

Abstract
Problem
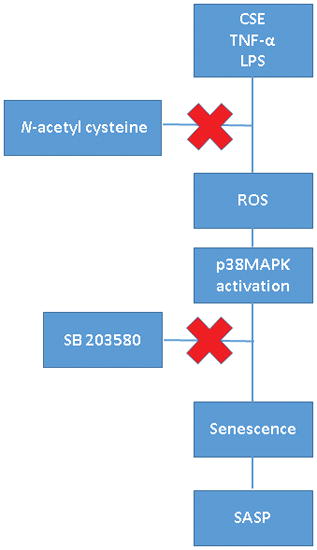
We investigated p38MAPK activation-induced fetal membrane cell senescence in response to inflammation (tumor necrosis factor-alpha [TNF-α]) and infection (lipopolysaccharide [LPS]), factors associated with spontaneous preterm birth.
Method of Study
Primary amnion epithelial cells (AECs) were exposed to TNF-α, 50ng/mL and LPS, 100ng/mL. Cigarette smoke extract (CSE), a known OS inducer, was used as positive control. AECs were co-treated with the antioxidant N-acetyl cysteine (NAC) and p38MAPK inhibitor SB203580 to determine the effect of OS and p38MAPK. Western blot analysis was performed for active (Phospho-p38MAPK) and total p38MAPK. Senescence was determined by flow cytometry and culture supernatants were tested for IL-6 using ELISA.
Results
TNF-α, but not LPS, increased p38MAPK activation compared to untreated cells (p=0.01). The number of senescent cells and senescence-associated IL-6 were higher in both TNF-α and LPS treated cells compared to control (p=0.001, p=0.01, respectively). Antioxidant NAC inhibited p38MAPK activation by TNF-α. p38MAPK inhibitor SB203580 reduced the development of senescence and IL-6 by TNF-α and LPS. CSE treatment validated our current data.
Conclusions
TNF-α caused OS-mediated p38MAPK induction, senescence and IL-6 increase from AECs. LPS also induced senescence and IL-6 increase. Inflammatory and infectious factors may cause premature fetal cell senescence contributing to preterm birth pathophysiology.
Keywords: fetal membranes, interleukin-6, lipopolysaccharide, p38MAPK, tumor necrosis factor-alpha
Graphical Abstract
INTRODUCTION
Recent years have witnessed a drop in the overall preterm birth rate from 12.8% in 2006 to 9.57% in 2014.1,2 This rate, however, increased slightly to 9.63% in 2015.1 These changes in the preterm birth rate are attributed to variations in the use of induction of labor and cesarean delivery at 34–36 completed weeks of gestation, while the rate of spontaneous preterm birth (PTB), with and without rupture of the fetal membranes remains unchanged.1,3 Management strategies using tocolytics, antibiotics, and progesterone have not been successful in reducing the PTB rate.4,5,6 Approximately 40–45% of all preterm births are preceded by spontaneous preterm labor and 25–30% are preceded by preterm premature rupture of membranes (pPROM).7 The exact cause of PTB and pPROM are unclear and often multifactorial.7,8 These include static risk factors such as epidemiologic and genetic factors (ethnicity/race, nutrition, behavior, socio-economic status, body habitus and prior history) and dynamic risk factors (infection, hydramnios, uterine over-distention, autoimmune disease, endocrine disease, physiologic stress and trauma).9 Complex interactions between various static and dynamic risk factors generate uterine oxidative stress (OS) and inflammation.9 OS and inflammation-associated markers are hallmarks of pathways leading to labor and delivery both at term (physiologic) and preterm (pathologic).10,11 However, generation of OS and inflammation in response to various etiologies of PTB and pPROM are still not fully elucidated.12
Multiple factors can generate OS and inflammation.9 In human normal term fetal membrane (amniochorion) tissues, we have reported OS-induced fetal membrane senescence, a mechanism associated with aging, as a factor contributing to inflammation associated with parturition.9 Increased OS at term due to increased feto-maternal metabolic demands, reduced substrate and antioxidant availability, is a natural and physiologic mechanism of normal term parturition. However, premature senescence activation and inflammation in response to OS-inducing risk factors can predispose pregnant women to PTB and pPROM.13 Senescence in decidual tissues and placenta, and its association with preterm birth and other pregnancy complications is also reported. This indicates that this phenomenon is not restricted to fetal membranes, although mechanistic mediators of senescence activation may differ in different tissues.14–18
Senescence is characterized by the irreversible arrest of cell growth.19 Fetal membrane senescence is mediated by OS-induced telomere shortening, and stress-signaler p38 mitogen-activated protein kinase (p38MAPK) activation.13,20,21 Senescence and senescence-associated secretory phenotype (SASP, sterile inflammation) were increased in term labor fetal membranes and amniotic fluid compared to term not in labor samples.20 In vitro primary amnion epithelial cell (AEC) culture models have recapitulated these findings.9,21 Primary AECs from term not in labor membranes exposed to OS (induced by cigarette smoke extract [CSE]) caused telomere dependent, p38MAPK-mediated senescence and SASP.9 p38MAPK inhibitor-regulated senescence and SASP production suggests that stress-signaler p38MAPK activation is a key mediator of this process in fetal cells. Similar findings were reported in murine pregnancy and parturition, where both placenta and membranes undergo senescence prior to murine parturition.22 These models suggest that the transition from pre-labor to labor status involves OS-induced activation of senescence and inflammation in fetal membranes.
