

Abstract
In tumour immunology, complement has traditionally been considered as an adjunctive component that enhances the cytolytic effects of antibody-based immunotherapies, such as rituximab. Remarkably, research in the past decade has uncovered novel molecular mechanisms linking imbalanced complement activation in the tumour microenvironment with inflammation and suppression of antitumour immune responses. These findings have prompted new interest in manipulating the complement system for cancer therapy. This Review summarizes our current understanding of complement-mediated effector functions in the tumour microenvironment, focusing on how complement activation can act as a negative or positive regulator of tumorigenesis. It also offers insight into clinical aspects, including the feasibility of using complement biomarkers for cancer diagnosis and the use of complement inhibitors during cancer treatment.
An enormous body of data produced by converging disciplines has provided unprecedented insight into the intricate and dynamic relationship that exists between developing tumours and the host immune system1–3. It is widely accepted that neoplastic transformation is a multifactorial process that the immune system can detect through genetic and epigenetic changes that alter the antigenic signatures of tumour cells. Control of tumour growth relies on both innate and adaptive mechanisms of immunosurveillance and is subject to the selective pressure exerted by distinct immunoregulatory pathways in the tumour microenvironment4–7. In this regard, cancer treatment has been revolutionized by new immunomodulatory approaches that elicit durable clinical responses in cancer patients by restoring antitumour immunity and potentiating standard therapeutic regimens, such as tumour radiotherapy5,8.
Elements of the innate immune response are integral components of antitumour effector mechanisms3,9. In many ways, tumour cells are perceived by the innate immune system as noxious, ‘non-self’ matter that must be disposed of10. Therefore, potent antitumour cytotoxic responses are elicited by innate immune cells, followed by elimination of opsonized tumour cells through the concerted action of tumour-directed antibodies and complement9,11,12. Complement is a phylogenetically conserved branch of the innate immune response that has traditionally been perceived as a network of proteins that rapidly respond to microbial intruders, triggering the release of inflammatory mediators, phagocytic responses and cell lysis13 (BOX 1). Growing evidence has indicated, however, that this versatile innate immune effector system mediates key homeostatic functions in processes ranging from early vertebrate development and tissue morphogenesis to tissue regeneration, central nervous system synaptic pruning, host–microbiota symbiosis and adaptive immune regulation14–17. In the context of cancer immunotherapy, complement can be readily triggered into action by damage-associated molecular patterns (DAMPs) exposed on the surface of tumour cells3,6. Although the role of complement as an effector mechanism that potentiates antibody-dependent tumour cytolysis has been long appreciated, clinical challenges, such as the upregulation of a wide spectrum of complement regulatory proteins, remain to be addressed12,18,19.
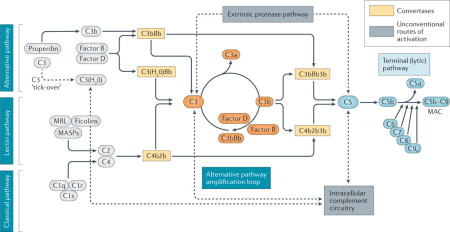
Box 1. An overview of the complement system.
The complement system comprises an extensive network of fluid-phase and membrane-bound glycoproteins, cofactors, receptors and regulatory proteins that engage in innate immune recognition, adaptive cell stimulation and pro-inflammatory effector responses126. Being a crucial mediator of tissue immunosurveillance, complement responds rapidly to molecular stress signals through a cascade of sequential proteolytic reactions initiated by the binding of pattern recognition molecules (for example, C1q, mannose-binding lectin (MBL), MBL-associated serine proteases (MASPs), ficolins and properdin (also known as Factor P)) to distinct structures on damaged cells, biomaterial surfaces or microbial intruders17. Whereas three ‘canonical’ pathways of activation have been described to date (that is, the classical, alternative and lectin pathways), mounting evidence indicates that complement can be activated via multiple routes, depending on the initiating triggers and the distinct microenvironment or pathophysiological context126. The classical pathway is initiated by binding to circulating or surface-bound immune complexes, while the lectin and alternative pathways are triggered by pathogen-associated molecular patters (PAMPs) or aberrant carbohydrate structures on damaged or necrotic cells. All activation pathways converge at the cleavage of C3, an abundant plasma protein that undergoes elaborate conformational changes upon activation, thereby exposing multiple sites of interaction with diverse immune effectors127. Notably, the spontaneous low-level hydrolysis of C3 keeps the alternative pathway in ‘standby’ mode to allow for rapid amplification upon microbial challenge128. Complement activation culminates in the assembly of short-lived multiprotein complexes with enzymatic activity termed ‘convertases’. These enzymes are responsible for the proteolytic activation of the central components C3 and C5 and the release of their respective bioactive fragments, C3a and C3b, and C5a and C5b. The rapid amplification of C3b deposition via the alternative pathway is a process known to underlie several clinical disorders associated with genetic or acquired complement dysregulation (for example, C3 glomerulopathy)17,129. Cleavage of C5 leads to the release of the potent inflammatory mediator C5a and initiates a sequence of protein–protein interactions that induces assembly of the membrane attack complex (MAC, C5b–C9), a multiprotein ‘pore’ with cell activating and cytolytic properties. Unconventional routes of complement activation include the deployment of proteolytic enzymes of the coagulation and fibrinolytic systems that can efficiently cleave both C3 and C5 into their bioactive fragments130. These non-canonical routes of activation, in conjunction with the intracellular complement circuitry38,131, define a broader pathophysiological base of triggering cues that can initiate homeostatic or disease-tailored complement responses.
Clearly, the role of complement as an effector of tumour cytotoxic responses remains relevant to antibody-mediated immunotherapy, guiding new therapeutic options. Intriguingly, however, accumulating evidence from studies published in the past decade has pointed to a fascinating paradigm shift: the realization that complement activation within the tumour microenvironment can serve a tumour-promoting role by perpetuating local T cell immunosuppression and chronic inflammation that promotes tumour immune escape, outgrowth and metastasis20–23. Diverse complement-derived effectors and downstream signalling partners have been implicated in processes ranging from tumour cell anchorage and proliferation to tumour-associated angiogenesis, matrix remodelling, migration, tissue invasiveness and metastasis6,19. With new insights being gained from a variety of tumour models, it is becoming clear that the contribution of complement to cancer pathophysiology is far more complex than originally thought and appears to be largely contextual, depending on several factors, such as the cellular origin of the tumour in question, its inherent capacity to produce autologous complement proteins, the nature of the tumour microenvironment and the magnitude of complement activation.
In this Review, we discuss current and emerging aspects of complement’s contribution to cancer elimination and progression and suggest therapeutic targets with clinical potential. We also place emphasis on clinical challenges faced in translating these findings to efficacious tumour cytotoxic modalities or combination immunotherapies and interrogate the feasibility of using complement-based biomarkers for cancer diagnosis, tumour staging and prognosis.
Complementing cancer immunotherapy
There is a growing appreciation that durable clinical responses in patients with cancer are more likely to be achieved when targeted therapies that ablate key oncogenic signalling pathways are combined with approaches that effectively reverse tumour-instigated immunosuppression and augment antitumour immunity — an example has been the success of immune checkpoint blockade5,8. Among the currently explored targeted therapies, monoclonal antibody (mAb)–based cancer immunotherapy has gained considerable traction as a viable therapeutic modality and has shown promising results, particularly in the treatment of certain haematological malignancies, such as B cell lymphomas, multiple myeloma and chronic lymphocytic leukaemia12,24–27.
How mAbs harness the complement system
The clinical benefit of mAb-based immunotherapies is attributed partially to their ability to evoke complement-dependent cytotoxicity (CDC) that culminates in tumour cell elimination12,18. Indeed, complement has long been considered a potent cytolytic ‘payload’ for tumour-specific antibodies, and seminal studies in the early 1970s laid the groundwork for what would later become a prevalent concept in the field28,29. Notably, various cancer immunotherapeutic protocols used today in the clinic rely on the two-pronged capacity of mAbs to halt oncogenic signalling and tumour cell growth and to simultaneously fix complement on the surface of the targeted tumour cells, thereby eliciting CDC18,24,25.
Despite modest attempts to harness the cytolytic capacity of complement in mAb-based cancer immunotherapy over the years, full realization of its tumoricidal potential came only with the US Food and Drug Administration (FDA) approval of rituximab, a chimeric anti-CD20 mAb developed for the treatment of B cell lymphomas12,24. A wide array of in vitro studies using cancer cell lines, primary tumour cells, mouse tumour models and blood profiling of patients with chronic lymphocytic leukaemia after mAb infusion have provided evidence for the auxiliary, tumour-destructive role of complement in the context of anti-CD20 immunotherapy12. mAbs targeting surface antigens such as CD20, CD38 and CD52 that are highly expressed on B cell- or T cell-derived tumours are among the most well-studied tumour-specific mAbs in terms of their ability to elicit CDC24,27,30.
