

Abstract
Purpose
Vandetanib is well-tolerated in patients with advanced medullary thyroid carcinoma (MTC). Long-term outcomes and mechanisms of MTC progression have not been reported previously.
Design
We monitored toxicities and disease status in patients taking vandetanib for hereditary, advanced MTC. Tumor samples were analyzed for molecular mechanisms of disease progression.
Findings
Seventeen patients (8 male, age 13 (9–17)* years) enrolled; 16 had a RET p.Met918Thr germline mutation. The duration of vandetanib therapy was 6.1 (0.1–9.7+)* years with treatment ongoing in nine patients. Best response was partial response (PR) in ten, stable disease (SD) in six, and progressive disease (PD) in one patient. Duration of response was 7.4 (0.6–8.7+)* and 4.9 (0.6–7.8+)* years in patients with PR and SD, respectively. Six patients died 2.0 (0.4–5.7)* years after progression. Median progression free survival (PFS) was 6.7 years (95% CI: 2.3 years-undefined) and 5-year overall survival (OS) was 88.2% (95% CI 60.6–96.9%). Of 16 patients with a RET p.Met918Thr mutation, progression free survival was 6.7 years (95% CI 3.1-undefined) and 5-year overall survival was 93.8% (95% CI 63.2–99.1%). No patients terminated treatment because of toxicity. DNA sequencing of tissue samples (n=11) identified an increase in copy number alterations across the genome as a potential mechanism of drug resistance.
Conclusion
This study demonstrates that vandetanib is safe and results in sustained responses in children and adolescents with hereditary MTC. Our preliminary molecular data suggest that an increase in copy number abnormalities may be associated with tumor progression in hereditary MTC patients treated with vandetanib.
*median (range)
INTRODUCTION
Multiple Endocrine Neoplasia (MEN) Type II includes two distinct genetic cancer predisposition syndromes, type IIA (MEN2A) and type IIB (MEN2B), caused by germline, activating mutations in the REarranged during Transfection (RET) proto-oncogene. MEN2A is associated with mutations in extracellular cysteine residues, and MEN2B is associated with the p.Met918Thr point mutation in the kinase domain of the RET protein(1). Nearly all patients with MEN2B develop medullary thyroid carcinoma (MTC) within the first few years of life and most succumb to metastatic disease as adolescents or young adults(2).
Based on an improvement in progression free survival in pivotal phase III studies RET-targeting tyrosine kinase inhibitors (TKIs), vandetanib and cabozantinib, have been approved for the treatment of patients with advanced MTC(3, 4). Vandetanib is a potent, orally bioavailable, small-molecule inhibitor of RET, VEGFR and other related receptor tyrosine kinases with inhibitory concentrations in the low nanomolar range(5).
Initial studies of vandetanib in adults with metastatic breast cancer(6), relapsed/refractory solid tumors(7), multiple myeloma(8), non-small cell lung cancer(9) and advanced MTC(10) provided a robust toxicity profile, maximum tolerated dose (MTD) of 100–300 mg/day, and pharmacokinetic characterization supportive of once daily dosing. In our phase I/II trial of vandetanib for children with hereditary MTC (NCT00514046) we showed that 100 mg/m2/day of vandetanib administered on a continuous dosing schedule induced partial disease remission(11).
Most patients who respond to treatment with vandetanib eventually develop progressive disease(12). Several studies have explored RET, RAS, and other genomic alterations in MTC(13–15), yet few address mechanisms of disease progression. Previously implicated mechanisms of disease progression in MTC include RET mutations that impair TKI binding(16, 17), activity of micro RNAs(18), mTOR pathway co-activation(19), and compound RET mutations(20).
Here, we present an updated analysis of a preliminary phase I/II trial with MEN2B and advanced MTC with a data cutoff of July 2017(11). We analyze the long-term outcomes of pediatric patients and identify clinical and biologic markers of response, progression, and TKI resistance. We performed genomic and transcriptomic analyses of tumor samples from patients who experienced progressive disease while on TKI therapy. Additionally, we performed an in-depth analysis on longitudinal tumor biopsies of a single patient who experienced progressive disease despite vandetanib and cabozantinib treatment.
METHODS
Patients
Vandetanib was provided by AstraZeneca for the phase I/II clinical trial. Sanofi/Genzyme continues to provide vandetanib to the ongoing protocol.
The design, methods, and objectives of this single institution phase I/II single-arm study (NCT00514046) were previously described(11) and are briefly summarized. The trial enrolled July 2007 to October 2012. Patients 5 to 18 years of age with measurable, locally advanced or metastatic, hereditary MTC were eligible for participation in the trial. Other eligibility criteria included recovery from toxic effects of prior therapy and adequate performance score and organ function. All patients were enrolled in a National Cancer Institute, Pediatric Oncology Branch MTC natural history study (NCT01660984) to allow for longitudinal follow up. The data in the primary report was censored July 2011.
Both the phase I/II and the natural history protocols conformed to the Declaration of Helsinki, Good Clinical Practice guidelines, and were approved by the NCI Institutional Review Board. All patients or their legal guardians signed a document of informed consent indicating their understanding of the investigational nature and risks of this study. Assent was obtained per institutional guidelines.
Clinical Trial Procedures
Patients received oral vandetanib at two dose levels within the 100–300 mg/m2/d dose range once daily, continuously (in 28-day cycles). A standard 3+3 dose escalation design was followed in age groups 13–18 years and 5–12 years(21). Dose was calculated based on body-surface area using a dosing nomogram. The recommended phase II dose in the absence of dose limiting toxicity was determined as 100 mg/m2/day(11). All patients in a subsequent expansion cohort were treated at this dose level.
Tumor response was quantified using RECIST v1.0(22) as the primary endpoint. Biomarker responses measuring serum levels of calcitonin and carcinoembryonic antigen (CEA) were secondary endpoints. Tumor response was assessed every 2 months (two cycles) until cycle 8 and every 4 months thereafter. In July 2013, after the FDA approval of vandetanib for adults with advanced MTC and after report of our initial trial results(11), we amended the protocol to increase interval tumor response measurements to every 6 months and to allow patients who experienced disease progression by RECIST after achieving a partial response to continue study treatment if there was ongoing clinical benefit (i.e. decrease in MTC-related diarrhea, constipation, and/or relief of pain). In some patients, alternative treatments were discussed by the investigators and offered if available.
The NCI CTEP Common Terminology Criteria for Adverse Events Version 3.0 was used to quantify severity of adverse events. Toxicity monitoring included physical exams, laboratory tests, and serial imaging to quantify growth plate volume. Patients were assessed weekly during cycle 1, biweekly until cycle 4, and then during each tumor response evaluation. The 2013 protocol amendment also included less stringent toxicity reporting criteria (to record only grade 3 or higher toxicities or those causing a medication hold) and allowed for flexibility in dosing schedule to accommodate grade 1/2 toxicities. Treatment limiting toxicity was defined as any adverse event possibly, probably, or definitely attributed to vandetanib therapy and required the dose be held, decreased, or stopped.
Endpoints included assessment of antitumor activity as measured by tumor response by RECIST, duration of response, progression-free survival (PFS), overall survival (OS), biomarker response (percent change in calcitonin and CEA from baseline and doubling time) and determination of the safety and tolerability of vandetanib. Tumor biopsies were optional. In addition, analysis of tumor samples from patients enrolled on the NCI Pediatric Oncology Branch MTC natural history study (NCT01660984) was performed.
DNA and RNA Sequencing
Formalin fixed paraffin embedded (FFPE) blocks or unstained slides were collected at the time of diagnosis, prior to therapy, and/or at disease progression when available. Coded samples were compiled and annotated with histological diagnosis and clinical information. Quality control was performed on all samples to ensure the match of tumor and normal pairs.
DNA and RNA samples from tumor and adjacent normal tissue (either unaffected thyroid or lymph node) were extracted from FFPE blocks or unstained slides as previously described(23). Approximately 200 nanograms of DNA was used for library preparation with the Kapa Hyper Prep Kit. Custom gene capture and paired end sequencing was performed as previously described(23). Approximately 500 nanograms of DNA and 500 nanograms of RNA from selected samples (patient #8) were subject to whole exome sequencing and whole transcriptome sequencing. FFPE exome libraries were prepared by Genomics Lab using Agilent SureSelectXT Human All Exon V5 plus UTR target enrichment kit. Exome samples were pooled and run on a HiSeq3000 with Illumina TruSeq V4 chemistry. RNA-seq libraries were prepared with the TruSeq Stranded Total RNA Sample Preparation kit and sequenced on one HiSeq2500 lane using Illumina TruSeq v4 chemistry.
Data analysis was accomplished using the Pipeliner tools package available from The Center for Cancer Research Collaborative Bioinformatics Resource (https://bioinformatics.cancer.gov/). After removal of duplicate reads, local realignment was performed with GATK version 3.5 and somatic point mutations, insertions, and deletions were called using the union of the calls of Strelka and Mutect version 2 bioinformatics platforms(24). To further refine the accuracy of the calls, raw variants were further filtered by requiring them to be nonsynonomous as well as restricted only to areas of the genome designed to be captured in the assay. For somatic calls, a minimum total coverage depth of 5 reads and a variant allele frequency of greater than 5% was required. DNA copy number (CN) discovery was performed using cnvKit and further visualized using NEXUS Copy Number Version 9.0 from BioDiscovery, Inc (25). Whole transcriptome sequencing files were assessed for quality and mapped using STAR(26). Resulting fastq files were analyzed by TopHat2 and Cufflinks using Partek Flow version 6.0 and samples were compared using fragments per kilobase of transcript per Million (FPKM) values(27, 28). DNA and RNA correlations were performed using Log2(Ratio) and Log2(FPKM) values, respectively.
