

Abstract
Post-translational modifications (PTMs) play important roles in regulating a variety of biological processes. To facilitate PTM studies, the genetic code expansion strategy has been utilized to co-translationally incorporate individual PTMs such as acetylation and phosphorylation into proteins at specific sites. However, recent studies have demonstrated that PTMs actually work together to regulate protein functions and structures. Thus, simultaneous incorporation of multiple distinct PTMs into one protein is highly desirable. In this study, we utilized the genetic incorporation systems of phosphoserine and acetyllysine to install both phosphorylation and acetylation into target proteins simultaneously in Escherichia coli. And we used this system to study the effect of coexisting acetylation and phosphorylation on malate dehydrogenase, demonstrating a practical application of this system in biochemical studies. Furthermore, we tested the mutual orthogonality of three widely-used genetic incorporation systems, indicating the possibility of incorporating three distinct PTMs into one protein simultaneously.
Keywords: genetic code expansion, post-translational modification, phosphorylation, acetylation, noncanonical amino acid
Table of Contents Use Only
Post-translational modifications (PTMs) play key roles in a wide range of biological processes across three domains of life.1 Proteomic studies have identified hundreds of types of PTMs in nature, such as phosphorylation, acetylation, glycosylation, amidation, hydroxylation, methylation, ubiquitylation, and sulfation.2, 3 However, limited studies have been reported to confirm and further characterize these mass spectrometry (MS)-identified PTM events. One challenge is to generate homogenously modified proteins at specific sites. For this purpose, the genetic code expansion strategy has been applied to site-specifically incorporate a number of PTMs into proteins,4 including lysine acetylation5–7 and serine phosphorylation.8–11 Different from PTMs which occur after translation, the genetic code expansion approach is able to co-translationally incorporate modified amino acids directly to produce homogeneously modified proteins at desired positions without knowing specific modifying enzymes such as kinases or acetyltransferases.
Although the genetic code expansion approach provide powerful tools to study biology,12–16 previous studies could only incorporate one type of PTM into proteins individually. However, it is known now that proteins commonly have multiple PTMs simultaneously, and different types of PTMs interact with each other to affect protein structures, functions, and interactions.17, 18 Thus, it is highly desirable to develop genetic incorporation systems to install multiple distinct PTMs in one protein simultaneously.
Scientists have been able to incorporate two distinct noncanonical amino acids (ncAAs) into one protein at different positions by using mutually orthogonal translation systems (OTSs), towards two different stop codons19–24 or one stop codon together with one four-base codon.25–27 In this study, we expanded this strategy to simultaneously incorporate two of the most abundant PTMs in nature,28 phosphoserine (Sep) and acetyllysine (AcK), into target proteins by using E. coli cells. We also demonstrated a practical application of this dual-PTM incorporation system in biochemical characterization of enzymes. Furthermore, we demonstrated that three widely used OTSs in E. coil: the Methanocaldococcus jannaschii tyrosyl-tRNA synthetase (mjTyrRS) system, the Methanosarcina species pyrrolysyl-tRNA synthetase (PylRS) system, and the Methanococcus maripaludis phosphoseryl-tRNA synthetase (SepRS) system are mutually orthogonal to each other, providing a solid base for installing three distinct PTMs simultaneously into one protein.
Results and Discussion
Codon selection
Three stop codons UAG, UGA, and UAA, as well as several four-base codons like AGGA have been used as signals to incorporate ncAAs by the genetic code expansion strategy.29–32 Thus, we firstly compared the incorporation efficiency and accuracy of different codons by the super-folder green fluorescent protein (sfGFP) reporter system (Figure 1). Because of the low selectivity of PylRS toward the tRNAPyl anticodon,33, 34 we used the recently optimized AcKRS which was derived from PylRS by phage-assisted protein evolution35 and the optimized tRNAPyl for better EF-Tu binding36 as the testing OTS towards UAG, UGA, UAA, and AGGA codons, individually.
Figure 1. The sfGFP readthrough assay towards various codons at different positions.
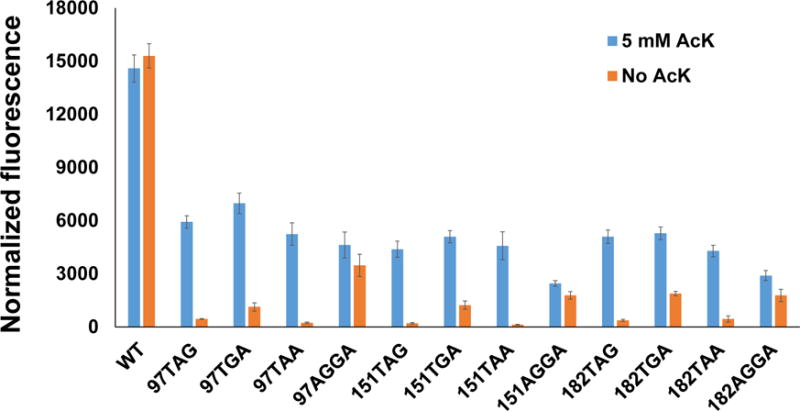
Normalized fluorescence intensities were calculated from fluorescence readings at 12 h divided by absorbance at 600 nm. Mean values and standard deviations were calculated from three replicates.
