAIT complex sufficient for complete inhibition of translation. Dynamic model simulations predicted an inhibitory GAIT‐element‐interacting, trans‐acting factor that might account for this conundrum. We identified the constitutive presence of low levels of a C‐terminus truncate of EPRS, termed EPRSN1, that contains the RNA‐binding domain, i.e., the upstream pair of WHEP domains, but not the downstream domain required for GAIT complex assembly.27 Thus, EPRSN1 shields a small amount of target transcripts, including VEGFA and ZIPK mRNAs, from GAIT complex‐mediated translation‐repression, thereby permitting continuous, leaky translation of the target transcripts at a very low rate that we termed a ‘translational trickle’ (Figure 6(v)). Investigation into the mechanism of EPRSN1 generation revealed an unexpected twist on alternative polyadenylation (APA). Polyadenylation normally occurs downstream of the coding region to terminate the 3′‐UTR. APA events most often alter 3′‐UTR lengths, but also can occur within introns to alter splicing events yielding an alternative coding region.102 We observed a cryptic polyadenylation signal within the EPRS coding sequence consisting of an UA cleavage site within an UAU codon specifying Tyr, an upstream AAUAAA polyadenylation element, and a downstream U‐rich element. During generation of EPRSN1, the UA cleavage site is polyadenylated to convert the UAU codon to the UAA stop codon.27 Thus, polyadenylation‐directed conversion of a Tyr codon to a stop codon (PAY*) results in a truncated, but translatable, transcript in which the newly generated stop codon is followed directly by a polyA tail without an intervening 3′‐UTR. The low efficiency of EPRSN1 mRNA generation might be due to sub‐optimal distances between the polyadenylation elements, or possibly to interference by downstream secondary sequences or structures that interfere with the PAY* process.103, 104 Global analyses have indicated several APA events within coding regions, however, they most often generate truncated transcripts lacking a stop codon, and are subject to nonstop mRNA decay.105, 106, 107 The PAY* process discovered in EPRSN1 not only reveals a novel cellular mechanism for regulating gene expression, but it also provides a new genetic mechanism for transcriptome and proteome expansion.
Additional human PAY* targets were sought by mining mRNA and expressed sequence tag databases using two criteria: (1) the presence of a coding RNA that has a poly(A) tail immediately following the first UAA stop codon and (2) this UAA stop codon replaces a Tyr‐encoding UAU or UAC in a larger transcript that extends past the truncated mRNA; this analysis revealed 7 candidate PAY* targets.27 Ribonucleotide reductase M1 (RRM1) mRNA was validated as an authentic PAY* target. Prediction based on classical APA signal elements and distance constraints between elements indicated many putative coding region APA events, comprising about ~0.7% of human mRNAs.106 The generation of EPRSN1 appears to be primate‐specific, but the PAY* mechanism is evolutionarily conserved. A bioinformatics analysis in yeast identified 28 mRNAs apparently generated by the PAY* mechanism.108 The in vivo significance of truncated versions of EPRS, or other protein products of the PAY* mechanism, is not known at this time. Possibly, the PAY* mechanism generates protein isoforms with alternate activity, including the dominant‐negative function observed for EPRSN1. As an example, conditional deletion of macrophage‐derived VEGFA markedly accelerates tumorigenesis.109 We speculate that the translational trickle directed by EPRSN1 maintain low levels of VEGF‐A, and other GAIT complex targets, which increases cell and organismal survival and well‐being.
PHYSIOLOGICAL FUNCTION OF THE GAIT SYSTEM
Inflammatory response during host defense and wound healing induces activation of macrophages to release multiple factors injurious to invasive organisms. However, sustained inflammation contributes importantly to initiation and progression of chronic diseases such as cancer, atherosclerosis, and pulmonary disease, among others.85, 110 Several transcriptional mechanisms have been described that limit expression of inflammatory proteins and accumulation of toxic products in the process termed ‘resolution of inflammation.’111 The delayed inhibition of expression of multiple inflammation‐related proteins by the GAIT complex suggests that this system represent a posttranscriptional off‐switch that also contributes to inflammation‐resolution.10 For example, accumulation of Cp oxidase activity is a risk factor for pathogenesis of atherosclerosis.112 Likewise, uncontrolled production of several GAIT targets, e.g., the angiogenic factor VEGF‐A and cytokines including CCL22 and CCR3, contributes to inflammatory disease progression.113, 114 Genetic or environmental defects in GAIT system proteins might contribute to disease progression. Indeed, defects in several GAIT system‐related genes including GAPDH, Cdk5 and its putative inhibitor, CDKAL1, DAPK, mTORC1, and S6K1 are associated with diseases in which inflammation has a critical function, e.g., type 2 diabetes and Alzheimer's.115, 116, 117, 118, 119, 120, 121, 122
Evidence to support the role of the GAIT system in inflammation‐resolution in vivo has been provided by several studies using genetically modified mice with a macrophage‐specific deletion of L13a.123 Unchallenged mice are healthy with no apparent phenotypic changes, consistent with the concept that the protein is dispensable for the canonical function of ribosomes, i.e., global protein synthesis. Upon challenge with LPS, the mice exhibited multiple symptoms of severe inflammation including widespread macrophage infiltration of organs, tissue injury, and reduced survival. Macrophages from Rpl13a‐null mice exhibited increased expression of several chemokines that are GAIT complex targets. A second mouse model took advantage of the propensity of apolipoprotein E (apoE)‐knockout mice fed a high‐fat (‘Western’) diet to develop severe atherosclerosis.124 Importantly, apoE‐deficient macrophages are a major contributor to atherosclerotic lesion formation.125 Myeloid cell‐specific Rpl13a knockout mice were bred with apoE‐null mice and fed a high‐fat diet.126 Double knockout mice exhibited significantly greater atherosclerosis as shown by increased lesion cross‐section in the aortic root and greater lesion area in the aortic tree. Elevated levels of plasma cytokines were also observed. In other experiments, the role of L13a in experimental colitis was investigated.127 Administration of dextran sodium sulfate to myeloid‐specific Rpl13a knockout mice exacerbated colitis.