All sequencing files reported were uploaded to dbGaP (https://www.ncbi.nlm.nih.gov/gap) to promote ongoing discovery.
Statistical Analysis
Time-to-event data were analyzed using the Kaplan-Meier method to estimate median event times, and are reported with two-sided 95% confidence intervals (CIs). Median duration of follow-up was estimated using the reverse Kaplan-Meier method. Duration of PR and duration of SD were determined using the Kaplan-Meier method. Duration of response for patients with a PR was measured from the date of PR until date of progression, censoring patients if they had not progressed as of July 1, 2017; duration of SD was measured from enrollment date until date of progression, censoring patients if they had not progressed by July 1, 2017. A time-varying covariate analysis was performed using a Cox proportional hazards model, along with a landmark analysis beginning at the date the last PR was noted, to assess the association between response and stable disease on overall survival. Rate of change (slope) and doubling time for calcitonin and CEA were calculated as previously reported(29, 30). Comparisons of baseline clinical values were made among groups of patients based on response using the Kruskal-Wallis test and the Jonckheere-Terpstra test for trend. Other reported values herein are medians, p-values were two-tailed, and reported without adjustment for multiple comparisons.
RESULTS
The first patient was enrolled on July 13, 2007 and the last patient was enrolled on October 17, 2012. For this report, the data cutoff was July 1, 2017. Seventeen patients (8 male, median age 13 years, range 9–17) enrolled. Sixteen had a RET p.Met918Thr germline mutation and one patient had a unique RET p.Ser836Ser/p.Gly691Ser/p.Ser904Ser/p.Leu769Leu mutation (Supplementary Table 1). The median duration of treatment was 6.1 years (range 0.1–9.7 years) and is ongoing in nine patients, seven of whom have not experienced disease progression.
All patients were evaluable for response. Ten (58.8%) achieved a partial response (PR) and six (35.3%) had stable disease (SD) as best response (Figure 1A). Seven patients maintained their best response (PR or SD) at the data cutoff (Figure 1A). The median time to first documented PR was 0.79 years (range, 0.45– 2.41 years) (Figure 1B). Responses were sustained for a median of 7.4 years (range, 0.6–8.7+) in patients achieving best response of PR and 4.9 years (range, 0.6–7.8+) in patients achieving best response of SD.
Figure 1. Response and survival analysis of patients on vandetanib.

A) Waterfall plot of best percent change in target lesions from baseline for 17 patients. Colors correspond to patient response at data cutoff and symbols indicate criteria for progression. B) Duration of initial response or stable disease and of ongoing vandetanib treatment in patients. Arrows indicate continued follow up at data cutoff. C) Kaplan-Meier plot of progression-free survival of 16 patients harboring the p.Met918Thr RET mutation. 8 patients were censored, 6 of whom remain in follow-up for progression-free survival. D) Kaplan-Meier plot of overall survival of 16 patients harboring the p.Met918Thr RET mutation. 11 patients were censored and remain in follow up on a natural history protocol. E) Kaplan-Meier landmark analysis from median time to PR of 16 patients harboring the p.Met918Thr RET mutation.
Median progression free survival (PFS) was 6.7 years (95% CI: 2.3 years-undefined) and 5-year overall survival (OS) was 88.2% (95% CI 60.6–96.9%). A single patient with a unique RET p.Ser836Ser/p.Gly691Ser/p.Ser904Ser/p.Leu769Leu mutation had rapid disease progression after one month of therapy. Of the 16 patients with the RET p.Met918Thr germline mutation, median follow-up was 8.0 years, the median PFS was 6.7 years (95% CI 3.1-undefined), and median OS was not reached (Figure 1C, 1D). The five-year OS probability was 93.8% (95% CI: 63.2–99.1%) and the seven-year OS probability was 72.3% (95% CI 41.5–88.7%) (Figure 1D). The hazard ratio from a time-varying covariate analysis was 0.64 (95% CI 0.10–3.93, p = 0.63) comparing patients achieving PR versus SD as best response, indicating no association between response and duration of survival. Landmark Kaplan-Meier analysis beginning 879 days (2.4 years) after enrollment, to reflect the time until the last patient was noted to respond, similarly did not show survival advantage in patients that achieved PR versus SD (p = 0.64, figure 1E). The single patient harboring a non-MEN2B, RET mutation (#3) experienced tumor progression and succumbed to disease.
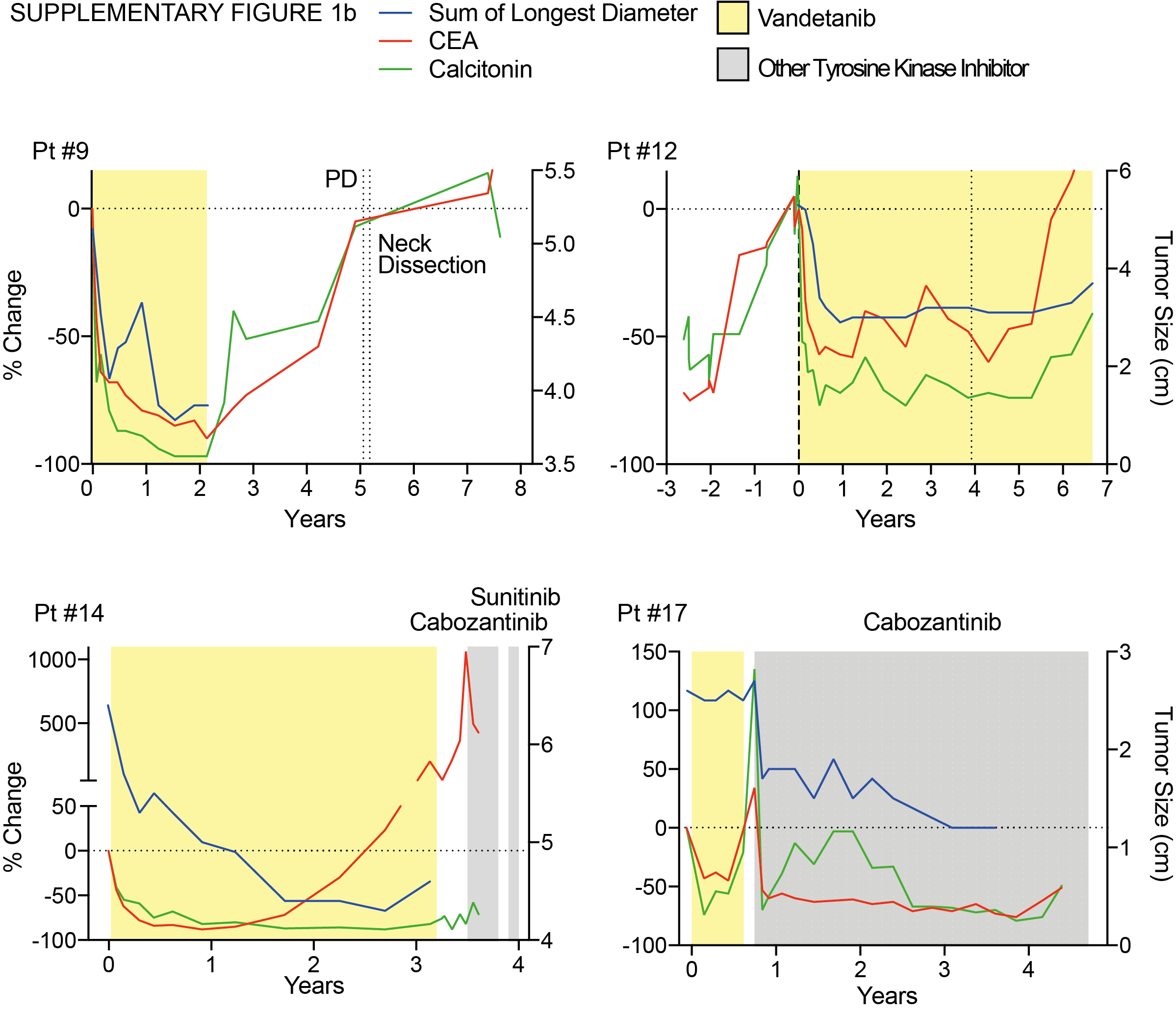
Ten patients developed progressive disease (PD) on vandetanib. Five patients experienced PD with increase in non-target lesions or previously unidentified lesions (#3, #4, #7, #8, #14), four patients experienced an increase in target lesions (#1, #6, #9, #12), and one patient experienced increase in tumor markers concerning for occult progression (#17). Sites of PD are illustrated in Figure 2 and show a new inguinal lesion (pt #7)(A–C), a prostate lesion (pt #8) (D–F), and increasing size of a lesion within the thyroid bed (pt #14) (G–I). Seven patients stopped vandetanib after experiencing PD and patient #9 discontinued due to personal preference (Table 1). Patient #6 discontinued vandetanib for a surgical procedure and did not resume therapy by the data cutoff and patient #12 continues vandetanib despite experiencing PD (Table 1). Treatments after stopping vandetanib are also summarized in Table 1. One patient did not receive subsequent medical therapy directed at the MTC (#3), five patients did not achieve objective responses on subsequent therapies (#1, #4, #7, #9, #14), two patients continued enrollment on the vandetanib protocol due to clinical benefit (#6, #12), and two patients (#8, #17) responded to a second TKI (Table 1, Supplementary Figure 1a–c). Four patients that experienced PD were alive after 0.4, 0.9, 3.0 and 4.1 years (#6, #9, #12, #17), respectively. Six patients with PD experienced death from metastatic disease after a median duration of 2.0 years (range 0.4–5.7 years). The Kaplan-Meier estimate of median OS from the date of progression for the 10 patients who progressed was 3.0 years (95% CI: 0.4–5.7 years).