It is known that ncAA incorporation efficiency varies at different positions even in the same protein.37 So we chose three different permissive sites in sfGFP (97, 151, and 182) for this experiment.37 The corresponding position in the gene of sfGFP was mutated to TAG, TGA, TAA, or AGGA, individually. The anticodon of tRNAPyl was correspondingly mutated to CUA, UCA, UUA, or UCCU, respectively. Here, we did not introduce the orthogonal ribosome for the quadruplet reading, though it could enhance ncAA incorporation.32 Firstly, the suppressor tRNA needs to be optimized for individual codons.26 And the efficiency of frameshift suppression might be another concern.20
The sfGFP readthrough assay showed that UGA suppression gave highest readings, but it also had high backgrounds if no AcK was supplemented in the medium, possibly due to near cognate suppression.38, 39 The four-base codon AGGA had the lowest efficiency and the highest backgrounds if no AcK added in the medium which might be caused by AGG decoding as Arg and then +1 frame shifting,39 So we chose UAG and UAA codons for later experiments which could provide high purity of ncAA incorporation without losing efficiency significantly.
Orthogonality among OTSs
It has been demonstrated that OTSs derived from the mjTyrRS/tRNATyr pair is orthogonal to OTSs engineered from the PylRS/tRNAPyl pair.19, 24 However, the orthogonality of Sep-OTS to these two systems has not been reported before. For this purpose, we used the sfGFP reporter system to test different combinations of AARSs and tRNAs. The permissive position 151 in sfGFP was selected for UAG or UAA codon suppression tests. AcK-OTS was chosen as a representative of the PylRS system, while pTyr-OTS was selected for the mjTyrRS system. tRNAs with CUA or UUA anticodon were tested for UAG or UAA suppression, respectively (Figure 2). Results showed that all the AARSs tested could only recognize their cognate tRNA suppressors. The change of tRNA anticodon from CUA to UUA decreased the efficiencies of both Sep-OTS and pTyr-OTS significantly. This is probably because both SepRS and mjTyrRS bind to anticodon regions of their cognate tRNAs, and they were engineered for optimal binding with the CUA anticodon originally.40, 41 On the other hand, the change of tRNAPyl anticodon did not affect the AcK-OTS efficiency as expected. Thus, we chose the UAG codon as the signal to incorporate Sep and the UAA codon for AcK incorporation in later experiments.
Figure 2. The orthogonality of different OTSs towards UAG or UAA codon.
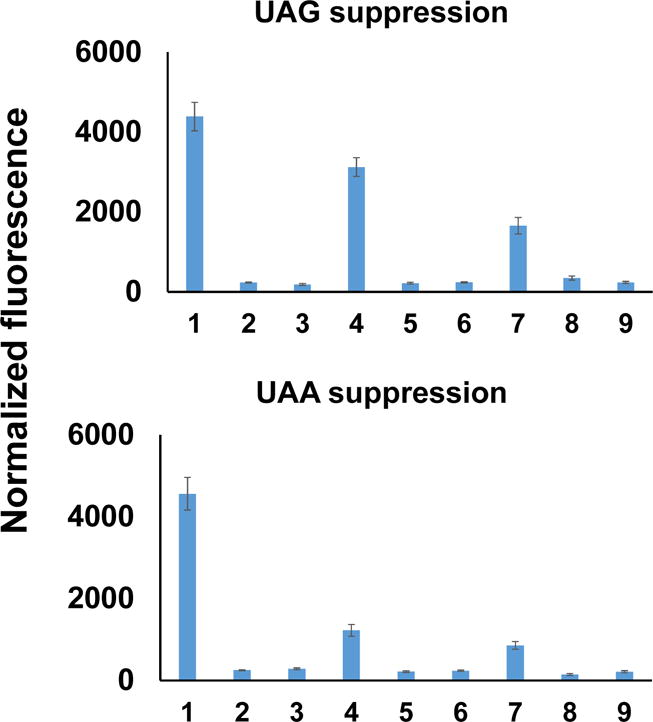
Lane 1: AcKRS/tRNAPyl; Lane 2: AcKRS/tRNATyr; Lane 3: AcKRS/tRNASep; Lane 4: SepRS-EFSep/tRNASep; Lane 5: SepRS-EFSep/tRNAPyl; Lane 6: SepRS-EFSep/tRNATyr; Lane 7: pTyrRS-EFpY/tRNATyr; Lane 8: pTyrRS-EF-pY/tRNAPyl; Lane 9: pTyrRS-EF-pY/tRNASep. The anticodons of tRNAs were mutated to UUA for UAA codon suppression. For ncAAs, 5 mM AcK, 2 mM Sep, or 10 mM pTyr was added in the medium, respectively. Normalized fluorescence intensities were calculated from fluorescence readings at 12 h divided by absorbance at 600 nm. Mean values and standard deviations were calculated from three replicates.
The mutual orthogonality of these three widely used OTSs indicates the possibility of incorporating three different PTMs into one protein at the same time. For example, phosphotyrosine can be incorporated by systems originally from the mjTyrRS system,42, 43 while phosphothreonine has been incorporated by the SepRS system derivative.44 So it is possible to generate a protein with tyrosine phosphorylation, threonine or serine phosphorylation, and lysine acetylation at controlled positions simultaneously. In this case, the UAG codon could be used for phosphoserine or phosphothreonine incorporation by the SepRS system or its derivative. The UAA codon could be used for phosphotyrosine incorporation since the mjTyrRS system has been used to readthrough the UAA codon.24 Due to the flexibility of codon recognition of tRNAPyl, either the UGA codon or a four-base codon could be selected for acetyllysine incorporation.