The results from experiments using myeloid cell‐specific Rpl13a knockout mice are consistent with the hypothesis that the GAIT system contributes to the resolution of inflammation, and limits chronic inflammatory pathology. However, there are important limitations to this approach as the L13a gene locus and L13a protein specify several functions. Depletion of Rpl13a will inhibit non‐GAIT‐related, alternate functions of L13a, e.g., rRNA methylation and cap‐independent translation of IRES‐containing mRNAs.76 Also, confounding the Rpl13a gene knockout approach is the presence of four snoRNAs encoded by box C/D genes within the introns of the Rpl13a locus. Deletion of essentially the entire Rpl13a gene, as was done in the experiments described here, might have also deleted the snoRNAs that have been shown to confer resistance to lipotoxic and oxidative stress in vitro.72 Importantly, deletion of the Rpl13a snoRNAs in a mouse model, while leaving the coding and regulatory regions intact, profoundly influenced mitochondrial metabolism, resulting in enhanced systemic glucose tolerance and protection against oxidative stress.128 In view of these potentially confounding functions of the Rpl13a gene locus and protein, the development of alternative in vivo models is required to confirm the role of the GAIT system in inflammation‐resolution. We have generated mice with Ser999‐to‐Ala mutation in the Eprs gene and thus defective in stimulus‐dependent phosphorylation required for EPRS activation and participation in the GAIT complex.17 Unexpectedly, these mice exhibit low body weight, reduced fat mass, and longer life span comparable to mice with defects in the upstream mTORC1‐S6K1 signaling axis that phosphorylates EPRS at Ser999. The phospho‐deficient mice do not exhibit an inflammatory phenotype as determined by cytokine levels in serum, but they have not yet been challenged with pro‐inflammatory stimuli. Phospho‐defective EPRS mice will be helpful for investigating GAIT system activity, however, the metabolic phenotype, and other noncanonical functions of EPRS, might confound interpretation of inflammation‐related parameters. Possibly, the development of phospho‐defective L13a mice will provide the most specific and reliable information on the role of the GAIT system in inflammatory responses.
EVOLUTION AND CONSERVATION OF THE GAIT SYSTEM
Both EPRS linker phosphorylation sites are conserved in mammals from opossum to primates (except for the Ser886 site that is absent in mice), and the essential Ser phosphorylation site (Ser77 in humans) in L13a is conserved from yeast to human.13, 18 However, a functional GAIT system has been experimentally validated only in mice and humans. The human and mouse GAIT systems differ as the latter lacks NSAP1 in the GAIT complex and its phosphorylation‐mediated binding site in the EPRS linker.13 This species‐specific difference is not completely unexpected, particularly in innate immunity systems. For example, macrophages and other immune cells in the two species, often respond differently to pro‐inflammatory stimuli.52, 129 These species might have acquired these disparate functional differences in both orthologous and paralogous genes due to (1) divergence ~65–75 million years ago and (2) more importantly, dissimilar ecological niches with distinct pathogenic challenges. The targets of the GAIT complex also reveal species differences. A pattern search for Cp‐like GAIT elements in an UTR database suggests their presence in multiple nonhuman mammalian species, e.g., sheep and baboons, but it appears to be absent in rodent Cp mRNA.14, 82 Likewise, the same search suggests that mouse VEGFA mRNA lacks a GAIT element. However, translational repression of VEGFA mRNA is observed in both mice and humans.99 These observations suggest that the mouse and human GAIT complexes recognize different structural elements, and our ability to bioinformatically detect GAIT element is limited due to the structural diversity.
In the human GAIT system, the upstream pair of EPRS WHEP domains are required for binding GAIT element‐bearing RNA targets. The presence of WHEP domains in EPRS is conserved from unicellular filasterean animal‐like organisms to multicellular mammals; the sole exception is the WHEP‐less EPRS found in several parasitic protostomes.44 One informative example is the stinging sea anemone N. vectensis which expresses three alternatively spliced mRNAs from the EPRS gene; two mRNAs encode EPRS bearing a single WHEP domain in the linker, whereas the third encodes a form bearing two adjacent WHEP domains.45 Binding studies of expressed N. vectensis EPRS linkers revealed one of the 1‐WHEP isoforms bound tRNA with high affinity, but the 2‐WHEP isoform bound human Cp GAIT element RNA with high affinity. A third isoform with a unique single WHEP domain binds both RNA classes with intermediate affinity. One can surmise the evolutionary history of EPRS function from the unusually complex splicing pattern that generated these isoforms. Possibly, a linked EPRS bearing a single WHEP domain evolved to insure the availability of tRNA to the adjacent synthetases for increased charging efficiency. Later duplication of the WHEP domain permitted additional RNA‐binding characteristics such as GAIT element‐binding, and perhaps other regulatory RNAs. Studies to test the role of these isoforms in regulating N. vectensis gene expression have not been done. However, it is tempting to speculate that an early version of the GAIT system originated in a closely related ancestor of N. vectensis and has persisted in metazoans for hundreds of millions of years. There is no experimental evidence for the presence of GAIT function in more complex, i.e., bilaterial, metazoans other than mice and humans. The urochordate sea squirt Ciona intestinalis exhibits a structural equivalent of a GAIT element in the 3′‐UTR of a CAP gene member of the cysteine‐rich secretory protein, antigen 5 and pathogenesis‐related 1 superfamily thought to be involved in modulation of host immune responses.130 Interestingly, formation of the GAIT is regulated, and only forms when the organism is challenged with LPS, consistent with a role in innate immunity. Genome sequencing of C. intestinalis revealed that EPRS mRNA contains two WHEP domains comparable to the RNA‐binding, upstream pair of WHEP domains in the human ortholog, suggesting a potential GAIT element‐binding activity of EPRS and GAIT‐mediated innate immune function.44 Finally, a GAIT‐like RNA sequence has been described in the 3′ terminus of the transmissible Gastroenteritis coronavirus genome, with a possible function in modulating innate immunity.131
CONCLUSIONS AND FUTURE PERSPECTIVES
Much progress has been made following the original observation of delayed inhibition of Cp mRNA translation in IFN‐γ‐treated monocytes in 1997 that led to the discovery of the GAIT system.132 However, it is likely that we are still near the ‘tip of the iceberg’ as many issues remain unexplored. Prominent among these are questions at the molecular level such as the mechanism of release of phospho‐EPRS and phospho‐L13a from their parental macromolecular complexes, and the specific role of phosphorylation in these events. Although, the IFN‐γ‐activated proximal kinases that phosphorylate EPRS and L13a have been elucidated, our knowledge of the upstream activation pathways that coordinate early and delayed phosphorylation of EPRS and L13a, respectively, is limited. Also, the unexpected requirement of both Cdk5/p35 and S6K1 (as well as mTORC1) in EPRS phosphorylation remains unclear.16, 17, 133 The early formation of the pre‐GAIT complex about 12 h before formation of the functional GAIT complex remains puzzling, and its function, if it has one, is completely undefined. We also have very limited structural information about the protein–protein interactions within the GAIT, HILDA and S100A8/A9‐iNOS complexes, and their interactions with targets. Additionally, the potential contribution of microRNAs and other noncoding RNAs to GAIT system activity has not been explored.