Figure 2. Radiographic responses and progression in children and adolescents with MEN2B and MTC.

A–C) Computed tomography (CT) of the pelvis of patient #7, a male who achieved SD and subsequently experienced PD in a new lesion. D–F) CT of the prostate of patient #8, a male who achieved SD and subsequently experienced PD in a previously unrecognized lesion. G–I) T2-weighted MRI of the neck of patient #14, a female whom achieved PR and subsequently experienced PD in the thyroid bed.
Table 1.
Patient Course and Management After Disease Progression
| Patient | Response | Site(s) of Progression |
Treatment(s) and duration (years) after Vandetanib |
Time after Stopping Vandetanib (years) |
|---|---|---|---|---|
| 3 | PD | Non-target- cervical spine lesion* | None | 0.4# |
| 7 | SD -> PD | Non-target– groin | Cabozantinib (0.44), Sunitinib (0.8), Sorafenib (1.35) | 3.1# |
| 8 | Previously unidentified–prostate* | Cabozantinib (0.79) | 1.1# | |
| 9 | Target- neck | Neck Dissection/Debulking (5.14) | 5.9 | |
| 17 | Elevation in CEA/Calcitonin | Cabozantinib (3.21) | 4.1 | |
| 1 | PR -> PD | Target - liver | Cabozantinib (1.34), Sunitinib (2.33) | 4.1# |
| 4 | Non-target– mediastinum | Sorafenib (0.15), Cabozantinib (0.22), Cabozantinib (0.61), Sunitinib (1.07) | 3.1# | |
| 6 | Target- neck, chest | None | 1.73 | |
| 12 | Target- mediastinum | None | 0ϕ | |
| 14 | Non-target– neck* | Sunitinib (0.13), Cabozantinib (0.15) | 0.7# |
Progressive Disease (PD), Stable Disease (SD), Partial Response (PR), Carcinoembryonic Antigen (CEA)
Present on baseline imaging,
Death from Disease,
Still taking vandetanib at data cutoff
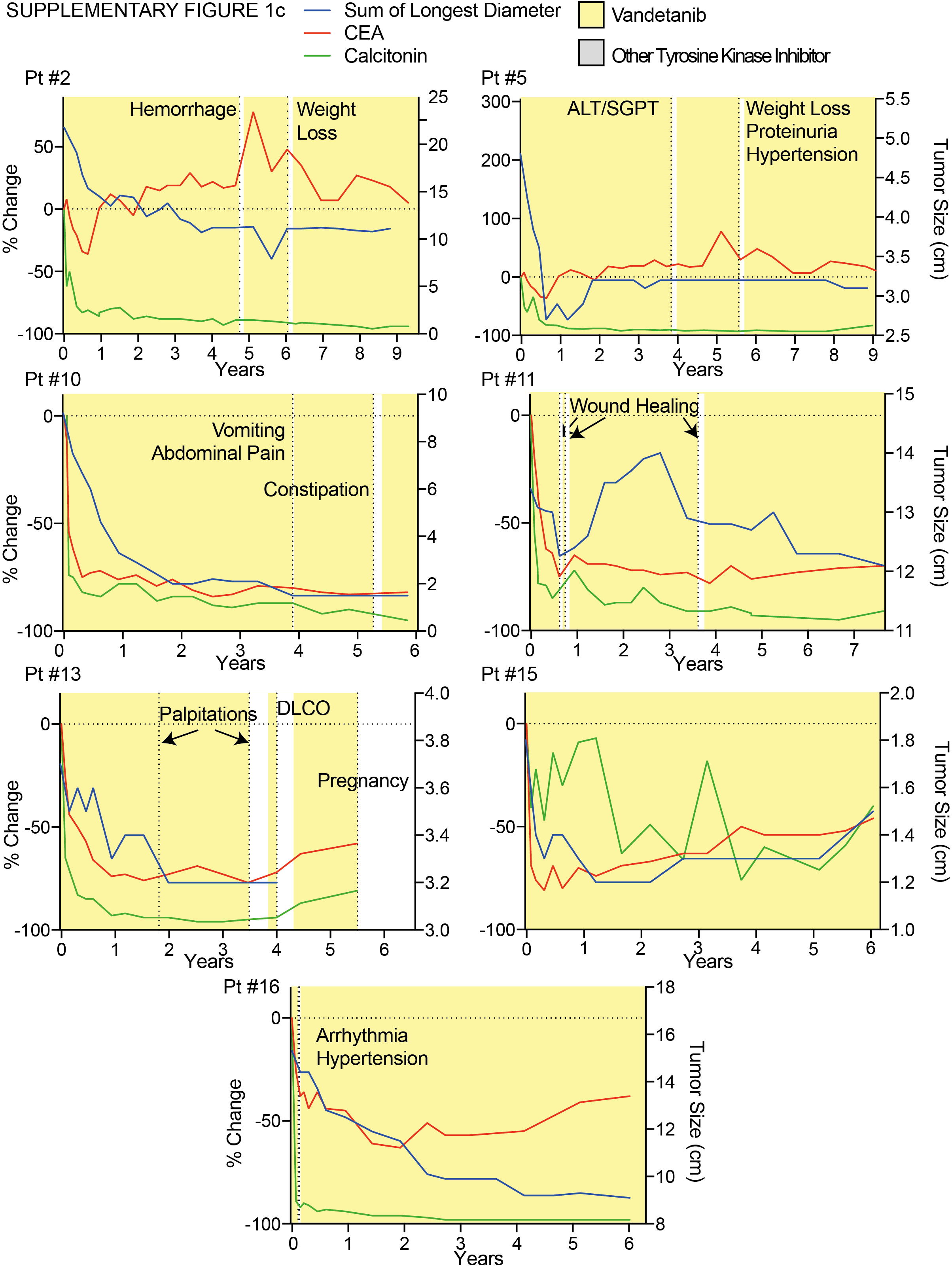
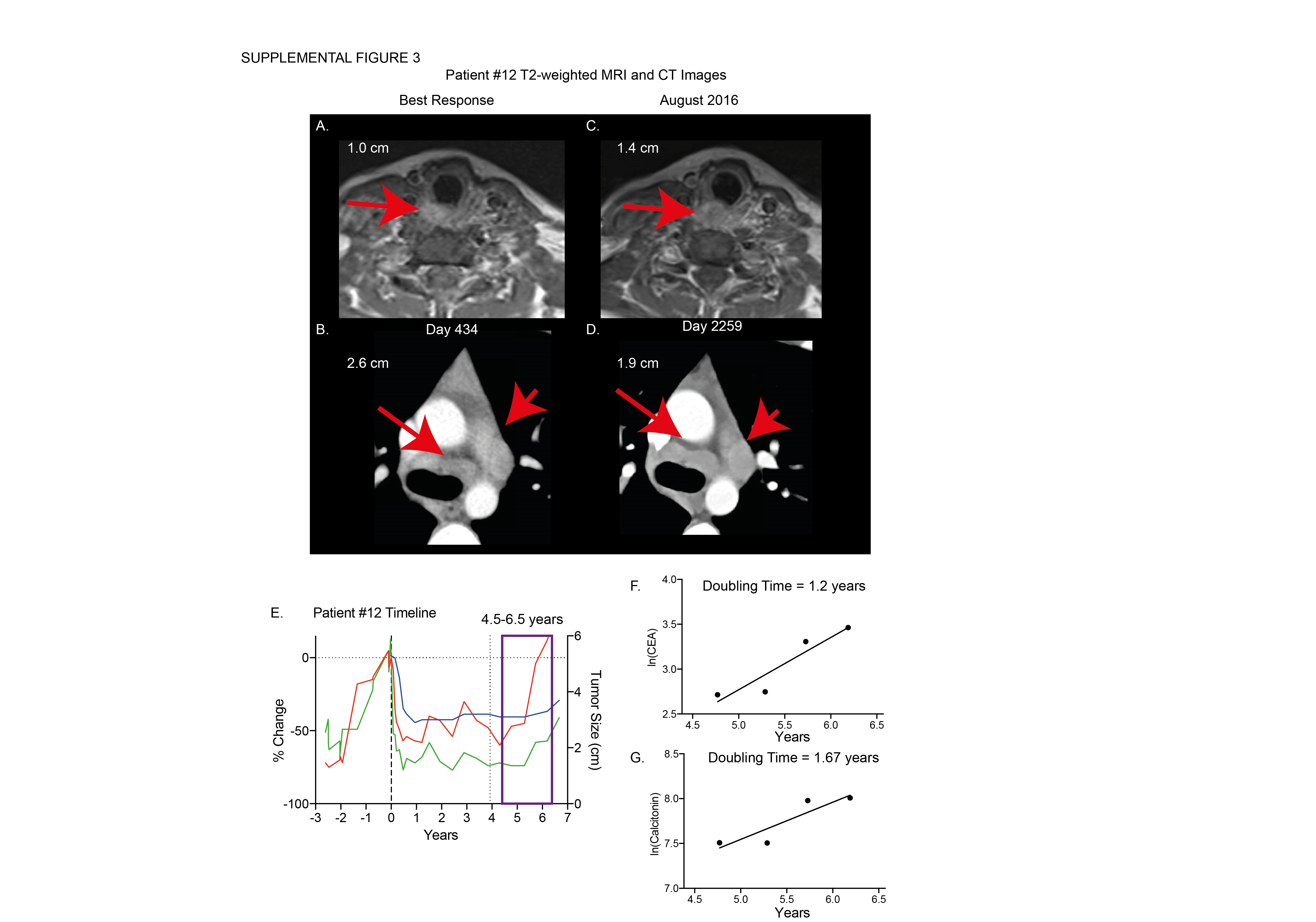
Patients with PR and SD at the data cutoff had similar baseline calcitonin levels, CEA levels, tumor size per RECIST, percent change in tumor size, and time to best response compared to patients with PD (Supplementary Table 2). Patients exhibited a trend towards higher baseline calcitonin with increasing degree of tumor response (Supplementary Table 2). No other measured baseline characteristics were associated with sustained response. Sixteen patients exhibited overall decline or stabilization in calcitonin and CEA during vandetanib therapy (Figure 3). Four of eight patients maintained calcitonin levels below baseline throughout therapy despite eventual disease progression. Increase in calcitonin and CEA coincided with drug holds as demonstrated by patients #2, #6, and #13 (Figure 3A–F, Supplemental Figure 1a–c). There was no difference between patients that eventually progressed on therapy in rate of change of tumor markers during the first four months of vandetanib treatment (Supplemental Figure 2A–B). Nine of ten patients that experienced progressive disease had increasing calcitonin or CEA levels corresponding to doubling times of less than five years prior to experiencing PD (Supplemental Figure 2E). Interestingly, for some patients, such as patient #12, the doubling times of calcitonin and CEA decreased one full year before the patient met RECIST criteria for progressive disease (Supplemental Figure 3).