The binding of mAbs to tumour antigens triggers the concerted activation of multiple effector pathways that operate in parallel to elicit cytolytic responses. These include CDC elicited via the classical pathway of complement activation and assembly of the membrane attack complex (MAC) (BOX 1); antibody-dependent cell-mediated cytotoxicity (ADCC), operating primarily through the engagement of Fc receptors on natural killer (NK) cells; and antibody-mediated phagocytosis12 (FIG. 1). Depending on the magnitude of the complement response on targeted cells, tumour cell elimination can occur either through MAC assembly and direct cell lysis or via recruitment of macrophages that take up opsonized tumour cells through the recognition of opsonic fragments (for example, C3b, iC3b and C3dg) by complement receptors CR1, CR3, CR4 or CRIg12 (BOX 2). Concomitant recognition of the Fc portion of tumour-specific mAbs by Fcγ receptors (FcγRs) on macrophages allows for synergistic clearance of tumour cells via complement CR3- and FcγR-dependent uptake (FIG. 1). Notably, the release of complement-derived inflammatory mediators, such as C5a (BOX 1), further potentiates the cytolytic activity of mAbs by promoting the recruitment of phagocytic cells and skewing the relative expression of activating and inhibitory FcγRs on neutrophils and macrophages31.
Figure 1. Complement mediates tumour cytolysis in the context of antibody-based immunotherapy.
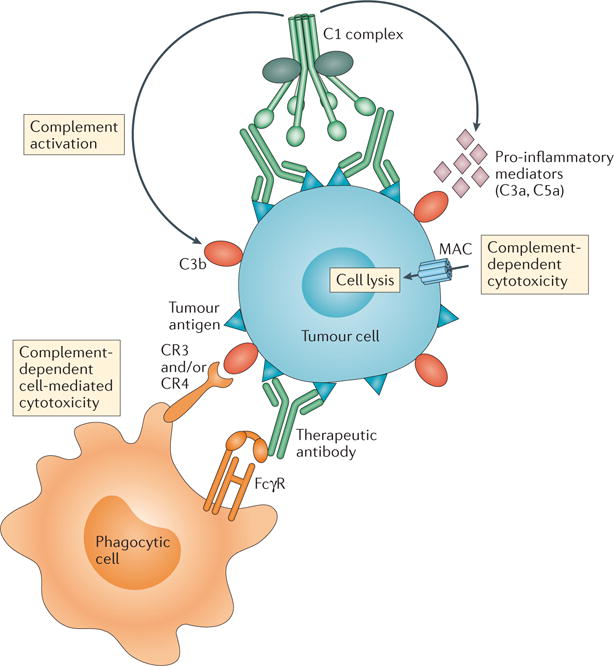
Schematic illustration of the basic humoral and cellular elements that evoke tumour cytolysis in the context of monoclonal antibody (mAb)-based cancer immunotherapy. Direct tumour cell elimination is achieved by (i) complement-dependent cytotoxicity (CDC) and (ii) antibody-dependent cell-mediated cytotoxicity (ADCC) and complement-dependent phagocytosis elicited upon targeting of tumour antigens by clinically available therapeutic mAbs. Tumour-targeted mAbs known to elicit complement-mediated cytolytic responses include the anti-CD20 mAbs rituximab and ofatumumab in B cell lymphomas and chronic lymphocytic leukaemia. Targeting of tumour cells by therapeutic mAbs triggers complement activation via the classical pathway. Binding of C1q to the Fc portion of these antibodies leads to the assembly of the active C1 complex (C1q, C1r and C1s), which acquires proteolytic activity over complement components, initiating the cascade. Complement activation leads to tumour cell opsonization by C3-derived opsonins (C3b, iC3b and C3dg) and the generation of potent pro-inflammatory mediators (C3a and C5a), which in turn recruit and activate immune cells with phagocytic properties (neutrophils and macrophages). Downstream activation of terminal complement components results in the assembly of the pore-forming membrane attack complex (MAC), or C5b–C9, on the tumour cell membrane. The anaphylatoxin C5a is known to upregulate activating Fcγ receptors (FcγRs) on phagocytic cells, priming them for enhanced phagocytosis and increasing the magnitude of the tumour cytolytic response. C3-derived fragments (C3b, iC3b and C3dg) on tumour cells bind to CR3 and/or CR4 complement receptors on phagocytes, thus augmenting the FcγR-dependent phagocytic uptake of opsonized tumour cells.
Box 2. Complement: numerous proteins, plenty of functions.
Soluble complement proteins are primarily produced in the liver (the exceptions being C1q and Factor D, which are secreted mainly by macrophages and adipocytes, respectively)55. Whereas the major ‘pool’ of complement is located in the blood, a variety of cell types, including fibroblasts, adipocytes and endothelial and immune cells, secrete complement proteins. A boost in complement production is often observed during inflammatory and pathologic conditions, so that virtually any tissue can be surrounded by complement activation products13. Such a notion of local complement is key to perceiving how imbalanced complement activation affects pathology in distinct tissues, from the skin to the kidneys, from the lung to the brain, and influences the fate of an assortment of tumour cells3,17,126. Essentially, the presence of a trigger or improper regulation results in the production of activation fragments (BOX 1) that can bind to their respective receptors on cell surfaces and elicit biological responses. Upon complement activation, C3 fragments (that is, C3b, iC3b and C3dg) that are associated with antigens bind to the complement receptors CR1 (CD35), CR3 (CD11b and CD18), CR4 (CD11c and CD18) and/or CRIg, inducing phagocytosis and modulating the function of antigen-presenting cells13. In turn, triggering of CR2 by iC3b and C3dg is associated with the activation of B cells35. Furthermore, signalling through the G protein-coupled anaphylatoxin receptors C3aR, in the case of C3a, or C5aR1 (also known as CD88), in the case of C5a and desarginated C5a (C5a-desArg), regulates cell activation, degranulation, proliferation and cytokine responses, which are essential for initiating and maintaining inflammatory responses36. In addition, adaptive immune responses are modulated when membrane cofactor protein (MCP, also known as CD46) is triggered on T cells132. Such an array of interactions between complement proteins and cell-surface receptors not only guarantees immune surveillance but also determines the outcome of immune responses, metabolic pathways and developmental processes. Proper control of the activation signals described above is required to prevent excessive activation that can lead to tissue damage and is achieved by an assortment of complement regulators (TABLE 1) that restrain certain steps of the complement cascade133.
It should be noted, however, that a wide array of tumour cells, especially in solid tumours, have evolved diverse mechanisms by which they can limit CDC on their surfaces. Overexpression of membrane-bound complement regulatory proteins (that is, CD55 (also known as complement decay-accelerating factor (DAF)), CD46 (also known as membrane cofactor protein (MCP)), and CD59) or sequestering of fluid-phase regulators (for example, Factor H or C4-binding protein (C4BP, also known as C4BPA)) (TABLE 1) by tumour cells has been shown to prevent CDC by limiting C3 opsonization to levels well below the threshold required for terminal pathway activation and MAC assembly32–34. In this regard, several strategies have been devised to overcome this impediment in mAb-based immunotherapy (discussed below).
Table 1.