In addition to these important mechanistic knowledge gaps, our understanding of the physiological role of the GAIT system remains uncertain. For example, the potential role of the GAIT complex in macrophage switching between classically‐ (M1) or alternatively‐ (M2) activated states remains unexplored despite its likely significance in clarifying the role of the GAIT system in inflammation‐resolution. Furthermore, the discovery of GAIT complex activation in other immune cells as well as in nonmyeloid cells by non‐IFN‐γ agonists would certainly alter our understanding of the significance of the system. Also, the identification and verification of new GAIT element‐bearing target mRNAs should provide additional insight. Additional genetic animal models should prove helpful. As described above, the important studies using myeloid‐specific, L13a‐deficient mice are consistent with an anti‐inflammatory role of the GAIT system, however, their interpretation is confounded by the other moonlighting activities of L13a, and by the presence of snoRNAs within the RPL13a gene itself.72, 73, 76, 128 Likewise, phospho‐deficient EPRS mice exhibit strong, potentially confounding phenotypes unrelated to inflammation.17 Possibly, a mouse with a mutation in the RPL13a gene that prevents release from the ribosome and GAIT complex formation, but still permits rRNA methylation, would be informative. A critical void is the absence of information on the role of the GAIT system in regulating inflammation and inflammatory disease in humans. Single nucleotide polymorphisms in genes that influence GAIT complex constituents or GAIT elements in target mRNAs have not been reported. Likewise, the roles of environmental effectors of GAIT system activity such as LDLox or hypoxia have not been investigated in vivo, either in humans or in mice. The regulation and dysregulation of posttranscriptional mechanisms contributing to the resolution of inflammation is a critical, under‐investigated area. We believe that a deeper understanding of the ‘off‐switch’ provided by the GAIT system, both at the mechanistic and physiological levels, will provide new avenues and opportunities for development of novel therapies against chronic inflammatory diseases.
ACKNOWLEDGMENTS
This work was supported by NIH grants P01HL029582, P01HL076491, R01GM086430, and R01GM115476 to P.L.F., by Wellcome Trust‐DBT India Alliance Intermediate Fellowship WT500139/Z/09/Z to P.S.R., and by AHA SDG 10SDG3930003 to A.A.
Conflict of interest: The authors have declared no conflicts of interest for this article.
REFERENCES
- 1. de Nadal E, Ammerer G, Posas F. Controlling gene expression in response to stress. Nat Rev Genet 2011, 12:833–845. [DOI] [PubMed] [Google Scholar]
- 2. Lackner DH, Bahler J. Translational control of gene expression from transcripts to transcriptomes. Int Rev Cell Mol Biol 2008, 271:199–251. [DOI] [PubMed] [Google Scholar]
- 3. Dever TE. Gene‐specific regulation by general translation factors. Cell 2002, 108:545–556. [DOI] [PubMed] [Google Scholar]
- 4. Gebauer F, Hentze MW. Molecular mechanisms of translational control. Nat Rev Mol Cell Biol 2004, 5:827–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Piccirillo CA, Bjur E, Topisirovic I, Sonenberg N, Larsson O. Translational control of immune responses: from transcripts to translatomes. Nat Immunol 2014, 15:503–511. [DOI] [PubMed] [Google Scholar]
- 6. Wilkie GS, Dickson KS, Gray NK. Regulation of mRNA translation by 5′‐ and 3′‐UTR‐binding factors. Trends Biochem Sci 2003, 28:182–188. [DOI] [PubMed] [Google Scholar]
- 7. Jia J, Yao P, Arif A, Fox PL. Regulation and dysregulation of 3′UTR‐mediated translational control. Curr Opin Genet Dev 2013, 23:29–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kafasla P, Skliris A, Kontoyiannis DL. Post‐transcriptional coordination of immunological responses by RNA‐binding proteins. Nat Immunol 2014, 15:492–502. [DOI] [PubMed] [Google Scholar]
- 9. Keene JD. RNA regulons: coordination of post‐transcriptional events. Nat Rev Genet 2007, 8:533–543. [DOI] [PubMed] [Google Scholar]
- 10. Anderson P. Post‐transcriptional regulons coordinate the initiation and resolution of inflammation. Nat Rev Immunol 2010, 10:24–35. [DOI] [PubMed] [Google Scholar]
- 11. Mukhopadhyay R, Jia J, Arif A, Ray PS, Fox PL. The GAIT system: a gatekeeper of inflammatory gene expression. Trends Biochem Sci 2009, 34:324–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sampath P, Mazumder B, Seshadri V, Gerber CA, Chavatte L, Kinter M, Ting SM, Dignam JD, Kim S, Driscoll DM, et al. Noncanonical function of glutamyl‐prolyl‐tRNA synthetase: gene‐specific silencing of translation. Cell 2004, 119:195–208. [DOI] [PubMed] [Google Scholar]
