

Abstract
Bacteria and archaea rely on CRISPR (clustered regularly interspaced short palindromic repeats) RNA-guided adaptive immune systems for sequence specific elimination of foreign nucleic acids. In Escherichia coli, short CRISPR-derived RNAs (crRNAs) assemble with Cas (CRISPR-associated) proteins into a 405-kilodalton multi-subunit surveillance complex called Cascade (CRISPR-associated complex for antiviral defense). Cascade binds foreign DNA complementary to the crRNA guide and recruits Cas3, a trans-acting nuclease-helicase required for target degradation. Structural models of Cascade have captured static snapshots of the complex in distinct conformational states, but conformational dynamics of the 11-subunit surveillance complex have not been measured. Here we use hydrogen-deuterium exchange coupled to mass spectrometry (HDX-MS) to map conformational dynamics of Cascade onto the three-dimensional structure. New insights from structural dynamics are used to make functional predictions about the mechanisms of the R-loop coordination and Cas3 recruitment. We test these predictions in vivo and in vitro. Collectively, we show how mapping conformational dynamics onto static 3D-structures adds an additional dimension to the functional understanding of this biological machine.
INTRODUCTION
The adaptive immune system in Escherichia coli (type I-E) consists of eight cas genes flanked by a CRISPR locus (Fig 1A). Five of the cas genes encode proteins (Cse11, Cse22, Cas76, Cas51, Cas6e1) that assemble with a 61-nucleotide CRISPR-derived RNA (crRNA) into a seahorse-shaped complex called Cascade (CRISPR-associated complex for antiviral defense), with subunits that resemble a head, backbone, belly, and tail (Fig 1B) 1–4. Cascade is a crRNA-guided surveillance complex that identifies foreign DNA and recruits a trans-acting nuclease-helicase (i.e. Cas3) for target degradation 5–7.
Figure 1. Binding of dsDNA increases the stability of Cascade.

(A) The type 1-E CRISPR system of E. coli consists of eight cas genes and a CRISPR locus. Five cas genes (colored) code for proteins that assemble with a crRNA into the Cascade complex. (B) Schematic representation of three stages of Cascade during the interference process. Subunits colored according to Figure 1A. (C) Deuterium uptake curves for the individual subunits of Cascade. Error bars show the standard deviation (n=3) and are smaller than the circular symbols used to plot the data. Both the unbound and dsDNA bound Cascade complexes were incubated in deuterated buffer for 0.5 to 77.5 minutes. Colors of the data points on the uptake curves correspond with the subunits of Cascade as colored in Figure 1A. Differences in deuterium uptake were determined by subtracting deuterium uptake of the unbound complex from the dsDNA bound complex at 77.5 minutes. The percentage change in uptake was mapped onto the dsDNA bound structure of Cascade (5H9F). Increasing blue hue indicates greater protection (i.e., less deuterium uptake) in the dsDNA bound form. Overall, the dsDNA bound form of Cascade takes up less deuterium and is more protected.
Target DNA recognition by Cascade relies on protein-mediated recognition of a three-nucleotide PAM (protostpacer adjacent motif) and a protospacer sequence that is complementary to the crRNA-guide 1, 8–10. Structures of E. coli Cascade before binding DNA 3, 4, 11, bound to ssDNA 12, bound to a partially duplexed dsDNA target 8, and recent structures of Cascade from Thermobifida fusca bound to a dsDNA target 13, provide snapshots of the complex poised at different stages of antiviral defense. Collectively, these structures explain how DNA binding triggers a conformational rearrangement in which the two belly subunits (i.e. Cse2) slide along the ridged Cas7 backbone via a molecular relay that relies on a series of conserved arginine residues 11. One of the Cse2 subunits abuts the C-terminal domain (CTD) of Cse1 (also called Cas8e), and the DNA-induced transition of Cse2 is thought to shove the CTD of Cse1 between the two strands of the DNA duplex 3, 4, 8, 12. While the CTD of Cse1 appears to function like a molecular pry bar that helps separate the two strands of the DNA duplex, three structural features (i.e. lysine finger, glycine rich loop and glutamine wedge) in the N-terminal domain of Cse1 are involved in PAM recognition 8. PAM recognition relies on sequence- and shaped-based recognition of the minor groove, resulting in a diverse combination of different PAMs that are recognized with varying affinities 7, 8, 14, 15.
While a variety of PAMs support target binding, different PAM and protospacer combinations have been shown to elicit distinct biological outcomes 14, 16, 17. Binding to a limited number of PAMs elicits an “interference response”, which is characterized by recruitment of a nuclease active form of Cas3 that processively degrades the target 7, 17–20. Alternatively, some PAMs or mutations at specific positions in the protospacer, reduce interference to levels below what is required for clearance of plasmids or virus elimination, and trigger a “priming response.” Priming is a positive feedback mechanism characterized by the rapid integration of new fragments of foreign DNA into the CRISPR locus, which are used to restore immunity 14, 16, 17, 21. Priming relies on all the of the CRISPR machinery (Cascade, Cas1, Cas2, and Cas3) in addition to a partial complementary target.
Recently, Förster resonance energy transfer (FRET) studies have been used to monitor the conformational state of Cascade in association with targets that elicit priming or interference. Xue et al. used structures of Cascade to position FRET dyes at specific locations in the tail of Cascade to monitor the conformational state of Cse1 in response to binding DNA targets that elicit priming or interference 19, 22. These studies demonstrated that Cascade adopts distinct conformational states based on sequence characteristics of the dsDNA target and that the conformational state of the Cse1 tail subunit correlates with the relative rates of interference and priming. While structure guided placement of FRET dyes provides a powerful means for measuring distances between two selected landmarks, this approach cannot provide a global understanding of the complex conformational dynamics of a large multi-subunit machine like Cascade.
Rather than examining movements of specific locations on Cascade, here we use hydrogen-deuterium exchange coupled to mass spectrometry (HDX-MS) to investigate protein interactions and dynamics of Cascade before and after DNA binding. HDX measures the stability of hydrogen bond networks and solvent accessibility on global and local scales 23, 24, enabling a mechanistic analysis of coordinated changes associated with target binding. To our knowledge, this is the first application of HDX-MS to a ribonucleoprotein of this complexity (i.e., 5 unique proteins totaling >1,450 amino acid residues). Peptide-resolution HDX-MS experiments reveal residues predicted to be involved in binding the displaced DNA strand (i.e. R-loop) and regions of the Cse1 subunit that are part of the Cas3 binding site. We test the function of these residues using in vivo and in vitro assays and show that HDX-guided mutations at specific locations eliminate Cas3-mediated DNA degradation without altering Cascade’s ability to bind dsDNA.
RESULTS
Structural studies have captured snapshots of Cascade before and after target binding, but these static structures provide limited information about the conformational dynamics of the complex, which are important for function 3, 4, 8, 12. To measure global changes in the stability and dynamics of Cascade upon target binding, we used intact protein HDX-MS to measure HD-exchange in all 11-protein subunits of Cascade, both before and after binding DNA. We diluted unbound Cascade and Cascade bound to a 72-base pair dsDNA target containing a 3′-TTC-5′ PAM into deuterated buffer and then analyzed both complexes by Liquid Chromatography Mass Spectrometry (LCMS) over 15 time points (Fig 1 and Supplementary Dataset 1). At each time point, dsDNA bound Cascade exhibited less deuterium uptake than the unbound form, though the difference was less pronounced at later time points. Overall, the intact protein HDX-MS revealed a net stabilization of each protein subunit in the complex upon dsDNA binding (Fig 1C). This stabilization was further confirmed using differential scanning fluorimetry, which showed that Cascade bound to a dsDNA target has a melting temperature that is approximately 10°C above the melting temperature of unbound Cascade (Supplemental Fig 1).