Regulators of complement activation
| Regulators | Gene | Main expression | Function | Connection with disease | Refs |
|---|---|---|---|---|---|
| C1 inhibitor (SERPING1) | SERPING1 | Liver | Regulates the CP and LP. Acts as an inhibitor for various proteases involved in the complement (C1r, C1s and MASPs), kinin, fibrinolytic and coagulation systems. | Hereditary angioedema. | 134 |
| sMAP (MAp19) | MASP2 | Liver | Form complexes with MBL. Reported to compete with MASP2, but physiologic function still controversial. | ND | 135 |
| MAP1 (MAp44) | MASP1 | Liver | Form complexes with MBL and ficolins. Reported to inhibit deposition of C4. | Mutations are associated with 3MC syndrome. | 136,137 |
| C4b-binding protein (C4BP) | C4BPA, C4BPB | Liver | Accelerates decay of CP and LP convertases. Cofactor for FI. Reported to bind apoptotic and necrotic cells and amyloid protein, preventing inflammation. | Mutations associated with aHUS and recurrent pregnancy loss. | 138,139 |
| CR1 (CD35) | CR1 | Kidney, bone marrow-derived cells, excluding platelets | Accelerates decay of AP, CP and LP convertases. Cofactor for FI. | Variants associated with susceptibility to SLE and resistance to malaria. Deficiency associated with glomerulonephritis. Low levels of CR1 on erythrocytes associated with poor outcome in PNH patients. | 140–143 |
| Factor H (FH) | CFH | Liver | Main regulator of the AP. Accelerates decay of AP convertases. Cofactor for FI. Recognizes self-surfaces, enabling local regulation of complement activation in cells and tissues. | Gene variants associated with aHUS, AMD and C3G. Deficiency associated with aHUS and increased susceptibility to infections. | 144–149 |
| FHL1 (Reconectin) | CFH | Liver | Accelerates convertase decay. Cofactor for FI. | Gene variants associated with aHUS, AMD and C3G. | 150,151 |
| Factor I (FI) | CFI | Liver | Protease that mediates degradation of C3b and C4b fragments, interrupting complement cascade activation. | Gene variants associated with aHUS and AMD. Deficiency associated with increased susceptibility to infections. | 152–154 |
| MCP (CD46) | CD46 | All nucleated cells | Cofactor for FI. | Gene variants associated with aHUS. Deficiency associated with male infertility. | 152,155 |
| DAF (CD55) | CD55 | Wide expression | Accelerates convertase decay. | Deficiency associated with PNH or Inab phenotype. | 156 |
| CD59 (Protectin) | CD59 | Wide expression | Binds to C8 and C9; prevents assembly of TCC. | Deficiency associated with PNH and neurological symptoms. | 157,158 |
| Vitronectin (S-protein, S40) | VTN | Liver | Binds to C5b–C9; prevents assembly of TCC. Also implicated in cell adhesion via binding to integrins. | ND | 159 |
| Clusterin (Apolipoprotein J, SP-40) | CLU | Reproductive organs | Binds to C7–C9; prevents assembly of TCC. Additionally, is a stress-activated chaperone associated with various physiologic processes. | ND | 160 |
| Carboxypeptidase N (Polypeptide 1) | CPN1 | Liver | Degrades C3a and C5a to their desArg forms. | Angioedema, chronic urticaria, asthma. | 161,162 |
Abbreviations: 3MC syndrome, a rare developmental disorder that unifies the overlapping autosomal recessive disorders previously known as Mingarelli, Malpuech, Michels and Carnevale syndromes; aHUS, atypical haemolytic uraemic syndrome; AMD, age-related macular degeneration; AP, alternative pathway; CP, classical pathway; CR1, complement receptor 1; DAF, decay accelerator factor; FH, Factor H; FI, Factor I; LP, lectin pathway; MBL, mannose-binding lectin; MCP, membrane cofactor protein; MASP, MBL-associated serine protease; ND, not determined; PNH, paroxysmal nocturnal haemoglobinuria; SLE, systemic lupus erythematosus; TCC, terminal complement complex.134–162
Beyond cytotoxicity
Growing evidence indicates that complement, in addition to providing an adjunctive mechanism for mAb-mediated tumour cytolysis, mediates a plethora of immunomodulatory functions that are pertinent to tumour immunosurveillance and antitumour immunity35,36. Fundamental processes such as tumour antigen processing and presentation by antigen-presenting cells (APCs), as well as B cell activation and T helper and effector T cell survival and differentiation, appear to be influenced by the intricate crosstalk of complement effector proteins with key cellular pathways that drive B cell and T cell responses37,38. Dendritic cells (DCs), for instance, express a wide array of complement proteins and receptors39. In this regard, C3 deficiency has been associated with a reduced antigen-presenting capacity of APCs (lower expression of MHC class II molecules and impaired co-stimulation via CD80 and CD86), with important implications for autoimmune conditions and antitumour immunity40. Notably, the absence of C3aR or C5aR1 stimulation at the APC–T cell interface has been shown to negatively affect the survival of CD4+ T cells41. Moreover, genetic or pharmacological ablation of C3aR or C5aR1 signalling has also been linked to the induction of an immunosuppressive phenotype in CD4+ T cells in a manner that could diminish the magnitude of antitumour effector T cell responses42.
Further insight into the fundamental role of complement in immune cell homeostasis has been provided by studies describing an intracellular complement circuitry in CD4+ T cells that modulates cell differentiation through its crosstalk with the NOD-, LRR- and pyrin domain-containing 3 (NLRP3) inflammasome and the generation of reactive oxygen species (ROS)38,43. Taken together, these studies support the presence of a constitutively active complement circuitry within lymphocytes that has a role in the maintenance of the T cell pool in the periphery and may also potentiate effector T cell responses in the context of tumour immunotherapy.
Complement and cancer vaccines
Cancer vaccines represent another promising modality in cancer immunotherapy, and several studies have indicated that complement activation may contribute to vaccine efficacy through enhanced antigen uptake and co-stimulatory signalling44. Indeed, C5a has been shown to potentiate antigen processing and presentation by human DCs45,46. In this context, a C5a-derived C5aR1 agonist fused to a peptide-based cancer vaccine has been tested in a preclinical model of melanoma, with encouraging results47. These studies point to a different therapeutic perspective whereby harnessing complement activation and triggering C5aR1-dependent signalling on tumour antigen-loaded DCs may help patients elicit potent antitumour immune responses. From a different standpoint, the function of the opsonin C3dg as a molecular adjuvant that lowers the activation threshold of B cells and the role of C3-derived fragments in promoting antigen retention by follicular DCs and boosting memory responses have long been appreciated as mechanisms bridging innate and adaptive immune responses48. However, this fundamental facet of complement biology has not been rigorously investigated in the context of cancer immunotherapy. These key immunoregulatory activities of complement may have important implications for tumour immunotherapy and may be worth exploring during the design of antitumour vaccines.
Complement and radiotherapy
Alongside surgical resection and chemotherapy, radiotherapy is also recognized as a clinical mainstay of cancer treatment, especially in the context of very aggressive tumours with poor prognosis49. Recent evidence supports a causative association between radiation-induced tumour cell damage, the tumour-derived pro-inflammatory milieu and the potentiation of antitumour immunity50,51. Interestingly, radiotherapy-induced cell death was recently shown to induce transient complement activation within solid tumours, with both tumour cells and infiltrating immune cells serving as sources of complement protein production52. More importantly, radiotherapy was found to upregulate the release of C3a and C5a within the tumour microenvironment, inducing potent DC responses and subsequently leading to pronounced infiltration of CD8+ effector T cells into the irradiated tumours52. Notably, an apparent discordance between these findings and those of a recent report showing that C3 inhibition potentiates antitumour immunity after fractionated radiotherapy53 could be attributed to the different radiotherapeutic regimens applied in each case and to the variable extent of tumour-associated inflammation elicited by these models. While these findings await corroboration in different tumour models, they provide fertile ground for considering complement activation as a beneficial component of radiotherapeutic protocols in various types of cancer.
Feeding inflammation and tumorigenesis
Undeniably, in the context of immunotherapy, complement potentiates tumour killing via its immune adjuvant properties and induction of cytolysis (BOX 2). Extensive investigation of the tumour microenvironment, however, has uncovered molecular mechanisms linking imbalanced complement activation and cancer progression. The tumour microenvironment is essentially composed of genetically abnormal cells surrounded by blood vessels, fibroblasts, immune cells, stem cells and extracellular matrix (ECM), and a dynamic crosstalk among these various components ultimately determines the fate of the tumour54 (FIG. 2). It is fascinating that tumours emerge from a single cell or a small number of cells bearing mutations in tumour suppressor genes and/or oncogenes. To become cancerous, however, these cells must acquire several mutations in specific genes that allow proliferation beyond the regular rate, as well as mutations that facilitate cell migration, survival in the circulation, invasion of tissues and adaptation to new tissue environments. In this context, it is particularly interesting that the past decade has uncovered dysregulated complement activation in the tumour microenvironment as a key link between inflammation, the suppression of antitumour immune responses and the promotion of tumorigenesis3.
Figure 2. Complement activation in the tumour microenvironment promotes tumorigenesis.
