

Abstract
Diabetes is a debilitating health care problem affecting 422 million people around the world. Diabetic patients suffer from multi-systemic complications that can cause mortality and morbidity. Recent advancements in high throughput next generation RNA-sequencing and computational algorithms led to the discovery of aberrant post-transcriptional gene regulatory programs in diabetes. However, very little is known about how these regulatory programs are mis-regulated in diabetes. RNA-binding proteins (RBPs) are important regulators of post-transcriptional RNA networks, which are also dysregulated in diabetes. Human genetic studies provide new evidence that polymorphisms and mutations in RBPs are linked to diabetes. Therefore, we will discuss the emerging roles of RBPs in abnormal post-transcriptional gene expression in diabetes. Questions that will be addressed are: Which post-transcriptional mechanisms are disrupted in diabetes? Which RBPs are responsible for such changes under diabetic conditions? How are RBPs altered in diabetes? How does dysregulation of RBPs contribute to diabetes? Can we target RBPs using RNA-based methods to restore gene expression profiles in diabetic patients? Studying the evolving roles of RBPs in diabetes is critical not only for a comprehensive understanding of diabetes pathogenesis, but also to design RNA-based therapeutic approaches for diabetic complications.
Keywords: RNA binding proteins, diabetes, therapy, RNA, post-transcriptional gene expression
Graphical/Visual Abstract
Role of RNA binding proteins in development of diabetes and diabetic complications
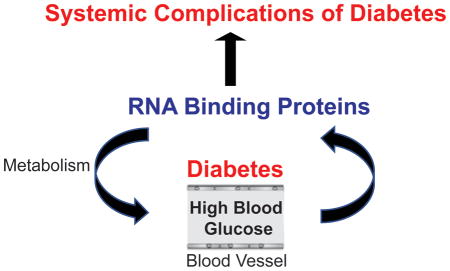
Introduction
Diabetes
Diabetes is a group of metabolic disorders in which blood glucose levels are abnormally high. In Type 1 diabetes (T1D), blood glucose levels rise due to the destruction of pancreatic beta islet cells via autoimmunity that causes low or no insulin production (Fig. 1). In Type 2 diabetes (T2D), the insulin production is unable to stimulate glucose uptake into target tissues liver, adipose and skeletal muscle, increasing blood glucose levels (Fig. 1). Defects in insulin-mediated uptake of glucose into these target tissues cause chronic hyperglycemia in the bloodstream and triggers pathogenic signals including inflammation, hypertension, mitochondrial dysfunction, oxidative stress, endoplasmic reticulum (ER) stress, and dyslipidemia. Chronic hyperglycemia leads to systemic complications and increases the risk for cancer, infections and Alzheimer’s disease 1, 2. Systemic complications of diabetes include: a) vascular disease that can lead to heart attack and/or stroke 3, 4, b) neuropathy which causes pain and numbness 5–7, c) retinopathy that can progress into blindness 7–9, d) nephropathy, the biggest contributor to end-stage renal disease among diabetics 5–7, e) cardiomyopathy that can result in heart failure 10–12 and f) skeletal muscle myopathy that induces muscle wasting and weakness13, 14 (Fig. 2).
Figure 1. Glucose homeostasis in Type 1 and Type 2 diabetes.

Pancreatic beta-islet cells secrete insulin to control glucose levels in the blood. Insulin allows glucose absorption into target tissues that include skeletal muscle, adipose tissue and liver to maintain normal glucose levels in the blood. In Type 1 diabetes, blood glucose levels increase because insulin is production is reduced due to the destruction of beta-islet cells in the pancreas. In Type 2 diabetes, insulin resistance prevents the efficient uptake of glucose into target tissues causing hyperglycemia.
Figure 2. Systemic complications of diabetes.

Diabetes increases the risk for coronary artery disease, which can block blood flow and cause heart attack and/or stroke 3, 4. Hyperglycemia damages nerve fibers causing neuropathies that can adversely affect the digestive tract, urinary tract, heart and blood vessels 5–7. Retinopathy is the leading cause of blindness in diabetic adults 7–9. Almost half of diabetic patients develop diabetic nephropathy 5–7. In most cases, patients develop kidney failure requiring dialysis 6. Under diabetic conditions, heart muscle structure and function are impaired leading to cardiomyopathy 15, 16. Diabetes also impacts skeletal muscle function causing muscle weakness and atrophy 13, 14.
Due to the multi-systemic manifestations, diabetes is a major health care problem, costing ~$322 billion per year in the US alone. Recent advancements in RNA-based technologies indicate that RNA-regulatory networks controlled by RNA binding proteins (RBPs) are modulated in diabetes and contribute to systemic manifestations of diabetes. Therefore, we will discuss how changes in RBPs: LIN28, RBFOX2, FTO,IGF2BP2, eIF4E, CELF1 and HuR disrupt post-transcriptional RNA programs in diabetes and play a central role in development of diabetes and diabetic complications. We will also review how RBPs can be targeted using RNA-centric therapies to treat diabetic complications, based on the current progress made in such therapeutics.
RNA binding proteins
There is increasing evidence that many RBPs and RBP-regulated RNA networks are disrupted under diabetic conditions. RBPs are critical for post-transcriptional gene expression. They bind to RNA based on sequence and/or RNA structure and form ribonucleoprotein (RNP) complexes 17–20. The interactions of RNA with different RBPs are highly dynamic and necessary for distinct steps of RNA biogenesis including 5′ capping, pre-mRNA splicing, 3′ mRNA cleavage and polyadenylation, mRNA export, mRNA editing and methylation, mRNA localization, mRNA decay and translation 17–20.
RBPs are important regulators of cell survival and function, because they have essential roles in fundamental cellular processes such as proliferation, differentiation, and apoptosis 17, 21, 22. Dysregulation of RBPs has been observed in a plethora of diseases including cancer, (reviewed in 17, 22), neurological (reviewed in 23, 24), and cardiovascular diseases (reviewed in 25–28). Several RBPs have been associated with the development of diabetes or with diabetic complications 29–36. RBPs that are implicated in diabetes are summarized in Table 1. For example, Elav-like protein 1 (Elavl1) (HuR), hnRNP K, hnRNP F, YBX1, Insulin like growth factor 2 mRNA-binding protein 2 (IGF2BP2/IMP2), and LIN28 are dysregulated in diabetic kidneys and have been associated with diabetic nephropathy 37–44. Quaking (QKI) 45, 46, tristetraprolin (TTP) 47, 48, ADAR1 49, hnRNP C 50, HuR 51 and calreticulin (CALR) 52 are linked to atherosclerosis and/or vascular endothelial dysfunction. CUG triplet repeat binding protein ELAV-like family member 1 (CUGBP1/CELF1), RNA binding protein Fox-1 homolog 1 (Rbfox1), and eukaryotic initiation factor 4E (eIF4E) have been associated with diabetic skeletal muscle myopathy 53–56. RNA binding protein Fox-1 homolog 2 (Rbfox2), CELF1, LIN28, HuR and QKI are implicated in diabetic cardiomyopathy 28, 57–62. RBPs eIF4E and HuR are involved in diabetic retinopathy 63–68. Polymorphisms in fat mass and obesity associated protein (FTO) are associated with obesity and kidney disease 33–36, 69.
Table 1.
RNA binding proteins-post-transcriptional networks affected in diabetes and mechanism of diabetes induced changes in RNA binding proteins.
| Post-transcriptional mechanisms affected in diabetes | RBPs altered in diabetes | Diabetes induced changes in RBPs | Mechanism(s) that affect RBPs in diabetes |
|---|---|---|---|
| mRNA translation microRNA biogenesis |
LIN28 |
Increased mRNA and protein levels (diabetic kidney) Decreased mRNA and protein levels (diabetic heart) |
Mediated by increased TGFB signaling and SMAD2/3 transcriptional activation (diabetic kidney) Unknown mechanisms (diabetic heart) |
| Alternative splicing | RBFOX2 |
Increased protein levels Low splicing activity |
Mediated likely by PKCα/βII phosphorylation Mediated by increased expression of a dominant negative isoform |
| RNA methylation | FTO | Increased mRNA levels | Unknown mechanisms |
| mRNA translation mRNA localization |
IGF2BP2 | Decreased mRNA and protein levels | Transcriptional downregulation by HMGA2 |
| mRNA translation | eIF4E | Decreased activity | Increase in protein levels and binding activity of 4E-BP1 that inhibits eIF4E mediated by increased O-GlcNAcylation and/or decreased phosphorylation |
| Alternative splicing | CELF1 | Increased protein levels | Mediated by PKCα/βII phosphorylation |
| Alternative splicing mRNA decay mRNA translation |
HuR |
Increased mRNA and protein levels (diabetic heart) Increased protein levels and localization to the cytoplasm (diabetic retina, kidney) |
Mediated by decreased miR-9 (diabetic heart) and miR23 Mediated by PKCβ phosphorylation (diabetic retina) |
In this review, we will only focus on seven of these RBPs: LIN28, Rbfox2, FTO, IG2BP2, CELF1 and HuR and the RNA regulatory processes they control (Table 1). We will start with the post-transcriptional mechanisms disrupted in diabetes, and follow up with RBPs that regulate these processes. Then, we will describe how these RBPs are dysregulated under diabetic conditions and their contribution to disease pathogenesis (Table 2). Finally, we will finish up by reviewing new therapies that target RBPs for treatment of human diseases and discuss their potential in the treatment of diabetic complications.
Table 2.
Contribution of RNA binding proteins to diabetes pathogenesis.
| RBPs | RBP target RNA(s) affected in diabetes | Diabetic complication(s) | Experimental Model(s) |
|---|---|---|---|
| LIN28A | Let7 microRNA family | Diabetic nephropathy and cardiac complications | Db/Db T2D mouse kidney and STZ T1D mouse kidney and heart |
| RBFOX2 | FXR1, MEF2A, global alternatively spliced transcripts | Diabetic cardiac complications | STZ T1D mouse and human T2D patient heart |
| FTO | Global changes in A6 methylated transcripts | Unknown | Human T2D patient and STZ T1D rat peripheral blood |
| IGF2BP2 | LAMB2 | Diabetic nephropathy | STZ T1D mouse kidney |
| eIF4E | VEGF and genome wide transcripts | Diabetic myopathy, retinopathy, and cardiac complications | Alloxan induced T1D rat skeletal muscle and heart, STZ T1D rat retina, STZ T1D mouse and Akita T1D mouse liver and retina |
| CELF1 | Global alternatively spliced transcripts | Diabetic myopathy and cardiac complications | NOD T1D mouse skeletal muscle and STZ T1D mouse heart |
| HuR | VEGF, CTGF, TGFB1, SNAI1, FOS, and NOD2 | Vascular disease, diabetic nephropathy, retinopathy, and cardiac complications | Human diabetic patient heart and kidney biopsies, human T2D patient kidney biopsies, human patient vascular tissue, and STZ T1D rat kidney and retina |
POST-TRANSCRIPTIONAL MECHANISMS ALTERED IN DIABETES
Alternative splicing (AS)
The alternative splicing regulators CELF1, Rbfox2, Rbfox1, QKI, and hnRNPC are implicated in diabetes by affecting alternative splicing decisions 28, 45, 46, 50, 53, 60–62. Splicing is a process by which the non-coding regions (introns) of the pre-mRNA are removed and coding regions (exons) are joined together. Splicing occurs co-transcriptionally and is mediated by the spliceosome complex composed of ~100 auxiliary proteins and 5 small nuclear RNAs (snRNAs) (reviewed in 70, 71). Although snRNAs catalyze the splicing reactions, RBPs bind to the pre-mRNA and help define exons and splice recognition sites, thereby facilitating removal of introns 70.
AS can directly affect gene expression and increase protein diversity by allowing differential inclusion/exclusion of exons in some cases parts of introns, generating multiple protein isoforms from a single gene 70–72. RBPs control AS decisions via binding to the pre-mRNA and communicating with the spliceosome components to promote or prevent exon inclusion 70. Regulation of AS by RBPs is highly specific and precisely regulated especially during organism development and the cell cycle 73–77.
AS plays a crucial role in the development of T1D 78. T1D is caused by autoimmune-mediated destruction of pancreatic beta-islet cells that produce insulin. AS impacts expression and function of the genes necessary for beta-islet cell function and auto-immunity 78–82. In this review, we will focus on the role of AS defects in diabetic complications as the contribution of AS dysregulation to islet cell dysfunction and increased inflammation in T1D has been recently reviewed 78.
Genome wide AS changes are identified in both the diabetic heart and retina (Table 1) 83. In diabetic retina, photoreceptor genes (arrestin, Impg2, and Trpm1) relevant to retina function are mis-spliced 83. In diabetic hearts, genes with important roles in macromolecular metabolism, calcium ion homeostasis and cell cycle are aberrantly spliced 28. We identified Rbfox2 as a major regulator of AS defects in diabetic hearts and a contributor to cardiac complications 28. Role of Rbfox2 in diabetic complications will be discussed in detail in the next section. According to our findings, among 967 AS changes in diabetic hearts, a subset of genes displayed fetal/newborn splicing patterns. Further studies have shown that one of the hallmarks of diabetes-induced AS changes is the reactivation of fetal/newborn-like splicing patterns in adult diabetic hearts and skeletal muscle 53, 62. Inappropriate expression of fetal/newborn spliced variants in adult tissues may contribute to muscle pathogenesis in diabetes.
One of the genes that is differentially spliced in diabetes is vascular endothelial growth factor (VEGF), a key component in vasculogenesis and angiogenesis and a central pathogenic factor in diabetes 8, 84, 85. AS of VEGF exon 8 determines the pro- or anti-angiogenic properties of VEGF 85–89. In the diabetic retina, VEGF switches to a pro-angiogenic spliced isoform and causes increased vascularization associated with retinopathy 89. AS of VEGF exon 7 affects its function by regulating its solubility. In the kidneys of T2D patients, AS of VEGF generates more soluble VEGF, which is associated with worse parameters of diabetic nephropathy 87. These studies suggest that mis-splicing of VEGF contribute to diabetic complications in the kidney and retina.