Simultaneously installing serine phosphorylation and lysine acetylation in the sfGFP
Firstly, we used the sfGFP as a validation reporter. Two permissive positions S2 and K101 were selected to incorporate Sep and AcK, respectively. The serine codon at position 2 was mutated to UAG, and the Sep-OTS was cloned into a pKD plasmid with the pBR322 origin and kanamycin resistance. The lysine codon at position 101 was mutated to UAA. Correspondingly, the anticodon of tRNAPyl was mutated to UUA, and the AcK-OTS was cloned in a pTech plasmid with the p15A origin and chloramphenicol resistance. The gene of sfGFP or its variant was cloned into a pCDF plasmid with the CloDF13 origin and streptomycin resistance. All the three plasmids were co-transformed into BL21(DE3) cells. The expression of sfGFP and its variant was analyzed by SDS-PAGE (Figure 3a). The yield of the sfGFP variant with both Sep and AcK incorporated was 52 mg per liter culture, while the yield of the wild-type sfGFP is 350 mg per liter culture under the same growth condition. If no AcK was supplemented in the medium, there was no detectable full-length sfGFP on the gel. Interestingly, when only AcK but no Sep was supplemented in the medium, there was a clear band of full-length sfGFP on the gel (Lane 4 in Figure 3a). There are three possible reasons for this unexpected result. Firstly, Sep is an intermediate in the serine biosynthesis pathway,45 so SepRS could charge tRNASep with free Sep in cells to read through the UAG codon. Secondly, due to the G:U wobble pairing, tRNAPyl with the UUA anticodon could also suppress the UAG codon partially.46 This was also found in the recent publication which used the cell-free translation system to incorporate two distinct ncAAs.24 Thirdly, the UAG codon could also be recognized by near-cognate tRNAs for the incorporation of canonical amino acids.38, 39 While 2 mM Sep was added to the medium, the expression of the sfGFP variant increased significantly. The simultaneous incorporation of Sep and AcK was confirmed with full-length MS analyses (Figure 3b, S1-2). The difference of molecular weights between the wild-type sfGFP and Sep/AcK-containing sfGFP variant was 122 Da, which equals to the expected value with both phosphorylation (80 Da) and acetylation (42 Da). The positions of Sep and AcK incorporation were confirmed by LC-MS/MS (Figure S3-4). We did not detect the incorporation of AcK in the UAG codon position by both full-length MS and LC-MS/MS, which was consistent with the previous study showing that the UAA suppressor did not readthrough the UAG codon when both ncAAs were supplemented into the medium.24
Figure 3. Expression of Sep/AcK-containing sfGFP.
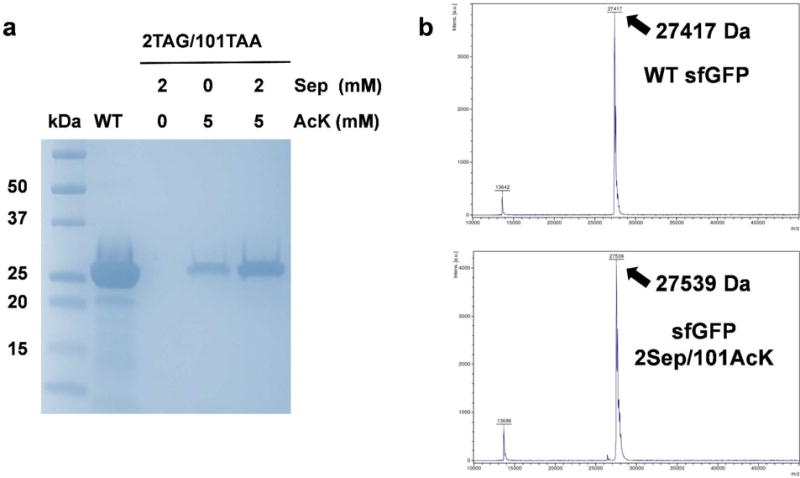
A) The Coomassie blue-stained SDS-PAGE gel of purified full-length sfGFP and its variant. The same volumes of elution fractions were loaded on the SDS-PAGE gel. B) MALDI-TOF mass spectra of purified full-length sfGFP (upper) and its variant with Sep incorporated at position 2 and AcK incorporated at position 101 (lower). Full spectra were listed as Figure S1 and S2.
Simultaneously incorporating AcK and Sep into malate dehydrogenase (MDH)
To demonstrate a practical application of our system in biochemical studies, the MDH in the citrate acid cycle was selected. Previous proteomic studies have shown that E. coli MDH has lysine acetylation at position 140 and serine phosphorylation at position 280.47, 48 By site-specifically incorporating AcK or Sep into MDH individually, we demonstrated that lysine acetylation at position 140 could increase the enzyme activity,49 while serine phosphorylation at position 280 impaired the enzyme activity.50 To explore the overall effect of these two PTMs, we used our dual-PTM incorporation system to produce simultaneously modified MDH. The serine codon at position 280 was mutated to UAG, and the lysine codon at position 140 was mutated to UAA. The same plasmid systems in sfGFP expression were used. The expression of MDH and its variant was analyzed by SDS-PAGE (Figure 4a). The yield of the MDH variant with both Sep and AcK incorporated was 6 mg per liter culture, while the yield of wild-type MDH is 35 mg per liter culture under the same growth condition. Again, if no AcK was supplemented in the medium, there was no detectable full-length MDH. When only AcK was supplemented in the medium, there was a faint band of MDH probably due to endogenous Sep in cells and wobble pairing of the UAG codon with the UUA anticodon in the UAA suppressor. Addition of 2 mM Sep in the medium could increase MDH expression. The simultaneous incorporation of Sep and AcK was confirmed by both western blotting (Figure 4b) and full-length MS analyses (Figure 4c, S5-6). The difference of molecular weights between the wild-type and Sep/AcK-containing MDH was 121 Da, which is close to the expected value with both phosphorylation (80 Da) and acetylation (42 Da). The positions of Sep and AcK incorporation were confirmed by LC-MS/MS (Figure S7-8). Again, we did not detect AcK incorporation at the UAG codon position when both AcK and Sep were added into the medium.
Figure 4. Expression of Sep/AcK-containing MDH.