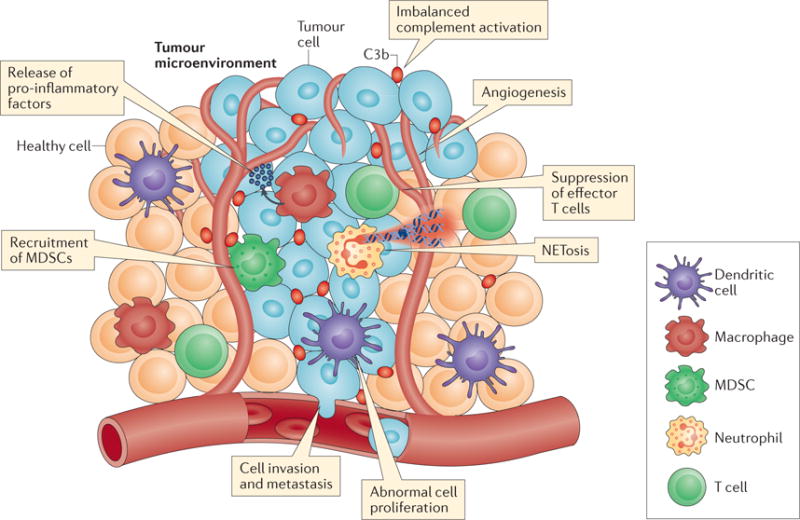
A schematic illustration of the fine interplay between complement effectors, tumour cells and innate and adaptive immune cells in the tumour microenvironment, highlighting the many means by which complement contributes to tumour progression. Complement proteins can be secreted by tumour cells or tumour-infiltrating immune cells or can enter the tumour stroma through the vasculature. Local complement imbalance and complement activation within the tumour vasculature can lead to the release of pro-inflammatory factors by both tumour cells and tumour-infiltrating myeloid cells (for example, macrophages and dendritic cells (DCs)). Deregulated complement activation perpetuates macrophage-sustained tumour inflammation, creating a favourable environment for tumour growth. Local release of distinct complement activation fragments, such as C5a, recruits myeloid-derived suppressor cells (MDSCs) into the solid tumour. MDSCs exert potent immunosuppressive effects by ablating effector CD8+ T cell responses and facilitating tumour growth. Complement proteins have also been shown to modulate CD4+ T helper cell responses within the tumour microenvironment, shifting the inflammatory milieu towards a pro-tumorigenic phenotype. Complement activation products have been shown to promote tumour-associated angiogenesis, enhancing cell invasion into adjacent tissues and tumour metastasis to distant organs. Moreover, complement activation has been associated with the induction of a neutrophil-driven hypercoagulative state in certain solid tumours that further promotes tumour progression. In this respect, complement activation triggers the accumulation of pro-tumorigenic neutrophils within solid tumours, potentiating their procoagulant responses (for example, NETosis) and thus establishing a link between complement-driven coagulation, inflammation and tumour progression.
Production and activation of complement in the tumour microenvironment
The local production of complement proteins by various tissues is well recognized55,56 (BOX 2). Furthermore, both tumour and stromal cells produce complement proteins, and evidence for local and systemic activation of complement is often observed in cancer settings57,58. In line with the notion that initiators of complement activation can sense DAMPs59, complement activation appears to occur in response to tumour-associated antigens, and increased deposition of complement activation fragments is commonly detected in tumour tissues. Indeed, the presence of C1q along the tumour vasculature has been detected in a rodent model of cervical cancer, and C1q has also been shown to bind directly (independent of antibodies) to phospholipids in lung tumour cell lines22,60. Evidence of classical pathway activation in cancer, such as increased plasma concentrations of the C4d fragment, has been obtained from patients with lung cancer, thyroid cancer, astrocytoma, lymphoma, leukaemia, or oropharyngeal cancer3,60. Moreover, signs of a deregulated alternative pathway of complement activation have been reported in patients with acute lymphoblastic leukaemia, Burkitt lymphoma or multiple myeloma, and a deregulated lectin pathway has been found in patients with T cell leukaemia61. Although evidence associating specific tumour antigens with differential activation of complement pathways is still scant, it is tempting to speculate that modified glycoproteins and glycolipids and aberrantly expressed mucins in transformed tumour cells can all act as DAMPs that are able to activate complement. Identification of such triggers would be of great value should anti-complement therapy become a reality in the adjunct treatment of cancer.
While tumour and stromal cells are active in producing complement proteins, and the local micro environment favours complement activation, complement regulates the activation status of the local cells. In the past decade, a wealth of research has provided compelling evidence regarding the role of complement in the modulation of immune cells (discussed above and BOX 2). In addition, in vitro data indicate that complement is linked to the differentiation of myeloid-derived suppressor cells (MDSCs), which are seemingly involved in immune responses to cancer22,62. Given that cancer development is strongly influenced by immune responses, one may assume that complement is a linking factor determining the quality and magnitude of the local antitumour immune response.
Does complement activation by infections affect tumorigenesis?
Our current understanding suggests a dual role of immunity in cancer, in which pro-inflammatory signals lead to DC activation, antigen presentation and consequent activation of effector T cells, while chronic inflammation, by contrast, promotes an immunosuppressive environment that supports tumour progression and metastasis63. Indeed, epidemiological data clearly show that tumour-associated inflammation is a hallmark of cancer, with close to 20% of the cancers worldwide being driven by microbial infection with organisms such as Helicobacter pylori, human papilloma virus (HPV), hepatitis virus, Epstein–Barr virus (EBV) and HIV. Although some of these infectious agents (for example, HPV and EBV) induce cell transformation directly, others promote tumorigenesis via local tissue inflammation64,65. Whether these infections trigger complement activation that fuels the inflammatory response in cancer is an interesting topic for future investigation.
Complement modulates immune responses in the tumour microenvironment
Animal models have contributed immensely to increasing our understanding of the participation of complement in the promotion of tumorigenesis (TABLE 2). Whereas a pioneering study demonstrated that the activation fragment C5a (BOX 1) recruits MDSCs to the tumour microenvironment, resulting in suppressed antitumour immune responses22, numerous other investigations have corroborated the view of imbalanced complement activation as a promoter of a pro-inflammatory environment that sustains tumorigenesis. MDSCs, for instance, are increasingly recognized to suppress effector T cells through deprivation of amino acids, production of nitric oxide (NO) and ROS, upregulated expression of programmed cell death 1 ligand 1 (PDL1) and secretion of angiogenic factors66. C5a-mediated activation of MDSCs has been associated with the induction of immunomodulators such as arginase 1 (ARG1), cytotoxic T lymphocyte antigen 4 (CTLA4), IL-6, IL-10, lymphocyte activation gene 3 protein (LAG3) and PDL1 in a murine model of lung cancer67. Interestingly, generation of C5a in this model appears to be independent of complement activation. Consistent with this observation, in vitro studies have indicated that C5a can be generated by the cleavage of C5 by serine proteases expressed on the surface of tumour cells68. In addition, a model of lung cancer has provided evidence to suggest that tissue factor-induced coagulation leads to C5 activation and tumour promotion, further supporting the notion of local complement activation by coagulation-related proteases in the tumour microenvironment69 (BOX 1). C5a has also been shown to participate in tumorigenesis by upregulating the expression of molecules that suppress local immune responses such as PDL1, transforming growth factor-β (TGFβ) and IL-10 (REFS 58,70).
Table 2.
Animal models targeting complement in cancer research
| Cancer model | Target protein | Observations | Refs |
|---|---|---|---|
| Cervical: syngeneic model (TC-1 cells) | C3, C5aR1 | C3 is deposited on engrafted tumours. Impaired tumour growth in C3−/− and C5aR−/− mice. Impaired tumour growth upon pharmacological inhibition of C5aR1. C5aR1-dependent migration of MDSCs to tumours. C5aR1-dependent production of ROS and RNS by MDSCs. | 22 |
| Ovarian: xenograft model (RMA-3CF4 cells) Lymphoma: syngeneic model (RMA-1474 cells) | C5aR1 | Tumour burden is associated with increased expression of C5a. C5a-induced inflammation correlates with decreased numbers of effector T cells. | 76 |
| Ovarian: genetic model (TgMISIIR-Tag) | C3, C5aR1 | Attenuated tumorigenesis in TgMISIIR-Tag mice that have a full or partial deficiency in C3 or C5aR1. C3-dependent production of pro-inflammatory cytokines. C5a-dependent production of pro-angiogenic factors. Impaired tumour growth upon pharmacological inhibition of C5aR1. | 85 |
| Ovarian: syngeneic model (ID8-VEGF) Ovarian: xenograft model (SKOV3ip1 cells) | C3, C5aR1 | Tumour cells secrete complement proteins that promote tumour growth in an autocrine fashion via induction of the PI3K–AKT pathway. Impaired tumour growth in C5aR−/− mice. | 57 |
| Breast: syngeneic, metastatic (4T1 cells) | C5aR1 | C5aR1-dependent recruitment of immature myeloid cells to the lung and consequent inhibition of effector T cells via production of TGFβ and IL-10. Pharmacologic or genetic ablation of C5aR1 attenuated lung metastasis. | 23 |
| Melanoma: syngeneic model (B16F10 cells transfected with gp33) | C3 | Robust antitumour response in C3-depleted mice (using CVF) via NK-mediated activation of gp33-specific CD8+ T cells. | 104 |
| Colon: syngeneic model (MC38 cells) | C3 | Robust antitumour response in C3-depleted mice (using CVF) via increased expression of CCL5, CXCL10 and CXCL11 and migration of CD8+ T cells. | 134 |
| Colon: syngeneic, metastatic (SL4, CT26 cells) Colon: xenograft model (HCT116, SW116 cells) | C5a | Increased production of C5a by colon cancer cell lines. Decreased infiltration of inflammatory cells and number of metastases in the liver of C5aR−/− mice. C5a-dependent production of CCL2 by macrophages via the AKT pathway. | 58 |