Figure 3. Biomarker Response.

The percent change from baseline in calcitonin (A–C) and CEA (D–F) per RECIST at data cutoff.
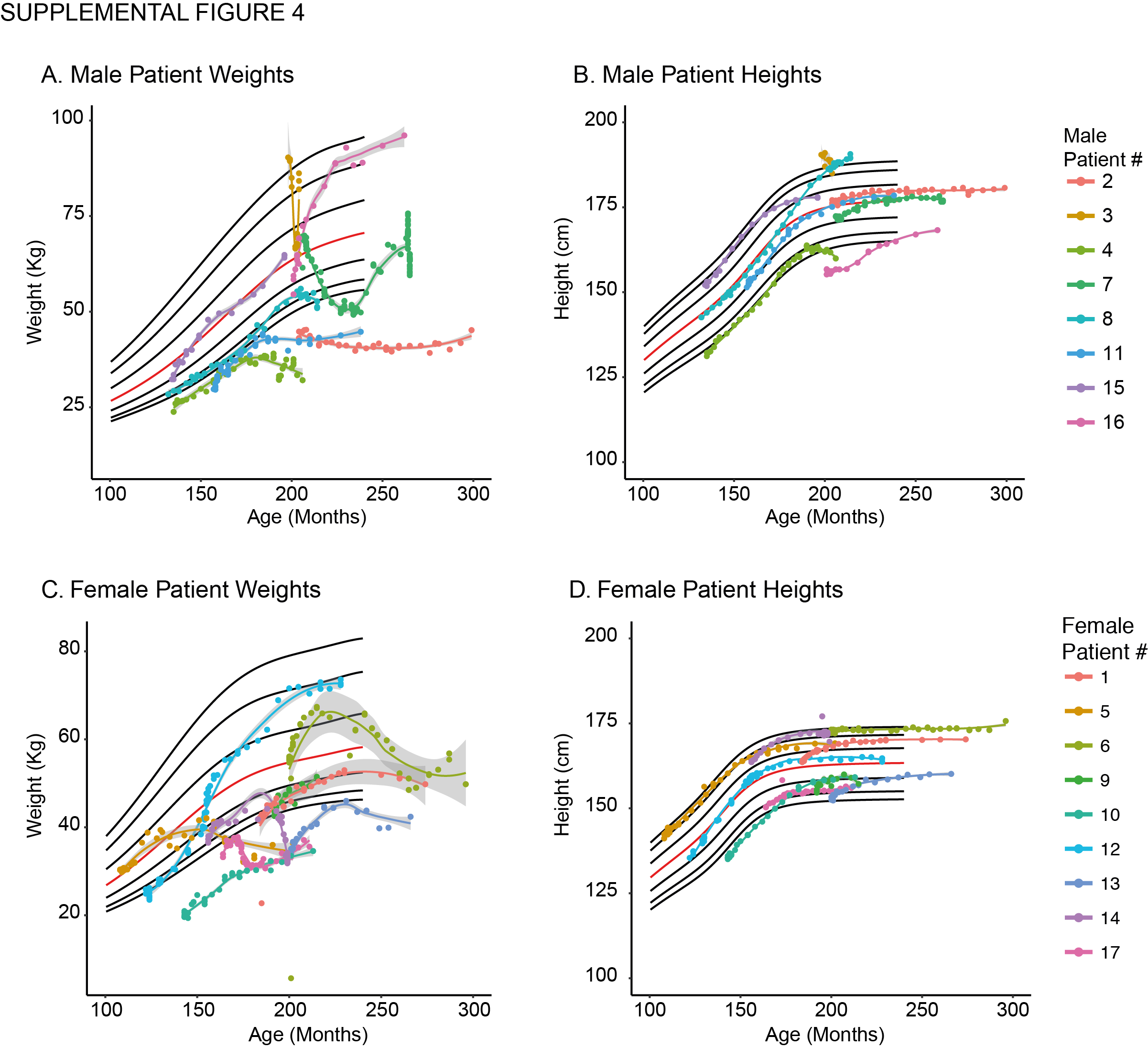
All patients experienced vandetanib related adverse events as described previously(11). Most adverse events were grade one or two and no patient was required to discontinue therapy due to drug toxicity. No patient experienced QT-prolongation mandating hold or discontinuation of therapy. Ten patients had adverse events grade three or higher, required drug holds, or dose reductions during the follow up period (Table 2). The most common reason for a drug hold was unrelated to vandetanib treatment, but was instituted as a precaution for elective surgeries to allow for wound healing. Ten patients experienced grade two hypertension that required medical management (Supplemental Table 2). Vandetanib-related adverse events included bleeding propensity, weight loss, hypertension, hepatotoxicity (elevated liver function tests), renal toxicity (elevated creatinine), pulmonary toxicity (abnormal pulmonary function tests), and gastrointestinal disturbances (diarrhea or constipation). Patient #12 and #16 experienced disproportionate weight gain while on vandetanib, however, this was not consistently observed (Supplemental Figure 4). No meaningful change in growth plate volumes was observed in patients treated with vandetanib (data not shown). Two patients continuing therapy had drug holds greater than one year due to recovery from scoliosis surgery (#6) and nine months due to pregnancy (#13). Both patients were holding vandetanib at the data cutoff. All patients adhered to greater than 95% of scheduled doses during the follow up period based on patient diary and pill counts. Individualized dose adjustments and schedules were common and the dose of vandetanib of patients continuing therapy (n = 9) at the data cutoff was 67–100 mg/m2 once daily.
Table 2.