Another example of a mis-spliced gene relevant to diabetes pathogenesis is myocardin. Myocardin is a transcriptional co-activator vital for contractile gene expression in vascular smooth muscle cells (VSMCs) 46. AS of myocardin is dysregulated in atherosclerosis, a common complication in diabetic patients 46. VSMCs isolated from atherosclerotic patient plaques expressed myocardin spliced isoforms that cause increased proliferation and lower contractility contributing to vascular pathology 46. Together, these findings show that AS patterns affecting essential gene function are disrupted in diabetes.
RNA methylation
RNA methylation was discovered in the 1970s 90. Discerning the functional consequences of this modification on RNA metabolism and gene expression are made possible by the recent technological advances in high throughput RNA sequencing combined with our ability to detect specific forms of methylated RNAs.
RNA methylation is a reversible modification that plays key roles in post-transcriptional gene regulation 91, 92. It is analogous to DNA epigenetic modifications such that it can be written, read, and erased 91, 92. The most abundant and highly conserved reversible mRNA modification in eukaryotes is N6-methylation of adenosine (m6A) 91, 92. Genome wide m6A RNA methylation analysis using m6A-specific methylated RNA immunoprecipitation followed by next-generation RNA sequencing (MeRIP-Seq) allowed the discovery of methylation in both mRNAs and non-coding RNAs 93. m6A modification of RNA involves methyltransferases as writers such as METTL3, METTL14, and WTAP; demethylases as erasers including FTO and ALKBH5, and readers such as YTHDF proteins. Reader proteins bind to the methylated RNA and influence splicing, mRNA stability, localization and translation 91, 92. Notably, RNA demethylase FTO polymorphisms are associated with obesity, which is a common feature of T2D 33–36. In addition, m6A methylation of mRNAs is decreased under diabetic conditions consistent with upregulation of FTO levels (Table 1) 94.
mRNA stability/degradation
mRNA stability is altered in diabetes via RBPs or microRNAs. In this review, we will address RBP-regulated mRNA decay in diabetes (Table 1). mRNA levels can be regulated post-transcriptionally via degradation as a quick mechanism to control gene expression in response to stimuli 95–97. Removal of the 5′ cap structure by decapping enzymes induces mRNA degradation 96. Similarly, removal of the 3′ poly(A) tail by deadenylases causes mRNA decay 96. There are several cis-acting elements within the mRNA that signals for degradation. These elements can be present in the 5′ or 3′ untranslated regions (UTRs) as well as within the coding region of the mRNAs 98. RBPs typically bind to these cis-acting elements within the mRNA to promote or prevent mRNA decay through interactions with decapping and/or deadenylation enzymes 95–97.
One of the well characterized cis-acting elements for regulation of mRNA stability are AU-rich elements (ARE) found in the 3′UTRs. mRNAs that contain ARE elements are rapidly degraded. RBPs such as HuR and TTP bind to AREs and control the degradation of ARE-containing mRNAs 98. Under diabetic conditions, mRNA levels of ARE-containing mRNAs such as NOD2, CTGF, TGFB1, SNAI1, TNF, FOS, EGR1, p21, and FOS are elevated due to changes in subcellular localization, protein levels and activities of ARE-binding proteins HuR and TTP 39, 40, 47, 48. HuR binding to the ARE elements stabilizes mRNAs 98. Increased levels of HuR in the cytoplasm is thought to contribute to the increase in levels of these ARE containing mRNAs genes. Another gene that is upregulated in obese mouse hearts via increased mRNA stability is FoxO1 60. FoxO1 is an important regulator of insulin signaling and contributor to ER stress in diabetic hearts. mRNA levels of FoxO1 are increased under diabetic conditions via QKI mediated changes in its mRNA stability. QKI is shown to promote FoxO1 mRNA degradation and changes in QKI in diabetic hearts are thought to cause increased levels of FoxO1. mRNA levels of glucose transporter-1 (GLUT1), which is necessary for glucose uptake by the cells, are altered in VSMCs under high glucose conditions 52. QKI is shown to control GLUT1 mRNA decay 52. Altogether, these studies suggest that mRNA stability is affected in diabetes via different RBPs. The role of HuR in mRNA stability and diabetes pathogenesis will be discussed in detail in the next section.
mRNA translation
Regulation of mRNA translation occurs mostly at the rate limiting initiation step 99–104. To start protein synthesis, the cap-binding protein eIF4E, binds the cap structure at the 5′ end of the mRNAs and helps recruit the small ribosomal subunit via interaction with other translation initiation factors eIF4G and eIF3 102, 104. Then, the large ribosomal subunit is recruited to the 5′ end of the mRNA with help from other translation initiation factors to start cap-dependent translation 102, 104. Alternatively, some eukaryotic mRNAs utilize internal ribosome entry sites (IRES) to load ribosomes onto the mRNA independent of 5′ cap and translate in a cap-independent manner 102, 105.
Under diabetic conditions, cap-dependent translation is inhibited at the initiation step due to low cap binding activity of eIF4E (Table 1). 4E-binding protein (4E-BP1) binds to eIF4E and blocks eIF4E interaction with the 5′ cap of mRNAs under diabetic conditions 54–56, 66–68, 106–111. Regulation of eIF4E by 4E-BP1 is affected under diabetic conditions and will be further discussed in the next section.
While cap-dependent translation is inhibited in diabetes, IRES-dependent translation of stress induced mRNAs is stimulated 112, 113. Since diabetic conditions affect mRNA translation preferentially favoring IRES dependent translation, future studies using ribosomal profiling followed by RNA-sequencing, are necessary to identify mRNAs that are actively being translated under diabetic conditions.
RNA BINDING PROTEINS IMPLICATED IN DIABETES
Diabetes are a group of metabolic disorders in which insulin signaling, glucose and fat metabolism are altered. Abnormal metabolism in diabetes activates pathogenic events such as inflammation, ER stress, oxidative stress, fibrosis and epithelial-mesenchymal transition (EMT) of cells. Figure 3 summarizes specific RBPs that contribute to some of the pathogenic events triggered in diabetes. In this part, we will discuss a) the roles of these specific RBPs: LIN28, Rbfox2, CELF1, FTO, eIF4E, and IGFBP2 in RNA metabolism, b) dysregulation of these RBPs in diabetes, and c) contribution of these RBPs to development of diabetes and diabetic complications.
Figure 3. RNA binding proteins involved in pathogenic events triggered in diabetes.

Chronic hyperglycemia initiates pathogenic signals that can cause mitochondrial dysfunction, fibrosis, inflammation and epithelial mesenchymal transition (EMT). RNA binding proteins that are implicated in diabetes associated pathogenic events are summarized.
LIN28
LIN28A/B is an RBP involved in microRNA biogenesis, RNA splicing, and mRNA translation 114–116. LIN28 binds looped RNA structures in pre-let7 RNA and blocks production of mature let7 microRNA, thereby affecting expression of let7 target genes. It has been shown that LIN28 can also bind to other RNAs with structural similarity to pre-let7 RNA that exhibit GGAG sequences 117.
LIN28 is important for embryonic development, pluripotency of stem cells, growth, and metabolism 114, 118, 119. LIN28 has a role in AS via interacting with hnRNPA1 115. In addition, LIN28 modulates AS by controlling mRNA translation of the splicing regulators hnRNPF, TIA-1, FUS/TLS and TDP-43 117. A new study shows that LIN28 binds to specific gene promoters and may regulate transcription 120.
Relevant to diabetes, LIN28 has a critical role in controlling glucose metabolism, mitochondrial function and insulin resistance 119, 121–123. More specifically, LIN28 increases glucose uptake by cells and prevents insulin resistance in high fat induced diabetic mice. This is mediated in part by its role in processing and maturation of let7 miRNA 123. On the other hand, muscle specific deletion of LIN28 increases insulin resistance independent of effects on let7 microRNA; indicating that other functions of LIN28 in RNA metabolism are important for insulin sensitivity. LIN28 is strongly linked to insulin resistance and T2D 38, 57, 58, 119, 121, 123. LIN28 regulates mRNA translation of HMGA genes 124, which are also genetically associated with insulin resistance and T2D 125. LIN28 regulates translation of mRNAs that are involved in oxidative phosphorylation 122. In addition, LIN28 helps control glycolysis in kidney cells 126 and in cancer cells and hepatocellular carcinoma 127.
LIN28 is implicated in kidney and cardiac complications of diabetes 38 (Table 2). LIN28 mRNA levels are increased in T1D and T2D mouse kidneys via transcriptional activation by SMAD2/3 (Table 1) 38. Upregulation of LIN28 leads to changes in Let7b target genes, which include Col1a2 and Col4a1 contributing to kidney fibrosis in T1D and T2D mouse kidneys 38. Conversely, LIN28 levels are decreased in T1D mouse hearts (Table 1) 57, 58. Low LIN28 levels correlate with cardiomyocyte apoptosis and decreased contractile function 58. In support of its role in diabetic hearts, LIN28 overexpression alleviates mitochondrial dysfunction and cardiomyocyte apoptosis preventing cardiomyopathy in T1D mice 58. LIN28 depletion exacerbates cardiac symptoms in diabetic mice 57, 58. These studies support a critical role for LIN28 in diabetic heart and kidney disease.
RNA binding protein Fox-1 homolog 2 (Rbfox2)
Rbfox2 is implicated in cardiac complications of diabetes. Rbfox2 belongs to a family of RBPs that include Rbfox1 and Rbfox3. Rbfox proteins bind the (U)GCAUG motif in RNA and regulate AS 128–134. While Rbfox1 and Rbfox2 are widely expressed in different tissues, Rbfox3 is neuron specific. Rbfox1 and Rbfox2 control motor neuron function and development of the brain cerebellum 135 as well as skeletal muscle and heart function 128, 130, 136, 137.
Rbfox2 has essential roles in AS regulation in the brain, skeletal muscle, heart and embryonic stem cells 130, 132, 135–137. In skeletal muscle, Rbfox2 controls fusion of myoblasts during skeletal muscle differentiation 128. In the heart, conditional deletion of Rbfox2 in mouse cardiomyocytes leads to dilated cardiomyopathy 130. In addition, Rbfox2 protein levels are downregulated in hearts undergoing pressure overload induced heart failure 130. Consistent with its important role in the brain and the heart, Rbfox2 mutations have been identified in patients with congenital neurodevelopmental and cardiac defects 138. Importantly, Rbfox2 contributes to genome wide gene expression changes observed in patients with congenital heart defects 27.
Recent studies defined new functions for Rbfox2 in regulating transcription via interactions with the polycomb complex 129 and in microRNA processing 139. Due to its importance, genome-wide RNA targets of Rbfox2 have been identified using CLIP-sequencing in several different cell types 128–134. Rbfox2 is a master regulator of post-transcriptional gene expression as it controls expression levels of other RBPs via AS-dependent nonsense-mediated mRNA decay 132–134.
Rbfox2 protein levels are upregulated in T2D human patient hearts and T1D mouse hearts, correlating with PKCα/βII activation in the diabetic heart (Tables 1 and 2) 28, 62, 140–142. Though Rbfox2 protein levels are elevated in the diabetic heart, Rbfox2 splicing function is adversely affected 28 due to increased expression of a spliced isoform with dominant negative activity 28, 143, 144. In T1D mouse hearts, Rbfox2 is a major contributor to genome-wide AS changes 28, 62. 69% of transcripts mis-spliced in diabetic hearts display Rbfox2 binding sites within alternative exons and/or flanking introns 28. Dysregulation of Rbfox2 impairs proper expression of cytoskeleton-associated genes and intracellular calcium handling in primary cardiomyocytes 28. These results indicate that dysregulation of Rbfox2 contributes to the cardiac complications of diabetes (Table 2).
A recent study has found that Rbfox2 is vital for insulin secretion and survival of beta islet cells in the pancreas 82. AS function of Rbfox2 is linked to EMT 145, 146, which is one of the pathogenic events that contribute to diabetic nephropathy, retinopathy and atherosclerosis. More studies are needed to determine whether Rbfox2 contributes to these diabetic complications.
Fat mass and obesity-associated protein (FTO)
FTO is an m6A specific demethylase that removes methyl groups in RNA which in turn controls the fate of the RNA in the cell 91, 92. FTO is implicated in DNA repair and cell survival 147. Genetic and molecular studies strongly associate FTO to obesity and adipogenesis 148 through FTO’s ability to regulate energy production by mitochondria 149 and its role in differentiation of fat cells 148.
In T2D patients and diabetic rats, FTO levels are elevated with a corresponding decrease in genome wide m6A modification of mRNAs 94 (Tables 1 and 2). Polymorphisms in FTO are associated with obesity in humans 33–36. FTO mutations are also linked to renal fibrosis in humans with chronic kidney disease 69. Therefore, FTO association to obesity may predispose individuals to T2D. Recent studies indicate that FTO may play a role in neurogenesis 150 and myogenesis 149, both of which are also affected in diabetes. Further studies are needed to characterize the contribution of FTO to diabetic complications including neuropathy, nephropathy and myopathy (Table 2).
Insulin like growth factor 2 mRNA-binding protein 2 (IGF2BP2/IMP2)
IGF2BP2 is an important regulator of insulin signalling. It binds target mRNAs and controls their localization, stability and translation. One of the well-known targets of IGF2BP2 is the insulin-like growth factor (IGF). IGF2BP2 binds to IGF mRNA and controls its translation mediated by an IRES element 151. Some other translationally-regulated targets include c-myc, SP1 and Igf1r 152. Importantly, IGF2BP2 is identified as a strong susceptibility gene for T2D in many human genetic studies 29–32. IGF2BP2 is also critical for skeletal muscle stem cell activation and proliferation 152, which are disturbed in diabetic skeletal muscle. In addition, IGF2BP2 levels are downregulated under diabetic conditions due to changes in its transcription mediated by transcription factor HMGA2, which is a risk allele for T2D (Table 1).
