

Abstract
An increasingly recognized component of resistance to tyrosine kinase inhibitors (TKI) involves persistence of a drug-tolerant subpopulation of cancer cells which survive despite effective eradication of the majority of the cell population. Multiple groups have demonstrated that these drug-tolerant persister cells undergo transcriptional adaptation via an epigenetic state change that promotes cell survival. Because this mode of TKI drug tolerance appears to involve transcriptional addiction to specific genes and pathways, we hypothesized that systematic functional screening of EGFR TKI/transcriptional inhibitor combination therapy would yield important mechanistic insights and alternative drug escape pathways. We therefore performed a genome-wide CRISPR/Cas9 enhancer/suppressor screen in EGFR-dependent lung cancer PC9 cells treated with erlotinib + THZ1 (CDK7/12 inhibitor) combination therapy,a combination previously shown to suppress drug tolerant cells in this setting. As expected, suppression of multiple genes associated with transcriptional complexes (EP300, CREBBP and MED1) enhanced erlotinib/THZ1 synergy. Unexpectedly, we uncovered nearly every component of the recently described ufmylation pathway in the synergy suppressor group. Loss of ufmylation did not affect canonical downstream EGFR signaling. Instead, absence of this pathway triggered a protective unfolded protein response (UPR) associated with STING upregulation, promoting pro-tumorigenic inflammatory signaling but also unique dependence on Bcl-xL. These data reveal that dysregulation of ufmylation and ER stress comprise a previously unrecognized TKI drug tolerance pathway that engages survival signaling, with potentially important therapeutic implications.
Keywords: Ufmylation, drug persister, ER stress, EGFR TKI, Bcl-xL
Introduction
Despite efficacy of targeted therapy with tyrosine kinase inhibitors (TKIs) in EGFR mutant lung adenocarcinoma and other cancers, acquired resistance limits durable clinical benefit (1, 2). An increasingly recognized reason for treatment failure involves drug tolerant persister (DTP) populations of cancer cells that survive and rapidly adapt to therapy (3–6). Understanding the pathways that facilitate DTP emergence is therefore critical to designing more effective combination therapies that can achieve cure.
Adaptive transcriptional responses have been well characterized to promote stress tolerance and cancer cell survival (5, 7, 8). We recently found that the CDK7/12 inhibitor THZ1 (9), which represses RNA polymerase II-mediated transcription and inhibits certain cancers (10), also synergizes with EGFR, ALK, and MEK inhibitors by eliminating DTPs (11). Similarly, others have reported synergy between the BRD4 inhibitor JQ1 and MEK inhibition to inhibit adaptive transcriptional responses (7). However, detailed mechanism and additional pathways that could buffer these cells against stress remain incompletely characterized.
The balance between pro-survival and pro-apoptotic BH3 proteins also modulates response to cancer chemo- and targeted therapies (12, 13). Regulation of this balance is particularly critical for cancer cells upon depletion of the addicted oncogenic signal in multiple cancer models (14, 15), such as changes in BIM levels following EGFR-TKI treatment of EGFR mutant lung cancer (16). Moreover, Bcl-xL and BCL-2 have been implicated specifically in EGFR TKI DTP cell survival (5). Activation of other post-transcriptional stress response pathways such as the UPR also regulates cell survival in diverse cancer models (17, 18). Although well described in other contexts, whether these pathways contribute to EGFR TKI DTP survival and how they might interface with apoptosis remains unknown.
Novel regulators of ER stress, such as protein ufmylation, have also been identified (19). Indeed, the enzymatic components of the ufmylation pathway were only recently characterized (20). This pathway is evolutionarily conserved in metazoans and thought to be important for ER homeostasis in several contexts including hematopoietic stem cells, and regulates the expression of the autophagy related protein SQSTM1 through modification of ER stress (19, 21–23). Genetic alterations of this pathway are occasionally found in several types of cancer including lung cancer (24) and can cause unique cancer dependencies (25). Theoretically, engagement of such mechanisms could bypass certain aspects of transcriptional inhibition and promote survival.
Unbiased genetic screens provide a powerful tool to probe biological mechanism in preclinical models of cancer (26, 27). To elucidate potentially novel pathways that regulate EGFR DTP cell survival we performed a genome-wide CRISPR/Cas9 enhancer/suppressor screen with the Avana sgRNA library (28), focusing on pathways that suppress the effect of erlotinib/THZ1 treatment on DTP eradication.
Materials and Methods
Cell lines and culture
PC9 cells and HCC827 cells were obtained from collaborating labs primarily in 2014 and authenticated by a short tandem repeat (STR) analysis. 293T/17 cell line was purchased from ATCC in 2016. PC9 cells and HCC827 cells were cultured in RPMI-1640 growth medium (Thermo Fishcer Scientific), supplemented with 10% fetal bovine serum (FBS) and 293T/17 cells was cultured in DMEM growth medium (Thermo Fishcer Scientific), supplemented with 10% FBS. All cell lines were cultured at 37 degree in a humidified 5% CO2 incubator. All cell lines were mycoplasma tested and negative
Genome scale CRISPR screen
Avana barcoded library contains 73687 barcoded sgRNAs targeting 18454 genes and 1000 non-targeting guides (28). For each screen, three infection replicates were performed with a sufficient number of cells per replicate that allowed to achieve 500 cells per guide following puromycin selection (4 × 107 surviving cells containing 74687 sgRNAs) 3 × 106 cells per well were seeded in 12-well plates and were infected with the amount of virus determined during optimization with a final polybrene concentration of 4 μg/mL. After 4 days of puromycin selection, 4 × 107 cells were stocked for the gDNA exraction at Day 0 (early time point sample), and 4 × 107 cells cells were treated with drugs (100 nM erlotinib or 100 nM erlotinib plus 50 nM THZ1). Cells were passaged or fresh drug-containing media was added every 4 days. Cells were harvested 18 days after initiation of treatment.
To harvest cells, cells were trypsinized, spun down, washed with PBS and resuspend in PBS. Then the cell pellets were frozen at −80°C. Genomic DNA was extracted using the Qiagen Blood and Cell Culture DNA Maxi Kit or Midi kit according to the manufacturer’s protocol. PCR and sequencing were performed as previously described (28). Samples were sequenced on a HiSeq2000 (Illumina). For analysis, the read counts were normalized to reads per million and then log2 transformed. The log2 fold-change of each sgRNAs was determined relative to the initial time point for each biological replicate.
Lentiviral infection
Lenti-Guide or Lenti-CRISPR v2 vectors were cloned as previously described (28). Briefly, HEK-293T/17 cells were transduced with lentiGuide puro and packaging plasmids using X-treme Gene 9 (Roche) according to the manufacturer’s instructions. On day 2, target cells (PC9 cells or HCC827 cells) were seeded, and allowed to adhere overnight and change the media of HEK293T/17 cells. On day 3 the supernatant of transduced HEK293T/17 cells was collected and added to the target cells through a 0.45 μm filter. On day 5, puromycin (1 mg/ml for PC9 cells and HCC827 cells) was added to select infected cells (for four days).
Oligonucleotides coding for guide RNAs that target UFM1, UBA5, UFC1, UFL1 and UFSP2 genes were chosen from the Avana sgRNA library or the Brunello sgRNA library (28). Non-targeting sgRNAs from the Avana library were used as Control sgRNA #1 or Control sgRNA #2. Each sgRNA target sequence is described in Supplementary Table S1.
Quantitative RT-PCR
Total cellular RNA was isolated from cells using an RNeasy Mini Ki (Qiagen) and 1.0 μg was then reverse transcribed to cDNA using the High Capacity RNA to c-DNA kit (Life Technologies). Quantitative PCR reactions were performed using SYBR green PCR Master Mix (Applied Biosystems) on an ABI Prism 7300 platform (Life Technologies). The relative expression was normalized with the expression of the housekeeping gene 36B4. The sequences of primers used have been listed in Supplementary Table S1.
RNA-seq analysis
RNA was isolated by RNeasy Mini kit (Qiagen) from the UFM1 knock out PC9 cells or control PC9 cells following treatment with DMSO, 50nM THZ1, 100nM erlotinib or 100nM erlotinib + 50nM THZ1 (combination therapy). Cells were harvested in the media 48hrs. Libraries were prepared using Illumina TruSeq Stranded mRNA sample preparation kits from 500 ng of purified total RNA according to the manufacturer’s protocol. The finished dsDNA libraries were quantified by Qubit fluorometer, Agilent TapeStation 2200, and qRT-PCR using the Kapa Biosystems library quantification kit according to manufacturer’s protocols. Uniquely indexed libraries were pooled in equimolar ratios and sequenced on an Illumina NextSeq500 with single-end 75bp reads by the Dana-Farber Cancer Institute Molecular Biology Core Facilities.
Principal component analysis
Principal component analysis (PCA) was performed for average FPKM values of each condition and resulting PCA scores were used to create Fig 4C. The single sample gene set enrichment analysis (ssGSEA) considering directions (positive and negative gene scores) was performed to derive in Fig 4C using GO gene sets (c5.all.v5.2.symbols.gmt) downloaded from MsigDB and gene scores defined by the loadings (i.e., the contributions to PC1 and PC2 of each genes) from the PCA results. Based on the normalized enrichment scores for the gene sets that were denoted as ‘stat’ in the Table, P-value was derived from 10,000 times gene permutations and FDR was calculated. The complete gene sets lists were included in Supplementary Table S2.