While CSE is an excellent inducer of OS and sterile inflammation, it is not representative of the intraamniotic inflammation and infection-associated inflammation often seen in PTB and pPROM. The objective of this study is to determine the role of infection (modeled by lipopolysaccharide [LPS]) and inflammation (represented by tumor necrosis factor alpha [TNF-α]) in causing p38MAPK-mediated AEC senescence and senescence-associated inflammation, and compare them to the effect produced by CSE.
MATERIALS AND METHODS
Placentas were obtained from John Sealy Hospital at The University of Texas Medical Branch (UTMB) at Galveston, TX, USA. No subjects were recruited or consented for this study as we used discarded placentas from normal term not in labor cesarean sections. The study protocol was submitted and approved by the Institutional Review Board at UTMB.
Placentas from women (18–40 years old) undergoing elective repeat cesarean delivery (between 37 and 41 weeks of gestation) prior to the onset of labor were included in the study. Women with prior history of preterm labor and delivery, pPROM, preeclampsia, placental abruption, intrauterine growth restriction, and gestational diabetes were excluded. Additionally, Group B streptococcus carriers, women treated for a urinary tract infection or a sexually transmitted disease, chronic infections like HIV or hepatitis, and women who smoked cigarettes, or reported drug or alcohol abuse, were also excluded.23
Isolation and culture of human amnion epithelial cells (AECs)
All reagents and media were warmed to 37°C prior to use. The amniotic membrane was processed as described previously.24–26 Briefly, the amnion membrane was manually peeled from normal, term not in labor caesarean section placental membranes, rinsed in saline and transferred to a petri dish containing Hanks Balanced Salt Solution (HBSS; Mediatech Inc., Manassas, VA). After cutting the amnion into 2 cm x 2 cm pieces, they were digested twice in 0.25% trypsin and 0.125% Collagenase A (Sigma–Aldrich, St. Louis, MO) in HBSS for 35 minutes at 37°C. After each digestion, the tissue was filtered through a 70 μm cell strainer (Thermo Fisher Scientific, Waltham, MA) and trypsin was inactivated using complete Dulbecco’s Modified Eagle Medium: Nutrient Mixture F-12 media (DMEM/F12; Mediatech Inc.) supplemented with 15% fetal bovine serum (FBS; Sigma-Aldrich), 10% Penicillin/Streptomycin (Mediatech Inc.) and 100 μg/mL epidermal growth factor (EGF; Sigma-Aldrich). The collected filtrate was centrifuged for 10 minutes at 3000 RPM and the pellet was resuspended in 3.0 mL complete DMEM/ F12. Once cells were counted, approximately 3–5 million cells per flask were cultured in T75 flasks containing complete DMEM/F12 media at 37°C, 5% CO2, and 95% air humidity to 70– 80% confluence. To ensure the purity of our primary AEC cultures, the epithelial nature of the cells was verified by immunofluorescent staining using an anti-human cytokeratin antibody as described by Sheller et al.24
Stimulation of AEC with TNF-α
Recombinant TNF-α expressed in E. coli (Sigma-Aldrich #T6674, St. Louis, MO) was reconstituted at a concentration of 0.1mg/mL. TNF-α concentrate was then diluted to 50 ng/mL in media prior to use. This concentration was chosen based on previously reported in vitro experiments on AECs and fetal membranes. 27–30 Once cells reached 70–80% confluence, the cells were passaged. After counting, 800,000 cells were cultured in T25 flasks for 48 hour treatments and 1 million cells for next day 1 hour treatments. A one hour serum starvation was done before each treatment with TNF- α containing media and incubated at 37°C, 5% CO2, and 95% air humidity for 1 hour or 48 hours.
Stimulation of AEC with LPS
LPS from E.coli 055:B5 (Sigma-Aldrich #L2880, St. Louis, MO) was reconstituted at a concentration of 1 mg/mL. LPS concentrate was then diluted to 100 ng/mL in media prior to use. This concentration is within the range of which is seen in the amniotic fluid of women with infection-associated pregnancy complications and used previously in our experiments.31 Once cells reached 70–80% confluence, the cells were passaged. After counting, 800,000 cells were cultured in T25 flasks for 48 hour treatments and 1 million cells for next day 1 hour treatments. A one hour serum starvation was done before each treatment with LPS containing media and incubated at 37°C, 5% CO2, and 95% air humidity for 1 hour or 48 hours.
