

Abstract
A growing body of evidence suggests that the inflammatory NFκB pathway is associated with the progression of ER+ tumors to more aggressive stages. However, it is unknown whether NFκB is a driver or a consequence of aggressive ER+ disease. To investigate this question, we developed breast cancer cell lines expressing an inducible, constitutively active form of IκB kinase β (CA-IKKβ), a key kinase in the canonical NFκB pathway. We found that CA-IKKβ blocked E2-dependent cell proliferation in vitro and tumor growth in vivo in a reversible manner, suggesting that IKKβ may contribute to tumor dormancy and recurrence of ER+ disease. Moreover, coactivation of ER and IKKβ promoted cell migration and invasion in vitro and drove experimental metastasis in vivo. Gene expression profiling revealed a strong association between ER and CA-IKKβ-driven gene expression and clinically relevant invasion and metastasis gene signatures. Mechanistically, the invasive phenotype appeared to be driven by an expansion of a basal/stem-like cell population rather than EMT. Taken together, our findings suggest that coactivation of ER and the canonical NFκB pathway promotes a dormant, metastatic phenotype in ER+ breast cancer and implicates IKKβ as a driver of certain features of aggressive ER+ breast cancer.
Keywords: Estrogen, breast cancer, dormancy, metastasis, IKKβ
INTRODUCTION
Breast cancer is the second leading cause of cancer-related mortalities in women of the US (1), and the majority of these deaths are the result of estrogen receptor α (ER) positive disease (2). ER positive tumors are typically dependent on estrogens for cell proliferation, survival, and disease progression. Endocrine therapies that target estrogen production or ER activity are generally effective; however, up to 50% of women receiving endocrine therapy will experience relapse (3, 4). A characteristic of ER positive breast cancer is an increased risk of late recurrence, which may occur after a period of dormancy ranging from 5 to more than 20 years after initial diagnosis and treatment (3). Moreover, when ER positive tumors recur, they tend to be metastatic with ~70% of all metastatic breast tumors arising from ER positive disease (2). Thus, there is an urgent need to better understand pathways and mechanisms contributing to prolonged dormancy and metastatic recurrence in ER positive breast cancer.
A growing body of evidence suggests that an inflammatory microenvironment and activation of the pro-inflammatory NFκB pathway are highly associated with the progression of ER positive tumors to more aggressive stages and therapy resistance (5-10). For example, studies have shown that the NFκB family members are active in ~15-25% of ER positive breast tumors, and that this activation is associated with increased risk of recurrence, metastasis, and poor overall survival (5, 7, 10). High DNA binding activity of NFκB subunits can also identify a subset of ER positive tumors with high-risk for relapse on endocrine therapy (10, 11), as well as poor response to chemotherapy (7). Several preclinical studies have shown that NFĸB inhibition restores sensitivity to endocrine therapy in resistant breast cancer cell lines (6, 8, 9) while a phase II clinical study suggests the combination of endocrine treatment and the protease inhibitor bortezomib, which blocks the NFκB pathway, slows disease progression in patients who developed metastatic disease during endocrine therapy (12). Mechanistically, previous work from our lab indicated that NFκB works cooperatively with ER to transcriptionally regulate genes associated with a Luminal B phenotype and poor response to tamoxifen (13). Despite this considerable body of evidence supporting a role for the NFκB pathway in aggressive ER positive breast tumors, it is not clear whether NFκB activation is a driver or a consequence of progression to aggressive ER positive disease.
To address this, we introduced a doxycycline (DOX)-inducible, constitutively active form of a key kinase in the canonical NFκB pathway, IKKβ (CA-IKKβ), into ER positive breast cancer cell lines. Canonical NFκB signaling is generally activated by stimuli such as pro-inflammatory cytokines, which activates the upstream IκB kinase complex, which includes IKKβ. Our focus on the canonical arm is based on the studies that report women who have ER positive tumors with constitutively active p50 and p65 represent a subset of patients with increased risk of recurrence, metastasis, and poor overall survival (7, 10). Also, IKKβ kinase activity has been reported in ER positive breast tumors (14). Moreover, multiple publically available gene expression profiling studies of human tumors suggest that IKKβ mRNA expression is associated with ER positive status (15-17).
Surprisingly, we found that the activation of IKKβ does not promote a more aggressive growth phenotype but rather inhibits E2-induced proliferation in vitro and tumor growth in vivo. However, this inhibition was reversible, indicative of a dormant or quiescent rather than a senescent phenotype. In contrast, ER and NFκB activation promoted cell survival, which is expected given that both are known survival factors. In addition, co-activation of both ER and the canonical NFκB pathway promoted cell migration and invasion in vitro, as well as experimental metastasis in vivo. Mechanistically, our studies suggest this is the result of an expansion of a cell population with stem-like and basal-like properties without accompanying epithelial-mesenchymal transition (EMT). Taken together, our findings suggest that activation of the NFκB pathway may contribute to tumor dormancy and recurrence through control of cell proliferation and survival, as well as to metastases, through cooperation with ER to enhance invasive properties.
MATERIALS AND METHODS
Reagents
17β-estradiol (E2) and doxycycline hyclate (DOX) were purchased from Sigma. TNFα was purchased from R&D Systems. ICI and IKK7 were purchased from Sigma (#I4409) and SelleckChem (#S2882), respectively. Antibodies for IKKβ (#2370), IKKα (#2682), p-IκBα (#2859), IκBα (#4814), p-p65 S536 (#3033), ERα (#8644), E-cadherin (#3195P), vimentin (#5741P), CK8/18 (#4546), GATA3 (#5852) and P63-α (#4892) were purchased from Cell Signaling. Antibodies for p65 (#sc-372) and Fibronectin (#sc-9068) were purchased from Santa Cruz. Antibodies for FoxA1 (#ab55178), and CK14 (#ab53115) were purchased from Abcam. Antibodies for CK5 (#CK5-L-CE) and β-actin (#A5441) were purchased from, Leica and Sigma, respectively.
Cell culture and cell lines
DOX-inducible, CA-IKKβ expressing cell lines were derived using the Retro-X™ Tet-On® Advanced Inducible Expression System (Clontech Laboratories). An IKKβ expression vector was purchased from InvivoGen and serine to glutamate mutations (S177E/S181E) were introduced using site-directed mutagenesis (Stratagene). CA-IKKβ was then subcloned into the puromycin resistant Tet-On vector pRetroX-Tight-Pur, which was co-transfected with the viral envelop expression vector, pVSV-G, into GP2-293 packaging cells to generate retrovirus. The retroviral supernatant was used to infect MCF-7-rtTA and T47D-rtTA cells, which are stably transduced with geneticin-resistant pRetroXTet-On Advanced vector encoding rtTA-Advanced transactivator. MCF-7-rtTA and T47D-rtTA cell lines were a gift from Dr. Debra Tonetti (UIC). Selection with Geneticin and Puromycin to select CA-IKKβ expressing cells was performed followed by limiting dilutions in 96-well plates. Multiple clones were derived from single cells. MCF-7-CA-IKKβ-LUC cells are MCF-7-CA-IKKβ cells transduced with the lentiviral expression vector for pFU-Luc2-eGFP (18), a gift from Dr. Michael Clarke (Stanford University). Transduced cells were isolated by fluorescence activating cell sorting using MoFlo Astrios Cell Sorter (Beckman Coulter) (UIC).