Having established that DNA binding globally decreases deuterium uptake and increases stability, we set out to map these changes to specific regions of the complex. To accomplish this, we digested Cascade and dsDNA-bound Cascade with pepsin (i.e., a low specificity protease), prior to LCMS. Deuterium uptake curves were generated by quenching the HDX reaction after 0, 0.16, 0.5, 2.5, 5.0, 10, 30 and 180 minutes. 255 peptides, representing more than 90% of all Cascade protein sequences, were analyzed (Supplemental Fig 2). Parent spectra (MS1) were used to determine the number of deuterons incorporated per peptide, and the difference in deuteron incorporation between peptides isolated from dsDNA bound and unbound states of Cascade were used to calculate changes in HD-exchange (Supplementary Dataset 1). Overall, 185 of the 255 peptides showed increased deuterium incorporation over the time course in either or both complexes, while 70 peptides did not. After binding dsDNA, 22 of the 255 peptides increased deuterium incorporation, while 119 peptides had decreased deuterium incorporation compared to the unbound complex.
HD-exchange profiles are not uniformly distributed across the complex or within an individual subunit. By analyzing the distribution of deuterium uptake over the time course, changes in the stability and dynamics of hydrogen bond networks upon dsDNA binding can be determined. Early time points report on changes in solvent accessibility upon dsDNA binding, whereas later time points provide information on the stability of hydrogen bonding networks and the participation of peptide backbone amides in protein secondary structures that are protected from deuterium exchange because of noncovalent interactions 25. Regions of the complex that displayed significant protection upon dsDNA binding (i.e. decreased deuterium exchange) were investigated as potential DNA binding sites (Fig 2A, blue). We also analyzed peptides that assimilate more deuterium (i.e. become less protected) after binding DNA (Fig 2A, red). An increase in exchange indicates greater solvent exposure or dynamics. These regions are anticipated to have functional roles, such as docking sites for the nuclease-helicase Cas3.
Figure 2. Peptide-resolution mapping of deuterium uptake reveals areas of Cascade that become more or less protected upon dsDNA binding.
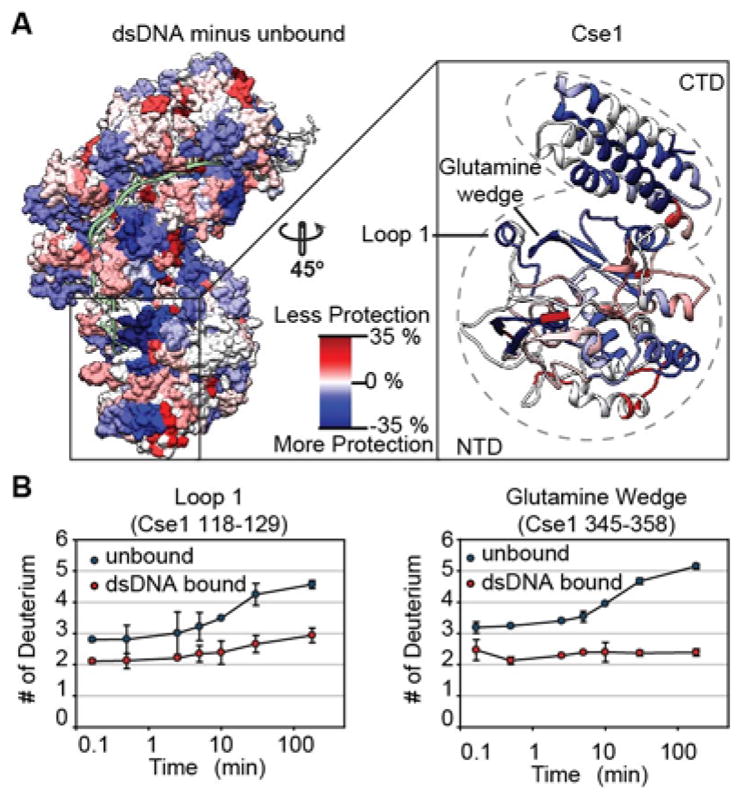
(A) The percentage difference in deuterium uptake between the unbound and dsDNA bound complex for individual peptides at the 180-minute time point was mapped onto the dsDNA bound structure (5H9F). Blue indicates less deuterium incorporation in the dsDNA bound form (protection) while the red indicates more deuterium incorporation in the dsDNA bound form (de-protection). White indicates regions where there is no difference in deuterium uptake or peptides where no data is available. The inset shows the ribbon structure of the Cse1 subunit with the glutamine wedge and Loop 1 showing increased protection. The Cse1 subunit consists of a four-helix bundle (CTD) and globular domain (NTD). (B) Deuterium uptake curves show increased protection in the dsDNA bound form for peptides in Loop 1 (p-value<0.01 for all time points) and the glutamine wedge (p-value<0.01 for all time points after 2.5 minutes). Error bars represent the standard deviation of three replicates.
DNA binding
To validate the HDX data we examined regions of the complex known to interact with DNA. The expectation is that DNA binding will stabilize the peptide backbone and physically occlude these regions from HD-exchange. Recognition of the PAM sequence takes place in the tail of the complex and involves three structural elements in the Cse1 subunit: lysine finger, glycine loop, and glutamine wedge 8. The glutamine wedge (Cse1 amino acids 352–358) forms a beta-hairpin that is positioned (i.e. wedged) between the complementary and non-complementary strands of the DNA target (Fig 2). In the dsDNA bound complex, a peptide corresponding to this feature (amino acids 345–358) exchanges less deuterium over time, than the same peptide in the unbound complex (Fig 2B). This indicates that the glutamine wedge is protected from exchange and is less dynamic when Cascade is bound to DNA. Similarly, a peptide covering the PAM sensing lysine finger (Cse1 amino acid 268) also exchanges less deuterium upon dsDNA binding (Supplemental Fig 3). Results for the glutamine wedge and lysine finger are consistent with the structural models, however the glycine loop (Cse1 amino acids 157–161) showed no increase in protection upon dsDNA binding, which was surprising since the crystal structure indicates that this feature interacts with the minor groove of the double-stranded PAM (Supplemental Fig 3). This suggests, that in solution, the glycine loop may be more dynamic after DNA binding than is indicated by the crystal structure.
The Cse1 subunit is critical for binding DNA and the conformational state of this subunit has been implicated in recruitment and allosteric regulation of Cas3 7, 19, 26, 27. In structures of Cascade without DNA or bound to dsDNA, the Cse1 subunit is tethered to Cas5 via a short loop (L1, amino acids 125–131) on Cse1 that is buried in a pore on Cas5 3. However, in the structure of Cascade bound to a complementary ssDNA target, the N-terminal domain (NTD) of Cse1 comes “unhitched” from the complex and L1 loop is disordered in the structure 12. Collectively, these structures demonstrate that the NTD of Cse1 adopts conformationally distinct positions and recent FRET experiments by Xue et al. suggest that NTD of Cse1 is conformationally dynamic 19. While the structures of Cascade without DNA suggest that Cse1 is in a “closed” conformation where L1 is buried in the Cas5 pore, our HDX data reveals that the L1 peptide of Cse1 (amino acids 118–129) is significantly more protected (less HD-exchange) in the dsDNA bound complex, as compared to the unbound complex (Fig 2B and Supplemental Fig 3). These data suggest that the Cse1 subunit is dynamic in the absence of DNA, sampling both the “open” and “closed” conformations.