- 13. Arif A, Chatterjee P, Moodt RA, Fox PL. Heterotrimeric GAIT complex drives transcript‐selective translation inhibition in murine macrophages. Mol Cell Biol 2012, 32:5046–5055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sampath P, Mazumder B, Seshadri V, Fox PL. Transcript‐selective translational silencing by gamma interferon is directed by a novel structural element in the ceruloplasmin mRNA 3′ untranslated region. Mol Cell Biol 2003, 23:1509–1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Arif A, Jia J, Mukhopadhyay R, Willard B, Kinter M, Fox PL. Two‐site phosphorylation of EPRS coordinates multimodal regulation of noncanonical translational control activity. Mol Cell 2009, 35:164–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Arif A, Jia J, Moodt RA, DiCorleto PE, Fox PL. Phosphorylation of glutamyl‐prolyl tRNA synthetase by cyclin‐dependent kinase 5 dictates transcript‐selective translational control. Proc Natl Acad Sci U S A 2011, 108:1415–1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Arif A, Terenzi F, Potdar AA, Jia J, Sacks J, China A, Halawani D, Vasu K, Li X, Brown JM, et al. EPRS is a critical mTORC1‐S6K1 effector that influences adiposity in mice. Nature 2017, 542:357–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mukhopadhyay R, Ray PS, Arif A, Brady AK, Kinter M, Fox PL. DAPK‐ZIPK‐L13a axis constitutes a negative‐feedback module regulating inflammatory gene expression. Mol Cell 2008, 32:371–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kim JW, Dang CV. Multifaceted roles of glycolytic enzymes. Trends Biochem Sci 2005, 30:142–150. [DOI] [PubMed] [Google Scholar]
- 20. Mazumder B, Sampath P, Seshadri V, Maitra RK, DiCorleto PE, Fox PL. Regulated release of L13a from the 60S ribosomal subunit as a mechanism of transcript‐specific translational control. Cell 2003, 115:187–198. [DOI] [PubMed] [Google Scholar]
- 21. Ray PS, Arif A, Fox PL. Macromolecular complexes as depots for releasable regulatory proteins. Trends Biochem Sci 2007, 32:158–164. [DOI] [PubMed] [Google Scholar]
- 22. Beadle GW, Tatum EL. Genetic control of biochemical reactions in neurospora. Proc Natl Acad Sci U S A 1941, 27:499–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Huberts DH, van der Klei IJ. Moonlighting proteins: an intriguing mode of multitasking. Biochim Biophys Acta 2010, 1803:520–525. [DOI] [PubMed] [Google Scholar]
- 24. Jeffery CJ. Proteins with neomorphic moonlighting functions in disease. IUBMB Life 2011, 63:489–494. [DOI] [PubMed] [Google Scholar]
- 25. Yang XL. Structural disorder in expanding the functionome of aminoacyl‐tRNA synthetases. Chem Biol 2013, 20:1093–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lo WS, Gardiner E, Xu Z, Lau CF, Wang F, Zhou JJ, Mendlein JD, Nangle LA, Chiang KP, Yang XL, et al. Human tRNA synthetase catalytic nulls with diverse functions. Science 2014, 345:328–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yao P, Potdar AA, Arif A, Ray PS, Mukhopadhyay R, Willard B, Xu Y, Yan J, Saidel GM, Fox PL. Coding region polyadenylation generates a truncated tRNA synthetase that counters translation repression. Cell 2012, 149:88–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ribas de Pouplana L, Schimmel P. Aminoacyl‐tRNA synthetases: potential markers of genetic code development. Trends Biochem Sci 2001, 26:591–596. [DOI] [PubMed] [Google Scholar]
- 29. Ibba M, Soll D. Aminoacyl‐tRNAs: setting the limits of the genetic code. Genes Dev 2004, 18:731–738. [DOI] [PubMed] [Google Scholar]
- 30. Rho SB, Lee JS, Jeong EJ, Kim KS, Kim YG, Kim S. A multifunctional repeated motif is present in human bifunctional tRNA synthetase. J Biol Chem 1998, 273:11267–11273. [DOI] [PubMed] [Google Scholar]
- 31. Guo M, Yang XL, Schimmel P. New functions of aminoacyl‐tRNA synthetases beyond translation. Nat Rev Mol Cell Biol 2010, 11:668–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Cho HY, Maeng SJ, Cho HJ, Choi YS, Chung JM, Lee S, Kim HK, Kim JH, Eom CY, Kim YG, et al. Assembly of multi‐tRNA synthetase complex via heterotetrameric glutathione transferase‐homology domains. J Biol Chem 2015, 290:29313–29328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Mirande M. The aminoacyl‐tRNA synthetase complex. Subcell Biochem 2017, 83:505–522. [DOI] [PubMed] [Google Scholar]
- 34. David A, Netzer N, Strader MB, Das SR, Chen CY, Gibbs J, Pierre P, Bennink JR, Yewdell JW. RNA binding targets aminoacyl‐tRNA synthetases to translating ribosomes. J Biol Chem 2011, 286:20688–20700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kyriacou SV, Deutscher MP. An important role for the multienzyme aminoacyl‐tRNA synthetase complex in mammalian translation and cell growth. Mol Cell 2008, 29:419–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lee EY, Lee HC, Kim HK, Jang SY, Park SJ, Kim YH, Kim JH, Hwang J, Kim JH, Kim TH, et al. Infection‐specific phosphorylation of glutamyl‐prolyl tRNA synthetase induces antiviral immunity. Nat Immunol 2016, 17:1252–1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Arif A, Jia J, Halawani D, Fox PL. Experimental approaches for investigation of aminoacyl tRNA synthetase phosphorylation. Methods 2017, 113:72–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Katsyv I, Wang M, Song WM, Zhou X, Zhao Y, Park S, Zhu J, Zhang B, Irie HY. EPRS is a critical regulator of cell proliferation and estrogen signaling in ER+ breast cancer. Oncotarget 2016, 7:69592–69605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Guo M, Schimmel P. Essential nontranslational functions of tRNA synthetases. Nat Chem Biol 2013, 9:145–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kim S, You S, Hwang D. Aminoacyl‐tRNA synthetases and tumorigenesis: more than housekeeping. Nat Rev Cancer 2011, 11:708–718. [DOI] [PubMed] [Google Scholar]