Cells were routinely maintained in Roswell Park Memorial Institute medium (RPMI) 1640 with phenol red (Life Technologies) supplemented with 10% fetal bovine serum, 1% penicillin-streptomycin, 1% non-essential amino acids, 2 mM L-glutamine and 6 ng/mL insulin. Cell line authentication is routinely performed for CA-IKKβ cell lines using short tandem repeat (STR) and routinely confirmed as mycoplasma negative. Before treatment, cells were cultured in phenol red-free RPMI 1640 medium supplemented with 5% charcoal-dextran-stripped FBS for 2-3 days. Unless otherwise noted, control cells (none) were left untreated. To test the role of NFκβ signaling, cells were infected with recombinant adenovirus IκBα-DN or GFP as a negative control (Vector Biolabs) at an MOI of 200 for 16 hr prior to treatment. To test the role of CK5 in invasion, cells were transfected with siRNA targeting cytokeratin 5 (siCK5) or non-targeting control siRNA (siNeg) (Thermo Fisher Scientific) using DharmaFECT 1 (Dharmacon).
Western blotting
Whole cell extracts were prepared and proteins were denatured, separated by SDS-PAGE, and transferred to nitrocellulose membranes. Membranes were blocked for 1hr in buffer containing 5% bovine serum albumin or 5% nonfat dry milk. Membranes were then incubated overnight at 4°C with the appropriate primary antibody followed by incubation in horseradish peroxidase conjugated secondary antibodies for 1 hr. The signal was visualized using Chemidoc XRS (Bio-Rad Laboratories) following incubation with the Pierce Supersignal West Pico Chemiluminescent Substrate (Thermo Fisher Scientific).
RT-Quantitative PCR (QPCR)
Total RNA was isolated using Trizol and quantitative PCR was performed and analyzed as described previously (13). Fold change was calculated using the ΔΔCt method with 36B4 serving as the internal control. Primer sequences are available upon request.
Proliferation assays
Cells were either trypsinized and counted using hemocytometer or incubated in Hoechst 33342 dye (Life Technologies) for 1 hr at 37°C. Fluorescence was read and cell numbers were extrapolated from cell number standard curves.
Cell cycle analysis
Cells were trypsinized and fixed with 1% p-formaldehyde followed by ice cold 70% ethanol, and incubated at 4°C overnight. Cells were centrifuged and resuspended in Propidium Iodide staining solution (40 μg/mL Propidium Iodide and 100 μg/mL RNAse A), and incubated for 30 min at 37°C. Cell cycle distribution was determined by flow cytometry using a BD LSR Fortessa flow cytometer (UIC).
Wound healing assay
Cells were seeded and incubated until they reached ~80% confluence. A wound was created using a 10μL pipette, then cells were rinsed with PBS and treated. Wounds were monitored for 96 hrs and wound gaps were measured using QCapture Pro and an inverted Nikon Eclipse Microscope and calculated as the percentage of the initial wound.
Transwell migration and invasion assays
Falcon transwell inserts (8.0 μm, Corning) were used for migration assays. The inserts were coated with 100 μL of 300 μg/mL phenol red free Matrigel (BD Biosciences) for invasion assays. Equal number of cells were seeded in the inserts and allowed to migrate or invade for 24 and 16 hrs (MCF-7-CA-IKKβ and T47D-CA-IKKβ respectively). Non-migrating/invading cells were removed from the inside of the inserts using cotton swabs. Cells on the outer side of the membrane were fixed and stained using 4% p-formaldehyde and 0.1% crystal violet. The total number of migrating/invading cells was counted manually using a 10× light microscope objective.
Animal experiments
Experiments were carried out at the UIC Biological Resources Laboratory and all procedures and studies were approved by the Animal Care and Use Committee according to institutional and national guidelines. Xenografts were established in female athymic nude mice (nu/nu) by injecting 5 × 106 cells orthotopically into the thoracic mammary fat pad of the animals. Silastic capsules (1 cm) containing 17β-estradiol (E2) were implanted at the same time of cell injection. Mice were examined for tumors by palpation using digital calipers and tumor cross-sectional area was calculated ((length/2) × (width/2) × π). When tumors reached a mean cross-sectional area of ~0.1 cm2 mice were randomized into 3 groups (n=7-8 tumors/group): 1) E2 only: received 5% sucrose in the drinking water, 2) E2 with 12 days DOX: received DOX (2 mg/mL in 5% sucrose) in the drinking water for 12 days, at which point it was withdrawn; 5% sucrose was administered throughout the rest of the study, 3) E2 positive with continuous DOX: received DOX continuously throughout the experiment (29 days). For experimental metastasis studies, cells were treated with DOX, E2 or E2+DOX in culture for 72 hrs prior to injection into the lateral tail vein (1×106 cells/animal). Two days prior cell injection, E2 silastic or mock capsules were implanted subcutaneously and DOX or sucrose was introduced to the animals in the drinking water and maintained for 5 more days post cell injection.
Animal imaging
Live animals were imaged using the IVIS spectrum, Xenogen (Caliper Life Sciences). D-Luciferin potassium salt (Biosynth Chemistry and Biology) was injected intraperitoneally (150 mg/kg body weight) and animals were anesthetized in an oxygen-rich induction chamber with 2.5% isofluorane. For ex vivo imaging, animals were injected intraperitoneally with D-Luciferin then euthanized with CO2 gas followed by cervical dislocation. Tissues of interest were excised, rinsed with DPBS, and soaked in D-Luciferin (300 μg/mL). The signal was measured as radiance (photons/second/square centimeter/steradian), and analyzed with Living Image Software (Caliper Life Sciences).
Microarray analysis
Cells were treated with E2, DOX or E2+DOX for 72 hrs (3 replicates per treatment). RNA was extracted using Trizol followed by purification with the RNeasy RNA Extraction Kit.
(Qiagen, Valencia, CA, USA). RNA integrity was determined and RINe ranged from 9.5 to 10 for all samples. Samples were reverse transcribed, amplified, fragmented, and biotin-labeled with the GeneChip® 3′ IVT Express Kit (Affymetrix, P/N 901229) according to the manufacturer’s protocol. Samples were then hybridized to GeneChip® PrimeViewTM Human Gene Expression Arrays (Affymetrix, P/N 901838) and washed and stained using the GeneChip® Hybridization, Wash, and Stain Kit (Affymetrix, P/N 900720) according to the manufacturer’s protocol. Arrays were hybridized in a GeneChip® Hybridization Oven 640, washed and stained on a GeneChip® Fluidics Station 450 and scanned on a GeneChip® Scanner 3000 7G. Analysis was performed in Core Genomics Facility at UIC. Data was normalized using Robust Multichip Average and statistical analysis was performed. Each of the three treatments was compared to control. Genes with fold change more than 1.5 and p<0.01, by t-test using log-transformed values, were identified and supervised clustering was performed as previously described (19). False Discovery Rates (FDRs) were estimated using the method of Storey and Tibshirani (20). Data are available on the Gene Expression Omnibus website (#GSE85683). The Molecular Signatures Database (MSigDB) gene set collections (version 4), made available by the Broad Institute (21), were evaluated for significance of overlap with the gene sets arising from our microarray analysis; significance of overlap was determined by one-sided Fisher’s exact tests.
Immunofluorescence and confocal microscopy
Cells were seeded on 0.1% gelatin coated or non-coated glass coverslips. After treatment, cells were fixed with 4% p-formaldehyde for 10 min and permeabilized using 0.1% Triton X-100 for 1 min. Cells were then blocked with 10% goat serum for 1 hr and incubated with the primary antibody in a humidified chamber at 4°C overnight. Coverslips were incubated with the secondary antibody for 1 hr and then mounted with ProLong Gold anti-fade reagent (Life Technologies). Images were acquired at ×63 magnification using a LSM710 confocal microscope (Carl Zeiss).
Cancer stem cell assays
The mammosphere (MS) assay was conducted using breast cancer cells seeded at single cell density (500 cells/well) on low attachment plates in media described by Dontu et al., supplemented with 1% methyl cellulose to prevent cellular aggregation (22). After 7 days, the diameter of MS was measured and MS ≥75μm in diameter were counted. MS forming efficiency (#MS ≥75μm /500 cells*100) and average MS diameter size are reported. Antibodies for CD44 and CD24 were purchased from Pharmingen. Cell labeling and flow cytometry was done according to Liu et al. (23). The aldefluor assay (Stem Cell technologies) and FACS analysis were conducted as previously reported by Charafe-Jauffret et al.(24).