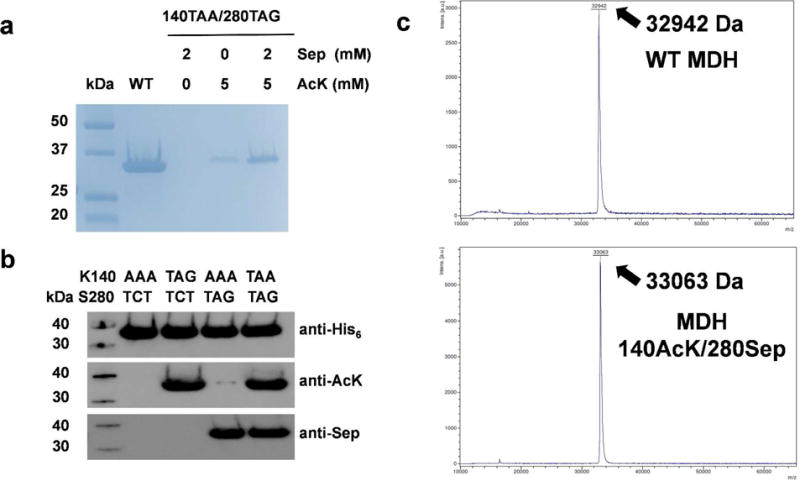
A) The Coomassie blue-stained SDS-PAGE gel of purified full-length MDH and its variant. The same volumes of elution fractions were loaded on the SDS-PAGE gel. B) Western blotting of purified full-length MDH and its variants. The codons used for positions 140 and 280 were listed. The same amount of proteins were loaded on the gels. C) MALDI-TOF mass spectra of purified full-length MDH (upper) and its variant with AcK incorporated at position 140 and Sep incorporated at position 280 (lower). Full spectra were listed as Figure S5 and S6.
In both sfGFP and MDH expression experiments, the yield of doubly modified proteins was about 15% of that of wild-type proteins (Figure 3a and 4a). Generally, the more stop codons were used, the lower efficiency of ncAA incorporation should be observed due to the competition with release factors, and the more mis-incorporation of canonical amino acids might also be detected because of near cognate suppression. Thus, both the protein yield and fidelity would be even lower when incorporating three different PTMs at the same time. To overcome these issues, the plasmid system should be optimized to increase the expression of different OTSs22, individual OTSs should be further engineered for higher efficiencies35, 51, and special strains could be used such as the strain with UAG codon reassignment which has the gene of the release factor-1 removed and all TAG codons in the genome replaced with TAA codons to eliminate the competition for the UAG codon with the release factor-1.51
Studying effects of acetylation and phosphorylation on MDH
Then, we measured the enzyme activities of wild-type, AcK only-containing, Sep only-containing, and AcK/Sep-containing MDHs (Figure 5a). Consistent with our previous studies, lysine acetylation at position 140 increased the enzyme activity by 3.5-fold, while serine phosphorylation at position 280 decreased the enzyme activity to only 30% of that of the wild-type enzyme. Interestingly, the combination of 140-AcK and 280-Sep had a similar activity with the wild-type enzyme, indicating that two PTMs work separately to affect MDH activity. We mapped these two residues onto the crystal structure of the MDH tetramer.52 Both residues are not at the active site but at the dimer-dimer interface (Figure 5B and 5C). From a zoom-in view of amino acids in the interface, K140 and S280 are at one side, and four hydrophobic residues (V189, P190, G191, and V192) are at the other side (Figure 5D). Our enzyme activity assays showed that acetylation (removal of the charge) of K140 increased the activity, while phosphorylation (addition of the charge) of S280 decreased the activity. So we proposed that better hydrophobic interaction in the dimer-dimer interface might increase the MDH activity. To test our assumption, we mutated S280 to alanine, valine, or leucine, separately. We also mutated K140 to glutamine which is traditionally used as a mimic of lysine acetylation. Then we measured activities of these MDH variants (Figure 5A). As expected, the S280V variant facilitated the reaction. However, the S280A variant had only a similar activity with the wild-type MDH which might be due to its smaller side chain, and the steric hindrance of leucine probably caused the impairment. Unexpectedly, the K140Q variant only had a slightly improved activity, indicating that glutamine may not always be an ideal mimic for lysine acetylation, while site-specific incorporation of acetyllysine could provide direct evidence of the acetylation impacts on proteins.
Figure 5. The structure and function of MDH.
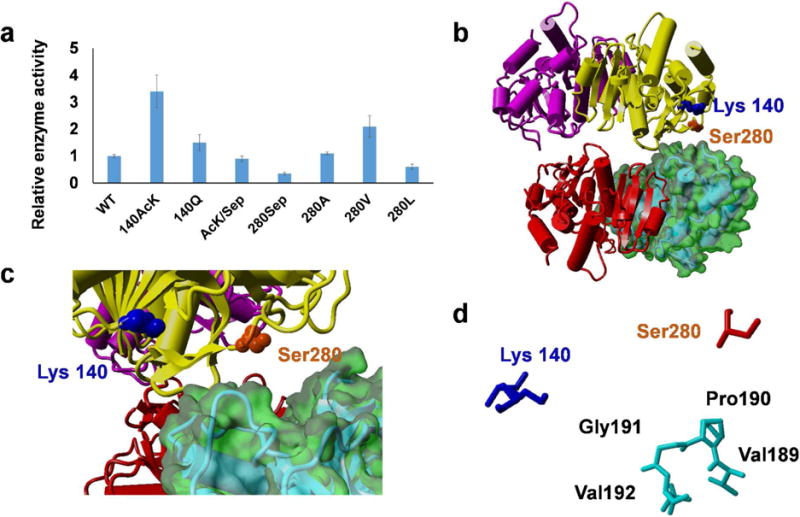
A) The relative enzyme activities of MDH and its variants. Mean values and standard deviations were calculated from three replicates. The enzyme activity of wild-type MDH was set as 1. B) The crystal structure of E. coli MDH tetramer (PDB ID: 3HHP). C) A zoom-in view of the dimer-dimer interface. D) A map of amino acids involved in the dimer-dimer interface.