Target recognition by Cascade relies on both specific- and non-sequence specific interactions with DNA. Recently, we showed that a lysine-rich helix (amino acid residues 137–144) on the fingers domain of two adjacent Cas7 subunits (i.e. Cas7.5 and Cas7.6) creates a lysine-rich vise that makes non-sequence specific interactions with dsDNA and that these interactions are essential for target binding 11. We predicted that dsDNA would result in less HD-exchange for peptides corresponding to the lysine-rich vise. To test this prediction, we examined a peptide covering the lysine-rich helices of Cas7 (amino acid residues 134–148). This peptide showed a small degree of protection upon dsDNA binding (Supplemental Fig 3 and 4). However, there are six copies of the Cas7 subunit in the surveillance complex and only two of these subunits (Cas7.5 and Cas7.6) interact with the phosphate backbone of the incoming DNA 11. The Cas7 peptide corresponding to the lysine-rich vise (residues 134–148) should have a different exchange rates depending on their position in the complex. Peptides from Cas7.5 and Cas7.6 should have less HD-exchange than the same peptides form Cas7.1–7.4, and this difference should result in a bimodal distribution of these peptides. In fact, we observe a bimodal behavior, which is consistent with a subset of the Cas7 subunits having increased protection upon DNA binding (Supplemental Fig 4).
Path of the Displaced DNA Strand
One of the outstanding questions about the mechanism of target binding is the location of the displaced DNA strand. Once Cascade binds to a dsDNA target, the complementary strand of the DNA target base pairs with the crRNA, while the non-complementary strand is displaced and forms an R-loop 2, 28. Structures of E. coli Cascade bound to dsDNA showed 10 to 12 nucleotides of the displaced DNA strand, but it remains unclear if this fragment reflects the path of the displaced strand when the complex binds to a full-length target duplex 8. In the structure, the partial displaced strand is stabilized by interactions with residues on the Cse1 subunit of Cascade and we expected that the remaining part of the displaced strand would interact with specific residues on Cas subunits as it traversed form the PAM proximal tail to the PAM distal head. These stabilizing interactions would shield specific amino acid residues from HD-exchange. To trace the path of the displaced strand, we used our HDX-MS data to identify peptides that show greater protection from exchange in the dsDNA bound form (Fig 3A).
Figure 3. Using HDX to identify residues involved in binding the displaced DNA strand.

(A) Differences in deuterium uptake between the unbound and dsDNA bound complex for individual peptides at the 180-minute time point are mapped onto the dsDNA bound structure . Only peptides that are more protected in the dsDNA bound complex (blue) are shown. The proposed path of the displaced strand is indicated (green). (B) Deuterium incorporation for peptides Cse2 6–27 and Cas7 296–311 at the 180-minute time point is displayed for Cascade without DNA (teal), ssDNA bound (green), and dsDNA bound complexes (red). The Cse2 peptide shows increased protection in the dsDNA bound complex, as compared to the unbound or the ssDNA bound complex (p<0.01). The Cas7 peptide shows the most protection in the dsDNA bound complex. Error bars represent the standard deviation of three replicates. (C) HDX-guided mutation of positively charged residues on Cse2 (R12E), Cse2 (R53D, K77D, K78D, R89D, R135D, R143D and K158D, named Cse2 charge swap), and Cas7 (K296E, K299E, K301E) were tested in a plasmid-curing assay to determine the biological impact of mutating these residues. The Cas7 mutant did not have a significant impact while the Cse2 mutants displayed a small, but significant defect. The single mutation of Cse2 R12E had an equal impact as mutation of seven Cse2 residues. The error bars represent the standard error of the mean of three replicates.
Analysis of the tail of Cascade revealed a peptide in the C-terminal domain (CTD) of Cse1 (amino acid residues 381–398) with significant protection (p<0.01) in the dsDNA bound form of the complex compared to the unbound complex (Fig 3A and Supplemental Fig 3) 24. This is consistent with the path of the displaced strand in the dsDNA bound structure from Hayes et al 8. However, to clarify the mechanism of protection at this location, we conducted an additional comparison by performing HDX-MS on Cascade bound to ssDNA (Supplemental Dataset 1). Comparison of the dsDNA and ssDNA bound complexes reveal regions of protection specifically resulting from the presence of the displaced strand. Importantly, this comparison showed equivalent levels of protection in the ssDNA bound and dsDNA bound form of Cascade, indicating that the increase in protection observed in the dsDNA bound structure is not necessarily due to direct interactions with the displaced strand, but likely an allosteric stabilization of the hydrogen-bonding network across this peptide. This result is consistent with a recent structure of T. fusca Cascade bound to a complete dsDNA target. In this structure, the R-loop is disordered in the region of the Cse1-CTD 13.
Once the displaced strand leaves the four-helix bundle, it must travel up the complex where it is expected to form interactions with the belly or backbone subunits of the complex. The HDX data revealed two distinct areas of increased protection that may accommodate the displaced strand (Fig 3A). The upper route travels across the top of the Cse2 subunits and includes a peptide that shows strong protection (residues 6–27). The lower path of protection is formed by peptides covering residues 296–311 of the Cas7 subunits. This lower route is part of the basic cleft that is formed by the Cse2 and Cas7 subunits and has previously been hypothesized to accommodate the displaced strand 3, 8, 12. Interestingly, the Cse2 peptide corresponding to the “upper path” (amino acid residues 6–27) showed comparable levels of exchange in the unbound and ssDNA bound forms, while it was more protected from exchange in the dsDNA bound form (p<0.001), suggesting that this peptide may interact with the displaced strand (Fig 3B and Supplemental Fig 3). Conversely, the Cas7 peptide corresponding to the “lower path” (amino acid residues 296–311) showed comparable levels of exchange in both the ssDNA and dsDNA bound states (p>0.05), suggesting that the enhanced stability measured by HDX upon dsDNA binding is not a result of stable interactions with the displaced strand (Fig 3B and Supplemental Fig 3). To test the role of these residues in stabilizing the displaced strand we mutated arginine 12 on Cse2 to glutamic acid (Cse2 R12E) and three lysine residues on Cas7 to glutamic acids (Cas7 K296E, K299E, and K301E). We then tested the impact of these mutations using a plasmid-curing assay that measures the in vivo efficiency of Cascade/Cas3 to detect and degrade a target plasmid (Fig 3C) 11, 29. The Cse2 mutant (i.e. Cse2 R12E) displayed small, but reproducible defects, while the Cas7 mutant did not result in a measurable defect. We compared the HDX-guided mutants to a mutant we previously made based on electrostatic surface potential, where we mutated seven positively charged residues on Cse2 that line the basic cleft (Cse2 R53D, K77D, K78D, R89D, R135D, R143D and K158D, named Cse2 charge swap) (Fig 3C) 3, 12. The charge swap of seven Cse2 residues resulted in a small defect comparable to the mutation of the single arginine 12 on Cse2. Collectively this suggests that precise coordination of the R-loop, may not be critical for immune system function.