Statistical analysis
Statistical analyses were performed using GraphPad Prism 6.0 software. One way or two way ANOVA (followed by Tukey or Sidak’s posttest) were used where appropriate. Data are presented as mean ± SEM or ± SD from at least two replicates. Metastatic incidence was analyzed using contingency tables and Chi Square test. All p-values were two-sided unless otherwise specified.
RESULTS
Constitutively active IKKβ reversibly inhibits E2-induced proliferation and tumor growth
To determine how NFκB pathway activation affects progression of ER positive breast cancer, we introduced DOX-inducible, CA-IKKβ into MCF-7 and T47D breast cancer cell lines (MCF-7-CA-IKKβ and T47D-CA-IKKβ). In both lines, DOX treatment induced a substantial increase in IKKβ expression and activity, as indicated by IκBα phosphorylation and degradation, p65 phosphorylation, and NFκB target gene expression (Fig. 1A and 1B). Since we hypothesized that NFκB pathway activation would promote more aggressive disease, we first examined the effect of CA-IKKβ on proliferation. Surprisingly, we found that DOX treatment prevented an E2-dependent increase in cell number in both cell lines expressing CA-IKKβ but not in control cells lacking CA-IKKβ (Fig. 2A and Supplemental Fig. 1). One possible explanation for the inhibitory effect of CA-IKKβ is that the NFκB pathway has been shown to down-regulate ER expression, which we confirmed on ERα mRNA and protein (Supplemental Fig. 2A and B). However, ER positive cells remain that co-express IKKβ and the ER remains transcriptionally active based on the up-regulation of canonical E2-target genes, such as pS2 and PR (Supplemental Fig. 2C and Supplemental 2D). This suggests that ER down-regulation is not likely to be the mechanism underlying inhibition of E2-induced proliferation by CA-IKKβ.
Figure 1. Doxycycline treatment activates the NFκB pathway in ER positive breast cancer cell lines expressing CA-IKKβ.
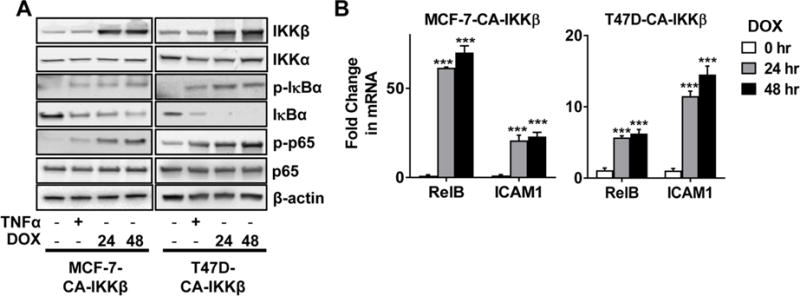
A. MCF-7-CA-IKKβ and T47D-CA-IKKβ cells were treated with DOX (1 μg/mL) for 24 or 48 hrs, or with TNFα (10 ng/mL) for 30 min as a positive control. Whole cell extracts were prepared and expression of IKKβ, IKKα, phospho-IκBα (Ser32), IκBα, phospho-p65 (Ser532), p65, and β-actin was detected by Western Blotting. B. MCF-7-CA-IKKβ and T47D-CA-IKKβ cells were treated with DOX for 24 hrs or 48 hrs. Expression of the NFκB target genes, RelB and ICAM1, was assessed by QPCR. (***p<0.001 vs. untreated control).
Figure 2. CA-IKKβ reversibly inhibits E2-induced proliferation and enhances cell survival.
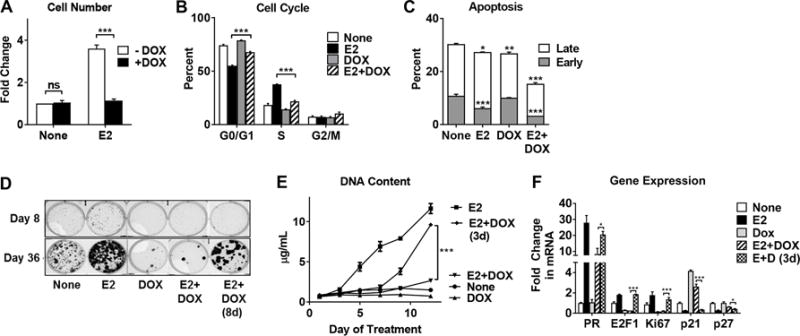
A-C, MCF-7-CA-IKKβ cells were treated with E2 (10 nM), DOX (1 μg/mL), or E2+DOX for 72 hrs. Cells were trypsinized and counted using a hemocytometer (A), fixed and stained with propidium iodide for cell cycle analysis (B), or fixed and stained with Annexin V-FITC and propidium iodide to assess apoptosis (C). D, MCF-7-CA-IKKβ cells were seeded as single cells and treated with E2, DOX, or E2+DOX for 36 days. On day 8, DOX was withdrawn from one E2+DOX group. Colonies were visualized following methanol fixation and crystal violet staining. E, MCF-7-CA-IKKβ cells were treated with E2, DOX, or E2+DOX for 12 days. After 3 days, DOX was withdrawn from one E2+DOX group. Cells were retreated every 2-3 days. DNA content was determined by Hoechst 33342 staining. F. To assess reversal of gene expression, MCF-7-CA-IKKβ cells were treated with E2, DOX, or both for 8 days and DOX was withdrawn from one group on day 3. RNA was isolated and gene expression was assessed by RT-QPCR for E2-regulated genes. (* P<0.05, ** P<0.01, *** P<0.001 and ns=non-significant).
The anti-proliferative effect of CA-IKKβ is the result of cell cycle arrest since a reduction of cells in the S phase and an accumulation of cells in the G0/G1 phase was observed with E2+DOX treatment (Fig. 2B and Supplemental Fig. 3A). Interestingly, apoptosis does not appear to contribute to the reduced cell number. In fact, cell survival is augmented by E2 and DOX co-treatment vs. E2 alone, as indicated by a reduction in both early and late apoptosis (Fig. 2C) and the synergistic up-regulation of the pro-survival gene BIRC3 (Supplemental Fig. 3B), which we have previously shown is necessary for the survival of ER positive breast cancer cells (25). DOX treatment (either alone or in combination with E2) suppressed clonogenic growth, an assay that measures colonies greater than 50 cells and reflects both the survival of colony initiating cells and subsequent proliferative outgrowth of colonies (Fig. 2D and Supplemental Fig. 3C).
To determine if CA-IKKβ may be inducing quiescence or senescence, we first investigated whether the anti-proliferative effects of CA-IKKβ were reversible. DOX withdrawal rescued E2-dependent proliferation, as observed by an increase in DNA content, as well as colony formation (Fig. 2D and 2E and Supplemental Figs. 3C and 3D). Next, regulation of a panel of genes associated with quiescence and senescence, as well as ER target genes, was investigated following prolonged DOX treatment (Fig. 2F). We found that continuous DOX treatment for 8 days, either alone or in combination with E2, suppressed expression of cell proliferation genes (E2F1 and Ki67) but increased expression of the cell cycle inhibitors and quiescence genes (p21 and p27). DOX withdrawal after 3 days of treatment reversed these effects. Expression of the senescence associated genes p16 and ARF were not detected under any of the treatment conditions (Supplemental Fig. 3E). In contrast, DOX treatment had minimal impact on the regulation of PR by E2 (Fig. 2F). Together, these results indicated that constitutive activation of IKKβ inhibits E2-stimulated proliferation by inducing a reversible quiescent phenotype, yet works together with E2 to promote cell survival.