Toxicities
| Patient | CTC Criteria | Grade of AE |
Time since Starting Vandetanib (years) |
Duration of Hold (days) |
Dose Change (mg/m2) |
|---|---|---|---|---|---|
|
| |||||
| 2 | Hemorrhage | 2 | 4.75 | 83 | |
|
| |||||
| Weight Loss | 2 | 6.04 | 0 | 150 -> 100 | |
|
| |||||
| 5 | ALT, SGPT | 3 | 3.83 | 11 | |
|
| |||||
| Weight Loss; Proteinuria; Hypertension | 2 | 5.56 | 13 | 100 -> 67 | |
|
| |||||
| 6 | Creatinine | 2 | 6.32 | 699 | 150 -> 100 |
|
| |||||
| Wound Healing# | N/A | 7.22 | 630* | ||
|
| |||||
| 8 | Wound Healing# | N/A | 3.47 | 24 | |
|
| |||||
| 3.9 | 33 | ||||
|
| |||||
| 4.62 | 49 | ||||
|
| |||||
| 9 | Patient Preference# | N/A | 1.93 | 2144* | discontinued |
|
| |||||
| 10 | Vomiting | 3 | 3.9 | 7 | |
| Abdominal Pain | 2 | ||||
|
| |||||
| Constipation | 3 | 5.27 | 51 | 150 -> 100 | |
|
| |||||
| 12 | Diarrhea | 2 | 3.92 | 0 | 150 -> 100 |
|
| |||||
| 13 | Palpitations | 2 | 1.81 | 28 | |
|
| |||||
| Palpitations | 2 | 3.49 | 112 | 150 -> 100 | |
|
| |||||
| DLCO; Pulmonary other; Pulmonary cysts by CT | 2 | 4 | 112 | ||
|
| |||||
| Pregnancy# | N/A | 5.57 | 363* | discontinued | |
|
| |||||
| 14 | Wound Healing# | N/A | 1.99 | 1 | |
|
| |||||
| 2.05 | 2 | ||||
|
| |||||
| Wound Healing# | N/A | 8 | |||
|
| |||||
| Oral Hemorrhage | 2 | 2.93 | 7 | ||
|
| |||||
| 16 | Supraventricular and nodal arrhythmia | 2 | 0.11 | 7 | 150 -> 100 |
|
| |||||
| Hypertension | 2 | 0.15 | 7 | ||
|
| |||||
| 17 | Diarrhea | 2 | 0.45 | 4 | 100 -> 67 |
Continues to hold at data cutoff,
unrelated to therapy
Custom-capture genome sequencing of 241 cancer implicated genes was performed on seven patients; 11 tumor biopsy samples, 10 with paired germline tissue. Seven samples were taken before patients started vandetanib and four were taken from patient tumors at the time of disease progression (Table 3). One patient (#8) had three longitudinal samples available. Except for the germline RET mutation, recurrent, nonsynonymous mutations were not found in this cohort (Table 3). Seven patients experienced few somatic mutations and no difference in the number of mutations was noted comparing samples before (n = 6) and after (n = 3) vandetanib therapy (Table 3, Figure 4A). There was no difference in percent change of genome in germline tissue. However, a trend towards increasing genome-wide, somatic CN changes was observed after vandetanib therapy (Figure 4B).
Table 3.
Genomic alterations identified by panel-sequencing of seven patients with MEN2B associated MTC
| Case# | Pt# | Age (years) |
Tumor Location |
Previous TKI |
Somatic Coding alterations |
CN Alterations (chromosome) |
Gene Breaks |
|---|---|---|---|---|---|---|---|
| 1# | 4 | 9 | Lymph Node | None | 1p del | BRCA1, TRAF7, PTEN | |
| 2 | 8 | 12 | Thyroid | None | |||
| 3 | 8 | 12 | Lymph Node | None | |||
| 4 | 17 | 9 | Thyroid | None | MUTYH p.Ala59Val | 1p del, 4 del, 9p del, 22 del | |
| 5 | 22λ | 15 | Thyroid | None | PRKAR1A sf | 1p del, 8 amp, 16 amp | CREBBP, RPTOR |
| 6 | 24λ | 12 | Thyroid | None | 1p del, 1q amp | ||
| 7 | 24λ | 16 | Prostate | None | SYNE1 sf | 1p del, 4 del | KMT2C |
| 8 | 7 | 19 | Lymph Node | vandetanib | EML4 p.Ile328Val, RET p.Leu790Phe | 14 del, 22 del, 1q amp, 16 amp | TRAF7 |
| 9* | 7 | 22 | Adrenal | vandetanib, sunitinib, sorafinib | 1q amp | TSC2, AXIN1 | |
| 10 | 8 | 16 | Prostate | vandetanib | SMARCA4 p.Leu783Arg | 13 del, 1q amp, 7 amp, 10 amp, 14 amp, 15 amp, 18 amp, 22 amp | KMT2C |
| 11 | 14 | 16 | Thyroid | vandetanib | 1p del, 3 del, 4 del, 9 del, 13 del, 14 del, 17p del, 1q amp, 17q amp | TRAF7, RPTOR |
Genomic alterations were predicted with computational methods. sf = sequence feature not in coding region, amp = Amplification, del = deletion.
normal sample unavailable,
patient enrolled on the MTC natural history study (NCT01660984),
sample with low tumor %
Figure 4. Sequencing of DNA from biopsies of patients with MEN2B and MTC.

A) Somatic genomic alterations found in a 241-gene panel in biopsy samples from unique patients before-vandetanib and after experiencing progressive disease on therapy. B) Percent change in genome from copy number (CN) alterations predicted by comparing DNA extracted from tumor lesions and germline tissue in unique patients before starting vandetanib and after experiencing progressive disease. Patient #8 and #24 had multiple tumor sample analyzed. C–D) CN gains and loss (C) and % genome and loss of heterozygosity (D) predicted in patient #8 before and after vandetanib, and after cabozantinib by whole exome sequencing. Representative H&E stains and genome-wide CN alterations from patient #8 are shown, and include lymph node (germline) (E), primary MTC (F), prostate metastasis (G), and skin metastasis (H).
Patient #8 had three samples collected from different time points during MTC therapy that were used for in-depth analysis by whole exome sequencing (WES) and whole transcriptome sequencing (RNA-Seq). Samples included a lymph node without evidence of MTC used as the germline sample, a primary MTC lesion taken before any TKI therapy, a prostate metastasis taken at disease progression on vandetanib (Metastasis 1), and a skin lesion taken after disease progression on cabozantinib (Metastasis 2). Molecular analysis on the longitudinal sample set demonstrated accumulation of whole-genome CN alterations and loss of heterozygosity throughout therapy at the DNA level (Figure 4C–D). Notably, the primary MTC sample (Figure 4F) and the prostate metastasis (Figure 4G) were separate samples taken from different regions of the same tumors used in the panel-sequencing (Case 2 and 10, respectively). Additional computationally predicted deleterious, driving, and cancer-implicated mutations were not found in the whole exome data compared to the panel-sequencing. Representative immunohistochemistry, CN alterations, and B-allele frequency plots are shown in Figure 4E–H. The SMARCA4 p.Leu783Arg somatic mutation found in Case 10 was also found in the WES data from metastasis one. Further, copy neutral, loss of heterozygosity was observed on chr5, chr8, chr13 and chr19 in both metastasis one and metastasis two (Figure 4G–H). Metastasis two was noted to have an RB1 p.Val654fs somatic mutation. Together, these data verified the chromosomal gains and loss as demonstrated by the targeted panel sequencing results.
Analysis of the full transcriptome identified 54 genes meeting computational cutoffs (false discovery rate < 0.05, p-value < 0.01) as the most differentially expressed in biopsies from different points during therapy (Figure 5A). An increase in the RET proto-oncogene was observed across time points. We examined the normalized RNA expression of other genes known to be related to RET and observed an increase in RNA expression of FLT4, coding for the vascular endothelial growth factor receptor (VEGFR3) protein, and Epidermal Growth Factor Receptor (EGFR) in the metastasis (Figure 5B). We compared the RNA expression with CN changes to determine if transcript changes were associated with genomic changes. RET (r2 = 0.985) and EGFR (r2 = 0.948) expression correlated with CN changes (Figure 5C). FLT4 had an increase RNA expression independent of DNA CN changes (Figure 5C). Further, we noted an increase in transcription across cell cycle genes when comparing the samples longitudinally from primary MTC, to metastasis one, to metastasis two (Supplemental Figure 5).
Figure 5. Gene expression changes across longitudinal biopsy specimens.

A) Gene expression heat map of the 54 most differentially expressed transcripts. Expression levels are colored based on Z-scores across individual genes. Genes are grouped by unsupervised k-means clustering. B) Normalized expression of RET-family genes by RNA-seq. C) Correlation of RET-family genes between RNA-seq and DNA CN across longitudinal biopsies. D) Model of disease progression in patient #8 (male, best response SD) despite vandetanib and cabozantinib therapy.
DISCUSSION
Long-term, continuous treatment with TKIs is beneficial in patients with advanced MTC. In this updated analysis, vandetanib was well tolerated and resulted in durable responses in pediatric patients with hereditary, metastatic MTC, with seven of 17 patients maintaining partial response or stable disease for median follow up over five years. Our study represents the largest prospectively followed cohort of patients with MEN2B and MTC and is consistent with other case reports and population-based studies characterizing the outcomes and lifespan of patients with MEN2B(31, 32).
While complete resection of the thyroid gland is the only curative therapy for patients with primary MTC, the presence of locally advanced or metastatic disease at the time of diagnosis limits the possibility of surgical cure(33). In this study, treatment with vandetanib led to durable responses in patients with disease not amenable to surgical cure. The median time to achieve a PR was 0.79 years with one patient taking as long as 2.41 years to achieve a response. Patients 14, 15, and 16 had stable disease at the time of the initial report of this study but later achieved a PR at 1.72, 1.21, and 2.41 years. This observation demonstrates the potential benefit of chronic TKI therapy for advanced MTC. Disease progression generally occurred in new, previously unrecognized, or non-target lesions (patients #3, #4, #7, #8, #14) and one patient (#8) developed progressive disease in the prostate. In this cohort, there was not a common location of progressive metastasis highlighting the importance of a careful imaging survey in patients with metastatic disease.