Recruitment of the nuclease-helicase Cas3
DNA binding triggers a conformational change in Cascade that recruits Cas3 to the Cse1 subunit 3, 4, 6, 8, 11, 30. We hypothesized that dsDNA binding exposes a Cas3 docking site on Cascade and that these residues would display greater HD-exchange after Cascade binds dsDNA. To test this hypothesis, we compared HD-exchange before and after dsDNA binding and identified a large beta-hairpin, termed Loop 3 (L3, amino acids 285–296) on Cse1 that is more flexible after DNA binding (Fig 4A). The increased exchange in L3 coincides with movement of a short loop called L4 (amino acids 316–326) (Supplemental Fig 5 and 6). L4 is positioned next to L3 in the unbound and ssDNA bound Cascade structures, but moves 11 Å away from L4 upon dsDNA binding (Supplemental Fig 6A). To assess the functional importance of L3 and L4 we compared the Cse1 protein structure from E. coli to homologous protein structures from Thermobifida fusca, Thermus thermophiles, and Acidimicrobium ferrooxidans 8, 11, 13, 31–33. While there is no detectable amino acid sequence conservation, L3 and L4 are conserved structural features (Supplemental Fig 5 and 6). Based on structural conservation and dynamic response to dsDNA binding, we hypothesized that L3 and L4 may participate in Cas3 recruitment. To test this hypothesis, we deleted L3 (L285-K296, ΔL3) and performed plasmid-curing assays. Deletion of L3 caused a plasmid-curing defect, indicating that L3 has an important functional role in DNA binding, Cas3 recruitment, or both (Fig 4B). To clarify the function of L3, we expressed and purified the Cse1ΔL3 deletion mutant of Cascade. Size exclusion chromatography and SDS-PAGE gel electrophoresis confirmed that the complex assembles and purifies like wild type Cascade (Supplemental Fig 7A). However, Electrophoretic Mobility Shift Assays (EMSAs) revealed that the L3 deletion results in a DNA binding defect (KD= 135.2 nM) compared to wild type Cascade (KD= 1.5 nM) (Fig 4C and Supplemental Fig 8), which indicates that defects in DNA binding are responsible for the immune defect observed in the plasmid-curing assays. To further investigate the function of L3, we focused on two conserved lysine residues (Cse1 K289 and K290) at the apex of L3. Alanine substitution of these two lysine residues (Cse1 K289A/K290A) resulted in a similar plasmid-curing defect as deletion of the entire 11 residue loop (Fig 4B). Like the ΔL3 mutant, the Cse1 K289A/K290A mutant expressed and purified similar to wild type (Supplemental Fig 7B). However, the Cse1 K289A/K290A mutation does not impair DNA binding (KD= 1.8 nM) (Fig 4C and Supplemental Fig 8). This suggested that the lysine residues might be involved in Cas3 recruitment or activation. To determine the role of the lysine residues, we performed an in vitro cleavage assay in which Cse1 K289A/K290A Cascade was pre-bound to dsDNA followed by the addition of Cas3. However, in contrast to our prediction, Cascade with the Cse1 K289A/K290A mutation results in a very modest DNA degradation defect (Fig 4D). We speculate that the discrepancy between the in vivo plasmid-curing assay (defect) and the in vitro DNA degradation assay (no defect) might be explained by high Cas3 concentrations (300 nM) and low of sensitivity in our DNA degradation assay. The mechanistic basis of this distinction requires further investigation, but the result illustrates the importance of performing both in vivo and in vitro experimentation.
Figure 4. Predicting the Cas3 interaction site using HDX.

(A) Ribbon representation of Cse1 colored according to the percentage difference in deuterium uptake of the dsDNA bound complex minus the unbound form of the Cascade complex at the 180-minute time point. Areas in red are more available for HDX after dsDNA binding. dsDNA is colored green. (B) Residues in three of the peptides (L3, H1 and H2) that exchange more deuterium after binding dsDNA and one peptide that exchanges less deuterium (H3) after binding dsDNA, were targeted for mutagenesis and tested using plasmid-curing assays. Deletion of Loop 3 (Cse1 ΔL285-K296, denoted ΔL3) or mutation of the lysine residues present at the tip of Loop 3 (Cse1 K289A/K290A, denoted KK) result in large immune system defects. Mutation of alpha helices H1 (Cse1 N379A/E380K), H2 (Cse1 N332A, Q333A, R335E) and H3 (Cse1 R194E/K197E) resulted in immune system defects for H1 and H3. Mutants showing defects were selected for further study. The error bars represent the standard error of the mean of three replicates. (C) Measurement of the binding affinity for 72 bp dsDNA containing a protospacer and interference PAM (3′-TAC-5′ or 3′-TTC-5′) using Electrophoretic Mobility Shift Assays (EMSAs). Only the deletion of Loop 3 resulted in a major binding defect. Error bars represent the standard deviation of three replicates. (D) Denaturing polyacrylamide gels showing time courses of Cascade/Cas3 mediated DNA degradation. 72 bp dsDNA containing P7 protospacer and 3′-TTC-5′ PAM was pre-bound to wild type Cascade or the Cascade mutants and incubated for various lengths of time with Cas3. Wild type Cascade facilitates destruction of the DNA by Cas3. The H1 mutant severely reduces the ability of Cas3 to degrade the DNA, indicating that this mutant blocks Cas3 recruitment or activation. The percentage of DNA degraded after 60 minutes is indicated with the error representing the standard deviation of three replicates.
In addition to L3, we identified other surface exposed peptides on Cse1 with changes in their HDX profile upon DNA binding. We made mutations in three distinct locations that cluster on an exposed surface of Cse1 that is anticipated to be involved in Cas3 recruitment (Fig 4A). Two alpha-helices (H1 and H2) that showed increased exchange upon dsDNA binding and one alpha helix (H3) with a slight decrease in HD-exchange were selected for mutational analysis. Polar residues with uncharged side chains were mutated to alanine residues, and residues with charged side chains were swapped for the opposite charge. The H1 mutation (Cse1 N379A/E380K) and the H3 mutation (Cse1 R194E/K197E) both result in pronounced defects in the plasmid-curing assay, while the H2 mutation (Cse1 N332A, Q333A and R335E) results in a modest immune system defect (Fig 4B). To clarify the functional importance of H1 and H3 we expressed and purified these two mutants. The expression levels and stoichiometry of the purified mutants were comparable to wild type Cascade (Supplemental Fig 7C). In vitro binding assays revealed that the H1 mutant binds dsDNA with wild type affinity (KD=0.86 nM), while the H3 mutant has a small defect (KD=4.87 nM) (Fig 4C and Supplemental Fig 8). To test the function of these residues in Cascade-mediated recruitment of Cas3, we performed DNA cleavage assays. The H1 mutant binds DNA like wild type Cascade, but does not facilitate Cas3-mediated DNA degradation, while the H3 mutant, which has a modest binding defect, degrades DNA similar to wild type (Fig 4D). A high concentration of Cascade (300 nM) was used in the DNA cleavage assays to compensate for the small DNA binding defect observed. These data indicate that residues in H1 are critical for Cas3 recruitment or activation, while the plasmid-curing defect of the H3 mutant is caused by the small DNA binding defect.
Discussion
Structures of Cascade have captured the complex in several distinct conformational states 3, 4, 6, 8, 11, 12, 34. Collectively, these structural models reveal a series of conformational changes that coincide with DNA recognition and Cas3 recruitment. Here, we add an additional dimension to this work by performing HDX-MS on Cascade before and after target recognition. HDX-MS analysis reveals specific regions of the complex that have more or less HD-exchange after binding DNA. Mapping this data onto 3D-structures of Cascade highlights regions of functional importance that have been previously identified, as well as functionally important regions that do not stand out in static structures.