We next investigated whether IKKβ could influence cellular response to tamoxifen, potentially through phosphorylation of ER on S305, as we previously reported with the proinflammatory cytokines TNFα and IL-1β (26). We first confirmed that IKKβ activation promotes ER phosphorylation on S305 (Supplemental Fig. 4A). However, IKKβ activation had no evident effect on the ability of tamoxifen to inhibit cell proliferation or ER target gene expression (Supplemental Figs. 4B-D). It is possible that the timing of IKKβ activation by DOX may be a confounding factor, since it takes 24 hr to induce S305 phosphorylation with DOX but 15-30 min with cytokines (26). Alternatively, S305 phosphorylation may be required but not sufficient on its own to cause resistance, suggesting that cytokines may utilize additional mechanisms to promote resistance beyond S305 phosphorylation.
In order to investigate how IKKβ affects primary tumor growth in vivo, MCF-7-CA-IKKβ-LUC cells were used to establish E2-dependent xenografts. DOX administration in the drinking water caused complete tumor regression within 12 days (Fig. 3A). However, viable tumor cells remained in the primary implantation site for up to 29 days based on bioluminescence imaging (Fig. 3B). In one group of animals where DOX was withdrawn on day 12, 7 of 8 tumors recurred, indicating the survival of cells capable of reseeding a recurrent tumor (Fig. 3A). These results indicated that constitutive activation of IKKβ inhibits E2-stimulated tumor growth in a reversible manner and suggests a role for IKKβ in dormancy and recurrence of ER positive tumors.
Figure 3. CA-IKKβ controls regression and recurrence of ER positive xenografts.
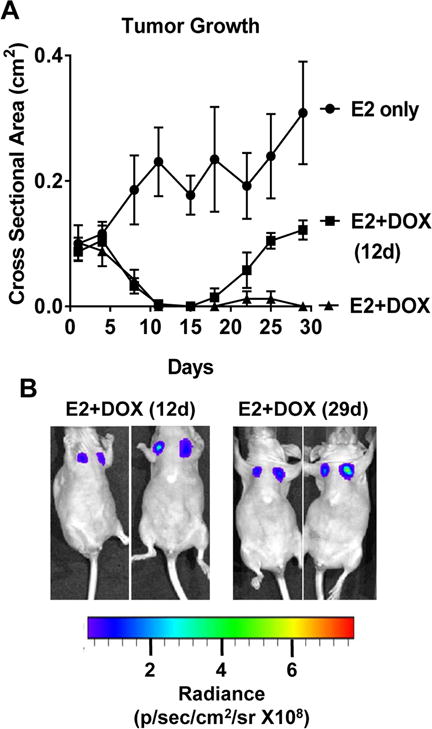
A, B. MCF-7-CA-IKKβ-LUC cells were injected into the mammary fat pad of female athymic nude mice (n=7-8/group) in which E2 silastic pellets were subcutaneously implanted. When tumors reached ~0.1 cm2 in area, animals were randomized into 3 groups: 1) E2, 2) E2+ DOX for 12 days in the drinking water, 3) E2+DOX continuously for 29 days. Tumor cross sectional area was monitored and plotted (A). Animals were injected with D-Luciferin (150 mg/kg) and bioluminescence was detected using IVIS spectrum on day 12 and 29 of DOX administration (B).
Constitutively active IKKβ induces a highly invasive phenotype in ER positive breast cancer cells and enhances ER positive breast cancer cell metastasis
Interestingly, in the course of our studies, we observed that cells adopted an elongated, fibroblastic morphology with cellular protrusions upon IKKβ activation (Supplemental Fig. 5A and 5B), suggestive of migratory behavior. Wound healing assays revealed that E2+DOX treatment significantly enhanced wound closure to a greater extent than either E2 or DOX alone (Fig. 4A). In contrast, wound closure was not different between E2+DOX and E2 alone in control cells lacking CA-IKKβ (Supplemental Fig. 6A). We confirmed that E2+DOX increases migration using a transwell migration assay (Fig. 4B and Supplemental Fig. 6B). Moreover, invasion was also significantly enhanced by co-treatment with E2 and DOX compared to either E2 or DOX alone (Fig. 4C and Supplemental Fig. 6C). Downstream of E2+DOX, both ER and NFκB activity were required for enhanced invasion, since the ER antagonist, ICI 182,780 and a dominant negative form of IκB (IκBα-DN), substantially reduced invasion (Fig. 4D and 4E). Of note, tamoxifen did not prevent the migratory phenotype stimulated by E2 + DOX, and may in fact stimulate migration on its own (Supplemental Fig. 6D), which is consistent with some reports in the literature (27, 28). Thus, these studies demonstrate that co-activation of ER and IKKβ switches ER positive breast cancer cells from a highly proliferative to a highly migratory and invasive phenotype.
Figure 4. E2 and CA-IKKβ work together to promote migration and invasion.
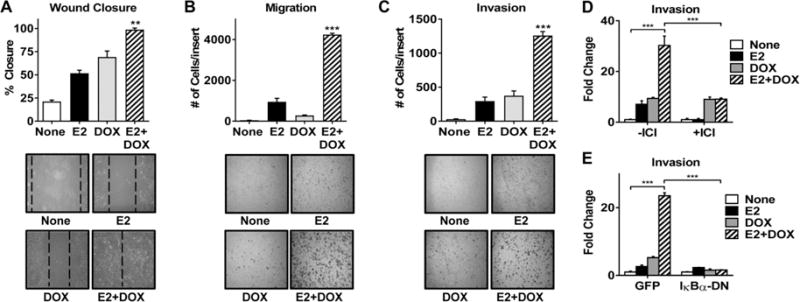
A. MCF-7-CA-IKKβ cells were grown to ~80% confluence and a wound was created. Cells were then washed and treated with E2, DOX, or E2+DOX for 96 hrs. Wound closure was measured as a percent of initial wound size (** P<0.01 vs all other groups). B, C. MCF-7-CA-IKKβ cells were treated with E2, DOX, or E2+DOX for 72 hrs. Equal numbers of cells were transferred to migration (B) or invasion (C) transwell inserts and allowed to migrate or invade for 24 hrs. Cells were fixed, stained and counted manually. (*** P<0.001 vs all other groups). D. MCF-7-CA-IKKβ cells were treated with E2+DOX in the presence or absence of 1 μM ICI 182,780 for 72 hr prior to conducting an invasion transwell assay. (***P<0.001). E. MCF-7-CA-IKKβ cells were infected with adenovirus expressing a dominant negative form of IκB (IκBα-DN) or GFP as a control prior to treatment and an invasion transwell assay. (***P<0.001).
To determine whether ER and IKKβ co-activation may affect metastasis in vivo, an experimental metastasis model was used. MCF-7-CA-IKKβ-LUC cells were pretreated in culture as indicated for 72 hrs, which corresponds to the time point of a migratory/invasive phenotype. Cells were then injected intravenously into the lateral tail vein of athymic nude mice bearing subcutaneously implanted mock or E2 pellets. DOX was administered in the drinking water for one week and then withdrawn in order to allow for the cells to resume proliferation at any metastatic sites. Live animal imaging revealed metastasis in 35% of animals injected with E2+DOX pretreated cells (n=6 of 17) and 23% animal injected with E2 pretreated cells (n=3 of 13), which was not significantly different between the groups (Fig. 5A and B, Supplemental Fig. 7A). Of note, no cases of metastases were detected by live animal imaging for groups injected with DOX pretreated (n=0 of 5) and control cells (n=0 of 5).
Figure 5. Estrogen and CA-IKKβ increase the metastatic potential of MCF-7 cells.