Six of ten patients experiencing PD ultimately died from disease (median of 2.0 (range, 0.4–5.7) years) after stopping vandetanib. Of the six patients that died, only patient #4 experienced a brief response of 0.79 years to a second, RET-targeting TKI. Interestingly, patient #17 had a prolonged, objective response to a subsequent RET-targeting TKI. Patient #17 was taken off vandetanib due to failure to meet secondary endpoints (increasing calcitonin and CEA) and was enrolled on a subsequent clinical trial (Supplementary Figure 1b). Discontinuation of vandetanib was allowed and recommended for this patient after the 2013 protocol amendment and based on healthcare provider experience with previous patients.
In some cases, the ability of RECIST criteria to capture disease burden may be limited in metastatic MTC due to the large number of lesions in a single patient, the presence of bony metastasis, and occult metastasis. This emphasizes the importance of biomarker characterization. Calcitonin and CEA doubling times are helpful in determining the rate of growth of MTC(34). Additionally, some studies have carefully examined these biomarkers in patients on TKI therapy(35–37). Consistent with previous findings, we observed that use of a single tumor biomarker may not accurately document the presence of progressive disease; however, by using a combination of calcitonin and CEA slopes and doubling times, we noted a correlation with progressive disease via RECIST (Supplemental Figure 2)(36). We examined the slope of biomarker change between patients one year prior to experiencing PD versus patients in the year prior to the data cutoff (Supplemental Figure 3). This comparison is inherently flawed by the dissimilarities in patients, time points, and logistical caveats such as drug holds. Furthermore, calcitonin levels may vary greatly due to the assay used, the hook effect with markedly elevated levels, and fed vs fasting sampling. Prospective, properly controlled studies are needed to truly validate use of biomarkers while on TKI therapy. Further, findings in this population may have limited applicability to adult patients with sporadic MTC. Despite the caveats, our data agree with the American Thyroid Association guidelines implying that patients with decreasing tumor marker doubling time seem to have a worse prognosis (38). All together we suggest that changes in tumor marker doubling time may be best interpreted in context with additional clinical correlates before therapy is altered.
Vandetanib was well tolerated and no patient discontinued therapy due to adverse events during the follow up period. Fourteen patients experienced hypertension that was amenable to medical therapy and three patients required more than one medication or dose adjustments to maintain normal blood pressures (Supplementary Table 3). No patients developed pheochromocytoma while on the study. Vandetanib was held in patients recovering from injuries or surgeries per previous recommendations(33). Almost all common adverse events occurred early in therapy; however, significant adverse events requiring drug holds such as elevated creatinine (patient #6) were discovered after six years on vandetanib, illustrating the need for continuous monitoring. The decrease in adverse events later in therapy may be related to dose reductions that occurred due to events earlier during treatment. Eight patients required a dose reduction during the follow up period. Despite dose reductions or discontinuing medication, many patients maintained responses for over one year. Patients who remain on vandetanib at the data cutoff continue a dose of 67–100 mg/m2/day, or incorporated an alternate day dosing scheme to minimize toxicities.
Continued adherence is of great concern for many patients on oral TKI therapy. We did not observe significant noncompliance in patient reported medication logs and study mandated pill counts. Additionally, the original phase I/II study quantified plasma drug levels suggesting sufficient levels for effect modulation(11). This suggests that lack of compliance may not be a mechanism of resistance in this clinical setting. We also noted instances of patients maintaining stable disease despite significant drug holds. Patient #9 discontinued vandetanib after achieving stable disease and was followed for over five years outside of the NIH before experiencing progressive disease (Supplemental Figure 1b). Patient #12 experienced repeated decrease in CEA and calcitonin without increase in tumor size despite two significant drug holds (Supplemental Figure 1b). These observations suggest that drug holidays may be useful in select patients without sacrificing efficacy.
Tumor samples were available for five patients before starting vandetanib (seven samples), three patients at progression on TKI (five samples), and one patient both pre-TKI and at progression (three samples). A uniform molecular mechanism of treatment failure was not observed in this small sample set. Pre-TKI samples exhibited few tumor specific mutations or CN alterations and four of five patients had loss of chromosome 1p. Copy number imbalances have been reported previously in both sporadic and hereditary MTC(39, 40). Marsh and colleagues also found loss of chromosome 1p in 21% of a cohort of MTC that included both hereditary and sporadic disease suggesting that loss of 1p may be involved in early tumorigenesis (39). We identified a number of genomic changes in progressive disease. Interestingly, progression for one patient was associated with acquisition of a previously unidentified p.Leu790Phe somatic mutation within the kinase domain of the RET gene. However, a pre-treatment sample was not available for this patient limiting the interpretation of this finding. Previous reports of a RET p.Leu790Phe mutation are as germline findings associated with MEN2A/FMTC and have not previously been reported in association with resistance to vandetanib(41, 42). Longitudinal data from patient #8 demonstrated acquisition of a SMARCA4 mutation in the first metastasis and an RB1 mutation in the second metastasis. Loss of heterozygosity and increase in CN variations were noted in four of five samples at tumor progression highlighting aneuploidy as the dominant genetic marker of progressive disease. Two patients had loss of chromosome 14 and three had gain of chromosome 1q.
Activation of the phosphatidylinositol 3-kinase (PI3K)/Akt/ mammalian target of rapamycin (mTOR) pathway has been reported in MTC(43, 44). Four patient samples including two samples obtained prior to TKI therapy and two samples following TKI therapy had breaks in genes coding for components of the PI3K signaling pathway including PTEN, RPTOR, and TSC2. It has been suggested that the oncogenic activity of RET may be mediated, at least partially, through the PI3K pathway(45, 46). Responses to the TORC1 inhibitor, everolimus, have been reported in patients with MTC(47). The presence of alterations in genes coding for PI3K pathway components is intriguing, however, the small number of patients in this study and the lack of uniform sample collection make assessment of the role of the PI3K pathway in oncogenesis and disease progression difficult.
Genome-wide CN alterations of rapidly progressive disease was recently documented in an autopsy case report of sporadic MTC(48). Such changes are recognized as a hallmark of many cancers, hereditary MTC is thought to be driven by a single oncogene(1, 49). The observed genome-wide CN alterations raise questions as to the underlying etiology, and whether CN alterations are induced by the TKI therapy or are selected cellular clones that occur in the natural course of MTC progression. As in other malignancies, the observed genomic aberrations are reflected at the transcriptional level(50). Regardless of underlying cause, the WES and RNA-seq data in this longitudinal set may represent a transition away from the single oncogenic driver mutation (RET p.Met918Thr) to a malignancy with multiple oncogenic drivers as proposed in Figure 5D. Currently, few options exist for patients that progress on RET-targeting medications. These data may provide evidence for use of non-specific agents and highlight the need for development of therapies that target cells with genomic instability in a progressive disease setting(33).
Increased RNA expression of cell cycle genes in the metastasis compared to the primary tumor in patient #8 may be expected in rapidly dividing lesions (Supplemental Figure 5). This finding was consistent with rapid clinical progression as well as the loss of RB1 function in metastasis two. We also observed increased RET and FLT4 expression throughout the longitudinal course of MTC disease suggesting potential gene upregulation in response to therapy.
Our data provide preliminary insights into mechanisms of vandetanib resistance in hereditary MTC. These data must be interpreted with caution and are best served as hypothesis generating rather than conclusive. It is notable that an additional RET mutation was only identified in one case of progressive disease. Further, a p.Leu790Phe mutation identified may not be expected to lead to vandetanib resistance. Other possible mechanisms of drug resistance including changes in cellular and systemic metabolism and aberrant activation of downstream targets of RET were not assessed. Pre-clinical evaluations and future trials that more completely investigate these factors may lead to a better understanding of vandetanib resistance and could inform treatment options for these patients. Limitations of these data are notable for the small sample size and the heterogeneity in timing and anatomical location of the available clinical samples, thus requiring independent confirmation of these observations. A prospective trial with defined points for clinical sample collection and the addition of pharmacokinetic analysis would help to better delineate mechanisms of resistance to vandetanib in this patient population.
In conclusion, most children and adolescents with MEN2B associated MTC have sustained clinical responses on vandetanib. We observed genomic instability in MTC metastasis after disease progression on TKI therapy, postulating a departure from the single driver mutation. Genome-wide CN alterations were correlated generally with RNA expression changes. These data highlight the importance of longitudinal sampling of tumor tissue either via direct biopsy or potentially through liquid biopsy as these technologies become available. While trials evaluating more potent RET inhibitors are already underway, future studies may also be warranted to assess the use of cytotoxic or non-RET pathway targeting therapies in patients that experience progressive disease in the face of standard RET-targeted therapy.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
STATEMENT OF TRANSLATIONAL RELEVANCE.
Vandetanib is well tolerated in adults and children with advanced MTC; however, long-term outcomes of patients on vandetanib have not been reported previously. This updated report of a phase I/II trial of vandetanib assessed the activity, safety, and outcome of children and adolescents with advanced, hereditary MTC and MEN2B treated with vandetanib. Tumor samples taken at disease progression were analyzed for genomic mechanisms of resistance. Patients exhibited durable partial responses and tolerated extended therapy with vandetanib. When disease progression occurred, tumor burden increased at an accelerated pace and subsequent therapies usually had limited activity. This study provides the longest follow-up on patients taking continuous vandetanib to date. Patients experiencing progressive disease on vandetanib may not respond to another RET-targeting TKI. This study provides new insights into the tumorigenesis of MTC in the era of tyrosine kinase inhibitor therapy by illustrating genome-wide copy number changes at MTC progression.