IGF2BP2 is an important factor in pathogenesis of diabetic nephropathy (Table 2). It binds to Laminin-β2 (LAMB2) mRNA and regulates its localization to the actin cytoskeleton and activates its translation 37. LAMB2 protein is important for proper kidney function and preventing proteinuria. In T1D rat kidneys, IGF2BP2 protein levels are reduced correlating with low LAMB2 protein levels 37. In sum, IGF2BP2 likely contributes to the development of diabetes due to its role in insulin signaling and to diabetic kidney manifestations (Table 2).
Eukaryotic initiation factor 4E (eIF4E)
eIF4E is a translation initiation factor that binds to the 5′cap structure and initiate translation via recruiting the ribosomal subunits to the 5′ end of eukaryotic mRNAs 102. eIF4E is necessary for cell survival and organism development 153, 154. As a downstream effector of the mTOR signaling pathway, eIF4E is implicated in diabetes. Recently, it has been found that the commonly used diabetes drug named metmorfin not only affects AMPK but also mTOR and ERK pathways and influences eIF4E dependent mRNA translation 155, 156.
eIF4E activity is controlled via phosphorylation by MAPK-activated protein kinase (Mnk) that increases its cap-binding activity 157. eIF4E cap binding activity is also tightly regulated via 4E-BP proteins, which bind eIF4E and inhibit its binding to the 5′ cap 102, 107. 4E-BP proteins are important regulators of eIF4E (Table 1). High glucose levels affect the 4E-BP proteins that blocks eIF4E binding to the cap leading to inhibition of cap dependent mRNA translation 111. It has been found that the mTOR/4E-BP1/eIF4E pathway is a regulatory network for proper cardiomyocyte function and survival 154 and for mitochondrial biogenesis 153.
In diabetes, 4E-BP1 is modulated by several different mechanisms. Many studies show that 4E-BP1 protein levels are increased under diabetic conditions 66, 67, 110. This increase in 4E-BP1 causes binding to eIF4E blocking its interaction with the 5′ cap of mRNAs. 4E-BP1 is also phosphorylated by mTOR leading to the release of eIF4E from 4E-BP1 157. In diabetes, decreased phosphorylation of 4E-BP1 by mTOR reduces eIF4E cap binding activity. 4E-BP1 also undergoes O-GlcNAcylation where N-Acetylglucosamine is added to Ser or Thr residues of 4E-BP1 66, 67, 110. Increased O-GlcNAcylation of 4E-BP1 under diabetic conditions strengthens binding to eIF4E, inhibiting cap dependent translation 66, 67, 110.
eIF4E/4E-BP is also important in the development of insulin resistance. Increased activation of 4E-BP1 protects against diet-and age-dependent insulin resistance 158. Furthermore, 4E-BP2 contributes to pancreatic beta cell loss in T1D 159. In the diabetic retina, eIF4E cap binding activity is repressed due to increased 4E-BP1 levels contributing to diabetic retinopathy 66, 68 (Table 2). This leads to a decrease in cap-dependent protein synthesis while IRES mediated translation of VEGF mRNA is stimulated 66–68, 110. Deletion of 4E-BP1 in the rodent retina inhibits hyperglycemia induced upregulation of VEGF 66 and delays diabetes induced blindness 67.
In T1D rat hearts and skeletal muscle, protein synthesis is decreased mostly due to low eIF4E activity 54–56 and this is thought to contribute to skeletal muscle myopathy and cardiomyopathy (Table 2). Notably, insulin treatment ameliorates eIF4E-dependent translation inhibition under diabetic conditions 54–56. Even though changes in eIF4E can block global translation, phosphorylation of eIF4E via TGFB1 signalling promotes translation of specific genes that promote EMT 160 contributing to diabetes pathogenesis.
It has been previously established that translation regulation is closely linked to metabolic dysfunction and diabetes. For instance, mutations in the eukaryotic translation initiation factor 2α kinase 3 that regulates translation initiation factor eIF2, cause Wolcott-Rallison syndrome. This genetic disorder is characterized by neonatal onset of diabetes 161. These genetic associations as well as molecular perturbations in translation factors and regulators of translation factors provide a strong link between translation regulation and diabetes.
CUG triplet repeat binding protein ELAV-like family member 1 (CUGBP1/CELF1)
CELF1 was identified as an RBP that binds to CUG triplet repeat RNA 162. Further studies showed that it could bind to GU- and UG-rich sequences 163–165. Genome wide RNA targets of CELF1 and its consensus binding sites were identified using CLIP-seq and Bind-n-seq 166, 167. CELF1 is a multi-functional RBP that has established roles in AS, mRNA stability and translation 164, 168–172. Its role in translation, mRNA stability and AS regulation have been implicated in the pathogenesis of Myotonic Dystrophy 171, 173–177. Conditional overexpression of CELF1 in mouse skeletal muscle causes skeletal muscle wasting reproducing several aspects of Myotonic Dystrophy muscle pathology 174. Ablation of CELF1 in mice affects growth, viability, and fertility 178. CELF1 also has an important role in the maturation of the heart after birth 179.
CELF1 has vital roles in glucose and energy metabolism. CELF1 regulates splicing of insulin receptor. Increased expression of CELF1 induces a splicing change in the insulin receptor that leads to expression of an isoform with lower signalling potential thereby contributing to insulin resistance in Myotonic Dystrophy 180, 181. Furthermore, CELF1 controls splicing of pyruvate kinase, which is necessary for glycolysis 182. Increased expression of CELF1 in pre-adipocytes impairs adipogenesis in a TNF-alpha dependent manner 183. CELF1 also controls mRNA translation of genes that promote EMT 163. CELF1 protein levels are regulated by PKC- and ERK-mediated phosphorylation 184, 185. Related to diabetes, CELF1 protein levels are elevated in T1D mouse skeletal and heart muscle (Tables 1 and 2) 53. Consistent with activation of ERK and PKC under diabetic conditions, protein levels of CELF1 are upregulated in the diabetic skeletal muscle and heart; correlating with abnormal AS patterns of its targets (Tables 1 and 2). 53, 62. Importantly, increased CELF1 levels in diabetes may impair glucose metabolism and insulin receptor signalling through regulation of AS of essential genes with roles in metabolism.
CELF1 overexpressing mice develop dilated cardiomyopathy and heart failure and display reactivation of embryonic splicing patterns 175. These phenotypic and molecular changes in CELF1 transgenic mice are similar to the changes observed in the diabetic skeletal muscle and heart. We have found that CELF1 protein levels are elevated in diabetic skeletal muscle and heart consistent with increased expression of fetal/newborn spliced isoforms of CELF1 targets 53, 62. Thus, increased CELF1 protein levels in diabetic heart and skeletal muscle might be an important factor in skeletal muscle weakness and atrophy and diabetic cardiomyopathy (Table 2). However, more studies are needed to further investigate this possibility.
Elav-like protein 1 (Elavl1) (HuR)
HuR is a multi-functional RBP implicated in many aspects of diabetes pathogenesis. It binds to polyU and AU-rich elements and regulates mRNA stability and translation 186–188. HuR shuttles between the nucleus and cytoplasm 189 and upon activation, translocates to the cytoplasm and controls target mRNA stability and translation 189. Phosphorylation by PKC controls mRNA binding activity and cytoplasmic localization of HuR 190.
HuR is a key regulator of genes involved in stress response, cell cycle, inflammation, and immune response 98, 191. Deletion of HuR in mice causes loss of hematopoietic stem cells and early postnatal death 192. HuR is implicated in apoptosis via controlling mRNA levels of the p53 regulator mdm2 192. Depletion of HuR impairs the inflammatory response in human umbilical cord endothelial cells in a interleukin 10 dependent manner 193, 194 and contributes to TNF-α mediated cell death and inflammation in primary human cardiomyocytes 59. As a regulator of mRNA decay, HuR affects the stability of m6A containing mRNAs and impacts pluripotency of embryonic stem cells 195. It binds and stabilizes Sirt1 mRNA 196, VEGF and TNF-alpha mRNAs 188. HuR also has roles in AS and polyadenylation 188, 189, 197. Its role in RNA splicing is linked to angiogenesis and mitochondrial function 198.
It has been shown by many groups that HuR levels are increased in diabetes via different mechanisms (Table 1) 40, 59, 65. First mechanism is the phosphorylation of HuR by PKC that leads to high HuR protein levels, and increased cytoplasmic localization of HuR in cells under diabetic conditions 51, 65. Second mechanism for HuR increase in diabetes is through micro-RNA mediated regulation. Although HuR is also controlled via many microRNAs, under diabetic conditions two specific microRNAs are identified as regulators of HuR 59. MiR-23 and miR9, which control HuR levels, are downregulated under diabetic conditions 59,199. Low levels of miR23 and miR9 lead to increased HuR protein levels in diabetic tissues.
HuR is a major contributor to diabetic manifestations including retinopathy and nephropathy (Table 2). Depletion of HuR improves retinal damage in diabetic mice 63. In diabetic patient kidneys, HuR is upregulated in the cytoplasm 40. The increase in HuR stabilized the mRNA levels of CTGF, TGFB1, FOS, and SNAIL, contributing to EMT and diabetic nephropathy 39. Silencing of HuR in T1D rats partly ameliorated kidney proteinuria, inflammation, and hypertrophy 40. Together these studies strongly support the hypothesis that HuR up-regulation contributes to the development of diabetic nephropathy and retinopathy. Upregulation of HuR in diabetic human hearts correlated with increased inflammatory markers that can lead to cardiomyocyte death 59. Silencing of HuR in mice reduces infarct size after myocardial infarction 193. These results suggest HuR may be a common mediator of diabetic complications in multiple tissues; and as such, a good target for systemic treatment options because it binds specific RNA sequences.
In summary, changes in these seven RBPs contribute to pathogenic events activated under diabetic conditions such as inflammation, EMT, mitochondrial dysfunction and fibrosis in diabetes (Fig. 3). Importantly, a majority of these RBPs have key roles in insulin signaling, and metabolism of fat and glucose, which influence the development of diabetes.
RNA-BASED THERAPEUTICS IN DIABETES
RBPs play a critical role in the development of T1D and T2D diabetes as well as in diabetic complications. Targeting the interaction between RBPs and their RNA targets could be an effective strategy to correct gene expression changes in diabetic patients. RNA-based therapies show promise in the treatment of several human diseases detailed below.
Many different RNA-based technologies have been used for therapeutic purposes. We will focus on anti-sense oligo (ASO) based therapy, which is successfully being used in clinics. ASOs are single-stranded deoxyribonucleotides that are usually modified, including the addition of methyl or fluoro groups for increased stability and uptake 200, 201. ASOs are highly flexible in that they can be designed to degrade or stabilize target RNA, inhibit or enhance translation of target RNAs, modulate splicing of a pre-mRNA, or modify RBP binding to RNA 201. Importantly, ASOs have reported mild side effects with almost no immune response, thus they are generally considered safe for the treatment of human diseases 202, 203.
An ASO based drug that blocks RBP binding to RNA is now used for the successful treatment of spinal muscular dystrophy (SMA). The SMN1 gene is mutated in patients with SMA and is the cause of progressive loss of motor neurons 204, 205. SMN1 mutations reduce SMN1 protein levels resulting in impaired motor neuron function. Therefore, children with this mutation suffer from severe muscular atrophy and weakness, which can lead to death 206. Recent clinical trials that used an ASO to restore SMN gene function improved motor function in patients with infantile-onset and late-onset SMA 207, 208.
SMA-specific ASO targets SMN2 gene, which is closely related to SMN1 but is normally degraded due to the exclusion of exon 7. SMN2 spliced isoforms including exon 7 can partially replace SMN1 protein function 209, 210. The ASO promotes SMN2 exon 7 inclusion by blocking the RBP hnRNP-A1/A2 binding site to the RNA, thereby generating more full-length SMN2 protein 209. Since ASOs work well in modulating splicing 211, 212, diabetes-induced AS changes may be corrected by modulating RBP binding to target RNAs using specific ASOs.
Modulation of eIF4E- and IRES-dependent translation by ASOs is actively being explored for cancer and HIV treatment (ClinicalTrials.gov). These translation-modulating ASOs could have important ramifications in diabetes since down-regulation of eIF4E-dependent translation 66–68, 106, 107 with an increase in IRES dependent translation 66, 68 is a common complication in diabetes. The clinical success of RNA-based therapeutics is encouraging for future applications as novel and effective therapy options for diabetic complications.
There are two completed clinical trials that used ASOs to improve insulin sensitivity and decrease dyslipidemia in diabetes patients 213, 214. One of the ASOs designed to degrade hepatic glucagon receptor (GCGR) mRNA significantly attenuated hepatic glucose production 213. Another ASO that targets degradation of apolipoprotein C-III mRNA resulted in lower plasma triglyceride levels and insulin resistance in T2D patients. Since RBP regulated RNA networks are disrupted in diabetes and contribute to diabetic complications, modulating RBP:RNA interactions in diabetic complications could be a promising therapy.
Some questions still remain about the tissue specificity of ASO treatments, but advances in tissue specific delivery are currently being investigated. The use of nanoparticles, exosomes, or direct injection to the muscle or lumbar region of the spine have proven efficacy for some tissue specificity 215, 216. In addition, targeted uptake of ASOs by the liver using specific lipid conjugates has been successful 217. Altogether, there are numerous opportunities in the diabetes field for RNA-centric therapies to improve patient health and lifespan.