| Sarcoma: induced by carcinogen (3-MCA) | PTX3 C5a | PTX3 regulates complement activation by interaction with C1q and FH. Increased complement activation in Ptx3−/− mice. Ptx3−/− mice are more susceptible to cancer development. Increased generation of C5a, CCL2 production and recruitment of tumour-promoting macrophages in Ptx3−/− mice | 21 |
| Colitis-induced colorectal cancer (azoxymethane plus dextran sulfate sodium) | C3, C5, C5aR1 | Decreased tumorigenesis in C3−/−, C5−/− or C5aR−/− mice. C5a signals on epithelial cells do not contribute to tumorigenesis. C5a signalling on neutrophils promotes IL-1β production. Complement inhibition results in decreased production of IL-1β and IL-17. | 79 |
| Melanoma: syngeneic model (B16F10 cells) | C3, C3aR, C5aR1 | Decreased tumorigenesis in C3−/−, C3aR−/− or C5aR−/− mice. Expression of C3aR and C5aR1 by 20% of melanoma TILs. C5a promotes secretion of IL-10 by effector CD8+ T cells. Complement inhibits antitumour immunity via a PDL1-independent pathway. Combined blockage of PDL1 and complement results in enhanced antitumour response. | 71 |
| Melanoma: syngeneic model (B16F10 cells) | C3aR | Decreased tumorigenesis in C3aR−/− mice. Increased numbers of neutrophils and CD4+ T cell subsets and decreased numbers of macrophages in the tumours of C3aR−/− mice. | 77 |
| Intestine: genetic model (ApcMin/+) | C3aR | Decreased systemic levels of C3a in aged ApcMin+/− mice. Reduced tumorigenesis in ApcMin/+C3aR−/− mice. C3a-induced hypercoagulation and NET formation leading to polarization of low-density neutrophils with a pro-tumorigenic phenotype. | 78 |
| Intestine: genetic model (ApcMin/+); diet-induced | C3, C5aR1 | Decreased tumorigenesis in C3−/− and C5aR−/− mice. Impaired tumour growth upon pharmacological inhibition of C5aR1. Tumorigenesis associated with increased levels of C5a and inflammation. Feeding with HFD using corn or coconut oil as a source of fat results in C5a generation, inflammation and tumorigenesis. Olive oil used as a source of fat does not promote complement activation or tumorigenesis. | 82 |
| Melanoma: syngeneic model (B16F10 cells) | C1q | C1q is expressed in the stroma and endothelium of various malignant tumours. Decreased tumorigenesis and metastasis in C1q-deficient mice. C1q promotes tumour growth independent of complement activation by inducing angiogenesis, cell adhesion and proliferation. | 86 |
| Squamous cells: xenograft model (cSCCIS cells) | Factor B, C3 | Decreased tumorigenesis in mice injected with cancer cells that had the genes encoding C3 or Factor B knocked down using siRNA. | 90 |
| Leptomeningeal metastasis (various breast cancer cell lines) | C3aR | C3 is upregulated in the cerebrospinal fluid in metastatic settings. C3 expression is correlated with poor disease outcome. C3aR is activated in the choroid plexus epithelium, resulting in the disruption of the blood–cerebrospinal fluid barrier. Ablation of C3aR signalling is beneficial in attenuating leptomeningeal metastasis. | 91 |
| Lung: syngeneic model (393P or LLC cells) in KrasLSL−G12D/+ mice | C5aR1 | Combined inhibition of C5a- and PD1-induced signalling pathways results in decreased tumorigenesis and metastasis via decreased numbers of MDSCs and increased numbers of effector T cells in the tumour. | 72 |
Abbreviations: APC, adenomatous polyposis coli; CCL, CC-chemokine ligand; CVF, cobra venom factor; CXCL, CXC-chemokine ligand; HFD, high-fat diet; MDSCs, myeloid-derived suppressor cells; NET, neutrophil extracellular traps; NK, natural killer; PD1, programmed cell death protein 1; PDL1, programmed cell death 1 ligand 1; PTX, pentraxin; RNS, reactive nitrogen species; ROS, reactive oxygen species; siRNA, small interfering RNA; TGFβ, transforming growth factor-β; TgMISIIR-Tag, transgenic mice that express the transforming region of Simian virus 40 under the control of the Mullerian inhibiting substance type II receptor and are prone to developing ovarian tumours; TILs, tumour-infiltrating lymphocytes.104,163
Findings from two independent groups, using models of melanoma and lung cancer, have uncovered a therapeutic potential for combined blockade of the immune checkpoint molecule PDL1 and complement71,72. Anti-PDL1 mAbs are currently used in the clinic for the treatment of melanoma and lung cancer. By binding to its receptor PD1 (also known as PDCD1) on activated T cells, PDL1 dampens T cell receptor (TCR)-mediated production of IL-2 and subsequent T cell proliferation. Therefore, instead of targeting tumour cells, these mAbs block inhibitory pathways that prevent effective antitumour responses73. Of note, mAbs that target immune checkpoints are designed to induce limited complement activation, CDC and ADCC. Combined ablation of C5a- and PDL1-induced signalling appears to be more efficient in restraining tumour growth than anti-PDL1 therapy alone71,72, thus indicating that complement inhibition increases the clinical benefit of immune checkpoint blockade. Additionally, the long pentraxin PTX3 has been shown to act as a tumour suppressor in skin, epithelial and mesenchymal carcinogenesis21. Evidence suggests that PTX3 is induced during carcinogenesis by IL-1 and that it regulates complement-derived chemokine production via recruitment of Factor H21 (TABLE 1). Most importantly, in three independent studies, the PTX3 gene was found to be methylated and silenced in selected human tumours74.
The immunosuppressive role of C5a in cancer settings is somewhat paradoxical, considering that it induces the maturation of DCs and the differentiation of T helper 1 (TH1) cells42,75. Such apparent contradictions were addressed in a study comparing tumour progression in mice injected with distinct murine lymphoma cell lines that secrete either low (RMA-1474 cells) or high (RMA-3CF4 cells) amounts of C5a. Notably, high production of C5a was associated with more prominent tumorigenesis accompanied by decreased numbers of interferon-γ (IFNγ)-producing CD8+ and CD4+ cells in the spleens and lymph nodes of the injected mice76. Consistent with these findings, in vitro polarization of CD4+ T cells in the presence of increasing amounts of C5a revealed a concentration-dependent polarization of T cells towards either the TH1 cell or regulatory T (Treg) cell lineage; high levels of C5a (500 ng/ml) favoured the polarization of CD4+ T cells to a Treg cell phenotype instead of a TH1 cell phenotype76. These observations nicely illustrate the dual role of complement in modulating immune responses. Whereas physiological concentrations of activated complement molecules can promote antitumour immune responses, imbalanced chronic complement activation, usually preceding pathologic conditions, is associated with tumour progression.
Supporting a role for complement in tumour promotion, the activation fragment C3a has also been implicated in tumorigenesis. Improved disease outcome in C3aR−/− mice has been observed in a model of melanoma; tumorigenesis was associated with increased numbers of neutrophils and a pro-inflammatory tumour microenvironment77. The deleterious role of neutrophils in cancer has been further confirmed in a model of spontaneous intestinal tumorigenesis in mice with a mutation in the adenomatous polyposis coli (Apc) gene (that is, ApcMin/+ mice). In this model, tumour growth was correlated with the accumulation of neutrophils having a pro-tumorigenic phenotype and the formation of neutrophil extracellular traps (NETs). Mechanistically, circulating lipopolysaccharide, likely leaking from the intestine, appears to induce complement activation and upregulate the expression levels of C3aR on neutrophils. C3aR-mediated signalling then triggers NET release, coagulation pathways and polarization of neutrophils towards a pro-tumorigenic phenotype78.
Complement in intestinal cancers
The role of complement in tumorigenesis is particularly interesting in the settings of intestinal and colorectal cancer because of its strong association with inflammatory bowel disease. Indeed, complement activation in a colitis-associated cancer model induced by combined administration of azoxymethane and dextran sulfate sodium has been shown to result in the induction of the pro-inflammatory cytokine IL-1β, mainly by infiltrating neutrophils and consequent production of IL-17, a major inducer of intestinal disease79. Supporting the correlation between imbalanced complement activation, inflammation and intestinal tumorigenesis, an elegant study using a model of diet-induced intestinal tumorigenesis in genetically susceptible ApcMin/+ mice has demonstrated that the source of dietary fat, and not obesity itself, triggers complement activation, inflammation, expression of oncogenes and intestinal polyposis; obesity and its associated metabolic changes had previously been associated with imbalanced complement activation, inflammation and cancer80,81. However, the ApcMin/+ study dissociated the direct link between obesity and cancer by showing that whereas obese mice showed increased tumorigenesis when fed a high-fat diet containing fat derived from corn and coconut oil, intestinal polyposis was not observed in obese mice who were fed a high-fat diet consisting of fat from olive oil82. In this model, tumour development was associated with increased amounts of C5a, both systemically and in the intestine. As no complement activation was observed in the mice fed the olive oil-based diet, these findings pointed to a novel concept of diet-induced complement activation and raised questions as to whether and how different sources of fat or other ‘trigger’ foods, such as sugar, dairy and gluten, trigger an imbalanced complement activation in the gastrointestinal tract that could be connected with pathological conditions.