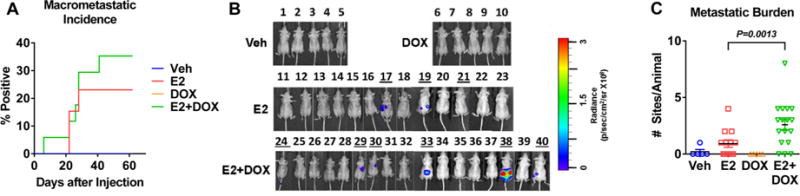
A. MCF-7-CA-IKKβ-LUC cells were injected into the tail vein of athymic mice and macro-metastatic incidence was determined by IVIS imaging over time. No significant difference was found between the groups. B. IVIS images taken for all experimental animals on final day of study. C. The total number of metastatic sites per animal, as detected by ex vivo imaging, is indicated as metastatic burden.
It is known that micrometastases are undetectable during live body imaging so we performed ex vivo imaging of spine, leg bones, and soft organs (brain, liver, spleen, kidney and adrenals) upon animal euthanasia. This approach revealed a significant difference between the groups (p<0.001) with ~82% of animals injected with E2+DOX pretreated cells and ~53% of animals injected with E2 treated cells have a detectable luminescent signal in at least one organ (Supplemental Figure 7A). Moreover, the overall metastatic burden, as determined by the number of metastatic sites per animal, was also significantly higher in the E2+DOX than the E2 only group (Fig. 5C). The major metastatic sites were bones, pelvic organs, and lungs for the E2+DOX group, and bones and pelvic organs but not lung for the E2 group (representative images shown in Supplemental Fig. 7B-D). The presence of cancer cells in metastatic and micrometastatic tumors was confirmed by H&E and immunohistochemistry for GFP and ERα (Supplemental Fig. 7E and 7F). These findings suggest that the combination of E2 and CA-IKKβ enhances the metastatic potential of ER positive breast cancer cells over E2 alone.
E2 and CA-IKKβ regulated gene expression is associated with invasive breast cancer
In order to assess the clinical relevance of the observed ER and CA-IKKβ phenotypes, a global gene expression study was performed using MCF-7-CA-IKKβ cells treated with E2, DOX, or E2+DOX for 72 hrs. More than 12,000 gene probes of 49,000 total were found to be differentially regulated by at least one treatment group vs. none (p<0.01 by t-test and fold change >1.5). Multiple discrete gene regulatory patterns were observed upon supervised clustering of 12271 differential gene probes, which represent 6248 unique genes (Fig. 6A and Supplemental Table 1). These include: Pattern 1) genes regulated by E2, not affected by CA-IKKβ alone, but the effect of E2 was reversed by CA-IKKβ, Pattern 2) genes regulated by CA-IKKβ, not affected by E2 alone, but the effect of CA-IKKβ was reversed by E2, Pattern 3) genes regulated by E2 and not affected by CA-IKKβ, Pattern 4) genes regulated by CA-IKKβ and not affected by E2, Pattern 5) genes similarly regulated by E2, CA-IKKβ, and E2+CA-IKKβ, and Pattern 6) genes that are oppositely regulated by E2 and CA-IKKβ. To understand the potential biological differences between the six gene regulatory patterns, pathway analysis of genes with high or low expression within each of the six major patterns was performed using the Molecular Signatures Database (MSigDB). Significantly enriched MSigDB gene sets included many involved in invasion or metastasis (Fig. 6B and Supplemental Table 2), suggesting that our finding of enhanced invasion upon E2 treatment and IKKβ activation may be of clinical relevance. Interestingly, gene signatures associated with proliferation were primarily enriched in Pattern 6, which showed opposite regulation by E2 and DOX and corresponded to the changes we observed in proliferation and cell cycle genes shown in Fig. 2F.
Figure 6. Global gene expression profiling reveals enrichment of invasive and metastatic, as well basal-like and stem-like, gene signatures following E2 treatment and activation of IKKβ.
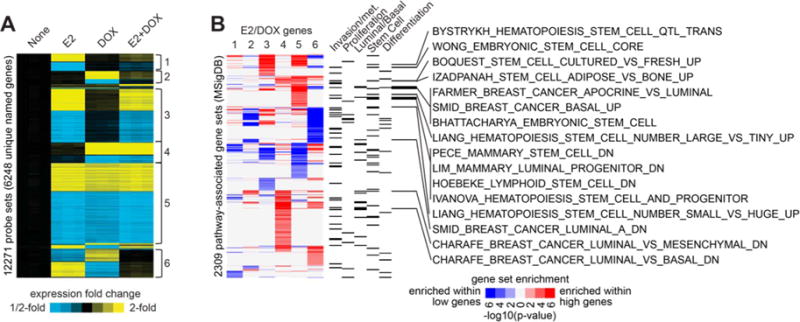
A. MCF-7-CA-IKKβ cells were treated with E2, DOX, or E2+DOX for 72 hrs. mRNA was purified, and samples were analyzed on an Affymetrix high-density oligonucleotide array. Statistical analysis (using cutoffs of p<0.01 by t-test, False Discovery Rate (FDR)<0.05, and fold change>1.5 for any experimental group relative to control) followed by supervised clustering was performed. Six major patterns of gene regulation were observed (labeled next to the expression heat map as 1 through 6), involving 12271 gene probes (representing 6248 unique named genes) showing differential expression out of ~49K probes assessed. B, Each of the gene sets represented by the six major patterns from panel A were examined for enrichment of pathway-associated genes sets from the Molecular Signature Database (MSigDB). For each pattern, “high” genes were evaluated separately from “low” genes (for pattern 6, high and low represents high and low in the DOX group); results for high and low genes shown in red or blue, respectively. P-values were determined by one-sided Fisher’s exact test. MSigDB gene sets with a significance level of FDR<0.1 (involving at least ten genes) for any one E2/DOX gene patterns are represented here (associations with FDR>0.1 are neutral color). Categories of interest for the MSigDB gene sets are indicated (according to key words “invasion” or “metastasis”, “proliferation”, “basal” or “luminal”, “stem cell”, or “differentiation”). MSigDB gene sets that were associated with either basal/luminal or stem cells, as well as enriched within the genes up in pattern 5, are listed individually.
E2 and IKKβ coactivation promotes a basal/stem-like cell phenotype
To understand how activation of ER and IKKβ might promote metastases, we originally hypothesized that EMT might be involved since this has been previously linked to NFkB signaling (29, 30). However, we found no changes in hallmark EMT markers (i.e. reduced E-cadherin or increased fibronectin or vimentin) were observed with E2+DOX treatment (Supplemental Figs. 8A-C), even after prolonged treatment (Supplemental Fig. 8D), suggesting EMT may not be the cellular mechanism underlying enhanced invasion.
Pathway analysis of gene expression data indicated an enrichment of genes associated with basal-like and stem-like cell populations (Fig. 6B), both of which have been associated with enhanced metastatic phenotypes (18, 24, 31, 32). Immunofluorescence studies revealed that E2, DOX, or E2+DOX co-treatment reduces the expression of the luminal markers CK8/18, Gata3, and FoxA1 (Fig. 7A). Expression of the basal markers CK14, p63-α, and CK5 were observed in subpopulations of cells in response to E2, DOX, or E2+DOX treatment, respectively (Fig. 7B and Supplemental Fig. 9A), suggesting that a luminal to basal de-differentiation response takes place upon ER and IKKβ activation. Interestingly, the increase in CK5+ cells required the co-treatment of E2+DOX (Fig. 7C). Although CK5+ cells represent a small percentage of the overall cell population (Fig. 7C), these cells are functionally relevant since knockdown of CK5 largely abrogated E2+DOX-stimulated invasion (Fig. 7D). In addition, MS forming efficiency, a functional stem cell endpoint, but not mammosphere size, was elevated in response to E2 and CA-IKKβ over either alone (Fig. 7E and Supplemental Fig. 9B). Similar results were observed for other markers of stem cell activity, including CD44+/CD24− phenotype and Aldefluor activity (Fig. 7F, 7G, and Supplemental Fig. 9C, representative chromatographs in Supplemental Figs. 9D and E). Both ER and IKKβ activity are required for the increase in the cancer stem-like cell population since the ER antagonist (ICI 182,780) and an IKK kinase inhibitor (IKK7) prevented the expansion of cells with a CD44+/CD24− phenotype (Fig. 7H). Together, these results suggest that the co-activation of ER and IKKβ may result in the expansion of basal-like or stem-like cells that can contribute to the enhanced invasive and metastatic capacity of these cells.