Conclusion
Recent studies demonstrate that RBPs play key roles in the development of diabetes and its systemic manifestations. The technological advancements in identification of global RNA targets of RBPs in a wide variety of tissues will help determine the major RNA regulatory programs disrupted under diabetic conditions. Many studies reviewed here indicate that modulating RBPs in rodent models of diabetes can improve or even prevent symptoms of diabetes. In the future, RBP function can be modulated for treatment of a variety of diabetic complications using modified oligonucleotides or ASOs that can control RBP interactions with RNA. However, caution is necessary for drug design targeting RBPs as the same RBP can have differing or even opposite roles in different tissues. In summary, post-transcriptional regulation by RBPs is emerging as a key mechanism in the development and pathogenesis of diabetes and has the potential to provide new therapeutic options for diabetic patients.
Acknowledgments
We thank Kuyumcu-Martinez lab members for reading the manuscript and Dr. Heather Lander for editing the manuscript. Graphics were downloaded from Servier Medical Art under a Creative Commons Attribution 3.0 Unported License. We apologize to authors whose works have not been cited due to the space constraints. This work was supported, in part, by an American Heart Association Grant [15GRNT22830010]; UTMB Department of Biochemistry and Molecular Biology Bridging funds; and a grant from the National Institutes of Health/National Heart Lung Blood Institute [1R01HL135031-01] to M.N.K-M. C.A.N. was supported by UTMB Kempner Fellowship.
Footnotes
The author listed above has no conflict of interest to report and have no financial gain in materials addressed in this manuscript.
The author whose name listed above has no conflict of interest to report and have no financial gain in materials addressed in this manuscript.
Contributor Information
Curtis A. Nutter, Department of Biochemistry and Molecular Biology, University of Texas Medical Branch, Galveston, Texas 77555, USA. New address: Department of Molecular Genetics and Microbiology, University of Florida, Gainesville, FL 32620, USA
Muge N. Kuyumcu-Martinez, Departments of Biochemistry and Molecular Biology, and Neuroscience and Cell Biology, and Institute for Translational Sciences, University of Texas Medical Branch, Galveston, Texas 77555, USA
References
- 1.Harcourt BE, Penfold SA, Forbes JM. Coming full circle in diabetes mellitus: from complications to initiation. Nat Rev Endocrinol. 2013;9:113–123. doi: 10.1038/nrendo.2012.236. [DOI] [PubMed] [Google Scholar]
- 2.Schneider AL, Kalyani RR, Golden S, Stearns SC, Wruck L, Yeh HC, Coresh J, Selvin E. Diabetes and Prediabetes and Risk of Hospitalization: The Atherosclerosis Risk in Communities (ARIC) Study. Diabetes Care. 2016;39:772–779. doi: 10.2337/dc15-1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shi Y, Vanhoutte PM. Macro- and microvascular endothelial dysfunction in diabetes. J Diabetes. 2017 doi: 10.1111/1753-0407.12521. [DOI] [PubMed] [Google Scholar]
- 4.Dong Y, Fernandes C, Liu Y, Wu Y, Wu H, Brophy ML, Deng L, Song K, Wen A, Wong S, et al. Role of endoplasmic reticulum stress signalling in diabetic endothelial dysfunction and atherosclerosis. Diab Vasc Dis Res. 2017;14:14–23. doi: 10.1177/1479164116666762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Conserva F, Gesualdo L, Papale M. A Systems Biology Overview on Human Diabetic Nephropathy: From Genetic Susceptibility to Post-Transcriptional and Post-Translational Modifications. J Diabetes Res. 2016;2016:7934504. doi: 10.1155/2016/7934504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chan GC, Tang SC. Diabetic nephropathy: landmark clinical trials and tribulations. Nephrol Dial Transplant. 2016;31:359–368. doi: 10.1093/ndt/gfu411. [DOI] [PubMed] [Google Scholar]
- 7.Sheetz MJ, King GL. Molecular understanding of hyperglycemia’s adverse effects for diabetic complications. JAMA. 2002;288:2579–2588. doi: 10.1001/jama.288.20.2579. [DOI] [PubMed] [Google Scholar]
- 8.Wilkinson-Berka JL, Miller AG. Update on the treatment of diabetic retinopathy. ScientificWorldJournal. 2008;8:98–120. doi: 10.1100/tsw.2008.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Aiello LP, Gardner TW, King GL, Blankenship G, Cavallerano JD, Ferris FL, 3rd, Klein R. Diabetic retinopathy. Diabetes Care. 1998;21:143–156. doi: 10.2337/diacare.21.1.143. [DOI] [PubMed] [Google Scholar]
- 10.Asghar O, Al-Sunni A, Khavandi K, Khavandi A, Withers S, Greenstein A, Heagerty AM, Malik RA. Diabetic cardiomyopathy. Clin Sci (Lond) 2009;116:741–760. doi: 10.1042/CS20080500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ferrannini E, Cushman WC. Diabetes and hypertension: the bad companions. Lancet. 2012;380:601–610. doi: 10.1016/S0140-6736(12)60987-8. [DOI] [PubMed] [Google Scholar]
- 12.Trachanas K, Sideris S, Aggeli C, Poulidakis E, Gatzoulis K, Tousoulis D, Kallikazaros I. Diabetic cardiomyopathy: from pathophysiology to treatment. Hellenic J Cardiol. 2014;55:411–421. [PubMed] [Google Scholar]
- 13.Andersen H, Gadeberg PC, Brock B, Jakobsen J. Muscular atrophy in diabetic neuropathy: a stereological magnetic resonance imaging study. Diabetologia. 1997;40:1062–1069. doi: 10.1007/s001250050788. [DOI] [PubMed] [Google Scholar]
- 14.Hernandez-Ochoa EO, Vanegas C. Diabetic Myopathy and Mechanisms of Disease. Biochem Pharmacol (Los Angel) 2015:4. doi: 10.4172/2167-0501.1000e179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Aneja A, Tang WH, Bansilal S, Garcia MJ, Farkouh ME. Diabetic cardiomyopathy: insights into pathogenesis, diagnostic challenges, and therapeutic options. Am J Med. 2008;121:748–757. doi: 10.1016/j.amjmed.2008.03.046. [DOI] [PubMed] [Google Scholar]
- 16.Boudina S, Abel ED. Diabetic cardiomyopathy, causes and effects. Rev Endocr Metab Disord. 2010;11:31–39. doi: 10.1007/s11154-010-9131-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gerstberger S, Hafner M, Tuschl T. A census of human RNA-binding proteins. Nat Rev Genet. 2014;15:829–845. doi: 10.1038/nrg3813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Moore MJ, Proudfoot NJ. Pre-mRNA Processing Reaches Back to Transcription and Ahead to Translation. Cell. 2009;136:688–700. doi: 10.1016/j.cell.2009.02.001. [DOI] [PubMed] [Google Scholar]
- 19.Marchese D, de Groot NS, Lorenzo Gotor N, Livi CM, Tartaglia GG. Advances in the characterization of RNA-binding proteins. Wiley Interdiscip Rev RNA. 2016;7:793–810. doi: 10.1002/wrna.1378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Heinrich S, Derrer CP, Lari A, Weis K, Montpetit B. Temporal and spatial regulation of mRNA export: Single particle RNA-imaging provides new tools and insights. Bioessays. 2017:39. doi: 10.1002/bies.201600124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brinegar AE, Cooper TA. Roles for RNA-binding proteins in development and disease. Brain Res. 2016;1647:1–8. doi: 10.1016/j.brainres.2016.02.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Castello A, Fischer B, Hentze MW, Preiss T. RNA-binding proteins in Mendelian disease. Trends Genet. 2013;29:318–327. doi: 10.1016/j.tig.2013.01.004. [DOI] [PubMed] [Google Scholar]
- 23.Cookson MR. RNA-binding proteins implicated in neurodegenerative diseases. Wiley Interdiscip Rev RNA. 2017:8. doi: 10.1002/wrna.1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Donlin-Asp PG, Rossoll W, Bassell GJ. Spatially and temporally regulating translation via mRNA-binding proteins in cellular and neuronal function. FEBS Lett. 2017 doi: 10.1002/1873-3468.12621. [DOI] [PubMed] [Google Scholar]
- 25.de Bruin RG, Rabelink TJ, van Zonneveld AJ, van der Veer EP. Emerging roles for RNA-binding proteins as effectors and regulators of cardiovascular disease. Eur Heart J. 2017 doi: 10.1093/eurheartj/ehw567. [DOI] [PubMed] [Google Scholar]
- 26.Xin H, Deng K, Fu M. Post-transcriptional gene regulation by RNA-binding proteins in vascular endothelial dysfunction. Sci China Life Sci. 2014;57:836–844. doi: 10.1007/s11427-014-4703-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Verma SK, Deshmukh V, Nutter CA, Jaworski EA, Jin W, Wadhwa L, Abata J, Ricci M, Lincoln J, Martin JF, et al. Rbfox2 function in RNA metabolism is impaired in hypoplastic left heart syndrome patient hearts. Sci Rep. 2016;6:30896. doi: 10.1038/srep30896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nutter CA, Jaworski EA, Verma SK, Deshmukh V, Wang Q, Botvinnik OB, Lozano MJ, Abass IJ, Ijaz T, Brasier AR, et al. Dysregulation of RBFOX2 Is an Early Event in Cardiac Pathogenesis of Diabetes. Cell Rep. 2016;15:2200–2213. doi: 10.1016/j.celrep.2016.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.van Hoek M, Dehghan A, Witteman JC, van Duijn CM, Uitterlinden AG, Oostra BA, Hofman A, Sijbrands EJ, Janssens AC. Predicting type 2 diabetes based on polymorphisms from genome-wide association studies: a population-based study. Diabetes. 2008;57:3122–3128. doi: 10.2337/db08-0425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Scott LJ, Mohlke KL, Bonnycastle LL, Willer CJ, Li Y, Duren WL, Erdos MR, Stringham HM, Chines PS, Jackson AU, et al. A genome-wide association study of type 2 diabetes in Finns detects multiple susceptibility variants. Science. 2007;316:1341–1345. doi: 10.1126/science.1142382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lyssenko V, Jonsson A, Almgren P, Pulizzi N, Isomaa B, Tuomi T, Berglund G, Altshuler D, Nilsson P, Groop L. Clinical risk factors, DNA variants, and the development of type 2 diabetes. N Engl J Med. 2008;359:2220–2232. doi: 10.1056/NEJMoa0801869. [DOI] [PubMed] [Google Scholar]
- 32.Rao P, Wang H, Fang H, Gao Q, Zhang J, Song M, Zhou Y, Wang Y, Wang W. Association between IGF2BP2 Polymorphisms and Type 2 Diabetes Mellitus: A Case-Control Study and Meta-Analysis. Int J Environ Res Public Health. 2016:13. doi: 10.3390/ijerph13060574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ben-Haim MS, Moshitch-Moshkovitz S, Rechavi G. FTO: linking m6A demethylation to adipogenesis. Cell Res. 2015;25:3–4. doi: 10.1038/cr.2014.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gerken T, Girard CA, Tung YC, Webby CJ, Saudek V, Hewitson KS, Yeo GS, McDonough MA, Cunliffe S, McNeill LA, et al. The obesity-associated FTO gene encodes a 2-oxoglutarate-dependent nucleic acid demethylase. Science. 2007;318:1469–1472. doi: 10.1126/science.1151710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chu X, Erdman R, Susek M, Gerst H, Derr K, Al-Agha M, Wood GC, Hartman C, Yeager S, Blosky MA, et al. Association of morbid obesity with FTO and INSIG2 allelic variants. Arch Surg. 2008;143:235–240. doi: 10.1001/archsurg.2007.77. discussion 241. [DOI] [PubMed] [Google Scholar]
- 36.Wood AR, Tyrrell J, Beaumont R, Jones SE, Tuke MA, Ruth KS, Yaghootkar H, Freathy RM, Murray A, et al. consortium G. Variants in the FTO and CDKAL1 loci have recessive effects on risk of obesity and type 2 diabetes, respectively. Diabetologia. 2016;59:1214–1221. doi: 10.1007/s00125-016-3908-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schaeffer V, Hansen KM, Morris DR, LeBoeuf RC, Abrass CK. RNA-binding protein IGF2BP2/IMP2 is required for laminin-beta2 mRNA translation and is modulated by glucose concentration. Am J Physiol Renal Physiol. 2012;303:F75–82. doi: 10.1152/ajprenal.00185.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Park JT, Kato M, Lanting L, Castro N, Nam BY, Wang M, Kang SW, Natarajan R. Repression of let-7 by transforming growth factor-beta1-induced Lin28 upregulates collagen expression in glomerular mesangial cells under diabetic conditions. Am J Physiol Renal Physiol. 2014;307:F1390–1403. doi: 10.1152/ajprenal.00458.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yu C, Xin W, Zhen J, Liu Y, Javed A, Wang R, Wan Q. Human antigen R mediated post-transcriptional regulation of epithelial-mesenchymal transition related genes in diabetic nephropathy. J Diabetes. 2015;7:562–572. doi: 10.1111/1753-0407.12220. [DOI] [PubMed] [Google Scholar]
- 40.Shang J, Wan Q, Wang X, Duan Y, Wang Z, Wei X, Zhang Y, Wang H, Wang R, Yi F. Identification of NOD2 as a novel target of RNA-binding protein HuR: evidence from NADPH oxidase-mediated HuR signaling in diabetic nephropathy. Free Radic Biol Med. 2015;79:217–227. doi: 10.1016/j.freeradbiomed.2014.12.013. [DOI] [PubMed] [Google Scholar]