Effects of complement on tumour cell proliferation and metastasis
In addition to fuelling inflammation, C3aR and C5aR1 expressed in tumour cell lines appear to signal in an autocrine manner, triggering the phosphoinositide 3-kinase (PI3K)–AKT pathway involved in tumour cell proliferation57. Similarly, sublytic levels of C5b–C9 deposited on cancer cells induce cell cycle progression by activating signal transduction pathways in cancer cells83,84. Furthermore, complement components C5a and C1q have been implicated in promoting angiogenesis in models of ovarian cancer and melanoma85,86. Interestingly, triggering of cell proliferation pathways by C3a and C5a is not necessarily deleterious and has been implicated in physiological processes such as liver and retina regeneration87,88.
Metastatic pathways are also triggered by imbalanced complement activation and inflammation. Various models of tumorigenesis, including colon cancer, pancreatic cancer, squamous cell carcinoma, lung cancer and breast cancer models, support this contention by showing increased production of complement proteins in metastatic settings, followed by modulation of immune responses and establishment of an environment that suppresses effector T cells23,58,89,90. C3aR has been further implicated in the dissemination of cancer to the nervous system by disrupting the blood–cerebrospinal fluid (CSF) barrier and permitting the access of plasma components and other mitogens to the CSF91.
Alongside the promotion of proliferation, angiogenesis and metastasis, hyperactivation of complement is associated with cancer cell motility and invasiveness. Aberrant expression of C5aR1 has been detected in squamous cell carcinoma, adenocarcinoma and transitional cell carcinoma from various organs, and vascular invasion appears to be correlated with the expression of C5aR1 (REFS 67,92,93). Cell invasiveness has been shown to occur in response to C5a-induced secretion of metalloproteinases from C5aR1-expressing cancer cells and degradation of ECM68. Interestingly, the basic helix– loop–helix transcription factor twist-related protein 1 (TWIST1), which is involved in epithelial–mesenchymal transition (EMT), binds to the promoter region of the C3 gene. C3 expression is then correlated with increased cell invasiveness through downregulation of E-cadherin on the surface of cancer cells and promotion of EMT94. In addition, leukaemia cell lines as well as clonogenic blasts derived from patients with chronic myeloid leukaemia or acute myeloid leukaemia show increased cell adhesion in response to C3a and C5a fragments95. In a model of gastric cancer, C5a promoted the invasiveness of human gastric cancer cell lines (the adenosquamous carcinoma cell line MKN1 and the tubular adenocarcinoma cell line MKN7) via conversion of RhoA-guanosine diphosphate (RhoA-GDP) to RhoA-guanosine triphosphate (RhoA-GTP), resulting in morphological cell changes and increased expression of stress fibres and filopodia92.
Collectively, the research described above points towards local production and increased activation of complement in the tumour microenvironment that is associated with modulation of antitumour immune responses and promotion of cancer development.
Challenges of translational research
The findings discussed above might seem controversial and unlikely to be incorporated into clinical practice, especially considering that rodent models of tumorigenesis do not fully recapitulate the diversity and course of human disease. Moreover, current clinical practices that rely on personalized protocols using immunotherapy, checkpoint inhibitors and vaccines are achieving favourable outcomes without considering complement responses. These concerns notwithstanding, it is remarkable how consistent the involvement of complement is in such a large variety of cancer models.
The apparent discrepancy between the ‘benign’ complement activation that potentiates immunotherapy and the ‘harmful’ complement activity that sustains inflammation and tumorigenesis is likely the consequence of a fine balance between physiological levels of and imbalanced complement activation. As mentioned above, differential concentrations of C5a in the tumour microenvironment seem to regulate the quality of immune responses and determine the fate of a tumour76. Furthermore, increased tumorigenesis, accompanied by early onset of lung metastasis and increased frequency of Treg cells and Her2 expression in the tumour microenvironment, has been observed in C3−/− mice transgenic for the Her2/neu oncogene neuT when compared with their C3-sufficient counterparts. Of particular interest is the observation that the transplantation of neuT-C3−/− malignant cells into syngeneic immunocompetent hosts results in milder pathology than does transplantation of tumour cells from neuT-C3+/+ tumours, suggesting that complement is a key factor in immunosurveillance mechanisms in the early stages of tumorigenesis and is also involved in the immunoediting of Her2-driven autochthonous mammary tumours96. Given that substantial evidence associates complement with both CDC and inflammation-mediated tumorigenesis, manipulation of the complement system as a means of supporting current anticancer therapies should be further explored.
Harnessing complement to enhance mAb-based immunotherapy
Immunotherapy based on mAbs, though a mainstream therapeutic option for patients with cancer, would further benefit from improvement in mAb-mediated effector functions and prevention of drug resistance. To this end, antibodies with increased propensity to form hexamers (HexaBodies) upon antigen (CD20 or CD38) binding have been engineered97. These HexaBodies have been shown to induce strong complement activation and CDC of primary chronic lymphocytic leukaemia cells, even in the presence of limiting amounts of C9 (REF. 98). Additionally, dose titration of the therapeutic mAb appears to be critical for optimal CDC. Evaluation of samples from patients under treatment with the anti-CD20 mAb ofatumumab indicates that doses currently used in the clinic may be excessive, leading to a rapid depletion of systemic complement and consequent exhaustion of complement-dependent effector functions. On the other hand, a lower mAb concentration in a more frequent dosing scheme has instead been accompanied by continuous deposition of C3b on malignant B cells and robust CDC99,100.
Importantly, the major mechanism responsible for the anti-CD20 response in patients, that is, the individual contributions of CDC, ADCC and phagocytosis, remains in debate101. In some therapeutic anti-CD20 mAbs (for example, obinutuzumab) ADCC appears to be dominant over CDC when compared with rituximab or other antibodies. Preclinical and in vitro data indicate that complement activation, and in particular C3b deposition, inhibits rituximab-induced NK cell activation and ADCC102–104. Further, anti-CD20 mAbs have been implicated in the induction of ROS by monocytes with consequent inhibition of NK cell-mediated cytotoxicity105. In light of these findings, a better understanding of the precise mechanisms triggered by therapeutic mAbs in the context of different tumours is imperative to determining the optimal strategy for clinical manipulation of complement in cancer patients.
Targeting complement regulators
In line with the idea of optimizing mAb effector functions, extensive research indicates that increased resistance to CDC results in part from the expression of complement regulators (TABLE 1) by tumour cells. Indeed, increased expression of complement regulators is often observed in cancer settings106–111, possibly representing a selective adaptation by tumours to escape from host immune responses. Notably, local inflammation has also been associated with an upregulation of the regulators CD55 and CD59 in hepatomas, again supporting a role for inflammation in tumour promotion106. It has been reported that although there is no direct correlation between lysis of tumour cells and expression of complement regulators, the ratio between the levels of CD20 and complement regulators on the membrane of mantle-cell lymphoma cells is indicative of their sensitivity to CDC112,113. Therefore, dual targeting of tumour antigen and complement regulators has been well investigated in in vitro models114–119.
Although a rational approach in theory, inhibition of complement regulators can be challenging because of the broad expression of regulators by normal host cells and the consequent pathology associated with the lack of proper regulation of complement on cell surfaces (TABLE 1). Nevertheless, a novel strain of NOD-scid IL2rγnull (NSG)-Hc1 mice with an intact complement system has been generated for use as a platform for testing mechanisms underlying CDC in vivo120. The parental strain, NSG, allows for engraftment of primary tumours from patients and investigation of ‘personalized’ therapy options but is deficient in the C5 component, making it impractical for in vivo evaluation of CDC. Collectively, these studies have taught us that the clinical efficacy of CDC in immunotherapeutic protocols largely depends on multiple factors, including tumour antigen expression, the overall tumour burden, the distinct expression profile of complement regulators and the surface density and tertiary conformation of tumour-deposited mAbs12. Issues related to the potential exhaustion of complement stores during the administration of high doses of therapeutic mAbs and the precise contribution of each individual effector mechanism to tumour cell killing warrant further consideration when mAb-based treatment regimens are being designed.
How is complement activated in tumours?