Methods
Chemicals and materials
Chemicals were purchased from Sigma-Aldrich (St. Louis, MO), BDH Chemicals (Radnor, PA) or CHEM-IMPEX (Wood Dale, IL). Primers were ordered from Integrated DNA Technologies (Coralville, IA). DNA sequencing were performed by Eurofins Scientific (Louisville, KY). Plasmid purification kits were purchased from Qiagen (Hilden, Germany). Cloning enzymes and site-directed mutagenesis kits were purchased from New England Biolabs (Ipswich, MA). Antibodies were purchased from Cell Signaling Technology (Danvers, MA).
Plasmid construction
DH5α cells were used for cloning purposes. The genes of aminoacyl-tRNA synthetases (AARSs) and their cognate tRNAs as well as target proteins were amplified from laboratory inventory, and inserted into corresponding vectors by DNA assembly. Mutations in genes of AARSs, tRNAs, and target proteins were made by site-directed mutagenesis. In the Sep-OTS, sequences of SepRS, EF-Sep, and tRNAsep were from the previous study.40 In the AcK-OTS, the sequence of optimized AcKRS is from a recent study,35 and the optimized tRNAPyl was from our previous study.36 In the phosphotyrosine (pTyr)-OTS, the sequence of pTyrRS was from our recent study,42 and the optimized tRNATyr was from the previous study.53 Strains used in this study are listed in Table S1.
The sfGFP readthrough assay
The protocol was modified slightly from the previous study.54 BL21(DE3) cells were used to express sfGFP and its variants. Strains harboring genes of sfGFP or its variants under the control of the lac promoter in the pCDF plasmid and OTSs in the pTech plasmid (AARSs are constitutively expressed by the lpp promoter, while tRNAs are constitutively expressed by the ProK promoter) were inoculated into 2 mL LB media, and grown at 37 °C overnight. For the pTyr-OTS testing, the specific phosphatase-removal strain and plasmids were used.42 The overnight culture was diluted with fresh LB medium to an absorbance of 0.15 at 600 nm, supplemented with corresponding ncAAs (2 mM Sep, 5 mM AcK, or 10 mM pTyr), 100 μg mL−1 streptomycin, and 50 μg mL−1 chloramphenicol. Then 0.05 mM Isopropyl-β-D-thiogalactoside (IPTG) was added to induce the expression of sfGFP or its variants. 200 μL culture of each strain was transferred to a 96-well clear bottom black microplate. Cells were grown at 37 °C with shaking. Fluorescence intensity (excitation 485 nm, emission 528 nm, bandwidths 20 nm) and optical density (absorbance at 600 nm) were read by the microplate-reader.
Protein expression, purification and characterization
Protocols were modified slightly from the previous study.55 The genes of target proteins were cloned into the pCDF-1b plasmid with a C-terminal His6-tag, and co-transformed into BL21(DE3) cells with corresponding vectors harboring different OTSs. The expression strain was grown in 1 L of LB medium supplemented with 100 μg mL−1 streptomycin, 50 μg mL−1 chloramphenicol, and 50 μg mL−1 kanamycin if needed at 37 °C to an absorbance of 0.6–0.8 at 600 nm. Then 0.05 mM IPTG, 2 mM Sep, 5 mM AcK, and 20 mM nicotinamine (NAM, a deacetylase inhibitor) were supplemented to the medium. Cells were incubated at 25 °C for an additional 16 h, and harvested by centrifugation at 3,000 × g for 15 min at 4 °C. The pellet was suspended in 15 mL lysis buffer (50 mM Na2HPO4, pH 8.0, 300 mM NaCl, 20 mM imidazole, 20 mM NAM, and 150 μL 100X phosphatase inhibitor cocktail [Sigma-Aldrich]), and broken by sonication. The crude extract was centrifuged at 18,000 × g for 20 min at 4 °C. The soluble fraction was loaded onto a column containing 3 mL Ni-NTA resin (Qiagen, Hilden, Germany) previously equilibrated with 20 mL lysis buffer. The column was washed with 20 mL washing buffer (50 mM Na2HPO4, pH 8.0, 300 mM NaCl, and 50 mM imidazole), and eluted with 2 mL elution buffer (50 mM Na2HPO4, pH 8.0, 300 mM NaCl, and 150 mM imidazole). The purified proteins were fractionated by SDS-PAGE with 4–20% gradient gels, and stained with Coomassie blue. For western blotting, the unstained SDS-PAGE gel was transferred onto a PVDF membrane, blocked by 20 mL blocking buffer (5% bovine serum albumin in Tris-buffered saline, 0.1% Tween 20) for 2 h, and then soaked with 1:1000 diluted horseradish peroxidase (HRP)-conjugated antibodies overnight at 4°C with gentle shaking. The membrane was prepared for detection by using Pierce ECL Western Blotting substrates (Thermo Scientific, Waltham, MA).