Acknowledgments
We thank the participating patients and their families. We recognize Dr. Samuel Wells and Dr. David Venzon for their critical review of the manuscript and advise on data analysis. We are indebted to Dr. Keith J. Killian and David Petersen for their advice and expertise in nucleic acid extraction and sequencing of FFPE specimens. Finally, we recognize the National Institutes of Health, Medical Research Scholars Program (https://clinicalcenter.nih.gov/training/mrsp/) for support in training the next generation of clinician scientists.
This research was, in part, supported by the National Cancer Institute, Center for Cancer Research. CDA is a contractor for Leidos Biomedical Research, Inc., NCI Campus at Frederick, Frederick, Maryland 21702. This project has also been funded in whole or in part with federal funds from the National Cancer Institute, National Institutes of Health, under Contract No. HHSN261200800001E. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
Footnotes
CONTRIBUTIONS
ILK performed literature searches, data collection, data analysis, interpreted findings, and wrote the manuscript. SA designed trial protocols, performed literature searches, data collection, data analysis, and interpreted findings. YZ and HL performed data analysis. CDA performed data collection, data analysis, and direct patient care. ED performed data collection, data analysis, and interpreted findings. SMS performed the statistical analysis. ML designed trial protocols, data collection, and interpret findings. SGW performed data collection and interpreted findings. OK performed data collection and data analysis. EF designed trial protocols, performed data collection, and data analysis. FMB designed trial protocols and performed data collection. MJM interpreted findings. PSM designed the genomic study, performed data collection, data analysis, and interpreted findings. JWG designed protocols, performed data collection, data analysis, interpreted findings, and helped draft the manuscript. JFS designed genomic study, performed data collection, data analysis, and interpreted findings. BCW designed trial protocols, performed data collection, data analysis, and interpreted findings. All authors provided critical review and feedback on the manuscript.
DECLARATION OF INTERESTS
AstraZeneca provided vandetanib for the phase I/II trial, Sanofi/Genzyme supports the ongoing trial. All authors report no financial or other competing interests.
References
- 1.Romei C, Ciampi R, Elisei R. A comprehensive overview of the role of the RET proto-oncogene in thyroid carcinoma. Nat Rev Endocrinol. 2016;12(4):192–202. doi: 10.1038/nrendo.2016.11. [DOI] [PubMed] [Google Scholar]
- 2.Dermody S, Walls A, Harley EH., Jr Pediatric thyroid cancer: An update from the SEER database 2007–2012. Int J Pediatr Otorhinolaryngol. 2016;89:121–6. doi: 10.1016/j.ijporl.2016.08.005. [DOI] [PubMed] [Google Scholar]
- 3.Wells SA, Jr, Robinson BG, Gagel RF, Dralle H, Fagin JA, Santoro M, et al. Vandetanib in patients with locally advanced or metastatic medullary thyroid cancer: a randomized, double-blind phase III trial. J Clin Oncol. 2012;30(2):134–41. doi: 10.1200/JCO.2011.35.5040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Elisei R, Schlumberger MJ, Muller SP, Schoffski P, Brose MS, Shah MH, et al. Cabozantinib in progressive medullary thyroid cancer. J Clin Oncol. 2013;31(29):3639–46. doi: 10.1200/JCO.2012.48.4659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hennequin LF, Stokes ES, Thomas AP, Johnstone C, Ple PA, Ogilvie DJ, et al. Novel 4-anilinoquinazolines with C-7 basic side chains: design and structure activity relationship of a series of potent, orally active, VEGF receptor tyrosine kinase inhibitors. J Med Chem. 2002;45(6):1300–12. doi: 10.1021/jm011022e. [DOI] [PubMed] [Google Scholar]
- 6.Miller KD, Trigo JM, Wheeler C, Barge A, Rowbottom J, Sledge G, et al. A multicenter phase II trial of ZD6474, a vascular endothelial growth factor receptor-2 and epidermal growth factor receptor tyrosine kinase inhibitor, in patients with previously treated metastatic breast cancer. Clin Cancer Res. 2005;11(9):3369–76. doi: 10.1158/1078-0432.CCR-04-1923. [DOI] [PubMed] [Google Scholar]
- 7.Holden SN, Eckhardt SG, Basser R, de Boer R, Rischin D, Green M, et al. Clinical evaluation of ZD6474, an orally active inhibitor of VEGF and EGF receptor signaling, in patients with solid, malignant tumors. Ann Oncol. 2005;16(8):1391–7. doi: 10.1093/annonc/mdi247. [DOI] [PubMed] [Google Scholar]
- 8.Kovacs MJ, Reece DE, Marcellus D, Meyer RM, Mathews S, Dong RP, et al. A phase II study of ZD6474 (Zactima, a selective inhibitor of VEGFR and EGFR tyrosine kinase in patients with relapsed multiple myeloma--NCIC CTG IND.145. Invest New Drugs. 2006;24(6):529–35. doi: 10.1007/s10637-006-9022-7. [DOI] [PubMed] [Google Scholar]
- 9.Arnold AM, Seymour L, Smylie M, Ding K, Ung Y, Findlay B, et al. Phase II study of vandetanib or placebo in small-cell lung cancer patients after complete or partial response to induction chemotherapy with or without radiation therapy: National Cancer Institute of Canada Clinical Trials Group Study BR.20. J Clin Oncol. 2007;25(27):4278–84. doi: 10.1200/JCO.2007.12.3083. [DOI] [PubMed] [Google Scholar]
- 10.Wells SA, Jr, Gosnell JE, Gagel RF, Moley J, Pfister D, Sosa JA, et al. Vandetanib for the treatment of patients with locally advanced or metastatic hereditary medullary thyroid cancer. J Clin Oncol. 2010;28(5):767–72. doi: 10.1200/JCO.2009.23.6604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fox E, Widemann BC, Chuk MK, Marcus L, Aikin A, Whitcomb PO, et al. Vandetanib in children and adolescents with multiple endocrine neoplasia type 2B associated medullary thyroid carcinoma. Clin Cancer Res. 2013;19(15):4239–48. doi: 10.1158/1078-0432.CCR-13-0071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fallahi P, Ferrari SM, Baldini E, Biricotti M, Ulisse S, Materazzi G, et al. The safety and efficacy of vandetanib in the treatment of progressive medullary thyroid cancer. Expert Rev Anticancer Ther. 2016;16(11):1109–18. doi: 10.1080/14737140.2016.1238764. [DOI] [PubMed] [Google Scholar]
- 13.Lian EY, Maritan SM, Cockburn JG, Kasaian K, Crupi MJ, Hurlbut D, et al. Differential roles of RET isoforms in medullary and papillary thyroid carcinomas. Endocr Relat Cancer. 2017;24(1):53–69. doi: 10.1530/ERC-16-0393. [DOI] [PubMed] [Google Scholar]
- 14.Kato S, Subbiah V, Marchlik E, Elkin SK, Carter JL, Kurzrock R. RET Aberrations in Diverse Cancers: Next-Generation Sequencing of 4,871 Patients. Clin Cancer Res. 2017;23(8):1988–97. doi: 10.1158/1078-0432.CCR-16-1679. [DOI] [PubMed] [Google Scholar]
- 15.Mancikova V, Montero-Conde C, Perales-Paton J, Fernandez A, Santacana M, Jodkowska K, et al. Multilayer OMIC Data in Medullary Thyroid Carcinoma Identifies the STAT3 Pathway as a Potential Therapeutic Target in RETM918T Tumors. Clin Cancer Res. 2017;23(5):1334–45. doi: 10.1158/1078-0432.CCR-16-0947. [DOI] [PubMed] [Google Scholar]
- 16.Carlomagno F, Guida T, Anaganti S, Provitera L, Kjaer S, McDonald NQ, et al. Identification of tyrosine 806 as a molecular determinant of RET kinase sensitivity to ZD6474. Endocr Relat Cancer. 2009;16(1):233–41. doi: 10.1677/ERC-08-0213. [DOI] [PubMed] [Google Scholar]
- 17.Carlomagno F, Guida T, Anaganti S, Vecchio G, Fusco A, Ryan AJ, et al. Disease associated mutations at valine 804 in the RET receptor tyrosine kinase confer resistance to selective kinase inhibitors. Oncogene. 2004;23(36):6056–63. doi: 10.1038/sj.onc.1207810. [DOI] [PubMed] [Google Scholar]
- 18.Chu YH, Lloyd RV. Medullary Thyroid Carcinoma: Recent Advances Including MicroRNA Expression. Endocr Pathol. 2016;27(4):312–24. doi: 10.1007/s12022-016-9449-0. [DOI] [PubMed] [Google Scholar]