With the exception of mAb-induced complement activation, initiators of the complement cascade in the tumour microenvironment are still elusive. Whereas complement activation in response to natural IgM antibodies against tumour antigens as well as direct cleavage of C3 and C5 by tumour-derived proteases have been detected in cancer settings121, novel insights into the mechanisms driving complement activation by specific cancer types are essential for rationalizing therapeutic approaches aimed at manipulating the complement system in cancer. A variety of complement inhibitors that target distinct stages and pathways of the complement cascade are under development. These compounds are in different phases of clinical evaluation and can be potentially employed in case imbalanced complement activation in the tumour environment results from classical (for example, C1 inhibitor or anti-C1s antibody), alternative (for example, C3 inhibitors, mini-Factor H or Factor B or Factor D inhibitors) or lectin (for example, anti-MASP antibodies) pathway activation or by targeting specific effector functions (for example, C5aR1 antagonists)122,123.
Complement proteins as biomarkers for cancer
Whatever the initiation route, evidence of excessive complement activation has been detected in a variety of clinical samples. In fact, several studies have suggested the use of complement proteins or activation fragment as biomarkers for disease diagnosis and prognosis (TABLE 3). By the use of proteomic techniques, increased levels of C3 fragments have been identified in the serum of patients with colorectal cancer, breast cancer, pancreatic cancer, lymphoma, prostate cancer, leukaemia, lung cancer and oesophageal cancer (TABLE 3). It is still questionable, however, whether complement biomarkers have clinical value and can discriminate between benign and malignant tumours or if they are reliable markers of disease prognosis. Increased levels of C3 fragments, for instance, are detected in a wide variety of clinical conditions, and biomarkers identified by surface enhanced laser desorption ionization time-of-flight (SELDI-TOF) mass spectrometry often represent abundant serum proteins (C3 is found at 1–2 mg/ml in the serum). Interesting findings based on samples from patients with ovarian cancer, however, have associated the expression of intra-tumoural C3 with disease prognosis124. In addition, increased levels of a previously uncharacterized C3 fragment have been detected in prostatic fluid from cancer patients. Of note, biochemical evaluation of this C3 cleavage fragment revealed a putative prostate-specific antigen (PSA) cleavage site. Indeed, purified PSA was able to cleave C3 and C5 in vitro, suggesting that PSA acts as an immunoregulatory protease that contributes to a pro-tumorigenic environment through the proteolysis of complement proteins125.
Table 3.
Complement markers as prognostic and diagnostic tools
| Cancer type | Markers | Refs |
|---|---|---|
| Acute leukaemia | C3f-desArg | 164 |
| B cell lymphocytic lymphoma | C3a, C4a | 165 |
| Bladder | C3f | 166 |
| Breast | C3 fragments, C4a, C5aR1 | 166–170 |
| Colorectal | C3a-desArg | 171,172 |
| Renal | C5a, C5aR1 | 173,174 |
| Oesophageal | C3a, C4a | 175 |
| Familial adenomatous polyposis | C3 and C4 fragments | 176 |
| Gastric | DAF | 177 |
| Glioblastoma | MBL | 178 |
| Non-Hodgkin lymphoma | C1RL | 179 |
| Lung | C3, CD55 | 180,181 |
| Ovarian | MBL, C4BP | 182,183 |
| Pancreatic | C3 fragments, Factor B | 184–186 |
| Prostate | C3 fragments | 125,187 |
| Thyroid | C3f | 188 |
Thus, whereas complement biomarkers confirm the occurrence of increased complement activation in cancer settings, the use of such markers to guide clinical decisions is still improbable. Further insights into tumour antigens that are able to activate complement and mAb-mediated complement effector functions are highly anticipated to influence current cancer therapy practices.
Conclusion
Given the multiple factors that shape the intricate interaction between complement, tumour cells and the tumour stroma, and the dynamic nature of this relationship throughout the various stages of cancer immunosurveillance and promotion (that is, tumour seeding at the primary site, immune escape, progression and metastasis), therapeutic manipulation of the complement system in cancer settings has great potential. Aside from further improvements in mAb-induced complement cytotoxicity, special consideration should be given to exploring approaches combining the targeting of complement and immune checkpoints.
Acknowledgments
We thank D. McClellan for editorial assistance. J.D.L. also thanks R. Weaver and S. Weaver for the generous endowment of his professorship. Given the broad scope of this review, we often refer to specialized review articles rather than primary literature, and we have been able to include only selected examples of the breadth of the transformative work in the field; we therefore want to thank all our colleagues who are not specifically cited for their contributions and their understanding. This work was supported by grants from the U.S. National Institutes of Health (AI068730, AI030040) and the National Science Foundation (grant No. 1423304) and by funding from the European Community’s Seventh Framework Programme, under grant agreement number 602699 (DIREKT).
Footnotes
Author contributions
E.S.R. researched the literature and wrote and edited the manuscript. D.C.M. researched the literature and wrote and edited the manuscript. D.R. edited the manuscript and contributed to discussions of the content. A.M. edited the manuscript and contributed to discussions of the content. J.D.L. wrote and edited the manuscript and contributed to discussions of the content. E.S.R. and D.C.M. contributed equally to writing the manuscript and to reviewing and editing the manuscript before submission.
Competing interests statement
The authors declare competing interests: see Web version for details.
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nature. 2008;454:436–444. doi: 10.1038/nature07205. [DOI] [PubMed] [Google Scholar]
- 2.Chen DS, Mellman I. Elements of cancer immunity and the cancer-immune set point. Nature. 2017;541:321–330. doi: 10.1038/nature21349. [DOI] [PubMed] [Google Scholar]
- 3.Berraondo P, et al. Innate immune mediators in cancer: between defense and resistance. Immunol Rev. 2016;274:290–306. doi: 10.1111/imr.12464. [DOI] [PubMed] [Google Scholar]
- 4.Mantovani A, Marchesi F, Malesci A, Laghi L, Allavena P. Tumour-associated macrophages as treatment targets in oncology. Nat Rev Clin Oncol. 2017;14:399–416. doi: 10.1038/nrclinonc.2016.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hughes PE, Caenepeel S, Wu LC. Targeted therapy and checkpoint immunotherapy combinations for the treatment of cancer. Trends Immunol. 2016;37:462–476. doi: 10.1016/j.it.2016.04.010. [DOI] [PubMed] [Google Scholar]
- 6.Pio R, Corrales L, Lambris JD. The role of complement in tumor growth. Adv Exp Med Biol. 2014;772:229–262. doi: 10.1007/978-1-4614-5915-6_11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Balkwill FR, Mantovani A. Cancer-related inflammation: common themes and therapeutic opportunities. Semin Cancer Biol. 2012;22:33–40. doi: 10.1016/j.semcancer.2011.12.005. [DOI] [PubMed] [Google Scholar]
- 8.Gotwals P, et al. Prospects for combining targeted and conventional cancer therapy with immunotherapy. Nat Rev Cancer. 2017;17:286–301. doi: 10.1038/nrc.2017.17. [DOI] [PubMed] [Google Scholar]
- 9.Woo SR, Corrales L, Gajewski TF. Innate immune recognition of cancer. Annu Rev Immunol. 2015;33:445–474. doi: 10.1146/annurev-immunol-032414-112043. [DOI] [PubMed] [Google Scholar]
- 10.Hernandez C, Huebener P, Schwabe RF. Damage-associated molecular patterns in cancer: a double-edged sword. Oncogene. 2016;35:5931–5941. doi: 10.1038/onc.2016.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Markiewski MM, Lambris JD. Is complement good or bad for cancer patients? A new perspective on an old dilemma. Trends Immunol. 2009;30:286–292. doi: 10.1016/j.it.2009.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Taylor RP, Lindorfer MA. Cytotoxic mechanisms of immunotherapy: harnessing complement in the action of anti-tumor monoclonal antibodies. Semin Immunol. 2016;28:309–316. doi: 10.1016/j.smim.2016.03.003. This review discusses the effectiveness of clinical mAbs in inducing CDC. [DOI] [PubMed] [Google Scholar]
- 13.Ricklin D, Hajishengallis G, Yang K, Lambris JD. Complement: a key system for immune surveillance and homeostasis. Nat Immunol. 2010;11:785–797. doi: 10.1038/ni.1923. This is a seminal review that offers a broad overview of complement functioning and its role in health and disease states. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stephan AH, Barres BA, Stevens B. The complement system: an unexpected role in synaptic pruning during development and disease. Annu Rev Neurosci. 2012;35:369–389. doi: 10.1146/annurev-neuro-061010-113810. [DOI] [PubMed] [Google Scholar]