- 41. Jia J, Arif A, Ray PS, Fox PL. WHEP domains direct noncanonical function of glutamyl‐prolyl tRNA synthetase in translational control of gene expression. Mol Cell 2008, 29:679–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kapasi P, Chaudhuri S, Vyas K, Baus D, Komar AA, Fox PL, Merrick WC, Mazumder B. L13a blocks 48S assembly: role of a general initiation factor in mRNA‐specific translational control. Mol Cell 2007, 25:113–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Castello A, Fischer B, Frese CK, Horos R, Alleaume AM, Foehr S, Curk T, Krijgsveld J, Hentze MW. Comprehensive identification of RNA‐binding domains in human cells. Mol Cell 2016, 63:696–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ray PS, Fox PL. Origin and evolution of glutamyl‐prolyl tRNA synthetase WHEP domains reveal evolutionary relationships within Holozoa. PLoS One 2014, 9:e98493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ray PS, Sullivan JC, Jia J, Francis J, Finnerty JR, Fox PL. Evolution of function of a fused metazoan tRNA synthetase. Mol Biol Evol 2011, 28:437–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Geuens T, Bouhy D, Timmerman V. The hnRNP family: insights into their role in health and disease. Hum Genet 2016, 135:851–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Bannai H, Fukatsu K, Mizutani A, Natsume T, Iemura S, Ikegami T, Inoue T, Mikoshiba K. An RNA‐interacting protein, SYNCRIP (heterogeneous nuclear ribonuclear protein Q1/NSAP1) is a component of mRNA granule transported with inositol 1,4,5‐trisphosphate receptor type 1 mRNA in neuronal dendrites. J Biol Chem 2004, 279:53427–53434. [DOI] [PubMed] [Google Scholar]
- 48. Svitkin YV, Yanagiya A, Karetnikov AE, Alain T, Fabian MR, Khoutorsky A, Perreault S, Topisirovic I, Sonenberg N. Control of translation and miRNA‐dependent repression by a novel poly(A) binding protein, hnRNP‐Q. PLoS Biol 2013, 11:e1001564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Park SM, Paek KY, Hong KY, Jang CJ, Cho S, Park JH, Kim JH, Jan E, Jang SK. Translation‐competent 48S complex formation on HCV IRES requires the RNA‐binding protein NSAP1. Nucleic Acids Res 2011, 39:7791–7802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Blanc V, Navaratnam N, Henderson JO, Anant S, Kennedy S, Jarmuz A, Scott J, Davidson NO. Identification of GRY‐RBP as an apolipoprotein B RNA‐binding protein that interacts with both apobec‐1 and apobec‐1 complementation factor to modulate C to U editing. J Biol Chem 2001, 276:10272–10283. [DOI] [PubMed] [Google Scholar]
- 51. Mourelatos Z, Abel L, Yong J, Kataoka N, Dreyfuss G. SMN interacts with a novel family of hnRNP and spliceosomal proteins. EMBO J 2001, 20:5443–5452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Mestas J, Hughes CC. Of mice and not men: differences between mouse and human immunology. J Immunol 2004, 172:2731–2738. [DOI] [PubMed] [Google Scholar]
- 53. Seidler NW. Basic biology of GAPDH. Adv Exp Med Biol 2013, 985:1–36. [DOI] [PubMed] [Google Scholar]
- 54. Zhang JY, Zhang F, Hong CQ, Giuliano AE, Cui XJ, Zhou GJ, Zhang GJ, Cui YK. Critical protein GAPDH and its regulatory mechanisms in cancer cells. Cancer Biol Med 2015, 12:10–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Donnelly RP, Finlay DK. Glucose, glycolysis and lymphocyte responses. Mol Immunol 2015, 68:513–519. [DOI] [PubMed] [Google Scholar]
- 56. Nicholls C, Li H, Liu JP. GAPDH: a common enzyme with uncommon functions. Clin Exp Pharmacol Physiol 2012, 39:674–679. [DOI] [PubMed] [Google Scholar]
- 57. Chang CH, Curtis JD, Maggi LB Jr, Faubert B, Villarino AV, O'Sullivan D, Huang SC, van der Windt GJ, Blagih J, Qiu J, et al. Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell 2013, 153:1239–1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Zheng L, Roeder RG, Luo Y. S phase activation of the histone H2B promoter by OCA‐S, a coactivator complex that contains GAPDH as a key component. Cell 2003, 114:255–266. [DOI] [PubMed] [Google Scholar]
- 59. Rodriguez‐Pascual F, Redondo‐Horcajo M, Magan‐Marchal N, Lagares D, Martinez‐Ruiz A, Kleinert H, Lamas S. Glyceraldehyde‐3‐phosphate dehydrogenase regulates endothelin‐1 expression by a novel, redox‐sensitive mechanism involving mRNA stability. Mol Cell Biol 2008, 28:7139–7155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Dollenmaier G, Weitz M. Interaction of glyceraldehyde‐3‐phosphate dehydrogenase with secondary and tertiary RNA structural elements of the hepatitis A virus 3′ translated and non‐translated regions. J Gen Virol 2003, 84:403–414. [DOI] [PubMed] [Google Scholar]
- 61. Bonafe N, Gilmore‐Hebert M, Folk NL, Azodi M, Zhou Y, Chambers SK. Glyceraldehyde‐3‐phosphate dehydrogenase binds to the AU‐rich 3′ untranslated region of colony‐stimulating factor‐1 (CSF‐1) messenger RNA in human ovarian cancer cells: possible role in CSF‐1 posttranscriptional regulation and tumor phenotype. Cancer Res 2005, 65:3762–3771. [DOI] [PubMed] [Google Scholar]