Figure 7. Regulation of luminal, basal, and cancer stem cell markers by E2 and CA-IKKβ.
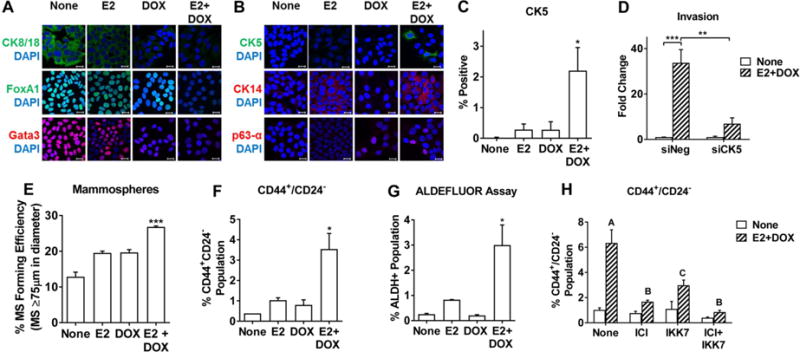
A, B. Expression of luminal markers (A) and basal markers (B) was examined by immunofluorescence staining and confocal microscopy following treatment of MCF-7-CA-IKKβ cells with E2, DOX, or E2+DOX for 72 hrs (scale bars=20μm). C. CK5 positive cells were visualized and quantified following treatment with E2, DOX, or both using VECTRA Automated Multispectral Image Analysis System and InForm software for 3 independent replicates (*P<0.05 vs.untreated control). D. MCF-7-CA-IKKβ cells were transfected with siNeg (control) or siCK5 24 hr prior to initiating treatments followed by an transwell invasion assay (**P<0.01 and ***P<0.001). E-G, Breast cancer stem cells were assessed by the ability of cells treated with E2, DOX, or both for 72 hr to form mammospheres or to express the CD44+/CD24- or ALDH+ phenotypes. (*P<0.05 and ***P<0.001 vs. all other groups). H. Cells were co-treated with E2+DOX and 1 μM ICI 182,780, 1 μM IKK7, or both prior to analysis of CD44 and CD24 cell surface markers by flow cytometry. The different letters above bars indicate significant difference between treatments, A vs B, P<0.001, A vs C, P<0.05.
DISCUSSION
NFκB activation, as well as ER/NFκB crosstalk, is highly associated with aggressive disease and poor patient outcome in women with ER positive breast cancer (7, 10, 13). However, it is not clear whether NFκB is a driver or a consequence of aggressive ER positive disease. In this study, we developed multiple ER positive cell lines that express an inducible, constitutively active form of the kinase IKKβ, which allowed us to specifically activate the canonical arm of the NFκB pathway in a controlled fashion. Using these cells, we found that CA-IKKβ enhances E2-dependent survival but blocks E2-dependent cell proliferation in vitro and tumor growth in vivo, in a reversible manner. An anti-proliferative effect of IKKβ is consistent with previous reports showing that CA-IKKβ inhibits the proliferation of normal human fibroblasts (33), and that immortalized mouse embryo fibroblasts derived from IKKβ knockout mice exhibit increased proliferation (34). Moreover, we found that co-activation of ER and the canonical NFκB pathway promotes cell migration and invasion in vitro and experimental metastasis in vivo potentially via the expansion of basal/stem-like cells. Together, these findings suggest that co-activation of ER and the canonical arm of the NFκB pathway may promote a dormant, metastatic phenotype in ER positive breast cancer.
Our finding that CA-IKKβ reversibly inhibits E2-induced cell proliferation and tumor growth, while enhancing cell survival, indicates that CA-IKKβ-induced growth arrest is due to quiescence rather than senescence. This is also supported by up-regulation of the quiescence-associated cell cycle inhibitors, p21 and p27, but the lack of expression of senescence markers. Mechanistically, it’s unclear how IKKβ may be promoting this phenotype but previous studies have shown that p21 derepression by ICI 182,780 leads to a loss of HDAC occupancy and results in histone acetylation and gene expression (35). It is possible that IKKβ may be capable of redirecting ER recruitment away from proliferation/quiescence associated genes to allow for their regulation in a reversible manner. Cellular quiescence has been proposed as one growth arrest mechanism underlying tumor dormancy, which can be defined as the presence of clinically undetectable cancer cells in specific organs that eventually give rise to recurrent tumors after a period of time (3, 36, 37). We found that primary tumors regress to undetectable levels upon DOX administration, suggesting cell death to reduce the bulk of the tumor. The explanation for regression could be that CA-IKKβ induces the expression of NFκB target genes, such as TNFα and MMP9, and reverses the tumor associated macrophages from the pro-tumorigenic state to the anti-tumorigenic state, resulting in tumor regression (38, 39). However, a bioluminescent signal was detectable at the site of the regressing tumors, indicating that a portion of the cells survived and were capable of seeding a recurrent tumor upon IKKβ inactivation. Based on our findings, we propose that these surviving cells may have a basal/ stem cell-like phenotype that could be responsible for the recurrent tumor.
Dormancy explains the long disease-free interval between the initial diagnosis and treatment of the disease and occurrence of relapse. Recurrence requires both a tumor initiating potential and a strong proliferative signal which results in the accumulation of a detectable tumor mass (3, 36, 37). Clinical studies report that tamoxifen treatment for 10 years reduces recurrence and mortality in patients compared to 5 years of tamoxifen only (40). Importantly, both the dormant and migratory phenotypes we observed are not affected by tamoxifen, suggesting that these dormant metastatic cells may survive long term in patients receiving endocrine therapy. It is proposed that the homing and survival of ER positive cells does not require E2 (41, 42); however, the pathways and mechanisms which allow homing of ER positive breast cancer cells and maintenance of both anti-proliferative and pro-survival pathways in these sites are unknown. Our animal study results indicate that turning off the activity of IKKβ, by withdrawing DOX from the drinking water of the animals, rescues the strong proliferative signal of E2, resulting in loco-regional recurrence of the tumor. Thus, we propose that CA-IKKβ may be a mediator of ER positive breast cancer dormancy by enhancing the survival of the cells and inhibiting the proliferative/growth signal of E2.
Activation of ER and CA-IKKβ proved to induce a highly invasive phenotype in ER positive breast cancer cells compared to E2 or CA-IKKβ alone. Furthermore, the incidence of metastasis was higher in tumors initiating from E2 and IKKβ activated cells versus E2 activated cells only. In fact, only a brief treatment to activate IKKβ, rather than a continuous activation of IKKβ in vivo, was sufficient to enhance metastasis compared to E2 treatment. This suggests that transient activation of IKKβ endows the cells with properties that persist long enough to induce a difference in metastasis between the two treatment groups. We suggest that CA-IKKβ and E2 may be expanding an invasive population, such as the stem/basal-like cell population, which enhances metastases (18, 24, 31, 32). This is supported by gene expression profiling and pathway analysis, as well as follow-up studies, showing an expansion of these populations in response to E2 and DOX treatment.