- 19.Heilmann AM, Subbiah V, Wang K, Sun JX, Elvin JA, Chmielecki J, et al. Comprehensive Genomic Profiling of Clinically Advanced Medullary Thyroid Carcinoma. Oncology. 2016;90(6):339–46. doi: 10.1159/000445978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Romei C, Casella F, Tacito A, Bottici V, Valerio L, Viola D, et al. New insights in the molecular signature of advanced medullary thyroid cancer: evidence of a bad outcome of cases with double RET mutations. J Med Genet. 2016 doi: 10.1136/jmedgenet-2016-103833. [DOI] [PubMed] [Google Scholar]
- 21.Storer BE. Design and analysis of phase I clinical trials. Biometrics. 1989;45(3):925–37. [PubMed] [Google Scholar]
- 22.Therasse P, Arbuck SG, Eisenhauer EA, Wanders J, Kaplan RS, Rubinstein L, et al. New guidelines to evaluate the response to treatment in solid tumors. European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J Natl Cancer Inst. 2000;92(3):205–16. doi: 10.1093/jnci/92.3.205. [DOI] [PubMed] [Google Scholar]
- 23.Killian JK, Miettinen M, Walker RL, Wang Y, Zhu YJ, Waterfall JJ, et al. Recurrent epimutation of SDHC in gastrointestinal stromal tumors. Sci Transl Med. 2014;6(268):268ra177. doi: 10.1126/scitranslmed.3009961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cibulskis K, Lawrence MS, Carter SL, Sivachenko A, Jaffe D, Sougnez C, et al. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol. 2013;31(3):213–9. doi: 10.1038/nbt.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Talevich E, Shain AH, Botton T, Bastian BC. CNVkit: Genome-Wide Copy Number Detection and Visualization from Targeted DNA Sequencing. PLoS Comput Biol. 2016;12(4):e1004873. doi: 10.1371/journal.pcbi.1004873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29(1):15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25(9):1105–11. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 2010;28(5):511–5. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Barbet J, Campion L, Kraeber-Bodere F, Chatal JF Group GTES. Prognostic impact of serum calcitonin and carcinoembryonic antigen doubling-times in patients with medullary thyroid carcinoma. J Clin Endocrinol Metab. 2005;90(11):6077–84. doi: 10.1210/jc.2005-0044. [DOI] [PubMed] [Google Scholar]
- 30.Gawlik T, d'Amico A, Szpak-Ulczok S, Skoczylas A, Gubala E, Chorazy A, et al. The prognostic value of tumor markers doubling times in medullary thyroid carcinoma - preliminary report. Thyroid Res. 2010;3(1):10. doi: 10.1186/1756-6614-3-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mathiesen JS, Kroustrup JP, Vestergaard P, Madsen M, Stochholm K, Poulsen PL, et al. Incidence and prevalence of multiple endocrine neoplasia 2B in Denmark: a nationwide study. Endocr Relat Cancer. 2017 doi: 10.1530/ERC-17-0122. [DOI] [PubMed] [Google Scholar]
- 32.Narayanan VK, Ronghe M, MacGregor FB, Bradshaw N, Davidson R, Welbury R, et al. Use of Vandetanib in Metastatic Medullary Carcinoma of Thyroid in a Pediatric Patient With Multiple Endocrine Neoplasia 2B. J Pediatr Hematol Oncol. 2016;38(2):155–7. doi: 10.1097/MPH.0000000000000432. [DOI] [PubMed] [Google Scholar]
- 33.Ernani V, Kumar M, Chen AY, Owonikoko TK. Systemic treatment and management approaches for medullary thyroid cancer. Cancer Treat Rev. 2016;50:89–98. doi: 10.1016/j.ctrv.2016.09.006. [DOI] [PubMed] [Google Scholar]
- 34.Meijer JA, le Cessie S, van den Hout WB, Kievit J, Schoones JW, Romijn JA, et al. Calcitonin and carcinoembryonic antigen doubling times as prognostic factors in medullary thyroid carcinoma: a structured meta-analysis. Clin Endocrinol (Oxf) 2010;72(4):534–42. doi: 10.1111/j.1365-2265.2009.03666.x. [DOI] [PubMed] [Google Scholar]
- 35.Hajje G, Borget I, Leboulleux S, Chougnet C, Al Ghuzlan A, Mirghani H, et al. Early changes in carcinoembryonic antigen but not in calcitonin levels are correlated with the progression-free survival in medullary thyroid carcinoma patients treated with cytotoxic chemotherapy. Eur J Endocrinol. 2013;168(2):113–8. doi: 10.1530/EJE-12-0771. [DOI] [PubMed] [Google Scholar]
- 36.Werner RA, Schmid JS, Muegge DO, Luckerath K, Higuchi T, Hanscheid H, et al. Prognostic Value of Serum Tumor Markers in Medullary Thyroid Cancer Patients Undergoing Vandetanib Treatment. Medicine (Baltimore) 2015;94(45):e2016. doi: 10.1097/MD.0000000000002016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kurzrock R, Atkins J, Wheler J, Fu S, Naing A, Busaidy N, et al. Tumor marker and measurement fluctuations may not reflect treatment efficacy in patients with medullary thyroid carcinoma on long-term RET inhibitor therapy. Ann Oncol. 2013;24(9):2256–61. doi: 10.1093/annonc/mdt177. [DOI] [PubMed] [Google Scholar]
- 38.Wells SA, Jr, Asa SL, Dralle H, Elisei R, Evans DB, Gagel RF, et al. Revised American Thyroid Association guidelines for the management of medullary thyroid carcinoma. Thyroid. 2015;25(6):567–610. doi: 10.1089/thy.2014.0335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Marsh DJ, Theodosopoulos G, Martin-Schulte K, Richardson AL, Philips J, Roher HD, et al. Genome-wide copy number imbalances identified in familial and sporadic medullary thyroid carcinoma. J Clin Endocrinol Metab. 2003;88(4):1866–72. doi: 10.1210/jc.2002-021155. [DOI] [PubMed] [Google Scholar]
- 40.Ye L, Santarpia L, Cote GJ, El-Naggar AK, Gagel RF. High resolution array-comparative genomic hybridization profiling reveals deoxyribonucleic acid copy number alterations associated with medullary thyroid carcinoma. J Clin Endocrinol Metab. 2008;93(11):4367–72. doi: 10.1210/jc.2008-0912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Berndt I, Reuter M, Saller B, Frank-Raue K, Groth P, Grussendorf M, et al. A new hot spot for mutations in the ret protooncogene causing familial medullary thyroid carcinoma and multiple endocrine neoplasia type 2A. J Clin Endocrinol Metab. 1998;83(3):770–4. doi: 10.1210/jcem.83.3.4619. [DOI] [PubMed] [Google Scholar]
- 42.Elisei R, Romei C, Cosci B, Agate L, Bottici V, Molinaro E, et al. RET genetic screening in patients with medullary thyroid cancer and their relatives: experience with 807 individuals at one center. J Clin Endocrinol Metab. 2007;92(12):4725–9. doi: 10.1210/jc.2007-1005. [DOI] [PubMed] [Google Scholar]
- 43.Tamburrino A, Molinolo AA, Salerno P, Chernock RD, Raffeld M, Xi L, et al. Activation of the mTOR pathway in primary medullary thyroid carcinoma and lymph node metastases. Clin Cancer Res. 2012;18(13):3532–40. doi: 10.1158/1078-0432.CCR-11-2700. [DOI] [PubMed] [Google Scholar]
- 44.Kouvaraki MA, Liakou C, Paraschi A, Dimas K, Patsouris E, Tseleni-Balafouta S, et al. Activation of mTOR signaling in medullary and aggressive papillary thyroid carcinomas. Surgery. 2011;150(6):1258–65. doi: 10.1016/j.surg.2011.09.022. [DOI] [PubMed] [Google Scholar]
- 45.Gild ML, Landa I, Ryder M, Ghossein RA, Knauf JA, Fagin JA. Targeting mTOR in RET mutant medullary and differentiated thyroid cancer cells. Endocr Relat Cancer. 2013;20(5):659–67. doi: 10.1530/ERC-13-0085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Manfredi GI, Dicitore A, Gaudenzi G, Caraglia M, Persani L, Vitale G. PI3K/Akt/mTOR signaling in medullary thyroid cancer: a promising molecular target for cancer therapy. Endocrine. 2015;48(2):363–70. doi: 10.1007/s12020-014-0380-1. [DOI] [PubMed] [Google Scholar]
- 47.Lim SM, Chang H, Yoon MJ, Hong YK, Kim H, Chung WY, et al. A multicenter, phase II trial of everolimus in locally advanced or metastatic thyroid cancer of all histologic subtypes. Ann Oncol. 2013;24(12):3089–94. doi: 10.1093/annonc/mdt379. [DOI] [PubMed] [Google Scholar]
- 48.Das S, Kelly D, Moran B, Han K, Mulligan N, Barrett C, et al. Postmortem Examination of an Aggressive Case of Medullary Thyroid Carcinoma Characterized by Catastrophic Genomic Abnormalities. JCO Precision Oncology. 2017;(1):1–7. doi: 10.1200/PO.16.00063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Negrini S, Gorgoulis VG, Halazonetis TD. Genomic instability--an evolving hallmark of cancer. Nat Rev Mol Cell Biol. 2010;11(3):220–8. doi: 10.1038/nrm2858. [DOI] [PubMed] [Google Scholar]
- 50.Buccitelli C, Salgueiro L, Rowald K, Sotillo R, Mardin BR, Korbel JO. Pan-cancer analysis distinguishes transcriptional changes of aneuploidy from proliferation. Genome Res. 2017;27(4):501–11. doi: 10.1101/gr.212225.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.