The lysine-rich vise of Cas7 and the PAM sensing domain of Cse1 are critical for DNA binding 8, 11. Slow deuteration of a peptide is indicative of low solvent exposure or extensive hydrogen bonding and we measure significant decreases in deuterium incorporation after dsDNA binding for peptides that cover the lysine-rich vise, and two the PAM sensing elements (i.e., lysine finger and glutamine wedge). However, the glycine-rich loop (amino acids 157–161), which has previously been shown to intercalate in the minor groove of the PAM duplex is not protected from HD-exchange in the dsDNA bound complex (Supplemental Fig 3). These results suggest that the glycine-rich loop may be more dynamic after binding than indicated by the crystal structure.
While the mechanism of PAM sensing and directional hybridization of the crRNA guide to the DNA target is relatively well understood, the location of the displayed strand has not been determined for the E. coli Cascade complex. Based on the structure of E. coli Cascade bound to a partially duplexed DNA target, we expected the displaced strand to traverse over the C-terminal four-helix bundle of Cse1 and continue along a basic cleft formed by the Cse2 and Cas7 subunits (Fig 3) 12. Indeed, peptides covering the C-terminal four-helix bundle of Cse1 (amino acids 381–398) incorporate significantly less deuterium after dsDNA binding. However, this same peptide is also protected (i.e. less deuterium incorporation) in the ssDNA bound complex, suggesting that the lack of deuterium uptake is due to changes in hydrogen bonding networks, rather than direct interactions with the displaced strand. This conclusion is consistent with the recent structure of T. fusca Cascade bound to a dsDNA target, which shows no density for the displaced DNA strand in this region 13.
Following the displaced strand beyond the four-helix bundle we predicted that the remaining portion of the R-loop would be coordinated by residues that form a positively charged cleft between Cse2 and Cas7 12. To test the role of this cleft in DNA binding, we flipped the charge on seven positively charged residues on Cse2 (i.e. R53D, K77D, K78D, R89D, R135D, R143D and K158D) and three positively charged residues on Cas7 (i.e. K296E, K299E and K301E). Remarkably, neither of these mutants resulted in a pronounced immune defect (Fig 3C). Using the HD-exchange data, rather than focusing strictly on electrostatic surface potential, we identified a peptide on Cse2 (amino acids 6–27) that exchanges significantly (p<0.001) less deuterium upon binding of dsDNA. Mutation of a single arginine (Cse2 R12) resulted in a small, but reproducible immune defect, which is consistent with a minor role for this peptide in stabilizing the displaced strand. In support of this conclusion, the recent T. fusca Cascade structure reveals an equivalently positioned arginine (R16) that is located ~4 Å from the phosphate backbone of the displaced strand 13. While we initially expected to identify an extensive network of non-sequence specific, protein-mediated interactions involved in R-loop stabilization, our results and recent structures of the Cascade complex from T. fusca reveal a poorly ordered displaced strand that makes few direct interactions with the Cas proteins. Collectively this suggests that precise coordination of the R-loop at positions distal to the PAM, may not be critical for immune system function.
In addition to DNA binding, Cse1 also plays a critical role in Cas3 recruitment 6, 7, 20. We hypothesized that regions of Cse1 that incorporate more deuterium after DNA binding, may be involved in Cas3 recruitment. To test this hypothesis, we designed HDX-guided mutations in L3, H1, and H2 regions of Cse1 (Fig 4). Mutations in L3 and H1 result in significant immune defects, while the H2 mutation has no measurable phenotype. We expected that the increase in HD-exchange would be indicative of regions involved in Cas3 recruitment; however, only the H1 mutant blocks Cas3-mediated DNA degradation, suggesting that this may be a docking site for Cas3.
Conformational dynamics are critical to the function of any molecular machine. Magnetic tweezers have been used to measure the dynamics of torque-dependent R-loop formation and DNA dissociation for Cascade 28, 35. These experiments reveal conformational changes in the DNA target and recent FRET studies have been used to identify distinct conformational states of Cascade that are controlled by the sequence of the DNA target 19, 22, 26. FRET has been used to show that binding to DNA targets containing specific PAM or protospacer mutations correspond with a rotation of the N-terminal domain in Cse1. This conformation is referred to as the “open” state, which does not effectively recruit nuclease-active Cas3, but recruits a nuclease-repressed form of Cas3, in the presence of Cas1 and Cas2 18. Collectively these studies suggest that the conformational state of Cascade is determined by the sequence of the DNA ligand, and that these conformations control the immune response by regulating the activity of Cas3 3, 18, 19.
While structure guided placement of FRET dyes provides a powerful means for measuring distances between two selected landmarks, this approach cannot provide a global understanding of the complex conformational dynamics of a large multi-subunit machine like Cascade. Here we demonstrate how the high-spatial resolution of FRET is complemented by the peptide-resolution of HDX-MS. In this paper, we validate the method by comparing our results to previous insights (e.g. PAM binding residues) and provide new information about the mechanism of DNA binding (e.g. coordination of the displaced strand), and Cas3 recruitment (e.g. role of H1). Future experiments performed using HDX-MS should focus on Cascade bound to DNA targets that have previously been shown to elicit interference or priming responses. These experiments will be critical to understanding how DNA-induced conformational states of Cascade control the downstream immune response.
Materials and Methods
Hydrogen-Deuterium Exchange coupled with Mass Spectrometry (HDX-MS)
Three forms of the Cascade complex were tested using HDX-MS: unbound Cascade, Cascade bound to a ssDNA substrate, and Cascade bound to a dsDNA bound substrate (Supplemental Dataset 1). First Cascade was expressed and purified (see below) and pre-bound to ssDNA or dsDNA substrate, by incubating unbound Cascade with an excess of the appropriate oligonucleotide at 37 °C for 15 minutes. Excess oligonucleotides were removed from the ssDNA and dsDNA bound Cascade preparations by gel filtration as described below (Supplemental Fig 6D). Cascade (10mg/mL) was diluted 10-fold into reaction buffer (10mM sodium acetate, 50mM NaCl, pH 7.0) which was made in D2O. Samples were removed at different time points and quenched to stop the exchange after 0, 0.16, 0.5, 2.5, 5.0, 10, 30 and 180 minutes. At each time point, 10 μL was withdrawn and placed into quenching/digestion solution at pH 2.5 containing 1% formic acid (FA, Sigma) and 0.2 mg/mL pepsin (Sigma) at 0°C. After one and half minutes of digestion, the tube was frozen in liquid N2 and stored at −80°C until liquid chromatography-mass spectrometry (LC-MS) analysis. For Intact protein HDX-MS the reaction started in the same manner as the pepsin-based HDX-MS. Time points were collected at 0.5, 6, 11.5, 17, 22.5, 28, 33.5, 39, 44.5, 50, 55.5, 61, 66.5, 72, and 77.5 minutes. The intact protein HDX-MS time points were quenched in the same manner; however, the intact protein quench solution did not contain pepsin, and tubes were immediately frozen in liquid N2 and stored at −80°C until liquid chromatography-mass spectrometry (LC-MS) analysis
LC-MS analysis of intact Cascade and Cascade peptides was completed on a 1290 UPLC series chromatography stack (Agilent Technologies) coupled directly to a 6538 UHD Accurate-Mass Q-TOF LC/MS mass spectrometer (Agilent Technologies). Before electrospray-time of flight (ESI-TOF) analysis, peptides were separated on a Reverse Phase (RP) column (Phenomenex Onyx Monolithic C18 column, 100 × 2 mm) at 1°C using a flow rate of 600 μL/min in the following conditions: 1min, 5% B; 1.0–9.0min, 5–45% B; 9.0–9.80 min, 95% B; 9.80–9.90 min, 5% B solvent A = 0.1% formic acid (FA, Sigma) in water (ThermoFisher) and solvent B = 0.1% FA in acetonitrile (ACN, ThermoFisher). Data was acquired at 2 Hz sec−1 over the scan range 50–1700 m/z in positive mode. Electrospray settings were: nebulizer set to 3.7 bar, drying gas 8.0 L/min, drying temperature 350°C, and capillary voltage 3.5 kV.