Figure 4. Absence of ufmylation induces inflammatory signaling and ER stress.
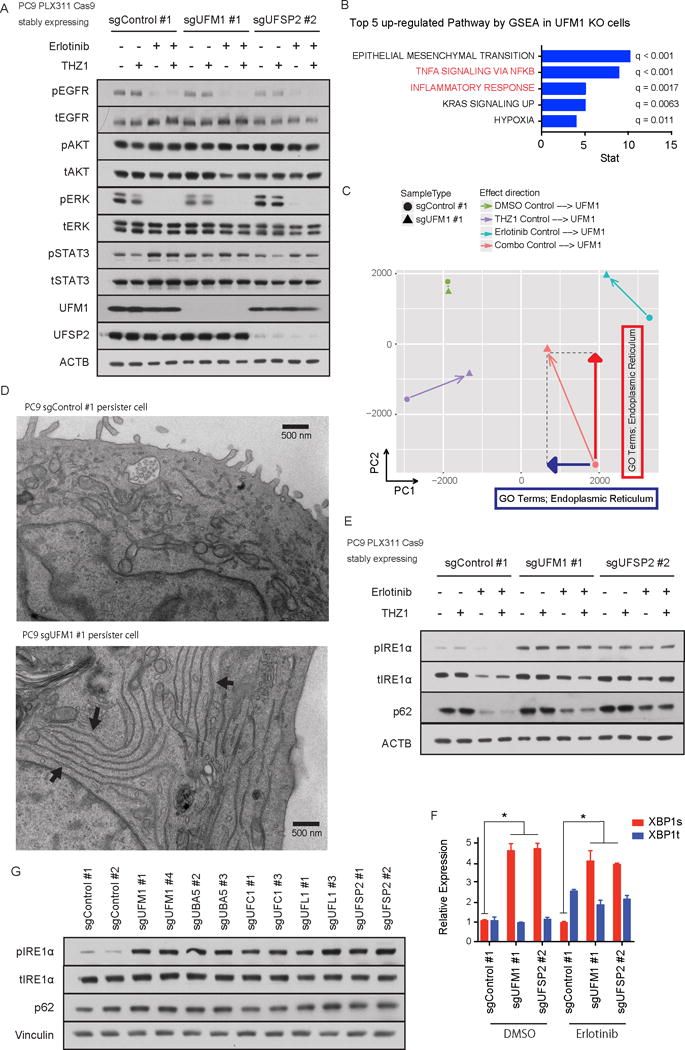
A) Immunoblotting of EGFR signaling pathway components. UFM1 or UFSP2 depleted PC9 cells or control PC9 cells were treated with DMSO, 50 nM THZ1, 100 nM erlotinib (Erlotinib) or 50 nM THZ1 plus 100 nM erlotinib (Combo) for 72 hr. B) Top 5 up-regulated pathways in UFM1 knock out PC9 cells compared with Control PC9 cells analyzed by GSEA analysis. C) Principal component analysis (PCA) was performed for average FPKM values of each condition and resulting PCA scores were used. GO term analysis was performed against each principal component and ER GO terms were highly ranked with the indicated directions. D) Representative pictures of electron microscopic experiments. Each arrow in UFM1 KO persister cells indicates accumulation of abnormal ER. E) Immunoblotting of unfolded protein response pathway components in UFM1 or UFSP2 depleted or control PC9 cells. Cells were incubated with DMSO, 50 nM THZ1, 100 nM erlotinib (Erlotinib) or 50 nM THZ1 plus 100 nM erlotinib (Combo) for 72 hr. F) qRT-PCR of spliced or total XBP1 in PC9 cells after 100 nM erlotinib treatment. PC9 cells were transduced with indicated sgRNAs. Each bar indicates mean ± SD. G) Immunoblotting analysis of ufmylation pathway genes in PC9 cells transduced with indicated sgRNAs.
Immunoblotting assay
For immunoblotting assay, cells were lysed in RIPA buffer containing 1× protease inhibitors (Roche) and Phosphatase Inhibitors Cocktails I and II (CalBioChem). Protein concentrations were determined using a Bradford assay (Bio-Rad). Proteins were separated by SDS gel electrophoresis using NuPAGE 4-12% Bis-Tris gels in MOPS buffer or 3-8% Tris-Acetate gels in Tris Acetate buffer (Life Technologies). Resolved protein was transferred to nitrocellulose membranes, blocked in 5% milk and probed with primary antibodies in HIKARI Signal Enhancer Solutions 1 (Nacalai USA). After incubation with the appropriate secondary antibody diluted in 2.5% Milk (Pierce anti-mouse IgG/IgM (31444, Thermo Scientific) and anti-rabbit IgG (31460, Thermo Scientific)), blots were imaged on film. For the information of primary antibodies used in this study see Supplementary Methods.
Cell viability assay
1500 cells were seeded in 96-well plates, allowed to attach overnight, and then incubated with growth media containing drugs as indicated for 96 hours. Values of CellTiter-Glo Luminescent Cell Viability assay (Promega) after 96 hours were normalized to vehicle treated cells. Plates were read on a Tecan Infinite M200 Pro plate reader and analysis was performed using Prism7 (GraphPad Software). All conditions were tested in triplicate. The values represent the average of three technical replicates and a representative experiment from at least two independent experiments (biological replicates).
Crystal violet staining
1 × 105 cells were seeded in 6-well type plates, allowed to attach overnight, and then incubated with media containing drugs as indicated for 18 days. Media and drugs were replaced every four days unless otherwise noted. After 18 days culture, cells were fixed with 1% paraformaldehyde (PFA) and then stained with 0.1% crystal violet as previously described (http://medicine.yale.edu/lab/kim/resources/protocols/cell/crystal_violet_stain.aspx). Plates were made duplicate and count the number of viable cells by Vi-cell counter (Beckman Coulter) for quantification.
In Vivo Treatment of EGFR Xenograft
All breeding, mouse husbandry, and in vivo experiments were performed with the approval of the NYU Langone Medical Center (NY, NY) Animal Care and Use Committee.
Nu/Nu mice were purchased from Charles River Laboratories International Inc. PC9-sgDummy and PC9-sgUFM1 cells were detected as pathogen free at Charles River Laboratories International Inc. and cultured in RPMI-1640 with 10% FBS. Cells were resuspended in serum-free medium mixed with an equal amount of Matrigel (BD Biosciences). Mice were injected with 2 million cells per shot and 2 locations per mouse in the flanks. The mice were randomly grouped (vehicle and THZ1 + Erlotinib combo groups for each cells). Each cohort included 5 mice. Treatment was started when tumor size reached 100 to 200 mm3. Tumor sizes were monitored weekly, and volumes were calculated with the following formula: (mm3) = length × width × width × 0.5.
THZ1 was dissolved in DMSO:5% Dextrose (1:10) and dosed as 25 mg/kg twice daily via intraperitoneal route. Erlotinib was prepared in 0.5% HPMC (Hydroxypropyl methylcellulose), and dosed as 25 mg/kg daily orally.
Electron Microscopy
PC9 cells were incubated with 100 nM erlotinib for 13 days then fixed the specimens with 2.5% Glutaraldehyde 1.25% Paraformaldehyde and 0.03% picric acid in 0.1 M sodium cacodylate buffer (pH 7.4). The cells were washed in 0.1 M sodium cacodylate buffer (pH 7.4), postfixed for 30 min in 1% Osmium tetroxide (OsO4)/1.5% Potassiumferrocyanide (KFeCN6), washed in 2× in water and 1× in Maleate buffer and incubated in 1% uranyl acetate in Maleate Buffer for 30min followed by 2 washes in water and subsequent dehydration in grades of alcohol (5min each; 50%, 70%, 95%, 2× 100%). The samples were subsequently embedded (on the aclar) in TAAB Epon (Marivac Canada Inc. St. Laurent, Canada) and polymerized at 60 degrees C for 48 hrs. After polymerization the aclar was peeled off and Ultrathin sections (about 80nm) were cut on a Reichert Ultracut-S microtome, picked up on to copper grids stained with lead citrate and examined in a TecnaiG2 Spirit BioTWIN and images were recorded with an AMT 2k CCD camera.
For additional information see Supplementary Methods.
Results
Genome wide CRISPR/Cas9 screen to identify genetic modifiers of Erlotinib and THZ1 treatment
To identify genes that, when deleted, enhance or suppress the impact of erlotinib/THZ1 combination therapy in EGFR dependent PC9 cells, we performed a genome-wide CRISPR screen. We first optimized treatment conditions for pooled screening (Supplementary Fig. S1A) in EGFR dependent PC9 or HCC827 cells, identifying erlotinib 100 nM ± THZ1 50 nM (for PC9 cells) or erlotinib 30 nM ± THZ1 50 nM (for HCC827 cells) as the minimum doses that inhibited cell viability and showed a differential impact between single and combination therapy. We focused the initial screen on PC9 and infected cells with the Cas9 expressing vector, PLX-311, which achieved high Cas9 activity (Supplementary Fig. S1B). Following infection of PC9 cells with the Avana sgRNA library (28), targeting 500 cells per each sgRNA with each replicate as a minimum representation, cells were incubated for 18 days with the indicated treatments (Fig. 1A). We isolated genomic DNA from these cell populations before (ETP: early time point sample) and after treatment, then analyzed sgRNA depletion or enrichment over time by next generation sequencing. Overall proliferation curves of Avana sgRNA library infected cells were similar with those of control EGFP vector infected PC9 cells under each treatment condition (Fig. 1B and Supplementary Fig. S2A). As the Avana sgRNA library contains 1000 non-targeting sgRNAs, we confirmed these non-targeting sgRNAs were distributed near the average position (Supplementary Fig. S2B and S2C). We also confirmed strong correlation between each treatment condition replicate (Supplementary Fig. S2D).