- 62. Jia J, Arif A, Willard B, Smith JD, Stuehr DJ, Hazen SL, Fox PL. Protection of extraribosomal RPL13a by GAPDH and dysregulation by S‐nitrosylation. Mol Cell 2012, 47:656–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Nomura M. Regulation of ribosome biosynthesis in Escherichia coli and Saccharomyces cerevisiae: diversity and common principles. J Bacteriol 1999, 181:6857–6864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Kim TS, Jang CY, Kim HD, Lee JY, Ahn BY, Kim J. Interaction of Hsp90 with ribosomal proteins protects from ubiquitination and proteasome‐dependent degradation. Mol Biol Cell 2006, 17:824–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Warner JR, McIntosh KB. How common are extraribosomal functions of ribosomal proteins? Mol Cell 2009, 34:3–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Robledo S, Idol RA, Crimmins DL, Ladenson JH, Mason PJ, Bessler M. The role of human ribosomal proteins in the maturation of rRNA and ribosome production. RNA 2008, 14:1918–1929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Ban N, Beckmann R, Cate JH, Dinman JD, Dragon F, Ellis SR, Lafontaine DL, Lindahl L, Liljas A, Lipton JM, et al. A new system for naming ribosomal proteins. Curr Opin Struct Biol 2014, 24:165–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Zhou X, Liao WJ, Liao JM, Liao P, Lu H. Ribosomal proteins: functions beyond the ribosome. J Mol Cell Biol 2015, 7:92–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Kim TH, Leslie P, Zhang Y. Ribosomal proteins as unrevealed caretakers for cellular stress and genomic instability. Oncotarget 2014, 5:860–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Ban N, Nissen P, Hansen J, Moore PB, Steitz TA. The complete atomic structure of the large ribosomal subunit at 2.4 Å resolution. Science 2000, 289:905–920. [DOI] [PubMed] [Google Scholar]
- 71. Ben‐Shem A, Garreau de Loubresse N, Melnikov S, Jenner L, Yusupova G, Yusupov M. The structure of the eukaryotic ribosome at 3.0 Å resolution. Science 2011, 334:1524–1529. [DOI] [PubMed] [Google Scholar]
- 72. Michel CI, Holley CL, Scruggs BS, Sidhu R, Brookheart RT, Listenberger LL, Behlke MA, Ory DS, Schaffer JE. Small nucleolar RNAs U32a, U33, and U35a are critical mediators of metabolic stress. Cell Metab 2011, 14:33–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Das P, Basu A, Biswas A, Poddar D, Andrews J, Barik S, Komar AA, Mazumder B. Insights into the mechanism of ribosomal incorporation of mammalian L13a protein during ribosome biogenesis. Mol Cell Biol 2013, 33:2829–2842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Wells SE, Hillner PE, Vale RD, Sachs AB. Circularization of mRNA by eukaryotic translation initiation factors. Mol Cell 1998, 2:135–140. [DOI] [PubMed] [Google Scholar]
- 75. Mazumder B, Seshadri V, Imataka H, Sonenberg N, Fox PL. Translational silencing of ceruloplasmin requires the essential elements of mRNA circularization: poly(A) tail, poly(A)‐binding protein, and eukaryotic translation initiation factor 4G. Mol Cell Biol 2001, 21:6440–6449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Chaudhuri S, Vyas K, Kapasi P, Komar AA, Dinman JD, Barik S, Mazumder B. Human ribosomal protein L13a is dispensable for canonical ribosome function but indispensable for efficient rRNA methylation. RNA 2007, 13:2224–2237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Gade P, Roy SK, Li H, Nallar SC, Kalvakolanu DV. Critical role for transcription factor C/EBP‐beta in regulating the expression of death‐associated protein kinase 1. Mol Cell Biol 2008, 28:2528–2548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Gade P, Manjegowda SB, Nallar SC, Maachani UB, Cross AS, Kalvakolanu DV. Regulation of the death‐associated protein kinase 1 expression and autophagy via ATF6 requires apoptosis signal‐regulating kinase 1. Mol Cell Biol 2014, 34:4033–4048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Bialik S, Kimchi A. The death‐associated protein kinases: structure, function, and beyond. Annu Rev Biochem 2006, 75:189–210. [DOI] [PubMed] [Google Scholar]
- 80. Ostareck‐Lederer A, Ostareck DH, Cans C, Neubauer G, Bomsztyk K, Superti‐Furga G, Hentze MW. c‐Src‐mediated phosphorylation of hnRNP K drives translational activation of specifically silenced mRNAs. Mol Cell Biol 2002, 22:4535–4543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Mazumder B, Seshadri V, Fox PL. Translational control by the 3′‐UTR: the ends specify the means. Trends Biochem Sci 2003, 28:91–98. [DOI] [PubMed] [Google Scholar]
- 82. Ray PS, Fox PL. A post‐transcriptional pathway represses monocyte VEGF‐A expression and angiogenic activity. EMBO J 2007, 26:3360–3372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Gilbert WV, Bell TA, Schaening C. Messenger RNA modifications: form, distribution, and function. Science 2016, 352:1408–1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Vyas K, Chaudhuri S, Leaman DW, Komar AA, Musiyenko A, Barik S, Mazumder B. Genome‐wide polysome profiling reveals an inflammation‐responsive posttranscriptional operon in gamma interferon‐activated monocytes. Mol Cell Biol 2009, 29:458–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Libby P, Ridker PM, Maseri A. Inflammation and atherosclerosis. Circulation 2002, 105:1135–1143. [DOI] [PubMed] [Google Scholar]
- 86. Meisinger C, Baumert J, Khuseyinova N, Loewel H, Koenig W. Plasma oxidized low‐density lipoprotein, a strong predictor for acute coronary heart disease events in apparently healthy, middle‐aged men from the general population. Circulation 2005, 112:651–657. [DOI] [PubMed] [Google Scholar]
- 87. Makedou KG, Mikhailidis DP, Makedou A, Iliadis S, Kourtis A, Vavatsi‐Christaki N, Papageorgiou GE. Lipid profile, low‐density lipoprotein oxidation and ceruloplasmin in the progeny of families with a positive history of cardiovascular diseases and/or hyperlipidemia. Angiology 2009, 60:455–461. [DOI] [PubMed] [Google Scholar]