Mass spectrometry (MS) analyses
The full-length MS analyses were performed by the University of Arkansas Statewide Mass Spectrometry Facility. Purified proteins were dialyzed with pure water, diluted to 0.05 mg mL−1, and analyzed by a Bruker UltraflexII TOF-TOF with a MALDI ionization source. The laser was an Azura Laser AG diode pumped solid state laser (Nd:YAG, 355nm). The matrix used was a saturated solution of sinipinic acid in water/acetonitrile. Spectra were obtained in the positive ion, linear mode. The LC-MS/MS analyses were performed by Yale University Keck Proteomics Facility. The protocol was the same as the previous study.56 Proteins were digested by trypsin with a standard in-gel digestion protocol, and analyzed by LC-MS/MS on an LTQ Orbitrap XL equipped with a nanoACQUITY UPLC system. The Mascot search algorithm was used to search for the appropriate noncanonical substitution (Matrix Science, Boston, MA).
The malate dehydrogenase (MDH) activity assay
The protocol was modified slightly from the previous study.49 10 ng purified MDH or its variants was added to a 200 μL reaction mixture, following the instruction provided by the malate dehydrogenase assay kit (BioAssay Systems, Hayward, CA). The reaction mixture was added to a 96-well microplate, and the absorbance at 565 nm was read by a microplate reader.
Supplementary Material
Acknowledgments
This work was supported by a grant from National Institutes of Health (National Institute of Allergy and Infectious Diseases; AI119813), the start-up fund from University of Arkansas, and the grant from Arkansas Biosciences Institute.
Footnotes
Author contribution: SV and CF designed experiments, analyzed data, and wrote the manuscript. All authors performed experiments and edited the manuscript.
Notes: All authors declare no competing financial interest.
Supporting Information
The list of strains and plasmids used in this work and mass spectra of modified proteins were supplied as Supporting Information.
References
- 1.Walsh CT. Posttranslational modification of proteins : expanding nature’s inventory. Roberts and Company Publishers; Englewood, Colorado: 2006. [Google Scholar]
- 2.Schweppe RE, Haydon CE, Lewis TS, Resing KA, Ahn NG. The characterization of protein post-translational modifications by mass spectrometry. Acc Chem Res. 2003;36:453–461. doi: 10.1021/ar020143l. [DOI] [PubMed] [Google Scholar]
- 3.Witze ES, Old WM, Resing KA, Ahn NG. Mapping protein posttranslational modifications with mass spectrometry. Nat Methods. 2007;4:798–806. doi: 10.1038/nmeth1100. [DOI] [PubMed] [Google Scholar]
- 4.Liu WR, Wang YS, Wan W. Synthesis of proteins with defined posttranslational modifications using the genetic noncanonical amino acid incorporation approach. Mol Biosyst. 2011;7:38–47. doi: 10.1039/c0mb00216j. [DOI] [PubMed] [Google Scholar]
- 5.Neumann H, Peak-Chew SY, Chin JW. Genetically encoding N(epsilon)-acetyllysine in recombinant proteins. Nat Chem Biol. 2008;4:232–234. doi: 10.1038/nchembio.73. [DOI] [PubMed] [Google Scholar]
- 6.Mukai T, Kobayashi T, Hino N, Yanagisawa T, Sakamoto K, Yokoyama S. Adding l-lysine derivatives to the genetic code of mammalian cells with engineered pyrrolysyl-tRNA synthetases. Biochemical and biophysical research communications. 2008;371:818–822. doi: 10.1016/j.bbrc.2008.04.164. [DOI] [PubMed] [Google Scholar]
- 7.Umehara T, Kim J, Lee S, Guo LT, Soll D, Park HS. N-acetyl lysyl-tRNA synthetases evolved by a CcdB-based selection possess N-acetyl lysine specificity in vitro and in vivo. FEBS Lett. 2012;586:729–733. doi: 10.1016/j.febslet.2012.01.029. [DOI] [PubMed] [Google Scholar]
- 8.Park HS, Hohn MJ, Umehara T, Guo LT, Osborne EM, Benner J, Noren CJ, Rinehart J, Soll D. Expanding the genetic code of Escherichia coli with phosphoserine. Science. 2011;333:1151–1154. doi: 10.1126/science.1207203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Heinemann IU, Rovner AJ, Aerni HR, Rogulina S, Cheng L, Olds W, Fischer JT, Soll D, Isaacs FJ, Rinehart J. Enhanced phosphoserine insertion during Escherichia coli protein synthesis via partial UAG codon reassignment and release factor 1 deletion. FEBS Lett. 2012;586:3716–3722. doi: 10.1016/j.febslet.2012.08.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rogerson DT, Sachdeva A, Wang K, Haq T, Kazlauskaite A, Hancock SM, Huguenin-Dezot N, Muqit MM, Fry AM, Bayliss R, Chin JW. Efficient genetic encoding of phosphoserine and its nonhydrolyzable analog. Nat Chem Biol. 2015;11:496–503. doi: 10.1038/nchembio.1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Oza JP, Aerni HR, Pirman NL, Barber KW, ter Haar CM, Rogulina S, Amrofell MB, Isaacs FJ, Rinehart J, Jewett MC. Robust production of recombinant phosphoproteins using cell-free protein synthesis. Nat Commun. 2015;6:8168. doi: 10.1038/ncomms9168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang L. Engineering the Genetic Code in Cells and Animals: Biological Considerations and Impacts. Acc Chem Res. 2017 doi: 10.1021/acs.accounts.7b00376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.O’Donoghue P, Ling J, Wang YS, Soll D. Upgrading protein synthesis for synthetic biology. Nat Chem Biol. 2013;9:594–598. doi: 10.1038/nchembio.1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hoesl MG, Budisa N. Recent advances in genetic code engineering in Escherichia coli. Curr Opin Biotechnol. 2012;23:751–757. doi: 10.1016/j.copbio.2011.12.027. [DOI] [PubMed] [Google Scholar]