Figure 1. Overview of pooled sgRNA screening.
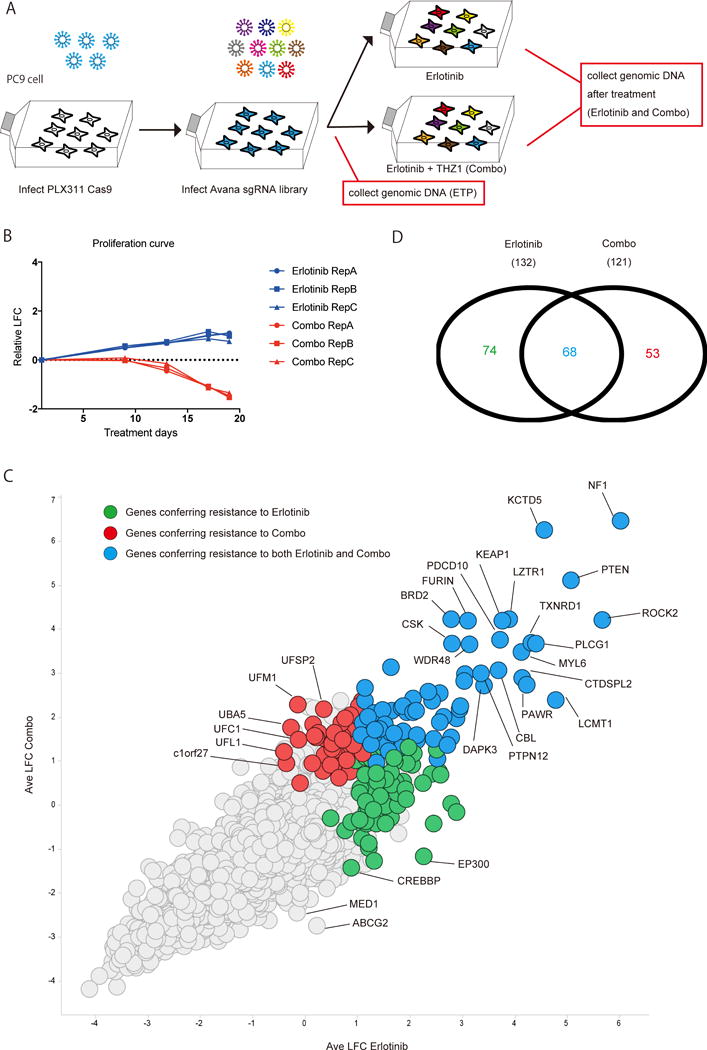
Overview of genome wide CRISPR screen with PC9 cells treated by erlotinib or combination therapy. A) A work flow of CRISPR screen. PC9 cells were infected with Cas9 expressing plasmid (PLX311), selected and then transduced with Avana sgRNA library. After antibiotic selection, PC9 cells were treated with 100nM erlotinib ± 50nM THZ1 for 18 days. Genomic DNA was isolated from each cell population before (ETP: early time point sample) and after treatment, then analyzed by next generation sequencing to see the enrichment or depletion of each sgRNA. B) Proliferation curves of PC9 cells infected with sgRNA library under the indicated treatment. C) Scatter plot for the result of CRISPR screen. Each dot indicates average of 4 sgRNAs for one target gene. Horizontal axis and vertical axis indicate log 2 fold change of sgRNA during erlotinib treatment or combination treatment compared with early time point sample. D) Venn diagram indicate the number of genes significantly enriched (FDR <0.25) after the treatments by the STARS algorithm (28).
To assess gene level effects we used the STARS algorithm (28) to rank genes according to the impact of multiple sgRNAs for each gene. We then distinguished between highly ranked genes whose deletion modified the impact of erlotinib treatment or erlotinib/THZ1 combination treatment, as well as genes shared across both treatments (erlotinib and erlotinib/THZ1) (Fig. 1C). When we used a False Discovery Rate (FDR) of < 0.25 as a cutoff, 142 genes were identified which rendered cells resistant to erlotinib treatment and 121 genes were found to render cells resistant to erlotinib/THZ1 treatment, with 68 genes shared between both groups (Fig. 1D). As expected the genes which rescued from both single and combination therapies contained previously reported erlotinib resistance related genes and genes which have scored in multiple prior screens across an array of cancer therapeutics (e.g. NF1, PTEN, KEAP1) (29–31). Because erlotinib/THZ1 treatment preferentially eradicates DTP cells (11), we instead focused on genes that uniquely modified the response to combination treatment, to isolate pathways especially important for the survival of this treatment refractory cell population.
Erlotinib/THZ1 synergy enhancer and suppressor genes
Examination of deleted genes that uniquely enhanced synergy of the erlotinib/THZ combination identified multiple components of the transcription initiation complex (e.g EP300, CREBBP, MED1) (Fig. 2A and Supplementary Table S3). These results are consistent with an on-target effect of THZ1 in mediating transcriptional inhibition and suppressing an adaptive transcriptional response to oncogene targeted therapy that would be further potentiated by genetic loss of one of these factors (11). Positive elongation factor complex b (P-TEFb) including CDK9 is necessary for the initiation and elongation of transcription by RNA polymerase II which is also regulated by the activity of the cyclin dependent kinases CDK7, 12 and 13. Inhibition of CDK7 and CDK12 by THZ1 can disrupt active enhancers and target transcriptionally addicted cancer cells (9, 10, 32). EP300 and MED1 are used as markers for active enhancer regions and are also considered to be functionally important (33, 34). Consistent with this we confirmed that the EP300 inhibitor SGC-CBP30 enhances the synergistic effect of combination therapy (Supplementary Fig. S2E). The drug efflux transporter ABCG2 was also highly ranked in this group, suggesting drug export as a potential mechanism of acquired resistance unique to THZ1 therapy (35). Together, these findings further validate the success of our CRISPR/Cas9 screen in uncovering biologically meaningful results.
Figure 2. Identified synergy enhancers and synergy suppressors.
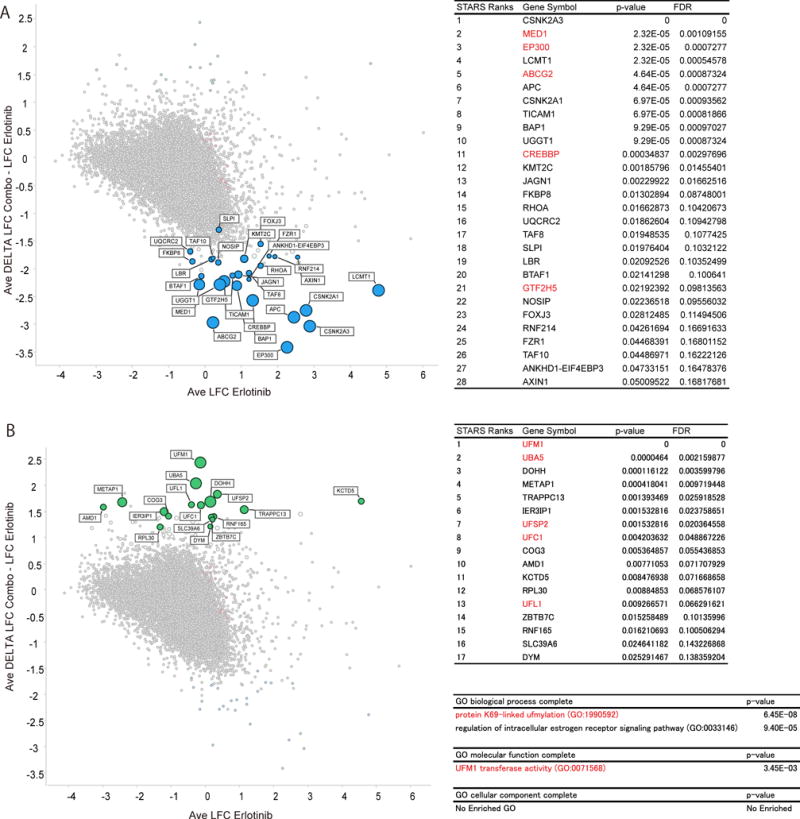
Genome wide CRISPR screen dissects synergistic effect of THZ1. A), B) Scatter plots for synergy enhancer genes (A) or synergy suppressor genes (B). Horizontal axis indicates average log 2 fold change (LFC) from early time point sample after erlotinib treatment. Vertical axis indicates delta log 2 fold change between erlotinib treatment and combination treatment. Tables indicate the most enriched (FDR <0.25) genes ranked by STARS algorithm for synergy enhancer genes (A) or synergy suppressor genes (B). GO analysis was performed with the enriched synergy suppressor genes (B).
Interestingly, analysis of the top ranked synergy suppressor genes unveiled nearly every component of the ufmylation pathway (UFM1, UFSP2, UBA5, UFC1, and UFL1) (Fig. 2B and Supplementary Table S3). We further confirmed this result by performing a confirmatory independent CRISPR screen with a customized mini sgRNA library to validate top hits in PC9 cells. Consistent with initial screen, most of these ufmylation pathway genes were re-identified as synergy suppressors (Supplementary Table S3). As mentioned above, this post-translational modification pathway is likely to play an important role in cell survival but overall very little is known about the function of ufmylation in cancer cells. Notably, while only a few target proteins have been identified in this pathway, two reported substrates CDK5RAP3 and DDRGK1 were also highly ranked as synergy suppressor genes (Supplementary Table S3).