It has been shown that E2 and IKKβ can each independently enhance stem cell activity in ER positive breast cancer cells (43, 44) while our results demonstrate that co-activation of these pathways significantly enhances stem cell activity compared to either alone. Stemness is linked to basal lineage (45), and each phenotype has been reported to enhance invasion and metastasis (18, 24, 31, 32). We report cells expressing invasion/metastasis-associated basal markers in ER positive cells activated with E2 and IKKβ. Moreover, we report a repression of luminal markers CK8/18, Gata3 and FoxA1, which has previously been associated with enhanced invasion and metastasis as well (46-48). We propose that E2 and activation of IKKβ can induce luminal-basal dedifferentiation that might underlie the observed metastatic phenotype.
In conclusion, our study presents a completely novel model whereby E2 and activation of IKKβ causes a major phenotypic switch from a highly proliferative to a highly invasive state, which is typical of a dormant metastatic disease. This implicates IKKβ as a driver of certain features of aggressive ER positive breast cancer and suggests IKKβ as both a potential biomarker and a novel therapeutic target in ER positive breast cancer.
Supplementary Material
Acknowledgments
We thank Sarah Baumgarten, Gergana Georgieva and Yasir Izhar for their technical support. This work was support by NIH R01 CA200669 and the Department of Defense #W81XWH-16-1-0044, both to J. Frasor. C.J. Creighton was supported in part by NIH CA125123. L. El- Shennawy was supported in part by the International Fulbright Science and Technology Award.
Financial Support: This work was support by NIH R01 CA200669 and the Department of Defense #W81XWH-16-1-0044, both to J. Frasor. C.J. Creighton was supported in part by NIH CA125123. L. El- Shennawy was supported in part by the International Fulbright Science and Technology Award.
Footnotes
Conflict of Interest Disclosure Statement: The authors have no financial conflicts to disclose
Reference List
- 1.Cancer Facts and Figures, 2016. Atlanta: American Cancer Society. American Cancer Society; 2016. [Google Scholar]
- 2.Kennecke H, Yerushalmi R, Woods R, Cheang MC, Voduc D, Speers CH, et al. Metastatic behavior of breast cancer subtypes. J Clin Oncol. 2010;28:3271–7. doi: 10.1200/JCO.2009.25.9820. [DOI] [PubMed] [Google Scholar]
- 3.Zhang XH, Giuliano M, Trivedi MV, Schiff R, Osborne CK. Metastasis dormancy in estrogen receptor-positive breast cancer. Clin Cancer Res. 2013;19:6389–97. doi: 10.1158/1078-0432.CCR-13-0838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Selli C, Dixon JM, Sims AH. Accurate prediction of response to endocrine therapy in breast cancer patients: current and future biomarkers. Breast Cancer Res. 2016;18:118. doi: 10.1186/s13058-016-0779-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Biswas DK, Shi Q, Baily S, Strickland I, Ghosh S, Pardee AB, et al. NF-kappa B activation in human breast cancer specimens and its role in cell proliferation and apoptosis. Proc Natl Acad Sci U S A. 2004;101:10137–42. doi: 10.1073/pnas.0403621101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.deGraffenried LA, Chandrasekar B, Friedrichs WE, Donzis E, Silva J, Hidalgo M, et al. NF-kappa B inhibition markedly enhances sensitivity of resistant breast cancer tumor cells to tamoxifen. Ann Oncol. 2004;15:885–90. doi: 10.1093/annonc/mdh232. [DOI] [PubMed] [Google Scholar]
- 7.Jones RL, Rojo F, A’Hern R, Villena N, Salter J, Corominas JM, et al. Nuclear NF-kappaB/p65 expression and response to neoadjuvant chemotherapy in breast cancer. J Clin Pathol. 2011;64:130–5. doi: 10.1136/jcp.2010.082966. [DOI] [PubMed] [Google Scholar]
- 8.Oida K, Matsuda A, Jung K, Xia Y, Jang H, Amagai Y, et al. Nuclear factor-kB plays a critical role in both intrinsic and acquired resistance against endocrine therapy in human breast cancer cells. Sci Rep. 2014;4:4057. doi: 10.1038/srep04057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Riggins RB, Zwart A, Nehra R, Clarke R. The nuclear factor kappa B inhibitor parthenolide restores ICI 182,780 (Faslodex; fulvestrant)-induced apoptosis in antiestrogen-resistant breast cancer cells. Mol Cancer Ther. 2005;4:33–41. [PubMed] [Google Scholar]
- 10.Zhou Y, Eppenberger-Castori S, Marx C, Yau C, Scott GK, Eppenberger U, et al. Activation of nuclear factor-kappaB (NFkappaB) identifies a high-risk subset of hormone-dependent breast cancers. Int J Biochem Cell Biol. 2005;37:1130–44. doi: 10.1016/j.biocel.2004.09.006. [DOI] [PubMed] [Google Scholar]
- 11.Kubo M, Kanaya N, Petrossian K, Ye J, Warden C, Liu Z, et al. Inhibition of the proliferation of acquired aromatase inhibitor-resistant breast cancer cells by histone deacetylase inhibitor LBH589 (panobinostat) Breast Cancer Res Treat. 2013;137:93–107. doi: 10.1007/s10549-012-2332-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Trinh XB, Sas L, Van Laere SJ, Prove A, Deleu I, Rasschaert M, et al. A phase II study of the combination of endocrine treatment and bortezomib in patients with endocrine-resistant metastatic breast cancer. Oncol Rep. 2012;27:657–63. doi: 10.3892/or.2011.1562. [DOI] [PubMed] [Google Scholar]
- 13.Frasor J, Weaver A, Pradhan M, Dai Y, Miller LD, Lin CY, et al. Positive cross-talk between estrogen receptor and NF-kappaB in breast cancer. Cancer Res. 2009;69:8918–25. doi: 10.1158/0008-5472.CAN-09-2608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Romieu-Mourez R, Landesman-Bollag E, Seldin DC, Traish AM, Mercurio F, Sonenshein GE. Roles of IKK kinases and protein kinase CK2 in activation of nuclear factor-kappaB in breast cancer. Cancer Res. 2001;61:3810–8. [PubMed] [Google Scholar]
- 15.Curtis C, Shah SP, Chin SF, Turashvili G, Rueda OM, Dunning MJ, et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature. 2012;486:346–52. doi: 10.1038/nature10983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lu X, Lu X, Wang ZC, Iglehart JD, Zhang X, Richardson AL. Predicting features of breast cancer with gene expression patterns. Breast Cancer Res Treat. 2008;108:191–201. doi: 10.1007/s10549-007-9596-6. [DOI] [PubMed] [Google Scholar]
- 17.Network CGA. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu H, Patel MR, Prescher JA, Patsialou A, Qian D, Lin J, et al. Cancer stem cells from human breast tumors are involved in spontaneous metastases in orthotopic mouse models. Proc Natl Acad Sci U S A. 2010;107:18115–20. doi: 10.1073/pnas.1006732107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Creighton CJ, Casa A, Lazard Z, Huang S, Tsimelzon A, Hilsenbeck SG, et al. Insulin-like growth factor-I activates gene transcription programs strongly associated with poor breast cancer prognosis. J Clin Oncol. 2008;26:4078–85. doi: 10.1200/JCO.2007.13.4429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Storey JD, Tibshirani R. Statistical significance for genomewide studies. Proc Natl Acad Sci U S A. 2003;100:9440–5. doi: 10.1073/pnas.1530509100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liberzon A, Subramanian A, Pinchback R, Thorvaldsdottir H, Tamayo P, Mesirov JP. Molecular signatures database (MSigDB) 3.0. Bioinformatics. 