Data processing was carried out in Agilent MassHunter Qualitative Analysis version 6.0. Unique peptides were identified by using Peptide Analysis Worksheet version 2000.6.8.0 (PAWs, ProteoMetrics LLC.) with an accuracy of 10 ppm. Peptide sequences were also confirmed by MS-MS data acquisition, with a scan range of 50–1700 m/z (auto MS-MS)) with an isolation width of 4 m/z and an acquisition rate of 1 spectrum/s. Different collision energy voltage (20V, 25V, and 30V) and linear gradient voltages were applied in auto MS-MS mode. MS-MS data were analyzed and mapped to confirm the corresponding sequence by using Peptide Shaker version 0.41.1 paired with Search GUI version 1.30.1 (Compomics) 36.
Deuterium incorporation in each peptide was calculated using the program HDExaminer (version 1.3.0 beta 6, Sierra Analytics Inc.) which measures the shift of the centroid peak of the peptides collected throughout the time course in different samples. The shift of the centroid was then compared to a non-deuterated control (no exchange) and a fully deuterated sample (24-hour reaction) to determine the extent of exchange using methods previously reported by Guttman et al. 23. Back-exchange during chromatography was estimated from a fully deuterated protein. The 24-hour time point was denoted as 100% (m100 ) exchange. The non-deuterated control was denoted as 0% (m0%) exchange. Percentage deuteration for each peptide at different time points was calculated by following formula.
mt = deuterium uptake at different times
Statistical analysis on deuterium uptake was performed using MEMHDX 24. P-values less than 0.05 were considered statistically significant. For the uptake curves (Fig 2,3 and Supplemental Fig 3), peptides that were the most representative of the data were displayed. Uptake information for all peptides can be found in Supplementary Data Set 1.
Nested or sister peptides that shared the same N-terminal or C-terminal residue and varied in length were manually resolved to determine the contributions of the different amides to the overall uptake of the peptides. The overlapping regions of the peptides were assumed to take up the same amount of deuterium and the non-overlapping regions were assumed to be responsible for the rest of the uptake in the longer peptide. For example, if the peptide covering residues 1–4 took up two deuterium and the peptide covering residues 1–6 took up three deuterium, it would be assumed that residues 5 and 6 together took up, on average, one deuterium. The uptake percentage of the resolved peptides of the unbound complex were then subtracted from the dsDNA bound complex and mapped onto the structural model of dsDNA bound Cascade (5H9F) using Chimera version 1.10.2 (UCSF Resource for Biocomputing, Visualization, and Informatics), to create the difference maps shown in Figures 1–4 37.
Cascade expression and purification
Cascade and Cascade mutants were expressed and purified using previously described methods 11. Briefly, cas genes and CRISPR RNAs were coexpressed in E. coli Bl21 (DE3) cells with Cse2 fused to an N-terminal Strep tag. Cells were grown in LB media at 37°C under antibiotic selection and induced at an OD600nm of 0.5 using 0.2 mM isopropyl-D-1-thiogalactopyranoside (IPTG). Cells were cultured overnight at 20 °C, pelleted by centrifugation and suspended in lysis buffer (100 mM Tris–HCl pH 8.0, 150 mM NaCl, 1 mM Ethylenediaminetetraaceticacid (EDTA), 1 mM tris (2-carboxyethyl) phosphine (TCEP) and 5% glycerol), and frozen at −80 °C. Cells were lysed by sonication and lysates were clarified by centrifugation. Cascade self-assembles in vivo and the complex was affinity purified on StrepTrap HP resin (GE) using N-terminal Strep-II tags on Cse2. Elution of Cascade was performed using the lysis buffer supplemented with 2.5 mM desthiobiotin. The Strep-II tag was removed with HRV-3C protease. Cascade was concentrated prior to gel filtration chromatography using 10/300 Superose6, 16/60 Superose6 or 26/60 Superose6 columns (GE Healthcare) equilibrated with 50 mM Tris–HCl pH 7.5, 100 mM NaCl, 1 mM EDTA, 1 mM TCEP and 5% glycerol (Supplemental Fig 6) and stored at −80 °C until use. Stoichiometry of the different Cascade mutants was analyzed by SDS-PAGE gel and the presence of crRNA was checked by phenol/chloroform extraction of the protein followed by denaturing polyacrylamide gel (Supplemental Fig 6).
Cas3 expression and purification
Cas3 was expressed and purified using previously described methods 5, 6. Briefly, the Cas3 gene fused to an N-terminal His6-MBP (maltose binding protein) tag was expressed from pHMGWA plasmid in E. coli BL21 (DE3) cells. Cells were grown in LB media at 20°C under antibiotic selection and expression was induced at an OD600 nm of 0.3 using 0.2 mM IPTG. Cells were pelleted by centrifugation and suspended in lysis buffer (20 mM TRIS-HCl pH 8.0, 200 mM NaCl, 1 mM TCEP and 10 % glycerol). Cells were lysed by sonication and lysates were clarified by centrifugation. Cas3 was affinity purified using Ni-NTA superflow resin (Qiagen), washed with lysis buffer supplemented with 5 mM Imidazole followed by a 1 M Nacl wash. Cas3 was eluted with lysis buffer supplemented with 250 mM Imidazole. Cas3 was concentrated prior to gel filtration chromatography using 16/60 Superoses6 column equilibrated with 20 mM TRIS-HCl pH 8.0, 200 mM NaCl, 1 mM TCEP and 5 % glycerol and stored at −80°C until use. Protein purity was checked by SDS-PAGE gel (Supplemental Fig 6E).
Oligoduplex preparation and DNA labeling
Oligonucleotides (Supplemental Table 1) were 5-end labeled with [γ-32P] ATP (PerkinElmer) using T4 polynucleotide kinase (NEB). Labeled oligonucleotides were purified by phenol/chloroform extraction followed by MicroSpin G-25 columns (GE Healthcare). Oligonucleotides were gel purified on a denaturing polyacrylamide gel and recovered. dsDNA was prepared by mixing labeled oligonucleotides with a 5 fold molar excess of the complementary oligonucleotide in hybridization buffer (20 mM HEPES pH 7.5, 100 mM KCl and 5 % glycerol). The mixture was heated to 95°C for 5 minutes and allowed to cool to room temperature. Duplexed oligonucleotides were purified on a native polyacrylamide gel and recovered in hybridization buffer. Duplexed oligonucleotides were used for EMSA and Cascade/Cas3 mediated DNA degradation assays.
Electrophoretic mobility shift assay (EMSA)
Electrophoretic mobility shift assays were carried out as described previously 11. The reported KDs are the average from three independent experiments and error bars represent the standard deviation. Oligonucleotides used for EMSAs are listed in Supplemental Table 1. The non-target DNA strand was labeled.