Since the screen identified the majority of known genes in this pathway, we focused on understanding how suppression of ufmylation might impact survival of drug tolerant persisters and rescue the synergistic effect of erlotinib/THZ1 combination therapy. First, we confirmed the efficiency of ufmylation pathway sgRNAs in the library by immunoblot, as compared with control non-targeting sgRNAs (Fig. 3A). We also analyzed protein levels of previously reported UFL1 complex members (36), and confirmed degradation of DDRGK1 and CDK5RAP3 specifically following UFL1 deletion. Deletion of ufmylation pathway genes in PC9 cells slightly inhibited their proliferation, possibly reflecting the dysregulation of ER homeostasis by ufmylation pathway deletion (Fig. 3B). However, in short-term drug sensitivity assays (96 hours), deletion of this pathway did not significantly affect the proliferation of PC9 cells in the context of THZ1 or erlotinib single therapy (Supplementary Fig. S3A). We next tested the impact of ufmylation pathway gene deletion on PC9 cell colony formation following erlotinib or erlotinib/THZ1 treatment. Consistent with results from the screen, UFM1 or UFSP2 deletion rescued day 18 colony growth despite eradication of erlotinib DTPs by THZ1 in control cells (Fig. 3C). Rescue of persister cell survival was also confirmed following deletion of multiple other ufmylation pathway genes (UBA5, UFC1, UFL1) (Supplementary Fig. S3B).
Figure 3. Absence of ufmylation pathway genes attenuates cell killing effect by THZ1 over erlotinib treatment.
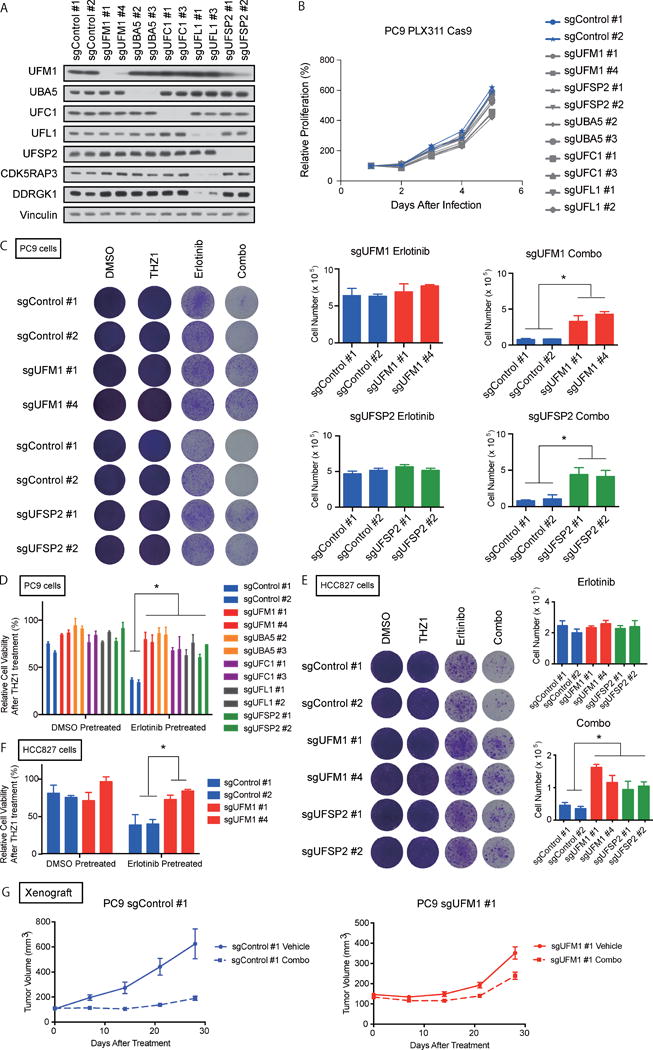
A) Immunoblotting analysis of ufmyation pathway genes. B) Cell proliferation assay of PC9 cells transduced indicated sgRNAs. C) Umylation pathway depleted or control PC9 cells were treated with 50 nM THZ1 (THZ1), erlotinib (Erlotinib) or 50 nM THZ1 + erlotinib (Combo) for 18 days, (100 nM erlotinib was used for PC9 cells and 30 nM erlotinib was used for HCC827, respectively). The viable number of the cells was counted by Vi-cell counter at day 18 to quantify the cell proliferation. Each bar indicates the mean and SD of triplicate. D) Ufmylation depleted PC9 cells were pre-treated with 100 nM erlotinib for 48 hrs. Then treated by 100 nM THZ1. Cells were incubated with THZ1 for 96 hr. Each bar indicate mean ± SD. E) UFM1 or UFSP2 depleted HCC827 cells were treated as in C. F) UFM1 depleted HCC827 cells were treated as in D. G) Xenograft experiments with PC9 cells transduced indicated sgRNAs. Detailed experimental procedures described in Materials and Methods in Supplemental information.
We previously showed that transcriptional adaptation to EGFR TKI treatment in PC9 cells begins as early as 48 hours, resulting in an enhancer landscape that is sustained for up to 7 days and promotes survival of DTP cells (11). We therefore pre-treated PC9 cells with erlotinib for 48 hours and monitored the impact of THZ1 treatment with control or ufmylation knockout. Consistent with prior results, as compared with DMSO pre-treatment, erlotinib pre-treatment enhanced THZ1 sensitivity in sgControl PC9 cells, which was significantly attenuated in sgUFM1, sgUBA5, sgUFC1, sgUFL1, or sgUFSP2 PC9 cells (Fig. 3D). UFM1 or UFSP2 deletion in another EGFR dependent cancer cell line (HCC827) also impaired the combinatorial effect of THZ1 relative to control cells by colony formation and in the pre-treatment assay (Fig. 3E, 3F, Supplementary Fig. S3C). We further examined the impact of erlotinib/THZ1 on tumor growth impairment of sgControl or sgUFM1 PC9 xenografts in vivo. Consistent with our in vitro data, while UFM1 knockout PC9 tumors grew more slowly than control tumors, the impact of the drug combination was significantly attenuated (Fig. 3G, Supplementary Fig. S3D). Finally, we assessed the specificity of these results relative to additional combination therapies that have been reported to overcome EGFR TKI resistance (37). Importantly, absence of ufmylation did not affect the synergistic impact of erlotinib plus trametinib treatment, revealing a specific impact on erlotinib/THZ1 synergy (Supplementary Fig S3E). Together, these data confirm that suppression of ufmylation pathway genes rescues DTP survival following transcriptional inhibition.
Disruption of ufmylation promotes inflammatory signaling and ER stress
To investigate the role of ufmylation in EGFR TKI DTP cell survival, we first interrogated canonical EGFR downstream signaling pathways in the presence or absence of ufmylation. We did not observe any clear differences in pERK, pAKT, or pSTAT3 levels between control cells and both UFM1 or UFSP2 knock out cells (Fig. 4A). To uncover potentially novel pathways activated by UFM1 knockout that promote survival, we performed RNA-seq and compared profiles of PC9 sgControl or sgUFM1 cells in the absence or presence of drug. Gene enrichment analysis (GSEA) revealed that UFM1 knockout upregulated pathways were associated with EMT and NF-κB activation (Fig. 4B, Supplementary Table S2), and consistent with increased IL-6 production in ufmylation deficient PC9 cells (Supplementary Fig. S4A,B). Next, to interrogate the impact of the different drug pertubations on these cells, and identify the unique features activated in erlotinib/THZ1 treated PC9 cells following UFM1 loss, we performed Principle Component Analysis (PCA). SgUFM1 PC9 cells were located in a similar position with control PC9 cells at basal status, but after erlotinib treatment both cells moved towards the positive direction in PC1. Interestingly UFM1 knock out PC9 cells were located in a completely different place to control PC9 cells after combination treatment. We therefore also performed GSEA to uncover features associated with PC1 and PC2, which revealed dominant GO terms associated with ER protein localization as the main factor for movement towards the negative direction of PC1 and positive direction of PC2 (Fig. 4C, Supplementary Table S2). Given this observation and the known link between loss of ufmylation and ER stress, we next performed electron microscopy of PC9 sgControl and sgUFM1 persister cells to assess for specific ultrastuctural differences. Indeed, following inhibitor pretreatment we noted that PC9 sgUFM1 cells in particular accumulated abnormal ER with elongated membranes (Fig. 4D).
We therefore assessed whether disruption of ufymylation induces well known markers of ER stress. CRISPR mediated deletion of either UFM1 or UFSP2 increased pIRE1α and splicing of XBP1 (Figs. 4E, F). We further confirmed this result with additional sgRNAs targeting UFM1 and other ufmylation pathway components (Fig. 4G). Since regulation of SQSTM1 (also known as p62) (21) by ufmylation was recently reported, we also examined levels of this autophagy adaptor in control versus UFM1 or UFSP2 knockout cells. Although we did not identify significant changes in baseline levels of p62 in PC9 cells in the absence of ufmylation (Fig. 4E, G), the decrease in p62 levels following erlotinib treatment (38) was blunted following UFM1 or UFSP2 knock out. In contrast, we failed to observe significant activation of PERK or its downstream signaling components, including DDT3 or ATF4 (Supplementary Fig. S4C,D). Morevover, whereas IRE1α was activated at baseline following UFM1 knockout, pPERK was only induced at high tunicamycin concentrations (Supplementary Fig. S4E). Additionally, we could not detect a clear impact of the PERK inhibitor GSK2606414 (39) on downstream signaling, in contrast to the IRE1α inhibitor KIRA6 (40), which suppressed XBP1s levels and impaired DTP cell viability, which correlated with sgRNA data from the screen (Supplementary Figs. S4F-I). Thus, ufmylation regulates specific post-transcriptional stress response pathways that could contribute to survival of erlotinib DTP cells.