2011;27:1739–40. doi: 10.1093/bioinformatics/btr260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dontu G, Abdallah WM, Foley JM, Jackson KW, Clarke MF, Kawamura MJ, et al. In vitro propagation and transcriptional profiling of human mammary stem/progenitor cells. Genes Dev. 2003;17:1253–70. doi: 10.1101/gad.1061803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu S, Ginestier C, Ou SJ, Clouthier SG, Patel SH, Monville F, et al. Breast cancer stem cells are regulated by mesenchymal stem cells through cytokine networks. Cancer Res. 2011;71:614–24. doi: 10.1158/0008-5472.CAN-10-0538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Charafe-Jauffret E, Ginestier C, Iovino F, Tarpin C, Diebel M, Esterni B, et al. Aldehyde dehydrogenase 1-positive cancer stem cells mediate metastasis and poor clinical outcome in inflammatory breast cancer. Clin Cancer Res. 2010;16:45–55. doi: 10.1158/1078-0432.CCR-09-1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stanculescu A, Bembinster LA, Borgen K, Bergamaschi A, Wiley E, Frasor J. Estrogen promotes breast cancer cell survival in an inhibitor of apoptosis (IAP)-dependent manner. Horm Cancer. 2010;1:127–35. doi: 10.1007/s12672-010-0018-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stender JD, Nwachukwu JC, Kastrati I, Kim Y, Strid T, Yakir M, et al. Structural and Molecular Mechanisms of Cytokine-Mediated Endocrine Resistance in Human Breast Cancer Cells. Mol Cell. 2017;65:1122–35e5. doi: 10.1016/j.molcel.2017.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Thompson EW, Reich R, Shima TB, Albini A, Graf J, Martin GR, et al. Differential regulation of growth and invasiveness of MCF-7 breast cancer cells by antiestrogens. Cancer Res. 1988;48:6764–8. [PubMed] [Google Scholar]
- 28.Borley AC, Hiscox S, Gee J, Smith C, Shaw V, Barrett-Lee P, et al. Anti-oestrogens but not oestrogen deprivation promote cellular invasion in intercellular adhesion-deficient breast cancer cells. Breast Cancer Res. 2008;10:R103. doi: 10.1186/bcr2206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Huber MA, Azoitei N, Baumann B, Grunert S, Sommer A, Pehamberger H, et al. NF-kappaB is essential for epithelial-mesenchymal transition and metastasis in a model of breast cancer progression. J Clin Invest. 2004;114:569–81. doi: 10.1172/JCI21358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li CW, Xia W, Huo L, Lim SO, Wu Y, Hsu JL, et al. Epithelial-mesenchymal transition induced by TNF-alpha requires NF-kappaB-mediated transcriptional upregulation of Twist1. Cancer Res. 2012;72:1290–300. doi: 10.1158/0008-5472.CAN-11-3123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cheung KJ, Gabrielson E, Werb Z, Ewald AJ. Collective invasion in breast cancer requires a conserved basal epithelial program. Cell. 2013;155:1639–51. doi: 10.1016/j.cell.2013.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shima H, Yamada A, Ishikawa T, Endo I. Are breast cancer stem cells the key to resolving clinical issues in breast cancer therapy? Gland Surg. 2017;6:82–8. doi: 10.21037/gs.2016.08.03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Araki K, Kawauchi K, Tanaka N. IKK/NF-kappaB signaling pathway inhibits cell-cycle progression by a novel Rb-independent suppression system for E2F transcription factors. Oncogene. 2008;27:5696–705. doi: 10.1038/onc.2008.184. [DOI] [PubMed] [Google Scholar]
- 34.Chen F, Lu Y, Castranova V, Li Z, Karin M. Loss of Ikkbeta promotes migration and proliferation of mouse embryo fibroblast cells. J Biol Chem. 2006;281:37142–9. doi: 10.1074/jbc.M603631200. [DOI] [PubMed] [Google Scholar]
- 35.Varshochi R, Halim F, Sunters A, Alao JP, Madureira PA, Hart SM, et al. ICI182,780 induces p21Waf1 gene transcription through releasing histone deacetylase 1 and estrogen receptor alpha from Sp1 sites to induce cell cycle arrest in MCF-7 breast cancer cell line. J Biol Chem. 2005;280:3185–96. doi: 10.1074/jbc.M408063200. [DOI] [PubMed] [Google Scholar]
- 36.Aguirre-Ghiso JA. Models, mechanisms and clinical evidence for cancer dormancy. Nat Rev Cancer. 2007;7:834–46. doi: 10.1038/nrc2256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sosa MS, Bragado P, Aguirre-Ghiso JA. Mechanisms of disseminated cancer cell dormancy: an awakening field. Nat Rev Cancer. 2014;14:611–22. doi: 10.1038/nrc3793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shime H, Matsumoto M, Oshiumi H, Tanaka S, Nakane A, Iwakura Y, et al. Toll-like receptor 3 signaling converts tumor-supporting myeloid cells to tumoricidal effectors. Proc Natl Acad Sci U S A. 2012;109:2066–71. doi: 10.1073/pnas.1113099109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Leifler KS, Svensson S, Abrahamsson A, Bendrik C, Robertson J, Gauldie J, et al. Inflammation induced by MMP-9 enhances tumor regression of experimental breast cancer. J Immunol. 2013;190:4420–30. doi: 10.4049/jimmunol.1202610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Davies C, Pan H, Godwin J, Gray R, Arriagada R, Raina V, et al. Long-term effects of continuing adjuvant tamoxifen to 10 years versus stopping at 5 years after diagnosis of oestrogen receptor-positive breast cancer: ATLAS, a randomised trial. Lancet. 2013;381:805–16. doi: 10.1016/S0140-6736(12)61963-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ganapathy V, Banach-Petrosky W, Xie W, Kareddula A, Nienhuis H, Miles G, et al. Luminal breast cancer metastasis is dependent on estrogen signaling. Clin Exp Metastasis. 2012;29:493–509. doi: 10.1007/s10585-012-9466-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ogba N, Manning NG, Bliesner BS, Ambler SK, Haughian JM, Pinto MP, et al. Luminal breast cancer metastases and tumor arousal from dormancy are promoted by direct actions of estradiol and progesterone on the malignant cells. Breast Cancer Res. 2014;16:489. doi: 10.1186/s13058-014-0489-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Storci G, Sansone P, Mari S, D’Uva G, Tavolari S, Guarnieri T, et al. TNFalpha up-regulates SLUG via the NF-kappaB/HIF1alpha axis, which imparts breast cancer cells with a stem cell-like phenotype. J Cell Physiol. 2010;225:682–91. doi: 10.1002/jcp.22264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sun Y, Wang Y, Fan C, Gao P, Wang X, Wei G, et al. Estrogen promotes stemness and invasiveness of ER-positive breast cancer cells through Gli1 activation. Mol Cancer. 2014;13:137. doi: 10.1186/1476-4598-13-137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Granit RZ, Slyper M, Ben-Porath I. Axes of differentiation in breast cancer: untangling stemness, lineage identity, and the epithelial to mesenchymal transition. Wiley Interdiscip Rev Syst Biol Med. 2014;6:93–106. doi: 10.1002/wsbm.1252. [DOI] [PubMed] [Google Scholar]
- 46.Fortier AM, Asselin E, Cadrin M. Keratin 8 and 18 loss in epithelial cancer cells increases collective cell migration and cisplatin sensitivity through claudin1 up-regulation. J Biol Chem. 2013;288:11555–71. doi: 10.1074/jbc.M112.428920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kouros-Mehr H, Bechis SK, Slorach EM, Littlepage LE, Egeblad M, Ewald AJ, et al. GATA-3 links tumor differentiation and dissemination in a luminal breast cancer model. Cancer Cell. 2008;13:141–52. doi: 10.1016/j.ccr.2008.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bernardo GM, Bebek G, Ginther CL, Sizemore ST, Lozada KL, Miedler JD, et al. FOXA1 represses the molecular phenotype of basal breast cancer cells. Oncogene. 2013;32:554–63. doi: 10.1038/onc.2012.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.