- 41.Kato M, Wang L, Putta S, Wang M, Yuan H, Sun G, Lanting L, Todorov I, Rossi JJ, Natarajan R. Post-transcriptional up-regulation of Tsc-22 by Ybx1, a target of miR-216a, mediates TGF-{beta}-induced collagen expression in kidney cells. J Biol Chem. 2010;285:34004–34015. doi: 10.1074/jbc.M110.165027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lo CS, Chang SY, Chenier I, Filep JG, Ingelfinger JR, Zhang SL, Chan JS. Heterogeneous nuclear ribonucleoprotein F suppresses angiotensinogen gene expression and attenuates hypertension and kidney injury in diabetic mice. Diabetes. 2012;61:2597–2608. doi: 10.2337/db11-1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Abdo S, Lo CS, Chenier I, Shamsuyarova A, Filep JG, Ingelfinger JR, Zhang SL, Chan JS. Heterogeneous nuclear ribonucleoproteins F and K mediate insulin inhibition of renal angiotensinogen gene expression and prevention of hypertension and kidney injury in diabetic mice. Diabetologia. 2013;56:1649–1660. doi: 10.1007/s00125-013-2910-4. [DOI] [PubMed] [Google Scholar]
- 44.Lo CS, Shi Y, Chang SY, Abdo S, Chenier I, Filep JG, Ingelfinger JR, Zhang SL, Chan JS. Overexpression of heterogeneous nuclear ribonucleoprotein F stimulates renal Ace-2 gene expression and prevents TGF-beta1-induced kidney injury in a mouse model of diabetes. Diabetologia. 2015;58:2443–2454. doi: 10.1007/s00125-015-3700-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.de Bruin RG, Shiue L, Prins J, de Boer HC, Singh A, Fagg WS, van Gils JM, Duijs JM, Katzman S, Kraaijeveld AO, et al. Quaking promotes monocyte differentiation into pro-atherogenic macrophages by controlling pre-mRNA splicing and gene expression. Nat Commun. 2016;7:10846. doi: 10.1038/ncomms10846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.van der Veer EP, de Bruin RG, Kraaijeveld AO, de Vries MR, Bot I, Pera T, Segers FM, Trompet S, van Gils JM, Roeten MK, et al. Quaking, an RNA-binding protein, is a critical regulator of vascular smooth muscle cell phenotype. Circ Res. 2013;113:1065–1075. doi: 10.1161/CIRCRESAHA.113.301302. [DOI] [PubMed] [Google Scholar]
- 47.Zhang H, Taylor WR, Joseph G, Caracciolo V, Gonzales DM, Sidell N, Seli E, Blackshear PJ, Kallen CB. mRNA-binding protein ZFP36 is expressed in atherosclerotic lesions and reduces inflammation in aortic endothelial cells. Arterioscler Thromb Vasc Biol. 2013;33:1212–1220. doi: 10.1161/ATVBAHA.113.301496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Patino WD, Kang JG, Matoba S, Mian OY, Gochuico BR, Hwang PM. Atherosclerotic plaque macrophage transcriptional regulators are expressed in blood and modulated by tristetraprolin. Circ Res. 2006;98:1282–1289. doi: 10.1161/01.RES.0000222284.48288.28. [DOI] [PubMed] [Google Scholar]
- 49.Stellos K, Gatsiou A, Stamatelopoulos K, Perisic Matic L, John D, Lunella FF, Jae N, Rossbach O, Amrhein C, Sigala F, et al. Adenosine-to-inosine RNA editing controls cathepsin S expression in atherosclerosis by enabling HuR-mediated post-transcriptional regulation. Nat Med. 2016;22:1140–1150. doi: 10.1038/nm.4172. [DOI] [PubMed] [Google Scholar]
- 50.Panchenko MP, Silva N, Stone JR. Up-regulation of a hydrogen peroxide-responsive pre-mRNA binding protein in atherosclerosis and intimal hyperplasia. Cardiovasc Pathol. 2009;18:167–172. doi: 10.1016/j.carpath.2008.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pullmann R, Jr, Juhaszova M, Lopez de Silanes I, Kawai T, Mazan-Mamczarz K, Halushka MK, Gorospe M. Enhanced proliferation of cultured human vascular smooth muscle cells linked to increased function of RNA-binding protein HuR. J Biol Chem. 2005;280:22819–22826. doi: 10.1074/jbc.M501106200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Totary-Jain H, Naveh-Many T, Riahi Y, Kaiser N, Eckel J, Sasson S. Calreticulin destabilizes glucose transporter-1 mRNA in vascular endothelial and smooth muscle cells under high-glucose conditions. Circ Res. 2005;97:1001–1008. doi: 10.1161/01.RES.0000189260.46084.e5. [DOI] [PubMed] [Google Scholar]
- 53.Nutter CA, Jaworski E, Verma SK, Perez-Carrasco Y, Kuyumcu-Martinez MN. Developmentally regulated alternative splicing is perturbed in type 1 diabetic skeletal muscle. Muscle Nerve. 2017 doi: 10.1002/mus.25599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Stirewalt WS, Wool IG, Cavicchi P. The relation of RNA and protein synthesis to the sedimentation of muscle ribosomes: effect of diabetes and insulin. Proc Natl Acad Sci U S A. 1967;57:1885–1892. doi: 10.1073/pnas.57.6.1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rampersad OR, Wool IG. Protein synthesis by ribosomes from heart muscle: effect of insulin and diabetes. Science. 1965;149:1102–1103. doi: 10.1126/science.149.3688.1102. [DOI] [PubMed] [Google Scholar]
- 56.Kelly FJ, Jefferson LS. Control of peptide-chain initiation in rat skeletal muscle. Development of methods for preparation of native ribosomal subunits and analysis of the effect of insulin on formation of 40 S initiation complexes. J Biol Chem. 1985;260:6677–6683. [PubMed] [Google Scholar]
- 57.Zhang M, Sun D, Li S, Pan X, Zhang X, Zhu D, Li C, Zhang R, Gao E, Wang H. Lin28a protects against cardiac ischaemia/reperfusion injury in diabetic mice through the insulin-PI3K-mTOR pathway. J Cell Mol Med. 2015;19:1174–1182. doi: 10.1111/jcmm.12369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sun S, Zhang M, Lin J, Hu J, Zhang R, Li C, Wei T, Sun D, Wei J, Wang H. Lin28a protects against diabetic cardiomyopathy via the PKA/ROCK2 pathway. Biochem Biophys Res Commun. 2016;469:29–36. doi: 10.1016/j.bbrc.2015.11.065. [DOI] [PubMed] [Google Scholar]
- 59.Jeyabal P, Thandavarayan RA, Joladarashi D, Suresh Babu S, Krishnamurthy S, Bhimaraj A, Youker KA, Kishore R, Krishnamurthy P. MicroRNA-9 inhibits hyperglycemia-induced pyroptosis in human ventricular cardiomyocytes by targeting ELAVL1. Biochem Biophys Res Commun. 2016;471:423–429. doi: 10.1016/j.bbrc.2016.02.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Guo W, Jiang T, Lian C, Wang H, Zheng Q, Ma H. QKI deficiency promotes FoxO1 mediated nitrosative stress and endoplasmic reticulum stress contributing to increased vulnerability to ischemic injury in diabetic heart. J Mol Cell Cardiol. 2014;75:131–140. doi: 10.1016/j.yjmcc.2014.07.010. [DOI] [PubMed] [Google Scholar]
- 61.Guo W, Shi X, Liu A, Yang G, Yu F, Zheng Q, Wang Z, Allen DG, Lu Z. RNA binding protein QKI inhibits the ischemia/reperfusion-induced apoptosis in neonatal cardiomyocytes. Cell Physiol Biochem. 2011;28:593–602. doi: 10.1159/000335755. [DOI] [PubMed] [Google Scholar]
- 62.Verma SK, Deshmukh V, Liu P, Nutter CA, Espejo R, Hung ML, Wang GS, Yeo GW, Kuyumcu-Martinez MN. Reactivation of fetal splicing programs in diabetic hearts is mediated by protein kinase C signaling. J Biol Chem. 2013;288:35372–35386. doi: 10.1074/jbc.M113.507426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Amadio M, Pascale A, Cupri S, Pignatello R, Osera C, D’Agata V, D’Amico AG, Leggio GM, Ruozi B, Govoni S, et al. Nanosystems based on siRNA silencing HuR expression counteract diabetic retinopathy in rat. Pharmacol Res. 2016;111:713–720. doi: 10.1016/j.phrs.2016.07.042. [DOI] [PubMed] [Google Scholar]
- 64.Amadio M, Osera C, Lupo G, Motta C, Drago F, Govoni S, Pascale A. Protein kinase C activation affects, via the mRNA-binding Hu-antigen R/ELAV protein, vascular endothelial growth factor expression in a pericytic/endothelial coculture model. Mol Vis. 2012;18:2153–2164. [PMC free article] [PubMed] [Google Scholar]
- 65.Amadio M, Bucolo C, Leggio GM, Drago F, Govoni S, Pascale A. The PKCbeta/HuR/VEGF pathway in diabetic retinopathy. Biochem Pharmacol. 2010;80:1230–1237. doi: 10.1016/j.bcp.2010.06.033. [DOI] [PubMed] [Google Scholar]
- 66.Schrufer TL, Antonetti DA, Sonenberg N, Kimball SR, Gardner TW, Jefferson LS. Ablation of 4E-BP1/2 prevents hyperglycemia-mediated induction of VEGF expression in the rodent retina and in Muller cells in culture. Diabetes. 2010;59:2107–2116. doi: 10.2337/db10-0148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Miller WP, Mihailescu ML, Yang C, Barber AJ, Kimball SR, Jefferson LS, Dennis MD. The Translational Repressor 4E-BP1 Contributes to Diabetes-Induced Visual Dysfunction. Invest Ophthalmol Vis Sci. 2016;57:1327–1337. doi: 10.1167/iovs.15-18719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Dennis MD, Kimball SR, Fort PE, Jefferson LS. Regulated in development and DNA damage 1 is necessary for hyperglycemia-induced vascular endothelial growth factor expression in the retina of diabetic rodents. J Biol Chem. 2015;290:3865–3874. doi: 10.1074/jbc.M114.623058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wang CY, Shie SS, Tsai ML, Yang CH, Hung KC, Wang CC, Hsieh IC, Wen MS. FTO modulates fibrogenic responses in obstructive nephropathy. Sci Rep. 2016;6:18874. doi: 10.1038/srep18874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sperling R. The nuts and bolts of the endogenous spliceosome. Wiley Interdiscip Rev RNA. 2017:8. doi: 10.1002/wrna.1377. [DOI] [PubMed] [Google Scholar]
- 71.Wang ET, Sandberg R, Luo S, Khrebtukova I, Zhang L, Mayr C, Kingsmore SF, Schroth GP, Burge CB. Alternative isoform regulation in human tissue transcriptomes. Nature. 2008;456:470–476. doi: 10.1038/nature07509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Keren H, Lev-Maor G, Ast G. Alternative splicing and evolution: diversification, exon definition and function. Nature Reviews Genetics. 2010;11:345–355. doi: 10.1038/nrg2776. [DOI] [PubMed] [Google Scholar]
- 73.Bland CS, Wang ET, Vu A, David MP, Castle JC, Johnson JM, Burge CB, Cooper TA. Global regulation of alternative splicing during myogenic differentiation. Nucleic Acids Research. 2010;38:7651–7664. doi: 10.1093/nar/gkq614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kalsotra A, Xiao X, Ward AJ, Castle JC, Johnson JM, Burge CB, Cooper TA. A postnatal switch of CELF and MBNL proteins reprograms alternative splicing in the developing heart. Proc Natl Acad Sci U S A. 2008;105:20333–20338. doi: 10.1073/pnas.0809045105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.David CJ, Manley JL. Alternative pre-mRNA splicing regulation in cancer: pathways and programs unhinged. Genes Dev. 2010;24:2343–2364. doi: 10.1101/gad.1973010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Chen M, Zhang J, Manley JL. Turning on a fuel switch of cancer: hnRNP proteins regulate alternative splicing of pyruvate kinase mRNA. Cancer Res. 2010;70:8977–8980. doi: 10.1158/0008-5472.CAN-10-2513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Dominguez D, Tsai YH, Weatheritt R, Wang Y, Blencowe BJ, Wang Z. An extensive program of periodic alternative splicing linked to cell cycle progression. Elife. 2016:5. doi: 10.7554/eLife.10288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Juan-Mateu J, Villate O, Eizirik DL. MECHANISMS IN ENDOCRINOLOGY: Alternative splicing: the new frontier in diabetes research. Eur J Endocrinol. 2016;174:R225–238. doi: 10.1530/EJE-15-0916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Nogueira TC, Paula FM, Villate O, Colli ML, Moura RF, Cunha DA, Marselli L, Marchetti P, Cnop M, Julier C, et al. GLIS3, a susceptibility gene for type 1 and type 2 diabetes, modulates pancreatic beta cell apoptosis via regulation of a splice variant of the BH3-only protein Bim. PLoS Genet. 2013;9:e1003532. doi: 10.1371/journal.pgen.1003532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ortis F, Naamane N, Flamez D, Ladriere L, Moore F, Cunha DA, Colli ML, Thykjaer T, Thorsen K, Orntoft TF, et al. Cytokines interleukin-1beta and tumor necrosis factor-alpha regulate different transcriptional and alternative splicing networks in primary beta-cells. Diabetes. 2010;59:358–374. doi: 10.2337/db09-1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Villate O, Turatsinze JV, Mascali LG, Grieco FA, Nogueira TC, Cunha DA, Nardelli TR, Sammeth M, Salunkhe VA, Esguerra JL, et al. Nova1 is a master regulator of alternative splicing in pancreatic beta cells. Nucleic Acids Res. 2014;42:11818–11830. doi: 10.1093/nar/gku861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Juan-Mateu J, Rech TH, Villate O, Lizarraga-Mollinedo E, Wendt A, Turatsinze JV, Brondani LA, Nardelli TR, Nogueira TC, Esguerra JL, et al. Neuron-enriched RNA-binding Proteins Regulate Pancreatic Beta Cell Function and Survival. J Biol Chem. 2017;292:3466–3480. doi: 10.1074/jbc.M116.748335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kandpal RP, Rajasimha HK, Brooks MJ, Nellissery J, Wan J, Qian J, Kern TS, Swaroop A. Transcriptome analysis using next generation sequencing reveals molecular signatures of diabetic retinopathy and efficacy of candidate drugs. Mol Vis. 2012;18:1123–1146. [PMC free article] [PubMed] [Google Scholar]