Stimulation of AECs with cigarette smoke extract (CSE)
As a positive control, as well as to compare and contrast the OS and p38MAPK response, we included CSE treatment of AECs in our experimental design with minor modifications.25 To produce water soluble cigarette smoke extract, smoke from a single lit commercial cigarette (unfiltered CamelTM, R.J. Reynolds Tobacco Co, Winston Salem, NC) was bubbled through 25 mL of media, consisting of DMEM/F12 supplemented with 15% FBS (System Biosciences, Mountain View, CA). The stock CSE was sterilized using 0.22 μm Steriflip filter unit (Millipore, Billerica, MA). CSE concentrate was diluted 1:10 (for 1 hour treatment) or 1:50 (for 48 hour treatment) in complete media prior to use. Once cells reached 70–80% confluence, the cells were passaged. After counting, 800,000 cells were cultured in T25 flasks for 48 hour treatments and 1 million cells for next day 1 hour treatments. A one hour serum starvation was done before each treatment with CSE containing media and incubated at 37°C, 5% CO2, and 95% air humidity for 1 hour or 48 hours.
Inhibition of p38MAPK activation by N-Acetyl Cysteine (NAC)
NAC (Sigma-Aldrich, St. Louis, MO), a potent antioxidant, was used to verify the influence of treatment-induced OS on p38MAPK activation. NAC (15mM) (#A7250-SG) was added to the treatment media of the primary amnion cells treated with the reagents as described above. The AECs were incubated at 37°C, 5% CO2, and 95% air humidity for 1 hour.
Inhibition of p38MAPK induced senescence by SB203580 (p38MAPK inhibitor)
SB203580 (Sigma-Aldrich), a p38MAPK inhibitor, was used in order to validate the influence of p38MAPK activation on AEC senescence. SB203580 (30 μM) (#S8307) was added to the treatment media of the primary amnion cells treated with the reagents as described above. For the 48 hour treatments, an additional SB203580 (30 μM) was added to the media after 24 hours to “refresh” the media. The AECs were incubated at 37°C, 5% CO2, and 95% air humidity for 1 hour or 48 hours.
Western blot analysis for P-p38MAPK
AECs (n=5) treated for one hour with control media, TNF- α, LPS and CSE were lysed in RIPA lysis buffer with freshly added protease and phosphatase inhibitors (0.01%). The lysate was collected after scraping the culture plate and the insoluble material was removed by centrifugation at 10,000 rpm for 20 min at 4°C. The concentration of protein in each lysate was determined by using the BCA protein assay kit (Pierce BCA Protein Assay Kit, Thermo Scientific, Waltham, MA, USA). Equal protein (24 μg) from each sample was loaded onto a 10% SDS-PAGE gel and electrophoresed at 120 V. The resolved proteins were transferred to a PVDF membrane using the iBlot transfer apparatus (Bio-Rad Laboratories, Hercules, CA, USA). The membranes were blocked in Tris-Buffered Saline (TBS) containing 0.1% Tween 20 (TBS-T) and 5% skim milk for 2h at room temperature. Blots were incubated separately with antibodies against total p38MAPK (Cell Signaling, Danvers, MA, USA, #9212) and phosphorylated (P)-p38MAPK (Cell Signaling, #9211) at 4°C and shaken overnight. Blots were washed three times with TBS-T and incubated with appropriate peroxidase-conjugated IgG secondary antibody for 1h at RT. All blots were developed using chemiluminescence reagents ECL Western Blotting Detection System (Amersham Piscataway, NJ, USA), in accordance with the manufacturer’s recommendations, followed by autoradiography. Densitometry was performed to normalize the data for statistical analysis.
Flow cytometry analysis for senescence
After 48 hours of incubation with treatment media as detailed above, the AECs (n=7) were analyzed for cell senescence using flow cytometry. Senescence was assessed with the commonly used biomarker, senescence associated-β-Galactosidase (SA-β-Gal), 32 which we have adapted for flow cytometry.33,34 Cells were incubated for one hour in growth medium supplemented with 100 nM bafilomycin A1 (baf A1). Bafilomycin A1 is a specific inhibitor of vacuolar-type H + ATPase and inhibits lysosomal acidification.35 We added 5-Dodecanoylaminofluorescein di-β-D-Galactopyranoside (C12FDG, eBioscience) for a final concentration of 6 μM and incubated the cells for one hour. C12FDG, a fluorogenic substrate for β-Galactosidase, is membrane permeable and non-fluorescent before hydrolysis. Once the compound is hydrolyzed by β-Galactosidase, it is no longer membrane permeable and emits green fluorescence upon excitation.35 Cells were collected, washed with PBS and resuspended in DNA Prep Stain containing propidium iodide (Beckman Coulter) to select for viable cells. Samples were run immediately on the CytoFlex flow cytometer (Beckman Coulter). AECs treated with DMSO only (vehicle) were used as negative controls for gating. After gating for single, viable cells, data analysis was performed using Cytexpert (Beckman Coulter). Percent positive cells for C12FDG fluorescence were calculated and compared to control.
Cytologic senescence-associated β-Galactosidase (SA β-Gal) assay
In addition to a flow cytometry based approach, SA β-gal staining was evaluated using a commercial histochemical staining assay, following the manufacturer’s instructions (Senescence Cells Histochemical Staining Kit; Sigma-Aldrich). Briefly, cells cultured in chamber slides were washed twice in PBS, fixed for 6–7 min with the provided Fixation Buffer, washed again in PBS and incubated for 3h at 37°C with fresh β-Gal solution. Following incubation, cells were evaluated using a standard light microscope.