ER-stress enhances STING induction in EGFR drug tolerant persister cells
We considered the possibility that STING, an ER resident protein with an increasingly recognized role in inflammation and cancer (41), might link increased inflammatory signaling with ER stress in erlotinib DTP cells. We therefore measured multiple markers of STING signaling in sgControl or sgUFM1 PC9 cells at baseline, or following erlotinib pre-treatment to enrich for DTP cell populations. We observed modest upregulation of STING levels in control DTP cells, which was significantly enhanced following UFM1 knockout (Fig. 5A, B). This induction of STING correlated with activation of NF-κB signaling as measured by p-p65 levels, as well increased expression of IL-6 (Fig. 5A, C). Importantly, while STING levels were increased, and primed to induce pIRF3 in response to the dsDNA mimic poly dAdT in UFM1 knockout PC9 DTP cells, we did not observe significant activation of pIRF3 at baseline (Supplementary Fig. S5A), suggesting preferential activation of pro-survival NF-kB signals. However, given a known role for STING-IRF3 signaling in triggering apoptosis (42, 43), and propensity of these cells to activate IRF3, these data also suggested a precarious balance associated with this cellular state.
Figure 5. STING induction and mitochondrial priming in erlotinib DTP cells.
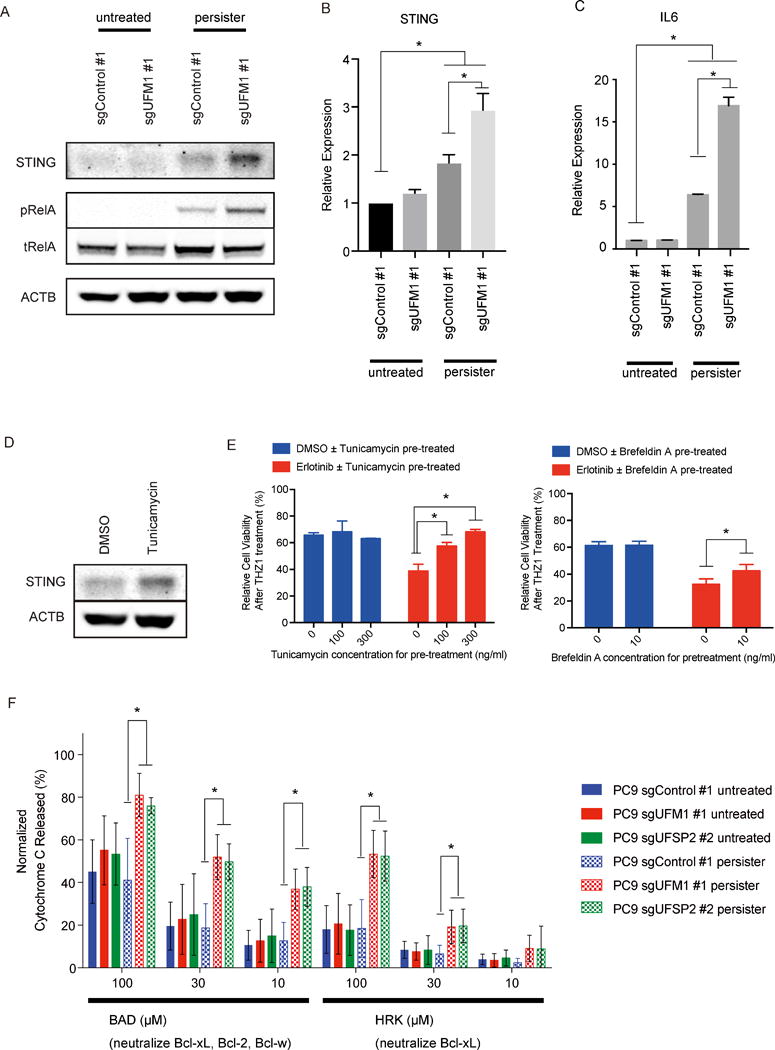
A) Immunobloting analysis of STING in parental or erlotinib DTP PC9 cells transduced with indicated sgRNA. B, C) qRT-PCR of IL6 UFM1 knockout PC9 cells or control PC9 cells. D) Immunobloting analysis of STING in PC9 cells after the incubation with 300 ng/ml of tunicamycin for 24 hr. E) UFM1 or UFSP2 deleted PC9 cells and control PC9 cells were pre-treated with Erlotinib or DMSO for 48 hr. Pre-treatment with ER stress inducer tunicamycin or brefeldin A could attenuate the enhance effect by for following 100 nM THZ1 treatment. F) BH3 profiling revealing alteration of mitochondrial permeability following incubation with the indicated BH3 peptides.
To confirm these results, we next examined the consequences of inducing ER stress in via alternative means. Indeed, pharmacologic treatment of PC9 persister cell populations with tunicamycin also increased STING levels as well as IL-6 (Fig. 5D, Supplementary Figs. S5B and S5C). Consistent with this data, low dose tunicamycin or brefeldin A treatment recapitulated the effect of UFM1 deletion on rescuing the impact of THZ1 treatment on erlotinib DTP cell viability (Fig. 5E, Suppementary Fig. S5D). Taken together, these data reveal that ER stress enhances STING induction in erlotinib DTP cells, with preferential activation of pro-survival NF-κB signals that could support stress tolerance and cell survival.
UFM1 loss also results in enhanced dependency on Bcl-xL
STING induction has also been linked to ER stress induced apoptosis (42, 44). Given the known role of BCL-2 family members in EGFR mediated survival, we wondered if loss of ufmylation in EGFR TKI drug tolerant persister cells might activate specific BCL-2 family members to counterbalance cell death. We first examined whether THZ1 treatment impacted the expression of any BH3 pro-apototic or anti-apoptic family members. We were unable to detect a consistent impact of single agent THZ1 or combined erlotinib/THZ1 treatment on either protein or mRNA levels of these family members in the presence or absence of ufmylation (Supplementary Figs. S5E and S5F).
We next utilized dynamic BH3 profiling as a more sensitive measure to determine whether loss of ufmylation might impact apoptotic priming and dependency on specific BCL-2 family members in persister cells at a post-transcriptional level. Activation of the intrinsic apoptotic pathway results in mitochondrial outer membrane permeability (MOMP) which is regulated by the BCL-2 family of proteins. The balance between anti- and pro-apoptotic BCL-2 family members determines how close to the apoptotic threshold a cell is, termed apoptotic priming, and is a more sensitive indicator as compared with gross changes in BCL2 family member levels or binding interactions (15). Briefly, this assay uses synthetic BH3 peptides derived from the BH3 domain of the pro-apoptotic BH3-only proteins to provoke a response from the mitochondria (12). Apoptotic priming is relative and perturbations to a cell can alter apoptotic priming as measured by promiscuously binding BH3 peptides such as BIM. Perturbations that alter anti-apoptotic dependencies can be determined by enhanced response to the more selective BH3 peptides such as HRK or NOXA which are specific Bcl-xL and MCL-1 antagonists respectively (13, 43, 45). We therefore performed BH3 profiling on UFM1 or UFSP2 knock out PC9 cells or control PC9 cells after 10 day incubation with 100 nM erlotinib (persister) or DMSO treatment, to elucidate whether activation of the UPR pathway alters mitochondrial priming. Interestingly, UFM1 or UFSP2 knock out PC9 cells displayed a unique increased dependency on Bcl-xL after erlotinib exposure, shown by a significant increase in cytochrome c released signal to the HRK BH3 peptide. This was also observed with the BAD BH3 peptide which binds Bcl-xL as well as BCL-2 and BCL-w (Fig. 5F). There was no significant change in dependency to MCL-1 measured by the MS1 peptide (specific MCL-1 antagonist (46)) or overall priming measured by the BIM BH3 peptide, confirming unique dependency on Bcl-xL (Supplementary Fig. S5G).
Bcl-xL inhibition triggers cell death following ER stress induction in EGFR TKI persister cells
To further validate the role of Bcl-xL in mediating survival downstream of UPR pathway activation, and to assess the potential therapeutic implications of this dependency, we next utilized the recently reported Bcl-xL inhibitor, A1331852 to disrupt this critical pro-survival signal (47). Loss of ufmylation in PC9 cells via UFM1 or UFSP2 knockout modestly enhanced sensitivity to A1331852 treatment across a range of doses regardless of erlotinib exposure, supporting a more general dependency on Bcl-xL for survival that becomes particularly critical in erlotinib DTP cells (Fig. 6A). HCC827 cells transduced with sgUFM1 also showed a modest increase in sensitivity to A1331852 treatment compared to sgControl cells (Supplementary Fig S6A). Importantly, treatment of UFM1 or UFSP2 deleted PC9 cells with the selective BCL-2 inhibitor ABT-199 or selective Mcl-1 inhibitor S63845 failed to show a significant difference compared with control PC9 cells, while the dual specificity BCL-2 and Bcl-xL inhibitor ABT-263 showed only a slight differential impact on survival in both PC9 and HCC827 cells (Fig. 6A, Supplementary Fig. S6A). Together, these results confirmed that BCL-XL becomes especially important for cell survival in the absence of ufmylation.