- 84.Rincon-Choles H, Kasinath BS, Gorin Y, Abboud HE. Angiotensin II and growth factors in the pathogenesis of diabetic nephropathy. Kidney Int Suppl. 2002:S8–11. doi: 10.1046/j.1523-1755.62.s82.3.x. [DOI] [PubMed] [Google Scholar]
- 85.Peiris-Pages M. The role of VEGF 165b in pathophysiology. Cell Adh Migr. 2012;6:561–568. doi: 10.4161/cam.22439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Xia X-b, Xiong S-q, Song W-t, Luo J, Wang Y-k, Zhou R-r. Inhibition of retinal neovascularization by siRNA targeting VEGF(165) Molecular Vision. 2008;14:1965–1973. [PMC free article] [PubMed] [Google Scholar]
- 87.Bortoloso E, Del Prete D, Dalla Vestra M, Gambaro G, Saller A, Antonucci F, Baggio B, Anglani F, Fioretto P. Quantitave and qualitative changes in vascular endothelial growth factor gene expression in glomeruli of patients with type 2 diabetes. Eur J Endocrinol. 2004;150:799–807. doi: 10.1530/eje.0.1500799. [DOI] [PubMed] [Google Scholar]
- 88.Robinson CJ, Stringer SE. The splice variants of vascular endothelial growth factor (VEGF) and their receptors. J Cell Sci. 2001;114:853–865. doi: 10.1242/jcs.114.5.853. [DOI] [PubMed] [Google Scholar]
- 89.Perrin RM, Konopatskaya O, Qiu Y, Harper S, Bates DO, Churchill AJ. Diabetic retinopathy is associated with a switch in splicing from anti- to pro-angiogenic isoforms of vascular endothelial growth factor. Diabetologia. 2005;48:2422–2427. doi: 10.1007/s00125-005-1951-8. [DOI] [PubMed] [Google Scholar]
- 90.Wei CM, Gershowitz A, Moss B. Methylated nucleotides block 5′ terminus of HeLa cell messenger RNA. Cell. 1975;4:379–386. doi: 10.1016/0092-8674(75)90158-0. [DOI] [PubMed] [Google Scholar]
- 91.Wei W, Ji X, Guo X, Ji S. Regulatory Role of N6 -Methyladenosine (m6 A) Methylation in RNA Processing and Human Diseases. J Cell Biochem. 2017 doi: 10.1002/jcb.25967. [DOI] [PubMed] [Google Scholar]
- 92.Li S, Mason CE. The pivotal regulatory landscape of RNA modifications. Annu Rev Genomics Hum Genet. 2014;15:127–150. doi: 10.1146/annurev-genom-090413-025405. [DOI] [PubMed] [Google Scholar]
- 93.Meyer KD, Saletore Y, Zumbo P, Elemento O, Mason CE, Jaffrey SR. Comprehensive analysis of mRNA methylation reveals enrichment in 3′ UTRs and near stop codons. Cell. 2012;149:1635–1646. doi: 10.1016/j.cell.2012.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Shen F, Huang W, Huang JT, Xiong J, Yang Y, Wu K, Jia GF, Chen J, Feng YQ, Yuan BF, et al. Decreased N(6)-methyladenosine in peripheral blood RNA from diabetic patients is associated with FTO expression rather than ALKBH5. J Clin Endocrinol Metab. 2015;100:E148–154. doi: 10.1210/jc.2014-1893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Feigerlova E, Battaglia-Hsu SF. Role of post-transcriptional regulation of mRNA stability in renal pathophysiology: focus on chronic kidney disease. FASEB J. 2017;31:457–468. doi: 10.1096/fj.201601087RR. [DOI] [PubMed] [Google Scholar]
- 96.Roy B, Jacobson A. The intimate relationships of mRNA decay and translation. Trends Genet. 2013;29:691–699. doi: 10.1016/j.tig.2013.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Suresh Babu S, Joladarashi D, Jeyabal P, Thandavarayan RA, Krishnamurthy P. RNA-stabilizing proteins as molecular targets in cardiovascular pathologies. Trends Cardiovasc Med. 2015;25:676–683. doi: 10.1016/j.tcm.2015.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Brennan CM, Steitz JA. HuR and mRNA stability. Cell Mol Life Sci. 2001;58:266–277. doi: 10.1007/PL00000854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Kasinath BS, Mariappan MM, Sataranatarajan K, Lee MJ, Ghosh Choudhury G, Feliers D. Novel mechanisms of protein synthesis in diabetic nephropathy--role of mRNA translation. Rev Endocr Metab Disord. 2008;9:255–266. doi: 10.1007/s11154-008-9091-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Chen G, Gharib TG, Huang CC, Taylor JM, Misek DE, Kardia SL, Giordano TJ, Iannettoni MD, Orringer MB, Hanash SM, et al. Discordant protein and mRNA expression in lung adenocarcinomas. Mol Cell Proteomics. 2002;1:304–313. doi: 10.1074/mcp.m200008-mcp200. [DOI] [PubMed] [Google Scholar]
- 101.Kasinath BS, Feliers D, Sataranatarajan K, Ghosh Choudhury G, Lee MJ, Mariappan MM. Regulation of mRNA translation in renal physiology and disease. Am J Physiol Renal Physiol. 2009;297:F1153–1165. doi: 10.1152/ajprenal.90748.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Sonenberg N, Hinnebusch AG. Regulation of translation initiation in eukaryotes: mechanisms and biological targets. Cell. 2009;136:731–745. doi: 10.1016/j.cell.2009.01.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Richter JD, Coller J. Pausing on Polyribosomes: Make Way for Elongation in Translational Control. Cell. 2015;163:292–300. doi: 10.1016/j.cell.2015.09.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Hinnebusch AG. The scanning mechanism of eukaryotic translation initiation. Annu Rev Biochem. 2014;83:779–812. doi: 10.1146/annurev-biochem-060713-035802. [DOI] [PubMed] [Google Scholar]
- 105.Martinez-Salas E, Lozano G, Fernandez-Chamorro J, Francisco-Velilla R, Galan A, Diaz R. RNA-binding proteins impacting on internal initiation of translation. Int J Mol Sci. 2013;14:21705–21726. doi: 10.3390/ijms141121705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Kimball SR, Jefferson LS, Fadden P, Haystead TA, Lawrence JC., Jr Insulin and diabetes cause reciprocal changes in the association of eIF-4E and PHAS-I in rat skeletal muscle. Am J Physiol. 1996;270:C705–709. doi: 10.1152/ajpcell.1996.270.2.C705. [DOI] [PubMed] [Google Scholar]
- 107.Pause A, Belsham GJ, Gingras AC, Donze O, Lin TA, Lawrence JC, Jr, Sonenberg N. Insulin-dependent stimulation of protein synthesis by phosphorylation of a regulator of 5′-cap function. Nature. 1994;371:762–767. doi: 10.1038/371762a0. [DOI] [PubMed] [Google Scholar]
- 108.Stirewalt WS, Wool IG. Protein synthesis by heart muscle ribosomes: an effect of insulin independent of substrate transport. Science. 1966;154:284–285. doi: 10.1126/science.154.3746.284. [DOI] [PubMed] [Google Scholar]
- 109.Wool IG, Rampersad OR, Moyer AN. Effect of insulin and diabetes on protein synthesis by ribosomes from heart muscle. Significance for theories of the hormone’s mechanism of action. Am J Med. 1966;40:716–723. doi: 10.1016/0002-9343(66)90152-5. [DOI] [PubMed] [Google Scholar]
- 110.Dennis MD, Schrufer TL, Bronson SK, Kimball SR, Jefferson LS. Hyperglycemia-induced O-GlcNAcylation and truncation of 4E-BP1 protein in liver of a mouse model of type 1 diabetes. J Biol Chem. 2011;286:34286–34297. doi: 10.1074/jbc.M111.259457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Dennis MD, Shenberger JS, Stanley BA, Kimball SR, Jefferson LS. Hyperglycemia mediates a shift from cap-dependent to cap-independent translation via a 4E-BP1-dependent mechanism. Diabetes. 2013;62:2204–2214. doi: 10.2337/db12-1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Core LJ, Waterfall JJ, Lis JT. Nascent RNA sequencing reveals widespread pausing and divergent initiation at human promoters. Science. 2008;322:1845–1848. doi: 10.1126/science.1162228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Sataranatarajan K, Lee MJ, Mariappan MM, Feliers D. PKCdelta regulates the stimulation of vascular endothelial factor mRNA translation by angiotensin II through hnRNP K. Cell Signal. 2008;20:969–977. doi: 10.1016/j.cellsig.2008.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Balzeau J, Menezes MR, Cao S, Hagan JP. The LIN28/let-7 Pathway in Cancer. Front Genet. 2017;8:31. doi: 10.3389/fgene.2017.00031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Yang J, Bennett BD, Luo S, Inoue K, Grimm SA, Schroth GP, Bushel PR, Kinyamu HK, Archer TK. LIN28A Modulates Splicing and Gene Expression Programs in Breast Cancer Cells. Mol Cell Biol. 2015;35:3225–3243. doi: 10.1128/MCB.00426-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Tsanov KM, Pearson DS, Wu Z, Han A, Triboulet R, Seligson MT, Powers JT, Osborne JK, Kane S, Gygi SP, et al. LIN28 phosphorylation by MAPK/ERK couples signalling to the post-transcriptional control of pluripotency. Nat Cell Biol. 2017;19:60–67. doi: 10.1038/ncb3453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Wilbert ML, Huelga SC, Kapeli K, Stark TJ, Liang TY, Chen SX, Yan BY, Nathanson JL, Hutt KR, Lovci MT, et al. LIN28 binds messenger RNAs at GGAGA motifs and regulates splicing factor abundance. Mol Cell. 2012;48:195–206. doi: 10.1016/j.molcel.2012.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Amen AM, Ruiz-Garzon CR, Shi J, Subramanian M, Pham DL, Meffert MK. A Rapid Induction Mechanism for Lin28a in Trophic Responses. Mol Cell. 2017;65:490–503. e497. doi: 10.1016/j.molcel.2016.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Zhu H, Shah S, Shyh-Chang N, Shinoda G, Einhorn WS, Viswanathan SR, Takeuchi A, Grasemann C, Rinn JL, Lopez MF, et al. Lin28a transgenic mice manifest size and puberty phenotypes identified in human genetic association studies. Nat Genet. 2010;42:626–630. doi: 10.1038/ng.593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Zeng Y, Yao B, Shin J, Lin L, Kim N, Song Q, Liu S, Su Y, Guo JU, Huang L, et al. Lin28A Binds Active Promoters and Recruits Tet1 to Regulate Gene Expression. Mol Cell. 2016;61:153–160. doi: 10.1016/j.molcel.2015.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Frost RJ, Olson EN. Control of glucose homeostasis and insulin sensitivity by the Let-7 family of microRNAs. Proc Natl Acad Sci U S A. 2011;108:21075–21080. doi: 10.1073/pnas.1118922109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Zhang J, Ratanasirintrawoot S, Chandrasekaran S, Wu Z, Ficarro SB, Yu C, Ross CA, Cacchiarelli D, Xia Q, Seligson M, et al. LIN28 Regulates Stem Cell Metabolism and Conversion to Primed Pluripotency. Cell Stem Cell. 2016;19:66–80. doi: 10.1016/j.stem.2016.05.009. [DOI] [PubMed] [Google Scholar]
- 123.Zhu H, Shyh-Chang N, Segre AV, Shinoda G, Shah SP, Einhorn WS, Takeuchi A, Engreitz JM, Hagan JP, Kharas MG, et al. The Lin28/let-7 axis regulates glucose metabolism. Cell. 2011;147:81–94. doi: 10.1016/j.cell.2011.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Lei XX, Xu J, Ma W, Qiao C, Newman MA, Hammond SM, Huang Y. Determinants of mRNA recognition and translation regulation by Lin28. Nucleic Acids Res. 2012;40:3574–3584. doi: 10.1093/nar/gkr1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Foti D, Chiefari E, Fedele M, Iuliano R, Brunetti L, Paonessa F, Manfioletti G, Barbetti F, Brunetti A, Croce CM, et al. Lack of the architectural factor HMGA1 causes insulin resistance and diabetes in humans and mice. Nat Med. 2005;11:765–773. doi: 10.1038/nm1254. [DOI] [PubMed] [Google Scholar]
- 126.Docherty CK, Salt IP, Mercer JR. Lin28A induces energetic switching to glycolytic metabolism in human embryonic kidney cells. Stem Cell Res Ther. 2016;7:78. doi: 10.1186/s13287-016-0323-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Ma X, Li C, Sun L, Huang D, Li T, He X, Wu G, Yang Z, Zhong X, Song L, et al. Lin28/let-7 axis regulates aerobic glycolysis and cancer progression via PDK1. Nat Commun. 2014;5:5212. doi: 10.1038/ncomms6212. [DOI] [PubMed] [Google Scholar]