ELISA for quantification of IL-6
Media were collected after treatment of AECs for 48 hours (n=5) as described above. Quantikine ELISA (R&D Systems, Minneapolis, MN) was performed on the cell-conditioned media for the cytokine IL-6 following manufacturer instructions. Standard curves were developed with duplicate samples of known quantities of recombinant proteins that were provided by the manufacturer. Sample concentrations were determined by relating the samples’ absorbance to the standard curve by linear regression analysis.
Statistical analysis
Data were analyzed using GraphPad Prism 7 (GraphPad Software, Inc., LaJolla, CA). Kruskal-Wallis ANOVA and the Tukey post-hoc test or Mann-Whitney U test were performed. A p ≤ 0.05 was considered significant.
RESULTS
TNF-α, but not LPS causes p38MAPK activation
p38MAPK activation was determined by western blot analysis using an antibody specific to the phosphorylated form (P-p38MAPK) and normalized to total p38MAPK (Figure 1). p38MAPK activation was higher in cells treated with TNF-α compared to untreated cells (control) (p=0.01; Figure 1A). Co-treatment with NAC reduced TNF-α-mediated p38MAPK activation (p = 0.02), suggesting that TNF-α-mediated activation of p38 MAPK is likely an OS response. p38MAPK activation after LPS treatment was not statistically significant compared to control (Figure 1B). Although an effect of NAC in reducing LPS-induced p38MAPK activation was seen in all our experiments, overall densitometric assessment did not reach significance.
Figure 1. p38MAPK activation.

(A) Amnion cells exposed to TNF-α demonstrated activation of p38MAPK. NAC inhibited this activation, confirming an OS-mediated effect. (B) LPS treatment did not activate p38MAPK to statistical significance. NAC decreased this effect; however, it did not reach statistical significance. (C) Amnion cells exposed to CSE had the highest activation of p38MAPK. NAC inhibited this activation, confirming an OS-mediated effect.
To provide validity to our p38MAPK-mediated senescence model, CSE experiments were conducted simultaneously with LPS and TNF-α experiments. CSE-treated cells activated p38MAPK compared to control (p = 0.006), which was inhibited by NAC (p = 0.05) (Figure 1C).
Both TNF-α and LPS cause AEC senescence
Flow cytometry analysis was used to determine senescence induced by TNF-α and LPS. We used CSE treatment as a positive control. As expected, CSE treatment produced a significant increase in senescent cells as detected by C12FDG fluorescence compared to untreated controls (p < 0.0001; Figure 2C).The number of senescent cells were increased by TNF-α compared to control (p=0.001; Figure 2A). Interestingly, LPS treatment also produced a significantly higher number of senescent cells compared to control (p=0.01; Figure 2B). Of note, LPS caused a lesser number of senescent cells compared to both TNF-α and CSE treatments. Senescence was further confirmed with cytologic staining (Figure 2D) that showed similar data as seen in the flow cytometry based analysis.
Figure 2. Senescence activation.

(A) C12FDG fluorescence was increased in TNF-α treated amnion cells. SB203580 co-treatment inhibited this change, confirming this p38-mediated effect. (B) LPS treatment also increased C12FDG fluorescence to a lesser degree, which was also inhibited by SB203580 co-treatment. (C) Amnion cells exposed to CSE had the largest increase in C12FDG fluorescence. SB203580 inhibited change, confirming a p38-mediated effect. (D) Senescence-associated β-Galactosidase staining was shown in TNF-α, LPS and CSE treated amnion cells, confirming the C12FDG fluorescence results.
To test whether senescence by TNF-α and LPS are mediated via p38MAPK activation, we co-treated our cells with p38MAPK functional inhibitor SB203580. In our system, SB203580 does not inhibit p38MAPK activation but prevents its functional effect, such as senescence activation similar to that reported by Kumar et al.36 A significant decrease in C12FDG fluorescence was seen when AECs were co-treated with TNF-α and SB when compared to cells treated with TNF-α alone (p=0.05; Figure 2A). Co-treatment of the AECs with LPS and SB significantly decreased C12FDG fluorescence when compared to cells treated with LPS alone (p=0.005; Figure 2B). This data is contradictory to our western blot analysis where we did not see a significant drop in p38MAPK activation (data not shown). This suggests that the trend to increase p38MAPK that we observed is likely sufficient to cause senescence activation after LPS treatment. As expected CSE had the highest activation of senescence that was reduced after SB treatment (p=0.01; Figure 2C).
We measured IL-6, a pro-labor and SASP marker using ELISA. IL-6 concentration was increased by both TNF-α and LPS (p=0.002, p=0.02 respectively; Figure 3), and this increase was subsequently down-regulated by SB (p=0.05, p < 0.05 respectively), confirming a p38MAPK-mediated pathway activation to increase cytokine production. In our CSE experiment, SB treatment produced a substantial decrease in IL-6 production (p < 0.05; Figure 3).
Figure 3. Senescence-associated inflammatory cytokine IL-6.

TNF-α treated amnion cells increased the inflammatory cytokine IL-6 when compared to untreated cells. This effect was down-regulated when co-treated with SB203580, confirming a p-38 mediated effect. LPS treatment also increased IL-6, to a lesser degree. This effect was reduced with SB203580 co-treatment. Amnion cells exposed to CSE increased the inflammatory cytokine IL-6; however, not significantly. Co-treatment with SB203580 decreased IL-6 production.