Figure 6. Enhanced dependency on Bcl-xL in the absence of ufmylation.
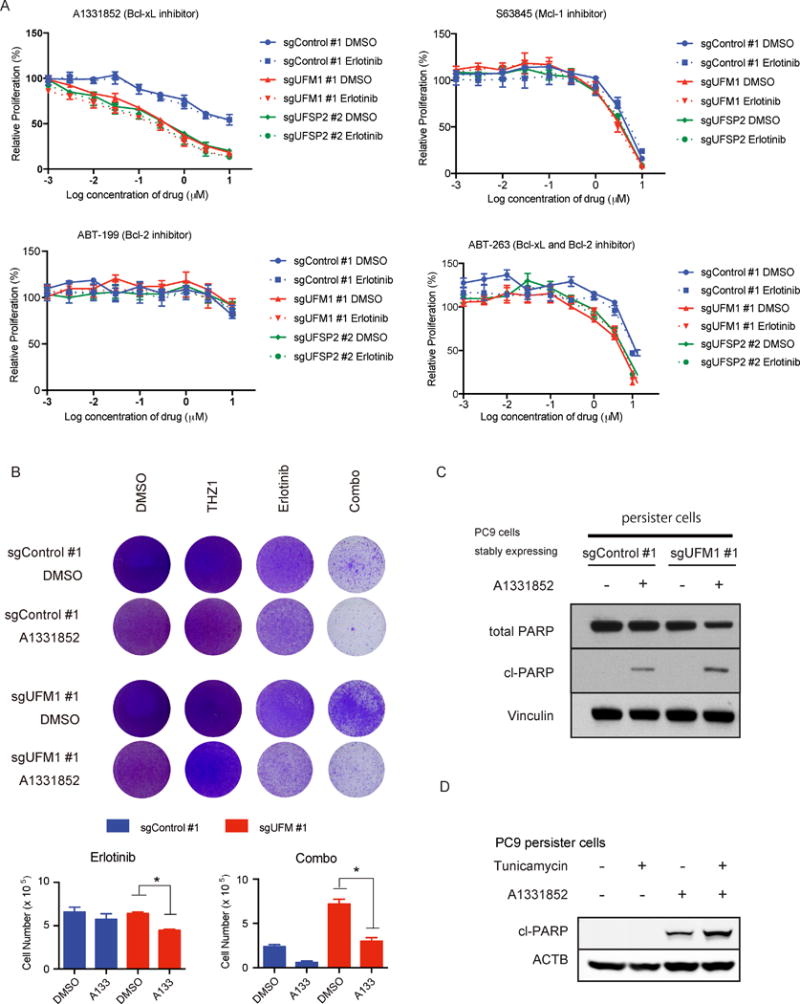
A) Drug sensitivity assay performed with UFM1 or UFSP2 depleted or control PC9 cells. Relative proliferation was measured by cell titer glo assay on day 4 after indicated treatment. B) Colony formation assay at day 18 ± 1 μM A1331852 treatment. PC9 cells transduced with indicated sgRNAs were incubated with DMSO, 50 nM THZ1, 100 nM erlotinib (Erlotinib) or 50 nM THZ1 plus 100 nM erlotinib (Combo). C) PARP immunobloting following 8 hr treatment with 1 μM A1331852 in parental or erlotinib DTP PC9 cells transduced with indicated sgRNAs. D) PARP immunoblot of parental PC9 cells and erlotinib persister PC9 cells were transduced with indicated sgRNAs. Cells were incubated with 300 ng/ml tunicamycin for 24 hr and 300 nM A1331852 for 3 hr before protein extraction.
We next measured the relative impact of A1331852 treatment on erlotinib DTP cells. A1331852 treatment partially suppressed viability of erlotinib only treated cells, while it robustly reversed the impact of UFM1 knockout on rescue of erlotinib/THZ1 combination therapy (Fig. 6B). Importantly selection of PC9 persisters by erlotinib pre-treatment also resulted in increased PARP cleavage following A1331852 exposure, which was enhanced by UFM1 deletion relative to control (Fig. 6C). We further confirmed these results using tunicamycin as a pharmacologic inducer of ER stress, as low dose tunicamycin treatment resulted in enhanced PARP cleavage (Fig. 6D) and viability impairment of PC9 and HCC827 erlotinib persister cells (Supplementary Fig S6B,C).
Thus, although loss of ufmylation and ER stress facilitate EGFR TKI drug tolerance and can bypass transcriptional inhibition, it also creates enhanced dependency on Bcl-xL for mitochondrial protection and cell survival.
Discussion
Despite the remarkable impact of EGFR targeted therapy in lung adenocarcinoma, drug resistance inevitably develops and few patients with advanced disease are cured. It has become increasingly apparent that a subpopulation of DTP cells contributes to relapse. Thus, understanding the multiple cellular adaptations that enable these cells to survive is an important step towards designing effective combination therapies that will enable durable disease control. Here we specifically interrogated mechanisms that promote EGFR TKI resistance of this subpopulation using genome-wide CRISPR/Cas9 enhancer/suppressor screening in EGFR dependent PC9 cells. By focusing on hits in cells treated with erlotinib combined with THZ1, we enriched for genes that specifically enhance or suppress the impact of transcriptional inhibition in drug tolerant persisters. As expected, deletion of multiple components of the transcriptional activation machinery enhanced perister cell loss, consistent with the importance of transcriptional adaptation and mechanism of THZ1 action as a CDK7/12 inhibitor (9). In contrast, in the synergy suppressor group we identified striking enrichment of gene deletion along the ufmylation enzymatic cascade. Mechanistically, ufmylation gene deletion failed to alter canonical downstream EGFR survival signals involving the MAPK or PI3K pathway axes. Instead, we found a key role for regulation of ER homeostasis. Specifically, depletion of this pathway resulted in moderate activation of the UPR and induction of STING, associated with increased pro-survival NF-κB signaling as well as a unique dependence on Bcl-xL mediated mitochondrial protection.
The role of adaptive transcription as a mechanism contributing to drug tolerant persister cell survival has been well described in the context of EGFR dependent PC9 cells. Indeed this mechanism was originally discovered in PC9 cells, which were shown to exhibit this state after erlotinib treatment through histone modification, which could be prevented by HDAC inhibition (3). Hata et al also recently reported a PC9 cell subpopulation of drug tolerant persister cells that express specific adaptive transcriptional responses when compared with parental cells and showed enhanced dependency on both BCL-2 and Bcl-xL, although the mechanistic basis for this phenomenon has remained unclear (5). Here we uncover an additional role for ER stress and activation of the UPR, which have also become increasingly appreciated to play important roles in pro-tumorigenic and immunomodulatory signaling over the last several years (18). While rapid or sustained ER stress induces cell death, tolerable levels of ER stress paradoxically promotes survival of tumor cells. For example, activation of the IRE1α-XBP1 pathway is a key component of tumor initiating cell survival in breast cancer (48). Adaptive activation of the UPR has also been implicated in tumor cell heterogeneity and escape from specific stress in the context of pancreatic cancer (17). However, while the UPR can be activated by intrinsic (e.g. genetic or epigenetic) or extrinsic (e.g. hypoxia, glucose deprivation or lactic acidosis) factors in vivo, further work will be needed to understand how this pathway becomes activated and the specific role of ufmylation in drug tolerant cells in actual tumors (18). Recently Liu et al described that one of the UFM1 adaptor proteins, DDRGK1 can bind and regulate one of the UPR sensor proteins, IRE1α (23). Furthermore Simsek et al found UFM1 can conjugate to some of ribosomal proteins suggesting their involvement in translation regulation (49). These reports highlight the potential that the ufmylation pathway may represent a more general regulatory component of protein quality and quantity, and potentially a novel checkpoint that, when deficient, engages low level ER stress and attempts to maintain cell survival. Furthermore, it is also possible that somatic genetic alterations in this pathway may be selected for in drug tolerant persister cells by emerging combination therapies such as erlotinib/THZ1, and promote acquired resistance.
Notably, we identified upregulation of the ER resident protein STING in PC9 persister cells, which was enhanced by UFM1 knockout or pharmacologic induction of ER stress with tunicamycin. A sensor of cytoplasmic DNA via the intermediate production of cyclic dinucleotides, STING activation is increasingly recognized as an important link between inflammation and carcinogenesis, as well as a potential target for cancer immunotherapy and auto-immune disease (50). Consistent with our findings STING activation has also been recently associated with ER stress in the context of innate immune sensing of specific microbes, promoting stress mediated autophagy of ER membranes (51). Although we also identified upreguation of p62, an autophagy adaptor, a definite link between UFM1 knockout and autophagy induction has remained inconclusive (21). Instead we found that STING and p62 induction co-incided with preferential activation of pro-survival NF-κB signals including IL-6. Furthermore, although we did not detect pIRF3 activation at baseline in these cells, stimulation of sgUFM1 PC9 persister cells with the dsDNA mimic poly dAdT strongly enhanced IRF3 phosphorylation. Taken together, these findings are consistent with a cellular state in which low level STING and p62 activation support ER stress tolerance via NF-κB, but are hypersensitive to excess stimulation by dsDNA and are potentially caught in a precarious balance with pro-death signals (52).