- 128.Singh RK, Xia Z, Bland CS, Kalsotra A, Scavuzzo MA, Curk T, Ule J, Li W, Cooper TA. Rbfox2-coordinated alternative splicing of Mef2d and Rock2 controls myoblast fusion during myogenesis. Mol Cell. 2014;55:592–603. doi: 10.1016/j.molcel.2014.06.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Wei C, Xiao R, Chen L, Cui H, Zhou Y, Xue Y, Hu J, Zhou B, Tsutsui T, Qiu J, et al. RBFox2 Binds Nascent RNA to Globally Regulate Polycomb Complex 2 Targeting in Mammalian Genomes. Mol Cell. 2016;62:875–889. doi: 10.1016/j.molcel.2016.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Wei C, Qiu J, Zhou Y, Xue Y, Hu J, Ouyang K, Banerjee I, Zhang C, Chen B, Li H, et al. Repression of the Central Splicing Regulator RBFox2 Is Functionally Linked to Pressure Overload-Induced Heart Failure. Cell Rep. 2015 doi: 10.1016/j.celrep.2015.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Weyn-Vanhentenryck SM, Mele A, Yan Q, Sun S, Farny N, Zhang Z, Xue C, Herre M, Silver PA, Zhang MQ, et al. HITS-CLIP and integrative modeling define the Rbfox splicing-regulatory network linked to brain development and autism. Cell Rep. 2014;6:1139–1152. doi: 10.1016/j.celrep.2014.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Yeo GW, Coufal NG, Liang TY, Peng GE, Fu XD, Gage FH. An RNA code for the FOX2 splicing regulator revealed by mapping RNA-protein interactions in stem cells. Nat Struct Mol Biol. 2009;16:130–137. doi: 10.1038/nsmb.1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Jangi M, Boutz PL, Paul P, Sharp PA. Rbfox2 controls autoregulation in RNA-binding protein networks. Genes Dev. 2014;28:637–651. doi: 10.1101/gad.235770.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Lovci MT, Ghanem D, Marr H, Arnold J, Gee S, Parra M, Liang TY, Stark TJ, Gehman LT, Hoon S, et al. Rbfox proteins regulate alternative mRNA splicing through evolutionarily conserved RNA bridges. Nat Struct Mol Biol. 2013;20:1434–1442. doi: 10.1038/nsmb.2699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Gehman LT, Meera P, Stoilov P, Shiue L, O’Brien JE, Meisler MH, Ares M, Jr, Otis TS, Black DL. The splicing regulator Rbfox2 is required for both cerebellar development and mature motor function. Genes Dev. 2012;26:445–460. doi: 10.1101/gad.182477.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Pedrotti S, Giudice J, Dagnino-Acosta A, Knoblauch M, Singh RK, Hanna A, Mo Q, Hicks J, Hamilton S, Cooper TA. The RNA-binding protein Rbfox1 regulates splicing required for skeletal muscle structure and function. Hum Mol Genet. 2015;24:2360–2374. doi: 10.1093/hmg/ddv003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Gallagher TL, Arribere JA, Geurts PA, Exner CRT, McDonald KL, Dill KK, Marr HL, Adkar SS, Garnett AT, Amacher SL, et al. Rbfox-regulated alternative splicing is critical for zebrafish cardiac and skeletal muscle functions. Developmental Biology. 2011;359:251–261. doi: 10.1016/j.ydbio.2011.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Homsy J, Zaidi S, Shen Y, Ware JS, Samocha KE, Karczewski KJ, DePalma SR, McKean D, Wakimoto H, Gorham J, et al. De novo mutations in congenital heart disease with neurodevelopmental and other congenital anomalies. Science. 2015;350:1262–1266. doi: 10.1126/science.aac9396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Chen Y, Zubovic L, Yang F, Godin K, Pavelitz T, Castellanos J, Macchi P, Varani G. Rbfox proteins regulate microRNA biogenesis by sequence-specific binding to their precursors and target downstream Dicer. Nucleic Acids Res. 2016;44:4381–4395. doi: 10.1093/nar/gkw177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Geraldes P, King GL. Activation of protein kinase C isoforms and its impact on diabetic complications. Circ Res. 2010;106:1319–1331. doi: 10.1161/CIRCRESAHA.110.217117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Inoguchi T, Battan R, Handler E, Sportsman JR, Heath W, King GL. Preferential elevation of protein kinase C isoform beta II and diacylglycerol levels in the aorta and heart of diabetic rats: differential reversibility to glycemic control by islet cell transplantation. Proc Natl Acad Sci U S A. 1992;89:11059–11063. doi: 10.1073/pnas.89.22.11059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Liu Y, Lei S, Gao X, Mao X, Wang T, Wong GT, Vanhoutte PM, Irwin MG, Xia Z. PKC beta inhibition with ruboxistaurin reduces oxidative stress and attenuates left ventricular hypertrophy and dysfuntion in rats with streptozotocin-induced diabetes. Clinical Science. 2012;122:161–173. doi: 10.1042/CS20110176. [DOI] [PubMed] [Google Scholar]
- 143.Nakahata S, Kawamoto S. Tissue-dependent isoforms of mammalian Fox-1 homologs are associated with tissue-specific splicing activities. Nucleic Acids Res. 2005;33:2078–2089. doi: 10.1093/nar/gki338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Damianov A, Black DL. Autoregulation of Fox protein expression to produce dominant negative splicing factors. RNA. 2010;16:405–416. doi: 10.1261/rna.1838210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Mauger DM, Lin C, Garcia-Blanco MA. hnRNP H and hnRNP F complex with Fox2 to silence fibroblast growth factor receptor 2 exon IIIc. Mol Cell Biol. 2008;28:5403–5419. doi: 10.1128/MCB.00739-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Shapiro IM, Cheng AW, Flytzanis NC, Balsamo M, Condeelis JS, Oktay MH, Burge CB, Gertler FB. An EMT-driven alternative splicing program occurs in human breast cancer and modulates cellular phenotype. PLoS Genet. 2011;7:e1002218. doi: 10.1371/journal.pgen.1002218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Xiang Y, Laurent B, Hsu CH, Nachtergaele S, Lu Z, Sheng W, Xu C, Chen H, Ouyang J, Wang S, et al. RNA m6A methylation regulates the ultraviolet-induced DNA damage response. Nature. 2017;543:573–576. doi: 10.1038/nature21671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Zhao X, Yang Y, Sun BF, Shi Y, Yang X, Xiao W, Hao YJ, Ping XL, Chen YS, Wang WJ, et al. FTO-dependent demethylation of N6-methyladenosine regulates mRNA splicing and is required for adipogenesis. Cell Res. 2014;24:1403–1419. doi: 10.1038/cr.2014.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Wang X, Huang N, Yang M, Wei D, Tai H, Han X, Gong H, Zhou J, Qin J, Wei X, et al. FTO is required for myogenesis by positively regulating mTOR-PGC-1alpha pathway-mediated mitochondria biogenesis. Cell Death Dis. 2017;8:e2702. doi: 10.1038/cddis.2017.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Li L, Zang L, Zhang F, Chen J, Shen H, Shu L, Liang F, Feng C, Chen D, Tao H, et al. Fat mass and obesity-associated (FTO) protein regulates adult neurogenesis. Hum Mol Genet. 2017 doi: 10.1093/hmg/ddx128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Dai N, Rapley J, Angel M, Yanik MF, Blower MD, Avruch J. mTOR phosphorylates IMP2 to promote IGF2 mRNA translation by internal ribosomal entry. Genes Dev. 2011;25:1159–1172. doi: 10.1101/gad.2042311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Li Z, Gilbert JA, Zhang Y, Zhang M, Qiu Q, Ramanujan K, Shavlakadze T, Eash JK, Scaramozza A, Goddeeris MM, et al. An HMGA2-IGF2BP2 axis regulates myoblast proliferation and myogenesis. Dev Cell. 2012;23:1176–1188. doi: 10.1016/j.devcel.2012.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Morita M, Gravel SP, Chenard V, Sikstrom K, Zheng L, Alain T, Gandin V, Avizonis D, Arguello M, Zakaria C, et al. mTORC1 controls mitochondrial activity and biogenesis through 4E-BP-dependent translational regulation. Cell Metab. 2013;18:698–711. doi: 10.1016/j.cmet.2013.10.001. [DOI] [PubMed] [Google Scholar]
- 154.Zhang D, Contu R, Latronico MV, Zhang J, Rizzi R, Catalucci D, Miyamoto S, Huang K, Ceci M, Gu Y, et al. MTORC1 regulates cardiac function and myocyte survival through 4E-BP1 inhibition in mice. J Clin Invest. 2010;120:2805–2816. doi: 10.1172/JCI43008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Bhat M, Yanagiya A, Graber T, Razumilava N, Bronk S, Zammit D, Zhao Y, Zakaria C, Metrakos P, Pollak M, et al. Metformin requires 4E-BPs to induce apoptosis and repress translation of Mcl-1 in hepatocellular carcinoma cells. Oncotarget. 2016 doi: 10.18632/oncotarget.10671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Gantois I, Khoutorsky A, Popic J, Aguilar-Valles A, Freemantle E, Cao R, Sharma V, Pooters T, Nagpal A, Skalecka A, et al. Metformin ameliorates core deficits in a mouse model of fragile X syndrome. Nat Med. 2017;23:674–677. doi: 10.1038/nm.4335. [DOI] [PubMed] [Google Scholar]
- 157.Pyronnet S, Imataka H, Gingras AC, Fukunaga R, Hunter T, Sonenberg N. Human eukaryotic translation initiation factor 4G (eIF4G) recruits mnk1 to phosphorylate eIF4E. EMBO J. 1999;18:270–279. doi: 10.1093/emboj/18.1.270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Tsai S, Sitzmann JM, Dastidar SG, Rodriguez AA, Vu SL, McDonald CE, Academia EC, O’Leary MN, Ashe TD, La Spada AR, et al. Muscle-specific 4E-BP1 signaling activation improves metabolic parameters during aging and obesity. J Clin Invest. 2015;125:2952–2964. doi: 10.1172/JCI77361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Blandino-Rosano M, Scheys JO, Jimenez-Palomares M, Barbaresso R, Bender AS, Yanagiya A, Liu M, Rui L, Sonenberg N, Bernal-Mizrachi E. 4E-BP2/SH2B1/IRS2 Are Part of a Novel Feedback Loop That Controls beta-Cell Mass. Diabetes. 2016;65:2235–2248. doi: 10.2337/db15-1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Robichaud N, del Rincon SV, Huor B, Alain T, Petruccelli LA, Hearnden J, Goncalves C, Grotegut S, Spruck CH, Furic L, et al. Phosphorylation of eIF4E promotes EMT and metastasis via translational control of SNAIL and MMP-3. Oncogene. 2015;34:2032–2042. doi: 10.1038/onc.2014.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Shi Y, Taylor SI, Tan SL, Sonenberg N. When translation meets metabolism: multiple links to diabetes. Endocr Rev. 2003;24:91–101. doi: 10.1210/er.2002-0018. [DOI] [PubMed] [Google Scholar]
- 162.Timchenko LT, Miller JW, Timchenko NA, DeVore DR, Datar KV, Lin L, Roberts R, Caskey CT, Swanson MS. Identification of a (CUG)n triplet repeat RNA-binding protein and its expression in myotonic dystrophy. Nucleic Acids Res. 1996;24:4407–4414. doi: 10.1093/nar/24.22.4407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Chaudhury A, Cheema S, Fachini JM, Kongchan N, Lu G, Simon LM, Wang T, Mao S, Rosen DG, Ittmann MM, et al. CELF1 is a central node in post-transcriptional regulatory programmes underlying EMT. Nat Commun. 2016;7:13362. doi: 10.1038/ncomms13362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Vlasova IA, Tahoe NM, Fan D, Larsson O, Rattenbacher B, Sternjohn JR, Vasdewani J, Karypis G, Reilly CS, Bitterman PB, et al. Conserved GU-rich elements mediate mRNA decay by binding to CUG-binding protein 1. Mol Cell. 2008;29:263–270. doi: 10.1016/j.molcel.2007.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Edwards J, Malaurie E, Kondrashov A, Long J, de Moor CH, Searle MS, Emsley J. Sequence determinants for the tandem recognition of UGU and CUG rich RNA elements by the two N--terminal RRMs of CELF1. Nucleic Acids Res. 2011;39:8638–8650. doi: 10.1093/nar/gkr510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Lambert N, Robertson A, Jangi M, McGeary S, Sharp PA, Burge CB. RNA Bind-n-Seq: quantitative assessment of the sequence and structural binding specificity of RNA binding proteins. Mol Cell. 2014;54:887–900. doi: 10.1016/j.molcel.2014.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Wang ET, Ward AJ, Cherone JM, Giudice J, Wang TT, Treacy DJ, Lambert NJ, Freese P, Saxena T, Cooper TA, et al. Antagonistic regulation of mRNA expression and splicing by CELF and MBNL proteins. Genome Res. 2015;25:858–871. doi: 10.1101/gr.184390.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Timchenko LT, Iakova P, Welm AL, Cai ZJ, Timchenko NA. Calreticulin interacts with C/EBPalpha and C/EBPbeta mRNAs and represses translation of C/EBP proteins. Mol Cell Biol. 2002;22:7242–7257. doi: 10.1128/MCB.22.20.7242-7257.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Timchenko NA, Welm AL, Lu X, Timchenko LT. CUG repeat binding protein (CUGBP1) interacts with the 5′ region of C/EBPbeta mRNA and regulates translation of C/EBPbeta isoforms. Nucleic Acids Res. 1999;27:4517–4525. doi: 10.1093/nar/27.22.4517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Moraes KC, Wilusz CJ, Wilusz J. CUG-BP binds to RNA substrates and recruits PARN deadenylase. RNA. 2006;12:1084–1091. doi: 10.1261/rna.59606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Philips AV, Timchenko LT, Cooper TA. Disruption of splicing regulated by a CUG-binding protein in myotonic dystrophy. Science. 1998;280:737–741. doi: 10.1126/science.280.5364.737. [DOI] [PubMed] [Google Scholar]