DISCUSSION
Inflammation in feto-maternal tissues throughout pregnancy is required for gestational tissue remodeling to accommodate fetal growth.28,37 The inflammatory process during gestation is a very balanced activity maintaining immune homeostasis; however, an inflammatory load increase in gestational tissues triggers labor and delivery associated changes.11,38,39 Inflammatory overload induced by pregnancy-associated risk factors is one of the trigger mechanisms for preterm labor.11 We have earlier reported that an inflammatory load increase at term and preterm can arise from either noninfectious (sterile) or infectious stimuli. Regardless of the initiation signals, both of these inflammatory processes have similar biomarker identities.13,20 As mentioned in the introduction, OS-induced fetal membrane senescence at term or preterm is one of the mechanisms by which sterile inflammation can emerge.20,40–42 OS-induced cell and organelle damage, not just OS-associated reactive oxygen radicals, initiate pathways leading to p38MAPK activation, senescence and sterile inflammation.42,43 Activation of this pathway in response to various risk factors (behavioral and environmental) have been reported;25,44 however, the potential of activation of this pathway in response to infectious or inflammatory stimuli are unknown.
Our study demonstrated that pathologic concentrations of TNF-α and LPS often associated with PTB and pPROM induced senescence and increased SASP marker IL-6; specifically for TNF-α, by activating p38MAPK. This in vitro experiment shows that inflammation (represented by TNF-α) and an infection-associated inflammatory response can cause amnion cell senescence and senescence-associated inflammatory biomarker production, that can likely contribute to the pathophysiologic pathways of PTB and pPROM.20 Of note, the extent of p38MAPK activation and senescence are not as pronounced as seen with CSE. Therefore, we postulate that senescence induction by infection/inflammation associated with PTB and pPROM are likely not as dominant as OS-inducing risk factors such as behavioral or environmental risks.25,44 NF-κB activation by OS is a well-established mechanism of activation of PTB and pPROM pathways.13 We have also shown that both LPS and CSE cause NF-κB activation, although CSE’s effect is not as dominant as the LPS-mediated effect in amnion cells.21 This is indicative of distinct ways of activation of similar inflammatory markers (e.g. IL-6) by different stimuli (infectious or sterile). IL-6 is a well-reported mediator of inflammation during pregnancy and parturition at term and preterm.45 Animal model studies have shown that IL-6 null mice have prolonged gestation, signifying the understanding of its generation by sterile and infectious inflammatory processes.46 Risk factors of PTB and pPROM have overlapping mechanisms of activation of inflammation, and this redundancy highlights the complexity of PTB and pPROM pathways.
Term and preterm labor are associated with a significant increase in the amniotic fluid concentration of TNF-α.30,47 TNF-α plays a role in both physiologic and pathologic parturition,47 and is a pro-inflammatory cytokine produced by the placenta in response to OS and other physiologic stressors that are separate from infection.48 Therefore, multiple risk factors can cause increased TNF-α production in fetal tissues that can lead to effects as reported here. However, the differential effects of TNF-α (i.e. development of p38MAPK mediated senescence or NF-κB activation independent of p38MAPK activation) may be dose dependent, which was not evaluated in this study. Noninfectious cellular stress and infection may produce different doses of TNF-α and therefore the pathway it can activate, and its effects may vary based on the type of exposure, as well as dose produced.
Similarly, intraamniotic infection by microbial invasion normally causes a marked increase in inflammation and OS.39,49 The pathways activated by OS and OS damage in response to infection are markedly different from what is normally observed in OS induced by other risk factors like cigarette smoke or obesity.21 LPS (an endotoxin from gram-negative bacteria) concentration is higher in intraamniotic infection-associated preterm labor compared to those without documented infection.31 Recently, maternal nanoparticle-based therapy using a dendrimer-NAC conjugate in a murine model found prevention of preterm birth, and a reduction of neuromotor deficits in offspring exposed to in utero inflammation (LPS).50 We have already reported that LPS produced a significantly lesser degree of OS than CSE from amnion cells.21 This is likely contributing to the differences in p38MAPK and senescence activation between LPS and CSE treatments. We did not see a statistically significant increase in p38MAPK activation by LPS in western blot analysis although the trend indicated an increase and NAC also downregulated this effect. This is likely due to sample size effect, as we may not have had an adequate number of samples to get the expected effect. A decrease in senescent cells after SB treatment, a mechanism inhibiting p38 function, does indicate that senescent pathways are functional through p38MAPK after LPS treatment, but the manifestation is not as strong as observed with CSE.