- 15.Neumann H. Rewiring translation - Genetic code expansion and its applications. FEBS Lett. 2012;586:2057–2064. doi: 10.1016/j.febslet.2012.02.002. [DOI] [PubMed] [Google Scholar]
- 16.Lemke EA. The exploding genetic code. Chembiochem. 2014;15:1691–1694. doi: 10.1002/cbic.201402362. [DOI] [PubMed] [Google Scholar]
- 17.Beltrao P, Bork P, Krogan NJ, van Noort V. Evolution and functional cross-talk of protein post-translational modifications. Mol Syst Biol. 2013;9:714. doi: 10.1002/msb.201304521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Soufi B, Soares NC, Ravikumar V, Macek B. Proteomics reveals evidence of cross-talk between protein modifications in bacteria: focus on acetylation and phosphorylation. Curr Opin Microbiol. 2012;15:357–363. doi: 10.1016/j.mib.2012.05.003. [DOI] [PubMed] [Google Scholar]
- 19.Wan W, Huang Y, Wang Z, Russell WK, Pai PJ, Russell DH, Liu WR. A facile system for genetic incorporation of two different noncanonical amino acids into one protein in Escherichia coli. Angew Chem Int Ed Engl. 2010;49:3211–3214. doi: 10.1002/anie.201000465. [DOI] [PubMed] [Google Scholar]
- 20.Hoesl MG, Budisa N. In vivo incorporation of multiple noncanonical amino acids into proteins. Angew Chem Int Ed Engl. 2011;50:2896–2902. doi: 10.1002/anie.201005680. [DOI] [PubMed] [Google Scholar]
- 21.Wu B, Wang Z, Huang Y, Liu WR. Catalyst-free and site-specific one-pot dual-labeling of a protein directed by two genetically incorporated noncanonical amino acids. Chembiochem. 2012;13:1405–1408. doi: 10.1002/cbic.201200281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chatterjee A, Sun SB, Furman JL, Xiao H, Schultz PG. A versatile platform for single- and multiple-unnatural amino acid mutagenesis in Escherichia coli. Biochemistry. 2013;52:1828–1837. doi: 10.1021/bi4000244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xiong H, Reynolds NM, Fan C, Englert M, Hoyer D, Miller SJ, Soll D. Dual Genetic Encoding of Acetyl-lysine and Non-deacetylatable Thioacetyl-lysine Mediated by Flexizyme. Angew Chem Int Ed Engl. 2016;55:4083–4086. doi: 10.1002/anie.201511750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ozer E, Chemla Y, Schlesinger O, Aviram HY, Riven I, Haran G, Alfonta L. In vitro suppression of two different stop codons. Biotechnol Bioeng. 2016;114:1065–1073. doi: 10.1002/bit.26226. [DOI] [PubMed] [Google Scholar]
- 25.Anderson JC, Wu N, Santoro SW, Lakshman V, King DS, Schultz PG. An expanded genetic code with a functional quadruplet codon. Proc Natl Acad Sci U S A. 2004;101:7566–7571. doi: 10.1073/pnas.0401517101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang K, Sachdeva A, Cox DJ, Wilf NM, Lang K, Wallace S, Mehl RA, Chin JW. Optimized orthogonal translation of unnatural amino acids enables spontaneous protein double-labelling and FRET. Nat Chem. 2014;6:393–403. doi: 10.1038/nchem.1919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Neumann H, Wang K, Davis L, Garcia-Alai M, Chin JW. Encoding multiple unnatural amino acids via evolution of a quadruplet-decoding ribosome. Nature. 2010;464:441–444. doi: 10.1038/nature08817. [DOI] [PubMed] [Google Scholar]
- 28.Khoury GA, Baliban RC, Floudas CA. Proteome-wide post-translational modification statistics: frequency analysis and curation of the swiss-prot database. Sci Rep 1. 2011 doi: 10.1038/srep00090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chin JW. Expanding and reprogramming the genetic code. Nature. 2017;550:53–60. doi: 10.1038/nature24031. [DOI] [PubMed] [Google Scholar]
- 30.Liu CC, Schultz PG. Adding new chemistries to the genetic code. Annu Rev Biochem. 2010;79:413–444. doi: 10.1146/annurev.biochem.052308.105824. [DOI] [PubMed] [Google Scholar]
- 31.Mukai T, Lajoie MJ, Englert M, Soll D. Rewriting the Genetic Code. Annu Rev Microbiol. 2017;71:557–577. doi: 10.1146/annurev-micro-090816-093247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang K, Schmied WH, Chin JW. Reprogramming the genetic code: from triplet to quadruplet codes. Angew Chem Int Ed Engl. 2012;51:2288–2297. doi: 10.1002/anie.201105016. [DOI] [PubMed] [Google Scholar]
- 33.Wan W, Tharp JM, Liu WR. Pyrrolysyl-tRNA synthetase: an ordinary enzyme but an outstanding genetic code expansion tool. Biochim Biophys Acta. 2014;1844:1059–1070. doi: 10.1016/j.bbapap.2014.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ambrogelly A, Gundllapalli S, Herring S, Polycarpo C, Frauer C, Soll D. Pyrrolysine is not hardwired for cotranslational insertion at UAG codons. Proc Natl Acad Sci U S A. 2007;104:3141–3146. doi: 10.1073/pnas.0611634104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bryson DI, Fan C, Guo LT, Miller C, Soll D, Liu DR. Continuous directed evolution of aminoacyl-tRNA synthetases. Nat Chem Biol. 2017 doi: 10.1038/nchembio.2474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fan C, Xiong H, Reynolds NM, Soll D. Rationally evolving tRNAPyl for efficient incorporation of noncanonical amino acids. Nucleic Acids Res. 2015;43:e156. doi: 10.1093/nar/gkv800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Albayrak C, Swartz JR. Cell-free co-production of an orthogonal transfer RNA activates efficient site-specific non-natural amino acid incorporation. Nucleic Acids Res. 2013;41:5949–5963. doi: 10.1093/nar/gkt226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gan Q, Fan C. Increasing the fidelity of noncanonical amino acid incorporation in cell-free protein synthesis. Biochim Biophys Acta. 2016;1861:3047–3052. doi: 10.1016/j.bbagen.2016.