- 88. Ramos MA, Kuzuya M, Esaki T, Miura S, Satake S, Asai T, Kanda S, Hayashi T, Iguchi A. Induction of macrophage VEGF in response to oxidized LDL and VEGF accumulation in human atherosclerotic lesions. Arterioscler Thromb Vasc Biol 1998, 18:1188–1196. [DOI] [PubMed] [Google Scholar]
- 89. Hess DT, Matsumoto A, Kim SO, Marshall HE, Stamler JS. Protein S‐nitrosylation: purview and parameters. Nat Rev Mol Cell Biol 2005, 6:150–166. [DOI] [PubMed] [Google Scholar]
- 90. Hara MR, Agrawal N, Kim SF, Cascio MB, Fujimuro M, Ozeki Y, Takahashi M, Cheah JH, Tankou SK, Hester LD, et al. S‐nitrosylated GAPDH initiates apoptotic cell death by nuclear translocation following Siah1 binding. Nat Cell Biol 2005, 7:665–674. [DOI] [PubMed] [Google Scholar]
- 91. Nakajima H, Amano W, Kubo T, Fukuhara A, Ihara H, Azuma YT, Tajima H, Inui T, Sawa A, Takeuchi T. Glyceraldehyde‐3‐phosphate dehydrogenase aggregate formation participates in oxidative stress‐induced cell death. J Biol Chem 2009, 284:34331–34341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Kornberg MD, Sen N, Hara MR, Juluri KR, Nguyen JV, Snowman AM, Law L, Hester LD, Snyder SH. GAPDH mediates nitrosylation of nuclear proteins. Nat Cell Biol 2010, 12:1094–1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Jia J, Arif A, Terenzi F, Willard B, Plow EF, Hazen SL, Fox PL. Target‐selective protein S‐nitrosylation by sequence motif recognition. Cell 2014, 159:623–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Amsterdam A, Sadler KC, Lai K, Farrington S, Bronson RT, Lees JA, Hopkins N. Many ribosomal protein genes are cancer genes in zebrafish. PLoS Biol 2004, 2:E139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Flygare J, Karlsson S. Diamond‐Blackfan anemia: erythropoiesis lost in translation. Blood 2007, 109:3152–3154. [DOI] [PubMed] [Google Scholar]
- 96. Ruvinsky I, Sharon N, Lerer T, Cohen H, Stolovich‐Rain M, Nir T, Dor Y, Zisman P, Meyuhas O. Ribosomal protein S6 phosphorylation is a determinant of cell size and glucose homeostasis. Genes Dev 2005, 19:2199–2211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Gazda HT, Sheen MR, Vlachos A, Choesmel V, O'Donohue MF, Schneider H, Darras N, Hasman C, Sieff CA, Newburger PE, et al. Ribosomal protein L5 and L11 mutations are associated with cleft palate and abnormal thumbs in Diamond‐Blackfan anemia patients. Am J Hum Genet 2008, 83:769–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Arcondeguy T, Lacazette E, Millevoi S, Prats H, Touriol C. VEGF‐A mRNA processing, stability and translation: a paradigm for intricate regulation of gene expression at the post‐transcriptional level. Nucleic Acids Res 2013, 41:7997–8010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Ray PS, Jia J, Yao P, Majumder M, Hatzoglou M, Fox PL. A stress‐responsive RNA switch regulates VEGFA expression. Nature 2009, 457:915–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Yao P, Potdar AA, Ray PS, Eswarappa SM, Flagg AC, Willard B, Fox PL. The HILDA complex coordinates a conditional switch in the 3′‐untranslated region of the VEGFA mRNA. PLoS Biol 2013, 11:e1001635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Breaker RR. Prospects for riboswitch discovery and analysis. Mol Cell 2011, 43:867–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Di Giammartino DC, Nishida K, Manley JL. Mechanisms and consequences of alternative polyadenylation. Mol Cell 2011, 43:853–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Ahmed YF, Gilmartin GM, Hanly SM, Nevins JR, Greene WC. The HTLV‐I Rex response element mediates a novel form of mRNA polyadenylation. Cell 1991, 64:727–737. [DOI] [PubMed] [Google Scholar]
- 104. Wu C, Alwine JC. Secondary structure as a functional feature in the downstream region of mammalian polyadenylation signals. Mol Cell Biol 2004, 24:2789–2796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. You L, Wu J, Feng Y, Fu Y, Guo Y, Long L, Zhang H, Luan Y, Tian P, Chen L, et al. APASdb: a database describing alternative poly(A) sites and selection of heterogeneous cleavage sites downstream of poly(A) signals. Nucleic Acids Res 2015, 43:D59–D67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Frischmeyer PA, van Hoof A, O'Donnell K, Guerrerio AL, Parker R, Dietz HC. An mRNA surveillance mechanism that eliminates transcripts lacking termination codons. Science 2002, 295:2258–2261. [DOI] [PubMed] [Google Scholar]
- 107. van Hoof A, Frischmeyer PA, Dietz HC, Parker R. Exosome‐mediated recognition and degradation of mRNAs lacking a termination codon. Science 2002, 295:2262–2264. [DOI] [PubMed] [Google Scholar]
- 108. Pelechano V, Wei W, Steinmetz LM. Extensive transcriptional heterogeneity revealed by isoform profiling. Nature 2013, 497:127–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Stockmann C, Doedens A, Weidemann A, Zhang N, Takeda N, Greenberg JI, Cheresh DA, Johnson RS. Deletion of vascular endothelial growth factor in myeloid cells accelerates tumorigenesis. Nature 2008, 456:814–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Nathan C. Points of control in inflammation. Nature 2002, 420:846–852. [DOI] [PubMed] [Google Scholar]
- 111. Schultze JL, Freeman T, Hume DA, Latz E. A transcriptional perspective on human macrophage biology. Semin Immunol 2015, 27:44–50. [DOI] [PubMed] [Google Scholar]