TNF-α induction of apoptosis and weakening of fetal membranes have been well reported.28,29,47 It has been shown by our lab that fetal membrane telomere-dependent senescence is a potential mechanism associated with pPROM.13,51 In vitro experiments have shown that LPS and CSE can increase proapoptotic p53 expression, but do not lead to p53 activation (phosphorylation).21 Therefore, it can be inferred that pPROM with infection and infection-associated inflammation (e.g. TNF-α increase) is not caused by fetal membrane apoptosis, but likely by fetal membrane senescence. Pathways of senescence and apoptosis often overlap, although one of the key differences is that senescence is always associated with inflammation regardless of infection, and apoptosis often lacks an inflammatory component.9,13
p38MAPK activation is higher in term labor and preterm fetal membranes,26 especially those complicated by pPROM. As seen in this study, TNF-α, which has a proven role in physiologic and pathologic parturition, can cause term not in labor amniotic cell senescence through the p38MAPK pathway. Antioxidant NAC inhibited p38MAPK activation and p38MAPK inhibitor SB203580 minimized development of senescence and SASP caused by TNF- α, confirming this pathway. These findings strengthen the postulation that the senescent changes of fetal membranes collected after preterm and term labor are not due to the labor process itself, but likely due to fetal membrane cell senescence that precedes parturition.
CONCLUSIONS
In summary, in vitro proxies of infection (modeled by LPS) and inflammation (TNF-α) induced amnion cell senescence and senescence-associated inflammation, leading us to believe that infectious and inflammatory factors may cause premature fetal cell senescence, contributing to pathways of preterm birth. Understanding these mechanisms is an integral step in discovering diagnostic tests and designing therapeutic strategies to combat preterm birth.
Acknowledgments
Funding: This study is supported by NIH grant NIH/NICHD - 1R01HD084532-01A1 and R03HD067446 to R Menon
Footnotes
The authors report no conflict of interest
References
- 1.Martin JA, Hamilton BE, Osterman MJ, Driscoll AK, Mathews TJ. Births: Final Data for 2015. Natl Vital Stat Rep. 2017;66(1):1. [PubMed] [Google Scholar]
- 2.Martin JA, Hamilton BE, Sutton PD, et al. Births: final data for 2007. Natl Vital Stat Rep. 2010;58(24):1–85. [PubMed] [Google Scholar]
- 3.Martin JA, Kirmeyer S, Osterman M, Shepherd RA. Born a bit too early: recent trends in late preterm births. NCHS Data Brief. 2009;(24):1–8. [PubMed] [Google Scholar]
- 4.Creasy RK. Preterm birth prevention: where are we? Am J Obstet Gynecol. 1993;168(4):1223–1230. doi: 10.1016/0002-9378(93)90373-q. [DOI] [PubMed] [Google Scholar]
- 5.Meis PJ, Klebanoff M, Thom E, et al. Prevention of recurrent preterm delivery by 17 alpha-hydroxyprogesterone caproate. N Engl J Med. 2003;348(24):2379–2385. doi: 10.1056/NEJMoa035140. [DOI] [PubMed] [Google Scholar]
- 6.Society for Maternal-Fetal Medicine Publications Committee waoVB. Progesterone and preterm birth prevention: translating clinical trials data into clinical practice. Am J Obstet Gynecol. 2012;206(5):376–386. doi: 10.1016/j.ajog.2012.03.010. [DOI] [PubMed] [Google Scholar]
- 7.Goldenberg RL, Culhane JF, Iams JD, Romero R. Epidemiology and causes of preterm birth. Lancet. 2008;371(9606):75–84. doi: 10.1016/S0140-6736(08)60074-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Muglia LJ, Katz M. The enigma of spontaneous preterm birth. N Engl J Med. 2010;362(6):529–535. doi: 10.1056/NEJMra0904308. [DOI] [PubMed] [Google Scholar]
- 9.Menon R. Oxidative stress damage as a detrimental factor in preterm birth pathology. Front Immunol. 2014;5:567. doi: 10.3389/fimmu.2014.00567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Smith R. Parturition. N Engl J Med. 2007;356(3):271–283. doi: 10.1056/NEJMra061360. [DOI] [PubMed] [Google Scholar]
- 11.Romero R, Espinoza J, Gonçalves LF, Kusanovic JP, Friel LA, Nien JK. Inflammation in preterm and term labour and delivery. Semin Fetal Neonatal Med. 2006;11(5):317–326. doi: 10.1016/j.siny.2006.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Romero R, Dey SK, Fisher SJ. Preterm labor: one syndrome, many causes. Science. 2014;345(6198):760–765. doi: 10.1126/science.1251816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dutta EH, Behnia F, Boldogh I, et al. Oxidative stress damage-associated molecular signaling pathways differentiate spontaneous preterm birth and preterm premature rupture of the membranes. Mol Hum Reprod. 2016;22(2):143–157. doi: 10.1093/molehr/gav074. [DOI] [PubMed] [Google Scholar]