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.O’Donoghue P, Prat L, Heinemann IU, Ling J, Odoi K, Liu WR, Soll D. Near-cognate suppression of amber, opal and quadruplet codons competes with aminoacyl-tRNAPyl for genetic code expansion. FEBS Lett. 2012;586:3931–3937. doi: 10.1016/j.febslet.2012.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lee S, Oh S, Yang A, Kim J, Soll D, Lee D, Park HS. A facile strategy for selective incorporation of phosphoserine into histones. Angew Chem Int Ed Engl. 2013;52:5771–5775. doi: 10.1002/anie.201300531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gan R, Perez JG, Carlson ED, Ntai I, Isaacs FJ, Kelleher NL, Jewett MC. Translation system engineering in Escherichia coli enhances non-canonical amino acid incorporation into proteins. Biotechnol Bioeng. 2017;114:1074–1086. doi: 10.1002/bit.26239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fan C, Ip K, Soll D. Expanding the genetic code of Escherichia coli with phosphotyrosine. FEBS Lett. 2016;590:3040–3047. doi: 10.1002/1873-3468.12333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Luo X, Fu G, Wang RE, Zhu X, Zambaldo C, Liu R, Liu T, Lyu X, Du J, Xuan W, Yao A, Reed SA, Kang M, Zhang Y, Guo H, Huang C, Yang PY, Wilson IA, Schultz PG, Wang F. Genetically encoding phosphotyrosine and its nonhydrolyzable analog in bacteria. Nat Chem Biol. 2017;13:845–849. doi: 10.1038/nchembio.2405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang MS, Brunner SF, Huguenin-Dezot N, Liang AD, Schmied WH, Rogerson DT, Chin JW. Biosynthesis and genetic encoding of phosphothreonine through parallel selection and deep sequencing. Nat Methods. 2017;14:729–736. doi: 10.1038/nmeth.4302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pizer LI. The Pathway and Control of Serine Biosynthesis in Escherichia Coli. J Biol Chem. 1963;238:3934–3944. [PubMed] [Google Scholar]
- 46.Odoi KA, Huang Y, Rezenom YH, Liu WR. Nonsense and sense suppression abilities of original and derivative Methanosarcina mazei pyrrolysyl-tRNA synthetase-tRNA(Pyl) pairs in the Escherichia coli BL21(DE3) cell strain. PLoS One. 2013;8:e57035. doi: 10.1371/journal.pone.0057035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Xu H, Zhou J, Lin S, Deng W, Zhang Y, Xue Y. PLMD: An updated data resource of protein lysine modifications. J Genet Genomics. 2017;44:243–250. doi: 10.1016/j.jgg.2017.03.007. [DOI] [PubMed] [Google Scholar]
- 48.Pan Z, Wang B, Zhang Y, Wang Y, Ullah S, Jian R, Liu Z, Xue Y. dbPSP: a curated database for protein phosphorylation sites in prokaryotes. Database (Oxford) 2015;2015:bav031. doi: 10.1093/database/bav031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Venkat S, Gregory C, Sturges J, Gan Q, Fan C. Studying the Lysine Acetylation of Malate Dehydrogenase. J Mol Biol. 2017;429:1396–1405. doi: 10.1016/j.jmb.2017.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gan Q, Lehman BP, Bobik TA, Fan C. Expanding the genetic code of Salmonella with non-canonical amino acids. Sci Rep. 2016;6:39920. doi: 10.1038/srep39920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Amiram M, Haimovich AD, Fan C, Wang YS, Aerni HR, Ntai I, Moonan DW, Ma NJ, Rovner AJ, Hong SH, Kelleher NL, Goodman AL, Jewett MC, Soll D, Rinehart J, Isaacs FJ. Evolution of translation machinery in recoded bacteria enables multi-site incorporation of nonstandard amino acids. Nat Biotechnol. 2015;33:1272–1279. doi: 10.1038/nbt.3372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zaitseva J, Meneely KM, Lamb AL. Structure of Escherichia coli malate dehydrogenase at 1.45 A resolution. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2009;65:866–869. doi: 10.1107/S1744309109032217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Guo J, Melancon CE, 3rd, Lee HS, Groff D, Schultz PG. Evolution of amber suppressor tRNAs for efficient bacterial production of proteins containing nonnatural amino acids. Angew Chem Int Ed Engl. 2009;48:9148–9151. doi: 10.1002/anie.200904035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Venkat S, Nannapaneni DT, Gregory C, Gan Q, McIntosh M, Fan C. Genetically encoding thioacetyl - lysine as a nondeacetylatable analog of lysine acetylation in Escherichia coli. FEBS Open Bio. 2017;7:1805–1814. doi: 10.1002/2211-5463.12320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Venkat S, Gregory C, Meng K, Gan Q, Fan C. A Facile Protocol to Generate Site-Specifically Acetylated Proteins in Escherichia Coli. J Vis Exp. 2017 doi: 10.3791/57061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Venkat S, Gregory C, Gan Q, Fan C. Biochemical Characterization of the Lysine Acetylation of Tyrosyl-tRNA Synthetase in Escherichia coli. Chembiochem. 2017;18:1928–1934. doi: 10.1002/cbic.201700343. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.