- 172.Dasgupta T, Ladd AN. The importance of CELF control: molecular and biological roles of the CUG-BP, Elav-like family of RNA-binding proteins. Wiley Interdiscip Rev RNA. 2012;3:104–121. doi: 10.1002/wrna.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Zhang L, Lee JE, Wilusz J, Wilusz CJ. The RNA-binding protein CUGBP1 regulates stability of tumor necrosis factor mRNA in muscle cells: implications for myotonic dystrophy. J Biol Chem. 2008;283:22457–22463. doi: 10.1074/jbc.M802803200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Ward AJ, Rimer M, Killian JM, Dowling JJ, Cooper TA. CUGBP1 overexpression in mouse skeletal muscle reproduces features of myotonic dystrophy type 1. Hum Mol Genet. 2010;19:3614–3622. doi: 10.1093/hmg/ddq277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Koshelev M, Sarma S, Price RE, Wehrens XHT, Cooper TA. Heart-specific overexpression of CUGBP1 reproduces functional and molecular abnormalities of myotonic dystrophy type 1. Human Molecular Genetics. 2010;19:1066–1075. doi: 10.1093/hmg/ddp570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Wang GS, Kearney DL, De Biasi M, Taffet G, Cooper TA. Elevation of RNA-binding protein CUGBP1 is an early event in an inducible heart-specific mouse model of myotonic dystrophy. J Clin Invest. 2007;117:2802–2811. doi: 10.1172/JCI32308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Timchenko NA, Patel R, Iakova P, Cai ZJ, Quan L, Timchenko LT. Overexpression of CUG triplet repeat-binding protein, CUGBP1, in mice inhibits myogenesis. J Biol Chem. 2004;279:13129–13139. doi: 10.1074/jbc.M312923200. [DOI] [PubMed] [Google Scholar]
- 178.Kress C, Gautier-Courteille C, Osborne HB, Babinet C, Paillard L. Inactivation of CUG-BP1/CELF1 causes growth, viability, and spermatogenesis defects in mice. Mol Cell Biol. 2007;27:1146–1157. doi: 10.1128/MCB.01009-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Giudice J, Xia Z, Wang ET, Scavuzzo MA, Ward AJ, Kalsotra A, Wang W, Wehrens XH, Burge CB, Li W, et al. Alternative splicing regulates vesicular trafficking genes in cardiomyocytes during postnatal heart development. Nat Commun. 2014;5:3603. doi: 10.1038/ncomms4603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Savkur RS, Philips AV, Cooper TA. Aberrant regulation of insulin receptor alternative splicing is associated with insulin resistance in myotonic dystrophy. Nat Genet. 2001;29:40–47. doi: 10.1038/ng704. [DOI] [PubMed] [Google Scholar]
- 181.Paul S, Dansithong W, Kim D, Rossi J, Webster NJ, Comai L, Reddy S. Interaction of muscleblind, CUG-BP1 and hnRNP H proteins in DM1-associated aberrant IR splicing. EMBO J. 2006;25:4271–4283. doi: 10.1038/sj.emboj.7601296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Gao Z, Cooper TA. Reexpression of pyruvate kinase M2 in type 1 myofibers correlates with altered glucose metabolism in myotonic dystrophy. Proc Natl Acad Sci U S A. 2013;110:13570–13575. doi: 10.1073/pnas.1308806110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Karagiannides I, Thomou T, Tchkonia T, Pirtskhalava T, Kypreos KE, Cartwright A, Dalagiorgou G, Lash TL, Farmer SR, Timchenko NA, et al. Increased CUG triplet repeat-binding protein-1 predisposes to impaired adipogenesis with aging. J Biol Chem. 2006;281:23025–23033. doi: 10.1074/jbc.M513187200. [DOI] [PubMed] [Google Scholar]
- 184.Kuyumcu-Martinez NM, Wang G-S, Cooper TA. Increased steady-state in levels of CUGBP1 in myotonic dystrophy 1 are due to PKC-mediated hyperphosphorylation. Molecular Cell. 2007;28:68–78. doi: 10.1016/j.molcel.2007.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Chettouh H, Fartoux L, Aoudjehane L, Wendum D, Claperon A, Chretien Y, Rey C, Scatton O, Soubrane O, Conti F, et al. Mitogenic insulin receptor-A is overexpressed in human hepatocellular carcinoma due to EGFR-mediated dysregulation of RNA splicing factors. Cancer Res. 2013;73:3974–3986. doi: 10.1158/0008-5472.CAN-12-3824. [DOI] [PubMed] [Google Scholar]
- 186.Peng SS, Chen CY, Xu N, Shyu AB. RNA stabilization by the AU-rich element binding protein, HuR, an ELAV protein. EMBO J. 1998;17:3461–3470. doi: 10.1093/emboj/17.12.3461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Fan XC, Steitz JA. Overexpression of HuR, a nuclear-cytoplasmic shuttling protein, increases the in vivo stability of ARE-containing mRNAs. EMBO J. 1998;17:3448–3460. doi: 10.1093/emboj/17.12.3448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Mukherjee N, Corcoran DL, Nusbaum JD, Reid DW, Georgiev S, Hafner M, Ascano M, Jr, Tuschl T, Ohler U, Keene JD. Integrative regulatory mapping indicates that the RNA-binding protein HuR couples pre-mRNA processing and mRNA stability. Mol Cell. 2011;43:327–339. doi: 10.1016/j.molcel.2011.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Govindaraju S, Lee BS. Adaptive and maladaptive expression of the mRNA regulatory protein HuR. World J Biol Chem. 2013;4:111–118. doi: 10.4331/wjbc.v4.i4.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Doller A, Schlepckow K, Schwalbe H, Pfeilschifter J, Eberhardt W. Tandem phosphorylation of serines 221 and 318 by protein kinase Cdelta coordinates mRNA binding and nucleocytoplasmic shuttling of HuR. Mol Cell Biol. 2010;30:1397–1410. doi: 10.1128/MCB.01373-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Lopez de Silanes I, Zhan M, Lal A, Yang X, Gorospe M. Identification of a target RNA motif for RNA-binding protein HuR. Proc Natl Acad Sci U S A. 2004;101:2987–2992. doi: 10.1073/pnas.0306453101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Ghosh M, Aguila HL, Michaud J, Ai Y, Wu MT, Hemmes A, Ristimaki A, Guo C, Furneaux H, Hla T. Essential role of the RNA-binding protein HuR in progenitor cell survival in mice. J Clin Invest. 2009;119:3530–3543. doi: 10.1172/JCI38263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Krishnamurthy P, Lambers E, Verma S, Thorne T, Qin G, Losordo DW, Kishore R. Myocardial knockdown of mRNA-stabilizing protein HuR attenuates post-MI inflammatory response and left ventricular dysfunction in IL-10-null mice. FASEB J. 2010;24:2484–2494. doi: 10.1096/fj.09-149815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Rhee WJ, Ni CW, Zheng Z, Chang K, Jo H, Bao G. HuR regulates the expression of stress-sensitive genes and mediates inflammatory response in human umbilical vein endothelial cells. Proc Natl Acad Sci U S A. 2010;107:6858–6863. doi: 10.1073/pnas.1000444107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Wang Y, Li Y, Toth JI, Petroski MD, Zhang Z, Zhao JC. N6-methyladenosine modification destabilizes developmental regulators in embryonic stem cells. Nat Cell Biol. 2014;16:191–198. doi: 10.1038/ncb2902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Abdelmohsen K, Pullmann R, Jr, Lal A, Kim HH, Galban S, Yang X, Blethrow JD, Walker M, Shubert J, Gillespie DA, et al. Phosphorylation of HuR by Chk2 regulates SIRT1 expression. Mol Cell. 2007;25:543–557. doi: 10.1016/j.molcel.2007.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Barnhart MD, Moon SL, Emch AW, Wilusz CJ, Wilusz J. Changes in cellular mRNA stability, splicing, and polyadenylation through HuR protein sequestration by a cytoplasmic RNA virus. Cell Rep. 2013;5:909–917. doi: 10.1016/j.celrep.2013.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Diaz-Munoz MD, Bell SE, Fairfax K, Monzon-Casanova E, Cunningham AF, Gonzalez-Porta M, Andrews SR, Bunik VI, Zarnack K, Curk T, et al. The RNA-binding protein HuR is essential for the B cell antibody response. Nat Immunol. 2015;16:415–425. doi: 10.1038/ni.3115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Li X, Zeng L, Cao C, Lu C, Lian W, Han J, Zhang X, Zhang J, Tang T, Li M. Long noncoding RNA MALAT1 regulates renal tubular epithelial pyroptosis by modulated miR-23c targeting of ELAVL1 in diabetic nephropathy. Exp Cell Res. 2017;350:327–335. doi: 10.1016/j.yexcr.2016.12.006. [DOI] [PubMed] [Google Scholar]
- 200.Bishop KM. Progress and promise of antisense oligonucleotide therapeutics for central nervous system diseases. Neuropharmacology. 2016 doi: 10.1016/j.neuropharm.2016.12.015. [DOI] [PubMed] [Google Scholar]
- 201.Lundin KE, Gissberg O, Smith CI. Oligonucleotide Therapies: The Past and the Present. Hum Gene Ther. 2015;26:475–485. doi: 10.1089/hum.2015.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Chi X, Gatti P, Papoian T. Safety of antisense oligonucleotide and siRNA-based therapeutics. Drug Discov Today. 2017 doi: 10.1016/j.drudis.2017.01.013. [DOI] [PubMed] [Google Scholar]
- 203.van Meer L, Moerland M, Gallagher J, van Doorn MB, Prens EP, Cohen AF, Rissmann R, Burggraaf J. Injection site reactions after subcutaneous oligonucleotide therapy. Br J Clin Pharmacol. 2016;82:340–351. doi: 10.1111/bcp.12961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Lefebvre S, Burglen L, Reboullet S, Clermont O, Burlet P, Viollet L, Benichou B, Cruaud C, Millasseau P, Zeviani M, et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cell. 1995;80:155–165. doi: 10.1016/0092-8674(95)90460-3. [DOI] [PubMed] [Google Scholar]
- 205.Lunn MR, Wang CH. Spinal muscular atrophy. Lancet. 2008;371:2120–2133. doi: 10.1016/S0140-6736(08)60921-6. [DOI] [PubMed] [Google Scholar]
- 206.Russman BS. Spinal muscular atrophy: clinical classification and disease heterogeneity. J Child Neurol. 2007;22:946–951. doi: 10.1177/0883073807305673. [DOI] [PubMed] [Google Scholar]
- 207.Finkel RS, Chiriboga CA, Vajsar J, Day JW, Montes J, De Vivo DC, Yamashita M, Rigo F, Hung G, Schneider E, et al. Treatment of infantile-onset spinal muscular atrophy with nusinersen: a phase 2, open-label, dose-escalation study. Lancet. 2016;388:3017–3026. doi: 10.1016/S0140-6736(16)31408-8. [DOI] [PubMed] [Google Scholar]
- 208.Corey DR. Nusinersen, an antisense oligonucleotide drug for spinal muscular atrophy. Nat Neurosci. 2017;20:497–499. doi: 10.1038/nn.4508. [DOI] [PubMed] [Google Scholar]
- 209.Chiriboga CA, Swoboda KJ, Darras BT, Iannaccone ST, Montes J, De Vivo DC, Norris DA, Bennett CF, Bishop KM. Results from a phase 1 study of nusinersen (ISIS-SMN(Rx)) in children with spinal muscular atrophy. Neurology. 2016;86:890–897. doi: 10.1212/WNL.0000000000002445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Cartegni L, Krainer AR. Disruption of an SF2/ASF-dependent exonic splicing enhancer in SMN2 causes spinal muscular atrophy in the absence of SMN1. Nat Genet. 2002;30:377–384. doi: 10.1038/ng854. [DOI] [PubMed] [Google Scholar]
- 211.Havens MA, Hastings ML. Splice-switching antisense oligonucleotides as therapeutic drugs. Nucleic Acids Res. 2016;44:6549–6563. doi: 10.1093/nar/gkw533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Lim KR, Maruyama R, Yokota T. Eteplirsen in the treatment of Duchenne muscular dystrophy. Drug Des Devel Ther. 2017;11:533–545. doi: 10.2147/DDDT.S97635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.van Dongen MG, Geerts BF, Morgan ES, Brandt TA, de Kam ML, Romijn JA, Cohen AF, Bhanot S, Burggraaf J. First proof of pharmacology in humans of a novel glucagon receptor antisense drug. J Clin Pharmacol. 2015;55:298–306. doi: 10.1002/jcph.396. [DOI] [PubMed] [Google Scholar]
- 214.Digenio A, Dunbar RL, Alexander VJ, Hompesch M, Morrow L, Lee RG, Graham MJ, Hughes SG, Yu R, Singleton W, et al. Antisense-Mediated Lowering of Plasma Apolipoprotein C-III by Volanesorsen Improves Dyslipidemia and Insulin Sensitivity in Type 2 Diabetes. Diabetes Care. 2016;39:1408–1415. doi: 10.2337/dc16-0126. [DOI] [PubMed] [Google Scholar]
- 215.Dowdy SF. Overcoming cellular barriers for RNA therapeutics. Nat Biotechnol. 2017;35:222–229. doi: 10.1038/nbt.3802. [DOI] [PubMed] [Google Scholar]
- 216.Huang Y. Preclinical and Clinical Advances of GalNAc-Decorated Nucleic Acid Therapeutics. Mol Ther Nucleic Acids. 2017;6:116–132. doi: 10.1016/j.omtn.2016.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Viney NJ, van Capelleveen JC, Geary RS, Xia S, Tami JA, Yu RZ, Marcovina SM, Hughes SG, Graham MJ, Crooke RM, et al. Antisense oligonucleotides targeting apolipoprotein(a) in people with raised lipoprotein(a): two randomised, double-blind, placebo-controlled, dose-ranging trials. Lancet. 2016;388:2239–2253. doi: 10.1016/S0140-6736(16)31009-1. [DOI] [PubMed] [Google Scholar]