Plasmid-curing assays
Plasmid-curing assays were carried out as described previously 11. Briefly, competent E. coli cells containing plasmids encoding Cascade, Cas3, and a crRNA targeting the pUC-P7-TAC plasmid were transformed with an equal molar mixture of pUC19 (non-target) and pUC-p7-TAC (target) plasmids. Expression of Cascade, Cas3, and the crRNA was induced using 0.2 mM isopropyl-D-1-thiogalactopyranoside (IPTG) and cells were plated on LB-agar plates. Forty individual colonies were screened using PCR designed to discriminate target from non-target plasmids based on size. Three replicates were carried out for each experiment and the average value of the ratio of target to non-target plasmid was calculated and plotted in the graphs (Fig 3,4). An active immune system will destroy the target plasmid and result in a value close to 0, while an inactive immune system will preserve the original 1:1 ratio of target to non-target plasmid and result in a value close to 1. The error bars represent standard error of the mean calculated from three independent experiments.
Cascade/Cas3 mediated DNA degradation
Duplexed oligonucleotides were [γ-32P] ATP labeled on the 5′end of the non-target DNA strand and incubated with 300 nM Cascade or Cascade mutants for 15 minutes at 37°C in reaction buffer (20 mM HEPES pH 7.5, 100 mM KCl, 10 mM MgCl2, 10 mM CaCl2, 100 μM NiSO4, 100 μM CoCl2 and 2 mM TCEP) supplemented with 2 mM ATP (Lanes marked No ATP did not receive ATP, lanes marked EDTA contained 17 mM EDTA, Fig 4D). Reactions were incubated on ice for 5 minutes prior to addition of Cas3 to a final concentration of 300 nM. After the addition of Cas3 reactions were incubated for 60 minutes at 37 °C except for the samples that were part of the time course (triangle in Fig 4D, incubated for 0, 15 and 60 minutes). Once the reactions were complete the samples were quenched by the addition of EDTA and Sodium Dodecyl Sulphate (SDS) to a final concentration of 8 mM EDTA and 1 % SDS. Samples were mixed with 2x Formamide loading buffer, heated to 95°C C for 5 minutes and run on a pre-run 7 M Urea, 15 % polyacrylamide (29:1) gel in Tris-Taurine-EDTA (TTE) buffer. Gels were dried, exposed to phosphor storage screens and scanned with a Typhoon phosphorimaginer. Degraded and undegraded DNA fractions were quantified using GelQuant.NET software and used to calculate the fraction of degraded DNA compared to the total DNA. Reported degradation percentages are the average of three independent experiments with the error representing the standard deviation.
Supplementary Material
Acknowledgments
Research in the Wiedenheft lab is supported by the National Institutes of Health (P20GM103500, P30GM110732-03, R01GM110270, and R01GM108888), the National Science Foundation EPSCoR (EPS-110134), the M. J. Murdock Charitable Trust, a young investigator award from Amgen, Montana University System Research Initiative, Gordon and Betty Moore Foundation, and the Montana State University Agricultural Experimental Station. Research in the Bothner lab is supported by National Institutes of Health (R01 AI081961, R21 AI126583). The Proteomics, Metabolomics, and Mass Spectrometry facility at MSU received support from the Murdock Charitable Trust and NIGMS of the National Institutes of Health (P20GM103474). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Author contributions: P.B.G.v.E., R.K., B.B. and B.W. designed the research; P.B.G.v.E., R.K., A.P., S.M.G. and B.L.F. performed experiments; P.B.G.v.E., R.K., A.P., L.B., R.N.J., J.C., B.B. and B.W. analyzed the data and P.B.G.v.E., A.P., L.B., B.B. and B.W. wrote the paper.
Supporting Information is available free of charge on the ACS Publications website. Figures S1–S8, and Supplemental dataset 1.
References
- 1.Brouns SJ, Jore MM, Lundgren M, Westra ER, Slijkhuis RJ, Snijders AP, Dickman MJ, Makarova KS, Koonin EV, van der Oost J. Small CRISPR RNAs guide antiviral defense in prokaryotes. Science. 2008;321:960–964. doi: 10.1126/science.1159689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jore MM, Lundgren M, van Duijn E, Bultema JB, Westra ER, Waghmare SP, Wiedenheft B, Pul U, Wurm R, Wagner R, Beijer MR, Barendregt A, Zhou K, Snijders AP, Dickman MJ, Doudna JA, Boekema EJ, Heck AJ, van der Oost J, Brouns SJ. Structural basis for CRISPR RNA-guided DNA recognition by Cascade. Nat Struct Mol Biol. 2011;18:529–536. doi: 10.1038/nsmb.2019. [DOI] [PubMed] [Google Scholar]
- 3.Jackson RN, Golden SM, van Erp PB, Carter J, Westra ER, Brouns SJ, Van Der Oost J, Terwilliger TC, Read RJ, Wiedenheft B. Crystal structure of the CRISPR RNA-guided surveillance complex from Escherichia coli. Science. 2014;345:1473–1479. doi: 10.1126/science.1256328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhao H, Sheng G, Wang J, Wang M, Bunkoczi G, Gong W, Wei Z, Wang Y. Crystal structure of the RNA-guided immune surveillance Cascade complex in Escherichia coli. Nature. 2014;515:147–150. doi: 10.1038/nature13733. [DOI] [PubMed] [Google Scholar]
- 5.Mulepati S, Bailey S. In Vitro Reconstitution of an Escherichia coli RNA-guided Immune System Reveals Unidirectional, ATP-dependent Degradation of DNA Target. J Biol Chem. 2013;288:22184–22192. doi: 10.1074/jbc.M113.472233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hochstrasser ML, Taylor DW, Bhat P, Guegler CK, Sternberg SH, Nogales E, Doudna JA. CasA mediates Cas3-catalyzed target degradation during CRISPR RNA-guided interference. Proc Natl Acad Sci U S A. 2014;111:6618–6623. doi: 10.1073/pnas.1405079111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Westra ER, van Erp PBG, Künne T, Wong SP, Staals RHJ, Seegers CLC, Bollen S, Jore MM, Semenova E, Severinov K, de Vos WM, Dameb RT, de Vries R, Brouns SJJ, van der Oost J. CRISPR immunity relies on the consecutive binding and degradation of negatively supercoiled invader DNA by Cascade and Cas3. Mol Cell. 2012;46:595–605. doi: 10.1016/j.molcel.2012.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hayes RP, Xiao Y, Ding F, van Erp PBG, Rajashankar K, Bailey S, Wiedenheft B, Ke A. Structural basis for promiscuous PAM recognition in type I–E Cascade from E. coli. Nature. 2016;530:499–503. doi: 10.1038/nature16995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Semenova E, Jore MM, Datsenko KA, Semenova A, Westra ER, Wanner B, van der Oost J, Brouns SJ, Severinov K. Interference by clustered regularly interspaced short palindromic repeat (CRISPR) RNA is governed by a seed sequence. Proc Natl Acad Sci U S A. 2011;108:10098–10103. doi: 10.1073/pnas.1104144108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jung C, Hawkins JA, Jones SK, Jr, Xiao Y, Rybarski JR, Dillard KE, Hussmann J, Saifuddin FA, Savran CA, Ellington AD, Ke A, Press WH, Finkelstein IJ. Massively Parallel Biophysical Analysis of CRISPR-Cas Complexes on Next Generation Sequencing Chips. Cell. 170:35–47.e13. doi: 10.1016/j.cell.2017.05.