- 112. Tang WH, Wu Y, Hartiala J, Fan Y, Stewart AF, Roberts R, McPherson R, Fox PL, Allayee H, Hazen SL. Clinical and genetic association of serum ceruloplasmin with cardiovascular risk. Arterioscler Thromb Vasc Biol 2012, 32:516–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Ferrara N, Gerber HP, LeCouter J. The biology of VEGF and its receptors. Nat Med 2003, 9:669–676. [DOI] [PubMed] [Google Scholar]
- 114. White GE, Iqbal AJ, Greaves DR. CC chemokine receptors and chronic inflammation—therapeutic opportunities and pharmacological challenges. Pharmacol Rev 2013, 65:47–89. [DOI] [PubMed] [Google Scholar]
- 115. Zeggini E, Weedon MN, Lindgren CM, Frayling TM, Elliott KS, Lango H, Timpson NJ, Perry JR, Rayner NW, Freathy RM, et al. Replication of genome‐wide association signals in UK samples reveals risk loci for type 2 diabetes. Science 2007, 316:1336–1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Saxena R, Voight BF, Lyssenko V, Burtt NP, de Bakker PI, Chen H, Roix JJ, Kathiresan S, Hirschhorn JN, Daly MJ, et al. Genome‐wide association analysis identifies loci for type 2 diabetes and triglyceride levels. Science 2007, 316:1331–1336. [DOI] [PubMed] [Google Scholar]
- 117. Scott LJ, Mohlke KL, Bonnycastle LL, Willer CJ, Li Y, Duren WL, Erdos MR, Stringham HM, Chines PS, Jackson AU, et al. A genome‐wide association study of type 2 diabetes in Finns detects multiple susceptibility variants. Science 2007, 316:1341–1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Li Y, Nowotny P, Holmans P, Smemo S, Kauwe JS, Hinrichs AL, Tacey K, Doil L, van Luchene R, Garcia V, et al. Association of late‐onset Alzheimer's disease with genetic variation in multiple members of the GAPD gene family. Proc Natl Acad Sci U S A 2004, 101:15688–15693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Li Y, Grupe A, Rowland C, Nowotny P, Kauwe JS, Smemo S, Hinrichs A, Tacey K, Toombs TA, Kwok S, et al. DAPK1 variants are associated with Alzheimer's disease and allele‐specific expression. Hum Mol Genet 2006, 15:2560–2568. [DOI] [PubMed] [Google Scholar]
- 120. Lee KY, Clark AW, Rosales JL, Chapman K, Fung T, Johnston RN. Elevated neuronal Cdc2‐like kinase activity in the Alzheimer disease brain. Neurosci Res 1999, 34:21–29. [DOI] [PubMed] [Google Scholar]
- 121. Weichhart T, Hengstschlager M, Linke M. Regulation of innate immune cell function by mTOR. Nat Rev Immunol 2015, 15:599–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell 2012, 149:274–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Poddar D, Basu A, Baldwin WM 3rd, Kondratov RV, Barik S, Mazumder B. An extraribosomal function of ribosomal protein L13a in macrophages resolves inflammation. J Immunol 2013, 190:3600–3612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Jawien J. The role of an experimental model of atherosclerosis: apoE‐knockout mice in developing new drugs against atherogenesis. Curr Pharm Biotechnol 2012, 13:2435–2439. [PubMed] [Google Scholar]
- 125. Fazio S, Babaev VR, Murray AB, Hasty AH, Carter KJ, Gleaves LA, Atkinson JB, Linton MF. Increased atherosclerosis in mice reconstituted with apolipoprotein E null macrophages. Proc Natl Acad Sci U S A 1997, 94:4647–4652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Basu A, Poddar D, Robinet P, Smith JD, Febbraio M, Baldwin WM 3rd, Mazumder B. Ribosomal protein L13a deficiency in macrophages promotes atherosclerosis by limiting translation control‐dependent retardation of inflammation. Arterioscler Thromb Vasc Biol 2014, 34:533–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Poddar D, Kaur R, Baldwin WM 3rd, Mazumder B. L13a‐dependent translational control in macrophages limits the pathogenesis of colitis. Cell Mol Immunol 2016, 13:816–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Lee J, Harris AN, Holley CL, Mahadevan J, Pyles KD, Lavagnino Z, Scherrer DE, Fujiwara H, Sidhu R, Zhang J, et al. Rpl13a small nucleolar RNAs regulate systemic glucose metabolism. J Clin Invest 2016, 126:4616–4625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Schroder K, Irvine KM, Taylor MS, Bokil NJ, Le Cao KA, Masterman KA, Labzin LI, Semple CA, Kapetanovic R, Fairbairn L, et al. Conservation and divergence in toll‐like receptor 4‐regulated gene expression in primary human versus mouse macrophages. Proc Natl Acad Sci U S A 2012, 109:E944–E953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Vizzini A, Bonura A, Longo V, Sanfratello MA, Parrinello D, Cammarata M, Colombo P. LPS injection reprograms the expression and the 3′ UTR of a CAP gene by alternative polyadenylation and the formation of a GAIT element in Ciona intestinalis. Mol Immunol 2016, 77:174–183. [DOI] [PubMed] [Google Scholar]
- 131. Marquez‐Jurado S, Nogales A, Zuniga S, Enjuanes L, Almazan F. Identification of a gamma interferon‐activated inhibitor of translation‐like RNA motif at the 3′ end of the transmissible gastroenteritis coronavirus genome modulating innate immune response. MBio 2015, 6:e00105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Mazumder B, Mukhopadhyay CK, Prok A, Cathcart MK, Fox PL. Induction of ceruloplasmin synthesis by IFN‐gamma in human monocytic cells. J Immunol 1997, 159:1938–1944. [PubMed] [Google Scholar]
- 133. Arif A. Extraneuronal activities and regulatory mechanisms of the atypical cyclin‐dependent kinase Cdk5. Biochem Pharmacol 2012, 84:985–993. [DOI] [PubMed] [Google Scholar]