- 14.Cox LS, Redman C. The role of cellular senescence in ageing of the placenta. Placenta. 2017;52:139–145. doi: 10.1016/j.placenta.2017.01.116. [DOI] [PubMed] [Google Scholar]
- 15.Hirota Y, Daikoku T, Tranguch S, Xie H, Bradshaw HB, Dey SK. Uterine-specific p53 deficiency confers premature uterine senescence and promotes preterm birth in mice. J Clin Invest. 2010;120(3):803–815. doi: 10.1172/JCI40051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cha J, Bartos A, Egashira M, et al. Combinatory approaches prevent preterm birth profoundly exacerbated by gene-environment interactions. J Clin Invest. 2013;123(9):4063–4075. doi: 10.1172/JCI70098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sultana Z, Maiti K, Aitken J, Morris J, Dedman L, Smith R. Oxidative stress, placental ageing-related pathologies and adverse pregnancy outcomes. Am J Reprod Immunol. 2017;77(5) doi: 10.1111/aji.12653. [DOI] [PubMed] [Google Scholar]
- 18.Maiti K, Sultana Z, Aitken RJ, et al. Evidence that fetal death is associated with placental aging. Am J Obstet Gynecol. 2017 doi: 10.1016/j.ajog.2017.06.015. [DOI] [PubMed] [Google Scholar]
- 19.Muñoz-Espín D, Serrano M. Cellular senescence: from physiology to pathology. Nat Rev Mol Cell Biol. 2014;15(7):482–496. doi: 10.1038/nrm3823. [DOI] [PubMed] [Google Scholar]
- 20.Behnia F, Taylor BD, Woodson M, et al. Chorioamniotic membrane senescence: a signal for parturition? Am J Obstet Gynecol. 2015;213(3):359e351–316. doi: 10.1016/j.ajog.2015.05.041. [DOI] [PubMed] [Google Scholar]
- 21.Behnia F, Sheller S, Menon R. Mechanistic Differences Leading to Infectious and Sterile Inflammation. Am J Reprod Immunol. 2016;75(5):505–518. doi: 10.1111/aji.12496. [DOI] [PubMed] [Google Scholar]
- 22.Bonney EA, Krebs K, Saade G, et al. Differential senescence in feto-maternal tissues during mouse pregnancy. Placenta. 2016;43:26–34. doi: 10.1016/j.placenta.2016.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fortunato SJ, Menon R, Swan KF, Lyden TW. Organ culture of amniochorionic membrane in vitro. Am J Reprod Immunol. 1994;32(3):184–187. doi: 10.1111/j.1600-0897.1994.tb01112.x. [DOI] [PubMed] [Google Scholar]
- 24.Sheller S, Papaconstantinou J, Urrabaz-Garza R, et al. Amnion-Epithelial-Cell-Derived Exosomes Demonstrate Physiologic State of Cell under Oxidative Stress. PLoS One. 2016;11(6):e0157614. doi: 10.1371/journal.pone.0157614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Menon R, Boldogh I, Urrabaz-Garza R, et al. Senescence of primary amniotic cells via oxidative DNA damage. PLoS One. 2013;8(12):e83416. doi: 10.1371/journal.pone.0083416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Menon R, Boldogh I, Hawkins HK, et al. Histological evidence of oxidative stress and premature senescence in preterm premature rupture of the human fetal membranes recapitulated in vitro. Am J Pathol. 2014;184(6):1740–1751. doi: 10.1016/j.ajpath.2014.02.011. [DOI] [PubMed] [Google Scholar]
- 27.Kumar D, Schatz F, Moore RM, et al. The effects of thrombin and cytokines upon the biomechanics and remodeling of isolated amnion membrane, in vitro. Placenta. 2011;32(3):206–213. doi: 10.1016/j.placenta.2011.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kumar D, Fung W, Moore RM, et al. Proinflammatory cytokines found in amniotic fluid induce collagen remodeling, apoptosis, and biophysical weakening of cultured human fetal membranes. Biol Reprod. 2006;74(1):29–34. doi: 10.1095/biolreprod.105.045328. [DOI] [PubMed] [Google Scholar]
- 29.Fortunato SJ, Menon R, Lombardi SJ. Support for an infection-induced apoptotic pathway in human fetal membranes. Am J Obstet Gynecol. 2001;184(7):1392–1397. doi: 10.1067/mob.2001.115434. discussion 1397–1398. [DOI] [PubMed] [Google Scholar]
- 30.Romero R, Manogue KR, Mitchell MD, et al. Infection and labor. IV. Cachectin-tumor necrosis factor in the amniotic fluid of women with intraamniotic infection and preterm labor. Am J Obstet Gynecol. 1989;161(2):336–341. doi: 10.1016/0002-9378(89)90515-2. [DOI] [PubMed] [Google Scholar]
- 31.Romero R, Roslansky P, Oyarzun E, et al. Labor and infection. II. Bacterial endotoxin in amniotic fluid and its relationship to the onset of preterm labor. Am J Obstet Gynecol. 1988;158(5):1044–1049. doi: 10.1016/0002-9378(88)90216-5. [DOI] [PubMed] [Google Scholar]
- 32.Harbo M, Koelvraa S, Serakinci N, Bendix L. Telomere dynamics in human mesenchymal stem cells after exposure to acute oxidative stress. DNA Repair (Amst) 2012;11(9):774–779. doi: 10.1016/j.dnarep.2012.06.003. [DOI] [PubMed] [Google Scholar]