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.van Erp Paul BG, Jackson RN, Carter J, Golden SM, Bailey S, Wiedenheft B. Mechanism of CRISPR-RNA guided recognition of DNA targets in Escherichia coli. Nucleic Acids Research. 2015;43:8381–8391. doi: 10.1093/nar/gkv793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mulepati S, Heroux A, Bailey S. Crystal structure of a CRISPR RNA-guided surveillance complex bound to a ssDNA target. Science. 2014;345:1479–1484. doi: 10.1126/science.1256996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xiao Y, Luo M, Hayes RP, Kim J, Ng S, Ding F, Liao M, Ke A. Structure Basis for Directional R-loop Formation and Substrate Handover Mechanisms in Type I CRISPR-Cas System. Cell. 2017;170:48–60.e11. doi: 10.1016/j.cell.2017.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fineran PC, Gerritzen MJ, Suarez-Diez M, Kunne T, Boekhorst J, van Hijum SA, Staals RH, Brouns SJ. Degenerate target sites mediate rapid primed CRISPR adaptation. Proc Natl Acad Sci U S A. 2014;111:1629–1638. doi: 10.1073/pnas.1400071111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Westra ER, Semenova E, Datsenko KA, Jackson RN, Wiedenheft B, Severinov K, Brouns SJ. Type I-E CRISPR-Cas Systems Discriminate Target from Non-Target DNA through Base Pairing-Independent PAM Recognition. PLoS Genet. 2013;9:e1003742. doi: 10.1371/journal.pgen.1003742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Datsenko KA, Pougach K, Tikhonov A, Wanner BL, Severinov K, Semenova E. Molecular memory of prior infections activates the CRISPR/Cas adaptive bacterial immunity system. Nat Commun. 2012;3:945. doi: 10.1038/ncomms1937. [DOI] [PubMed] [Google Scholar]
- 17.Richter C, Dy RL, McKenzie RE, Watson BN, Taylor C, Chang JT, McNeil MB, Staals RH, Fineran PC. Priming in the Type I-F CRISPR-Cas system triggers strand-independent spacer acquisition, bi-directionally from the primed protospacer. Nucleic Acids Res. 2014;42:8516–8526. doi: 10.1093/nar/gku527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Redding S, Sternberg Samuel H, Marshall M, Gibb B, Bhat P, Guegler Chantal K, Wiedenheft B, Doudna Jennifer A, Greene Eric C. Surveillance and Processing of Foreign DNA by the Escherichia coli CRISPR-Cas System. Cell. 2015;163:854–865. doi: 10.1016/j.cell.2015.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xue C, Whitis NR, Sashital DG. Conformational Control of Cascade Interference and Priming Activities in CRISPR Immunity. Molecular Cell. 2016;64:826–834. doi: 10.1016/j.molcel.2016.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xue C, Seetharam AS, Musharova O, Severinov K, Brouns JSJ, Severin AJ, Sashital DG. CRISPR interference and priming varies with individual spacer sequences. Nucleic Acids Research. 2015;43:10831–10847. doi: 10.1093/nar/gkv1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Swarts DC, Mosterd C, van Passel MW, Brouns SJ. CRISPR Interference Directs Strand Specific Spacer Acquisition. PLoS One. 2012;7:e35888. doi: 10.1371/journal.pone.0035888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Blosser TR, Loeff L, Westra ER, Vlot M, Kunne T, Sobota M, Dekker C, Brouns SJ, Joo C. Two distinct DNA binding modes guide dual roles of a CRISPR-Cas protein complex. Mol Cell. 2015;58:60–70. doi: 10.1016/j.molcel.2015.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Guttman MWD, Engen JR, Lee KK. Analysis of overlapped and noisy hydrogen/deuterium exchange mass spectra. J Am Soc Mass Spectrom. 2013;24:1906–1912. doi: 10.1007/s13361-013-0727-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hourdel VVS, O’Brien DP, Chenal A, Chamot-Rooke J, Dillies MA, Brier S. MEMHDX: An interactive tool to expedite the statistical validation and visualization of large HDX-MS datasets. 2016 doi: 10.1093/bioinformatics/btw420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Engen JR. Analysis of Protein Conformation and Dynamics by Hydrogen/Deuterium Exchange MS. Analytical Chemistry. 2009;81:7870–7875. doi: 10.1021/ac901154s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jackson RN, van Erp PBG, Sternberg SH, Wiedenheft B. Conformational regulation of CRISPR-associated nucleases. Current Opinion in Microbiology. 2017;37:110–119. doi: 10.1016/j.mib.2017.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sashital DG, Jinek M, Doudna JA. An RNA induced conformational change required for CRISPR RNA cleavage by the endonuclease Cse3. Nat Struct Mol Biol. 2011;18:680–687. doi: 10.1038/nsmb.2043. [DOI] [PubMed] [Google Scholar]
- 28.Szczelkun MD, Tikhomirova MS, Sinkunas T, Gasiunas G, Karvelis T, Pschera P, Siksnys V, Seidel R. Direct observation of R-loop formation by single RNA-guided Cas9 and Cascade effector complexes. Proc Natl Acad Sci U S A. 2014;111:9798–9803. doi: 10.1073/pnas.1402597111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Almendros C, Guzman NM, Diez-Villasenor C, Garcia-Martinez J, Mojica FJ. Target Motifs Affecting Natural Immunity by a Constitutive CRISPR-Cas System in Escherichia coli. PLoS One. 2012;7:e50797. doi: 10.1371/journal.pone.0050797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wiedenheft B, Sternberg SH, Doudna JA. RNA-guided genetic silencing systems in bacteria and archaea. Nature. 2012;482:331–338. doi: 10.1038/nature10886. [DOI] [PubMed] [Google Scholar]
- 31.Mulepati S, Orr A, Bailey S. Crystal structure of the largest subunit of a bacterial RNA-guided immune complex and its role in DNA target binding. J Biol Chem. 2012;287:22445–22449. doi: 10.1074/jbc.C112.379503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sashital DG, Wiedenheft B, Doudna JA. Mechanism of foreign DNA selection in a bacterial adaptive immune system. Mol Cell. 2012;48:606–615. doi: 10.1016/j.molcel.2012.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tay M, Liu S, Yuan YA. Crystal structure of Thermobifida fuscaCse1 reveals target DNA binding site. Protein Science. 2015;24:236–245. doi: 10.1002/pro.2609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wiedenheft B, Lander GC, Zhou K, Jore MM, Brouns SJJ, van der Oost J, Doudna JA, Nogales E. Structures of the RNA-guided surveillance complex from a bacterial immune system. Nature. 2011;477:486–489. doi: 10.1038/nature10402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rutkauskas M, Sinkunas T, Songailiene I, Tikhomirova MS, Siksnys V, Seidel R. Directional R-Loop Formation by the CRISPR-Cas Surveillance Complex Cascade Provides Efficient Off-Target Site Rejection. Cell Rep. 2015 doi: 10.1016/j.celrep.2015.01.067. [DOI] [PubMed] [Google Scholar]
- 36.Vaudel M, Burkhart JM, Zahedi RP, Oveland E, Berven FS, Sickmann A, Martens L, Barsnes H. PeptideShaker enables reanalysis of MS-derived proteomics data sets. Nat Biotech. 2015;33:22–24. doi: 10.1038/nbt.3109. [DOI] [PubMed] [Google Scholar]
- 37.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera—A visualization system for exploratory research and analysis. Journal of Computational Chemistry. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.