Indeed, to further understand how the UPR promotes survival of in UFM1 deleted PC9 drug tolerant persister cells, we performed BH3 profiling and observed enhanced dependency on Bcl-xL, without an increase of Bcl-xL mRNA or protein levels. We also confirmed that ER stress induced by tunicamycin both rescued erlotinib/THZ1 persister cell depletion and promoted Bcl-xL dependence. Mechanistically, the inability of THZ1 to inhibit BcL-xL transcripts may explain the ability of this state to rescue erlotinib/THZ1 combination therapy. Although others have reported that CDK7 can regulate expression of BCL-2 family members, our findings suggest this to be cell type and context dependent (53). Interestingly, STING-IRF3 signaling has been previously implicated in ER stress mediated hepatocyte apoptosis via direct activation of BAX, potentially explaining this dependency on Bcl-xL (42, 54). Moreover, BCL-2 family members are known to localize not only to mitochondria but also the ER, and these ER localized BCL-2 family member proteins are reported to modulate apoptotic signals through ER mitochondria calcium signaling (55). Importantly leakage of mitochondrial DNA into the cytoplasm is a well known trigger STING induced IRF3 activation and interferon signaling (56), suggesting that increased mitochondrial permeablization following Bcl-XL inhibitions may tip the balance towards IRF3 activation, promoting a feedforward signal that leads towards cell death. Further work will be necessary to dissect in greater detail how these signaling networks directly converge in EGFR drug tolerant persister cells.
The discovery of Bcl-xL as a unique anti-apoptotic effector of this adaptive response may have important clinical implications. A clinical trial combining the non-selective BCL-2/Bcl-xL inhibitor ABT-263 with the 3rd generation EGFR TKI osimerinib is ongoing in EGFR TKI resistant lung cancer patients (NCT02520778) (5). Our findings suggest that more selective targeting of Bcl-xL may provide more optimal on-target activity with an improved therapeutic window. Furthermore, given the renewed focus on eliminating drug tolerant persister cells with transcriptional inhibitors, incorporating intermittent treatment with a selective Bcl-xL inhibitor or driving excessive IRF3 activation with STING agonsists may ultimately represent the most effective way in which to successfully eradicate these tumor subclones and achieve durable clinical activity.
Supplementary Material
Acknowledgments
We are grateful to P. Choi, Y. Mitsuishi, for providing materials, technical instruction and useful discussion, and Molecular Biology Core Facilities at Dana-Farber Cancer Institute for the preparation and sequencing of RNA-seq.
Grant support
This work was supported by NCI-P01 CA154303 (P.S. Hammerman), NCI-R01 CA19632-02 (P.S. Hammerman), NCI-R01 CA190394-01 (D.A. Barbie), the Schaubert, Gloria T. Maheu and Heerwagen Family Funds for Lung Cancer Research (D.A. Barbie), NCI-R01 CA179483 (N.S. Gray) and Lilly oncology fellowship (H. Terai).
Footnotes
Conflict of Interest Statement: D.A.B. is a consultant for N-of-One.
References
- 1.Holohan C, Van Schaeybroeck S, Longley DB, Johnston PG. Cancer drug resistance: an evolving paradigm. Nature reviews Cancer. 2013;13:714–26. doi: 10.1038/nrc3599. [DOI] [PubMed] [Google Scholar]
- 2.Stewart EL, Tan SZ, Liu G, Tsao MS. Known and putative mechanisms of resistance to EGFR targeted therapies in NSCLC patients with EGFR mutations-a review. Transl Lung Cancer Res. 2015;4:67–81. doi: 10.3978/j.issn.2218-6751.2014.11.06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sharma SV, Lee DY, Li B, Quinlan MP, Takahashi F, Maheswaran S, et al. A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell. 2010;141:69–80. doi: 10.1016/j.cell.2010.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lee HJ, Zhuang G, Cao Y, Du P, Kim HJ, Settleman J. Drug resistance via feedback activation of Stat3 in oncogene-addicted cancer cells. Cancer cell. 2014;26:207–21. doi: 10.1016/j.ccr.2014.05.019. [DOI] [PubMed] [Google Scholar]
- 5.Hata AN, Niederst MJ, Archibald HL, Gomez-Caraballo M, Siddiqui FM, Mulvey HE, et al. Tumor cells can follow distinct evolutionary paths to become resistant to epidermal growth factor receptor inhibition. Nature medicine. 2016;22:262–9. doi: 10.1038/nm.4040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Obenauf AC, Zou Y, Ji AL, Vanharanta S, Shu W, Shi H, et al. Therapy-induced tumour secretomes promote resistance and tumour progression. Nature. 2015 doi: 10.1038/nature14336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zawistowski JS, Bevill SM, Goulet DR, Stuhlmiller TJ, Beltran AS, Olivares-Quintero JF, et al. Enhancer Remodeling during Adaptive Bypass to MEK Inhibition Is Attenuated by Pharmacologic Targeting of the P-TEFb Complex. Cancer discovery. 2017;7:302–21. doi: 10.1158/2159-8290.CD-16-0653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Johannessen CM, Johnson LA, Piccioni F, Townes A, Frederick DT, Donahue MK, et al. A melanocyte lineage program confers resistance to MAP kinase pathway inhibition. Nature. 2013;504:138–42. doi: 10.1038/nature12688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kwiatkowski N, Zhang T, Rahl PB, Abraham BJ, Reddy J, Ficarro SB, et al. Targeting transcription regulation in cancer with a covalent CDK7 inhibitor. Nature. 2014;511:616–20. doi: 10.1038/nature13393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bradner JE, Hnisz D, Young RA. Transcriptional Addiction in Cancer. Cell. 2017;168:629–43. doi: 10.1016/j.cell.2016.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rusan M, Li K, Li Y, Christensen CL, Abraham BJ, Kwiatkowski N, et al. Suppression of adaptive responses to targeted cancer therapy by transcriptional repression. Cancer discovery. 2017 doi: 10.1158/2159-8290.CD-17-0461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ryan J, Montero J, Rocco J, Letai A. iBH3: simple, fixable BH3 profiling to determine apoptotic priming in primary tissue by flow cytometry. Biol Chem. 2016;397:671–8. doi: 10.1515/hsz-2016-0107. [DOI] [PubMed] [Google Scholar]
- 13.Certo M, Del Gaizo Moore V, Nishino M, Wei G, Korsmeyer S, Armstrong SA, et al. Mitochondria primed by death signals determine cellular addiction to antiapoptotic BCL-2 family members. Cancer cell. 2006;9:351–65. doi: 10.1016/j.ccr.2006.03.027. [DOI] [PubMed] [Google Scholar]
- 14.Barbie DA, Tamayo P, Boehm JS, Kim SY, Moody SE, Dunn IF, et al. Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature. 2009;462:108–12. doi: 10.1038/nature08460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Potter DS, Letai A. To Prime, or Not to Prime: That Is the Question. Cold Spring Harb Symp Quant Biol. 2016 doi: 10.1101/sqb.2016.81.030841. [DOI] [PubMed] [Google Scholar]
- 16.Costa DB, Halmos B, Kumar A, Schumer ST, Huberman MS, Boggon TJ, et al. BIM mediates EGFR tyrosine kinase inhibitor-induced apoptosis in lung cancers with oncogenic EGFR mutations. PLoS medicine. 2007;4:1669–79. doi: 10.1371/journal.pmed.0040315. discussion 80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Genovese G, Carugo A, Tepper J, Robinson FS, Li L, Svelto M, et al. Synthetic vulnerabilities of mesenchymal subpopulations in pancreatic cancer. Nature. 2017;542:362–6. doi: 10.1038/nature21064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cubillos-Ruiz JR, Bettigole SE, Glimcher LH. Tumorigenic and Immunosuppressive Effects of Endoplasmic Reticulum Stress in Cancer. Cell. 2017;168:692–706. doi: 10.1016/j.cell.2016.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang Y, Zhang M, Wu J, Lei G, Li H. Transcriptional regulation of the Ufm1 conjugation system in response to disturbance of the endoplasmic reticulum homeostasis and inhibition of vesicle trafficking. PloS one. 2012;7:e48587. doi: 10.1371/journal.pone.0048587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tatsumi K, Sou YS, Tada N, Nakamura E, Iemura S, Natsume T, et al. A novel type of E3 ligase for the Ufm1 conjugation system. The Journal of biological chemistry. 2010;285:5417–27. doi: 10.1074/jbc.M109.036814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.DeJesus R, Moretti F, McAllister G, Wang Z, Bergman P, Liu S, et al. Functional CRISPR screening identifies the ufmylation pathway as a regulator of SQSTM1/p62. Elife. 2016;5 doi: 10.7554/eLife.17290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cai Y, Pi W, Sivaprakasam S, Zhu X, Zhang M, Chen J, et al. UFBP1, a Key Component of the Ufm1 Conjugation System, Is Essential for Ufmylation-Mediated Regulation of Erythroid Development. PLoS Genet. 2015;11:e1005643. doi: 10.1371/journal.pgen.1005643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu J, Wang Y, Song L, Zeng L, Yi W, Liu T, et al. A critical role of DDRGK1 in endoplasmic reticulum homoeostasis via regulation of IRE1alpha stability. Nature communications. 2017;8:14186. doi: 10.1038/ncomms14186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wei Y, Xu X. UFMylation: A Unique & Fashionable Modification for Life. Genomics Proteomics Bioinformatics. 2016;14:140–6. doi: 10.1016/j.gpb.2016.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang T, Yu H, Hughes NW, Liu B, Kendirli A, Klein K, et al. Gene Essentiality Profiling Reveals Gene Networks and Synthetic Lethal Interactions with Oncogenic Ras. Cell. 2017;168:890–903.e15. doi: 10.1016/j.cell.2017.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Berns K, Bernards R. Understanding resistance to targeted cancer drugs through loss of function genetic screens. Drug Resist Updat. 2012;15:268–75. doi: 10.1016/j.drup.2012.10.002. [DOI] [PubMed] [Google Scholar]