- 33.Southcombe J, Tannetta D, Redman C, Sargent I. The immunomodulatory role of syncytiotrophoblast microvesicles. PLoS One. 2011;6(5):e20245. doi: 10.1371/journal.pone.0020245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Atay S, Gercel-Taylor C, Kesimer M, Taylor DD. Morphologic and proteomic characterization of exosomes released by cultured extravillous trophoblast cells. Exp Cell Res. 2011;317(8):1192–1202. doi: 10.1016/j.yexcr.2011.01.014. [DOI] [PubMed] [Google Scholar]
- 35.Atay S, Gercel-Taylor C, Suttles J, Mor G, Taylor DD. Trophoblast-derived exosomes mediate monocyte recruitment and differentiation. Am J Reprod Immunol. 2011;65(1):65–77. doi: 10.1111/j.1600-0897.2010.00880.x. [DOI] [PubMed] [Google Scholar]
- 36.Kumar S, Jiang MS, Adams JL, Lee JC. Pyridinylimidazole compound SB 203580 inhibits the activity but not the activation of p38 mitogen-activated protein kinase. Biochem Biophys Res Commun. 1999;263(3):825–831. doi: 10.1006/bbrc.1999.1454. [DOI] [PubMed] [Google Scholar]
- 37.Fortunato SJ, Menon R, Lombardi SJ. MMP/TIMP imbalance in amniotic fluid during PROM: an indirect support for endogenous pathway to membrane rupture. J Perinat Med. 1999;27(5):362–368. doi: 10.1515/JPM.1999.049. [DOI] [PubMed] [Google Scholar]
- 38.Romero R, Mazor M, Wu YK, et al. Infection in the pathogenesis of preterm labor. Semin Perinatol. 1988;12(4):262–279. [PubMed] [Google Scholar]
- 39.Menon R, Taylor RN, Fortunato SJ. Chorioamnionitis--a complex pathophysiologic syndrome. Placenta. 2010;31(2):113–120. doi: 10.1016/j.placenta.2009.11.012. [DOI] [PubMed] [Google Scholar]
- 40.Menon R. Human fetal membranes at term: Dead tissue or signalers of parturition? Placenta. 2016;44:1–5. doi: 10.1016/j.placenta.2016.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Romero R, Miranda J, Chaiworapongsa T, et al. Prevalence and clinical significance of sterile intra-amniotic inflammation in patients with preterm labor and intact membranes. Am J Reprod Immunol. 2014;72(5):458–474. doi: 10.1111/aji.12296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bredeson S, Papaconstantinou J, Deford JH, et al. HMGB1 promotes a p38MAPK associated non-infectious inflammatory response pathway in human fetal membranes. PLoS One. 2014;9(12):e113799. doi: 10.1371/journal.pone.0113799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Polettini J, Behnia F, Taylor BD, Saade GR, Taylor RN, Menon R. Telomere Fragment Induced Amnion Cell Senescence: A Contributor to Parturition? PLoS One. 2015;10(9):e0137188. doi: 10.1371/journal.pone.0137188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Behnia F, Peltier MR, Saade GR, Menon R. Environmental Pollutant Polybrominated Diphenyl Ether, a Flame Retardant, Induces Primary Amnion Cell Senescence. Am J Reprod Immunol. 2015;74(5):398–406. doi: 10.1111/aji.12414. [DOI] [PubMed] [Google Scholar]
- 45.Prins JR, Gomez-Lopez N, Robertson SA. Interleukin-6 in pregnancy and gestational disorders. J Reprod Immunol. 2012;95(1–2):1–14. doi: 10.1016/j.jri.2012.05.004. [DOI] [PubMed] [Google Scholar]
- 46.Robertson SA, Christiaens I, Dorian CL, et al. Interleukin-6 is an essential determinant of on-time parturition in the mouse. Endocrinology. 2010;151(8):3996–4006. doi: 10.1210/en.2010-0063. [DOI] [PubMed] [Google Scholar]
- 47.Maymon E, Ghezzi F, Edwin SS, et al. The tumor necrosis factor alpha and its soluble receptor profile in term and preterm parturition. Am J Obstet Gynecol. 1999;181(5 Pt 1):1142–1148. doi: 10.1016/s0002-9378(99)70097-9. [DOI] [PubMed] [Google Scholar]
- 48.Leblanc S, Ouellet A, Giguère Y, Forest JC, Moutquin JM, Aris A. A positive correlation between hydrogen peroxide and soluble TNF-alpha receptor 2 early in maternal blood and at term in placenta of pregnant women with preeclampsia. Hypertens Pregnancy. 2012;31(3):357–366. doi: 10.3109/10641955.2010.525281. [DOI] [PubMed] [Google Scholar]
- 49.Gomez R, Romero R, Edwin SS, David C. Pathogenesis of preterm labor and preterm premature rupture of membranes associated with intraamniotic infection. Infect Dis Clin North Am. 1997;11(1):135–176. doi: 10.1016/s0891-5520(05)70347-0. [DOI] [PubMed] [Google Scholar]
- 50.Lei J, Rosenzweig JM, Mishra MK, et al. Maternal dendrimer-based therapy for inflammation-induced preterm birth and perinatal brain injury. Sci Rep. 2017;7(1):6106. doi: 10.1038/s41598-017-06113-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Menon R, Yu J, Basanta-Henry P, et al. Short fetal leukocyte telomere length and preterm prelabor rupture of the membranes. PLoS One. 2012;7(2):e31136. doi: 10.1371/journal.pone.0031136. [DOI] [PMC free article] [PubMed] [Google Scholar]