- 27.Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, Mikkelsen TS, et al. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science. 2014;343:84–7. doi: 10.1126/science.1247005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Doench JG, Fusi N, Sullender M, Hegde M, Vaimberg EW, Donovan KF, et al. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nature biotechnology. 2016;34:184–91. doi: 10.1038/nbt.3437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Krall EB, Wang B, Munoz DM, Ilic N, Raghavan S, Niederst MJ, et al. KEAP1 loss modulates sensitivity to kinase targeted therapy in lung cancer. Elife. 2017;6 doi: 10.7554/eLife.18970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kokubo Y, Gemma A, Noro R, Seike M, Kataoka K, Matsuda K, et al. Reduction of PTEN protein and loss of epidermal growth factor receptor gene mutation in lung cancer with natural resistance to gefitinib (IRESSA) Br J Cancer. 2005;92:1711–9. doi: 10.1038/sj.bjc.6602559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.de Bruin EC, Cowell C, Warne PH, Jiang M, Saunders RE, Melnick MA, et al. Reduced NF1 expression confers resistance to EGFR inhibition in lung cancer. Cancer discovery. 2014;4:606–19. doi: 10.1158/2159-8290.CD-13-0741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang T, Kwiatkowski N, Olson CM, Dixon-Clarke SE, Abraham BJ, Greifenberg AK, et al. Covalent targeting of remote cysteine residues to develop CDK12 and CDK13 inhibitors. Nature chemical biology. 2016;12:876–84. doi: 10.1038/nchembio.2166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Visel A, Blow MJ, Li Z, Zhang T, Akiyama JA, Holt A, et al. ChIP-seq accurately predicts tissue-specific activity of enhancers. Nature. 2009;457:854–8. doi: 10.1038/nature07730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Whyte WA, Orlando DA, Hnisz D, Abraham BJ, Lin CY, Kagey MH, et al. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell. 2013;153:307–19. doi: 10.1016/j.cell.2013.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yang Gao TZ, Terai Hideki, Ficarro Scott B, Kwiatkowski Nicholas, Hao Ming-Feng, Sharma Bandana, Christensen Camilla L, Chipumuro Edmond, Wong Kwok-kin, Marto Jarrod A, Hammerman Peter S, Gray Nathanael S, George Rani E. Overcoming resistance to the THZ series of covalent transcriptional CDK inhibitors. Cell Chemical Biology. 2017 doi: 10.1016/j.chembiol.2017.11.007. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wu J, Lei G, Mei M, Tang Y, Li H. A novel C53/LZAP-interacting protein regulates stability of C53/LZAP and DDRGK domain-containing Protein 1 (DDRGK1) and modulates NF-kappaB signaling. The Journal of biological chemistry. 2010;285:15126–36. doi: 10.1074/jbc.M110.110619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tricker EM, Xu C, Uddin S, Capelletti M, Ercan D, Ogino A, et al. Combined EGFR/MEK Inhibition Prevents the Emergence of Resistance in EGFR-Mutant Lung Cancer. Cancer discovery. 2015;5:960–71. doi: 10.1158/2159-8290.CD-15-0063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wei Y, Zou Z, Becker N, Anderson M, Sumpter R, Xiao G, et al. EGFR-mediated Beclin 1 phosphorylation in autophagy suppression, tumor progression, and tumor chemoresistance. Cell. 2013;154:1269–84. doi: 10.1016/j.cell.2013.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jiang X, Wei Y, Zhang T, Zhang Z, Qiu S, Zhou X, et al. Effects of GSK2606414 on cell proliferation and endoplasmic reticulum stressassociated gene expression in retinal pigment epithelial cells. Mol Med Rep. 2017;15:3105–10. doi: 10.3892/mmr.2017.6418. [DOI] [PubMed] [Google Scholar]
- 40.Ghosh R, Wang L, Wang ES, Perera BG, Igbaria A, Morita S, et al. Allosteric inhibition of the IRE1alpha RNase preserves cell viability and function during endoplasmic reticulum stress. Cell. 2014;158:534–48. doi: 10.1016/j.cell.2014.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dou Z, Ghosh K, Vizioli MG, Zhu J, Sen P, Wangensteen KJ, et al. Cytoplasmic chromatin triggers inflammation in senescence and cancer. Nature. 2017;550:402–6. doi: 10.1038/nature24050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Petrasek J, Iracheta-Vellve A, Csak T, Satishchandran A, Kodys K, Kurt-Jones EA, et al. STING-IRF3 pathway links endoplasmic reticulum stress with hepatocyte apoptosis in early alcoholic liver disease. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:16544–9. doi: 10.1073/pnas.1308331110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Letai A, Bassik MC, Walensky LD, Sorcinelli MD, Weiler S, Korsmeyer SJ. Distinct BH3 domains either sensitize or activate mitochondrial apoptosis, serving as prototype cancer therapeutics. Cancer cell. 2002;2:183–92. doi: 10.1016/s1535-6108(02)00127-7. [DOI] [PubMed] [Google Scholar]
- 44.Iracheta-Vellve A, Petrasek J, Gyongyosi B, Satishchandran A, Lowe P, Kodys K, et al. Endoplasmic Reticulum Stress-induced Hepatocellular Death Pathways Mediate Liver Injury and Fibrosis via Stimulator of Interferon Genes. The Journal of biological chemistry. 2016;291:26794–805. doi: 10.1074/jbc.M116.736991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kuwana T, Bouchier-Hayes L, Chipuk JE, Bonzon C, Sullivan BA, Green DR, et al. BH3 domains of BH3-only proteins differentially regulate Bax-mediated mitochondrial membrane permeabilization both directly and indirectly. Molecular cell. 2005;17:525–35. doi: 10.1016/j.molcel.2005.02.003. [DOI] [PubMed] [Google Scholar]
- 46.Foight GW, Ryan JA, Gulla SV, Letai A, Keating AE. Designed BH3 peptides with high affinity and specificity for targeting Mcl-1 in cells. ACS chemical biology. 2014;9:1962–8. doi: 10.1021/cb500340w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Leverson JD, Phillips DC, Mitten MJ, Boghaert ER, Diaz D, Tahir SK, et al. Exploiting selective BCL-2 family inhibitors to dissect cell survival dependencies and define improved strategies for cancer therapy. Sci Transl Med. 2015;7:279ra40. doi: 10.1126/scitranslmed.aaa4642. [DOI] [PubMed] [Google Scholar]
- 48.Chen X, Iliopoulos D, Zhang Q, Tang Q, Greenblatt MB, Hatziapostolou M, et al. XBP1 promotes triple-negative breast cancer by controlling the HIF1alpha pathway. Nature. 2014;508:103–7. doi: 10.1038/nature13119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Simsek D, Tiu GC, Flynn RA, Byeon GW, Leppek K, Xu AF, et al. The Mammalian Ribo-interactome Reveals Ribosome Functional Diversity and Heterogeneity. Cell. 2017;169:1051–65.e18. doi: 10.1016/j.cell.2017.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Barber GN. STING: infection, inflammation and cancer. Nat Rev Immunol. 2015;15:760–70. doi: 10.1038/nri3921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Moretti J, Roy S, Bozec D, Martinez J, Chapman JR, Ueberheide B, et al. STING Senses Microbial Viability to Orchestrate Stress-Mediated Autophagy of the Endoplasmic Reticulum. Cell. 2017;171:809–23.e13. doi: 10.1016/j.cell.2017.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gaidt MM, Ebert TS, Chauhan D, Ramshorn K, Pinci F, Zuber S, et al. The DNA Inflammasome in Human Myeloid Cells Is Initiated by a STING-Cell Death Program Upstream of NLRP3. Cell. 2017 doi: 10.1016/j.cell.2017.09.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cayrol F, Praditsuktavorn P, Fernando TM, Kwiatkowski N, Marullo R, Calvo-Vidal MN, et al. THZ1 targeting CDK7 suppresses STAT transcriptional activity and sensitizes T-cell lymphomas to BCL2 inhibitors. Nature communications. 2017;8:14290. doi: 10.1038/ncomms14290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Warner JD, Irizarry-Caro RA, Bennion BG, Ai TL, Smith AM, Miner CA, et al. STING-associated vasculopathy develops independently of IRF3 in mice. J Exp Med. 2017 doi: 10.1084/jem.20171351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Marchi S, Giorgi C, Oparka M, Duszynski J, Wieckowski MR, Pinton P. Oncogenic and oncosuppressive signal transduction at mitochondria-associated endoplasmic reticulum membranes. Mol Cell Oncol. 2014;1:e956469. doi: 10.4161/23723548.2014.956469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.West AP, Khoury-Hanold W, Staron M, Tal MC, Pineda CM, Lang SM, et al. Mitochondrial DNA stress primes the antiviral innate immune response. Nature. 2015;520:553–7. doi: 10.1038/nature14156. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.