- 15.Mastellos DC, Deangelis RA, Lambris JD. Complement-triggered pathways orchestrate regenerative responses throughout phylogenesis. Semin Immunol. 2013;25:29–38. doi: 10.1016/j.smim.2013.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hajishengallis G, Abe T, Maekawa T, Hajishengallis E, Lambris JD. Role of complement in host-microbe homeostasis of the periodontium. Semin Immunol. 2013;25:65–72. doi: 10.1016/j.smim.2013.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ricklin D, Reis ES, Lambris JD. Complement in disease: a defence system turning offensive. Nat Rev Nephrol. 2016;12:383–401. doi: 10.1038/nrneph.2016.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Derer S, Beurskens FJ, Rosner T, Peipp M, Valerius T. Complement in antibody-based tumor therapy. Crit Rev Immunol. 2014;34:199–214. doi: 10.1615/critrevimmunol.2014009761. [DOI] [PubMed] [Google Scholar]
- 19.Mamidi S, Hone S, Kirschfink M. The complement system in cancer: ambivalence between tumour destruction and promotion. Immunobiology. 2017;222:45–54. doi: 10.1016/j.imbio.2015.11.008. [DOI] [PubMed] [Google Scholar]
- 20.Pio R, Ajona D, Lambris JD. Complement inhibition in cancer therapy. Semin Immunol. 2013;25:54–64. doi: 10.1016/j.smim.2013.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bonavita E, et al. PTX3 is an extrinsic oncosuppressor regulating complement-dependent inflammation in cancer. Cell. 2015;160:700–714. doi: 10.1016/j.cell.2015.01.004. This article uncovers a role for PTX3 as a suppressor of tumorigenesis via regulating complement and inflammatory responses. [DOI] [PubMed] [Google Scholar]
- 22.Markiewski MM, et al. Modulation of the antitumor immune response by complement. Nat Immunol. 2008;9:1225–1235. doi: 10.1038/ni.1655. This is the first article that shows evidence of complement as a promoter of tumorigenesis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vadrevu SK, et al. Complement c5a receptor facilitates cancer metastasis by altering T-cell responses in the metastatic niche. Cancer Res. 2014;74:3454–3465. doi: 10.1158/0008-5472.CAN-14-0157. [DOI] [PubMed] [Google Scholar]
- 24.Reff ME, et al. Depletion of B cells in vivo by a chimeric mouse human monoclonal antibody to CD20. Blood. 1994;83:435–445. [PubMed] [Google Scholar]
- 25.Maloney DG, et al. IDEC-C2B8 (Rituximab) anti-CD20 monoclonal antibody therapy in patients with relapsed low-grade non-Hodgkin’s lymphoma. Blood. 1997;90:2188–2195. [PubMed] [Google Scholar]
- 26.Lokhorst HM, et al. Targeting CD38 with daratumumab monotherapy in multiple myeloma. N Engl J Med. 2015;373:1207–1219. doi: 10.1056/NEJMoa1506348. [DOI] [PubMed] [Google Scholar]
- 27.de Weers M, et al. Daratumumab, a novel therapeutic human CD38 monoclonal antibody, induces killing of multiple myeloma and other hematological tumors. J Immunol. 2011;186:1840–1848. doi: 10.4049/jimmunol.1003032. [DOI] [PubMed] [Google Scholar]
- 28.Irie K, Irie RF, Morton DL. Evidence for in vivo reaction of antibody and complement to surface antigens of human cancer cells. Science. 1974;186:454–456. doi: 10.1126/science.186.4162.454. [DOI] [PubMed] [Google Scholar]
- 29.Okada H, Baba T. Rosette formation of human erythrocytes on cultured cells of tumour origin and activation of complement by cell membrane. Nature. 1974;248:521–522. doi: 10.1038/248521a0. [DOI] [PubMed] [Google Scholar]
- 30.Zent CS, et al. Direct and complement dependent cytotoxicity in CLL cells from patients with high-risk early-intermediate stage chronic lymphocytic leukemia (CLL) treated with alemtuzumab and rituximab. Leuk Res. 2008;32:1849–1856. doi: 10.1016/j.leukres.2008.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Karsten CM, Kohl J. The immunoglobulin, IgG Fc receptor and complement triangle in autoimmune diseases. Immunobiology. 2012;217:1067–1079. doi: 10.1016/j.imbio.2012.07.015. [DOI] [PubMed] [Google Scholar]
- 32.Holmberg MT, Blom AM, Meri S. Regulation of complement classical pathway by association of C4b-binding protein to the surfaces of SK-OV-3 and Caov-3 ovarian adenocarcinoma cells. J Immunol. 2001;167:935–939. doi: 10.4049/jimmunol.167.2.935. [DOI] [PubMed] [Google Scholar]
- 33.Ajona D, et al. Expression of complement factor H by lung cancer cells: effects on the activation of the alternative pathway of complement. Cancer Res. 2004;64:6310–6318. doi: 10.1158/0008-5472.CAN-03-2328. [DOI] [PubMed] [Google Scholar]
- 34.Zipfel PF, Skerka C. Complement regulators and inhibitory proteins. Nat Rev Immunol. 2009;9:729–740. doi: 10.1038/nri2620. [DOI] [PubMed] [Google Scholar]
- 35.Carroll MC, Isenman DE. Regulation of humoral immunity by complement. Immunity. 2012;37:199–207. doi: 10.1016/j.immuni.2012.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schmudde I, Laumonnier Y, Kohl J. Anaphylatoxins coordinate innate and adaptive immune responses in allergic asthma. Semin Immunol. 2013;25:2–11. doi: 10.1016/j.smim.2013.04.009. [DOI] [PubMed] [Google Scholar]
- 37.Freeley S, Kemper C, Le Friec G. The “ins and outs” of complement-driven immune responses. Immunol Rev. 2016;274:16–32. doi: 10.1111/imr.12472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liszewski MK, et al. Intracellular complement activation sustains T cell homeostasis and mediates effector differentiation. Immunity. 2013;39:1143–1157. doi: 10.1016/j.immuni.2013.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li K, et al. Expression of complement components, receptors and regulators by human dendritic cells. Mol Immunol. 2011;48:1121–1127. doi: 10.1016/j.molimm.2011.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Reis ES, Barbuto JA, Kohl J, Isaac L. Impaired dendritic cell differentiation and maturation in the absence of C3. Mol Immunol. 2008;45:1952–1962. doi: 10.1016/j.molimm.2007.10.031. [DOI] [PubMed] [Google Scholar]
- 41.Strainic MG, et al. Locally produced complement fragments C5a and C3a provide both costimulatory and survival signals to naive CD4+ T cells. Immunity. 2008;28:425–435. doi: 10.1016/j.immuni.2008.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Strainic MG, Shevach EM, An F, Lin F, Medof ME. Absence of signaling into CD4+ cells via C3aR and C5aR enables autoinductive TGF-beta1 signaling and induction of Foxp3+ regulatory T cells. Nat Immunol. 2013;14:162–171. doi: 10.1038/ni.2499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Arbore G, et al. T helper 1 immunity requires complement-driven NLRP3 inflammasome activity in CD4+ T cells. Science. 2016;352:aad1210. doi: 10.1126/science.aad1210. This article reveals a role for complement-mediated NLRP3 activity in the differentiation of T H1 cells. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Morgan EL, et al. Enhancement of in vivo and in vitro immune functions by a conformationally biased, response-selective agonist of human C5a: implications for a novel adjuvant in vaccine design. Vaccine. 2009;28:463–469. doi: 10.1016/j.vaccine.2009.10.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hegde GV, Meyers-Clark E, Joshi SS, Sanderson SD. A conformationally-biased, response-selective agonist of C5a acts as a molecular adjuvant by modulating antigen processing and presentation activities of human dendritic cells. Int Immunopharmacol. 2008;8:819–827. doi: 10.1016/j.intimp.2008.01.031. [DOI] [PubMed] [Google Scholar]
- 46.Hung CY, et al. An agonist of human complement fragment C5a enhances vaccine immunity against Coccidioides infection. Vaccine. 2012;30:4681–4690. doi: 10.1016/j.vaccine.2012.04.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Floreani AA, et al. Novel C5a agonist-based dendritic cell vaccine in a murine model of melanoma. Cell Cycle. 2007;6:2835–2839. doi: 10.4161/cc.6.22.4899. [DOI] [PubMed] [Google Scholar]
- 48.Dempsey PW, Allison ME, Akkaraju S, Goodnow CC, Fearon DT. C3d of complement as a molecular adjuvant: bridging innate and acquired immunity. Science. 1996;271:348–350. doi: 10.1126/science.271.5247.348. [DOI] [PubMed] [Google Scholar]
- 49.Prise KM, O’Sullivan JM. Radiation-induced bystander signalling in cancer therapy. Nat Rev Cancer. 2009;9:351–360. doi: 10.1038/nrc2603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gupta A, et al. Radiotherapy promotes tumor-specific effector CD8+ T cells via dendritic cell activation. J Immunol. 2012;189:558–566. doi: 10.4049/jimmunol.1200563. [DOI] [PubMed] [Google Scholar]
- 51.Sharma A, et al. Radiotherapy of human sarcoma promotes an intratumoral immune effector signature. Clin Cancer Res. 2013;19:4843–4853. doi: 10.1158/1078-0432.CCR-13-0352. [DOI] [PubMed] [Google Scholar]
- 52.Surace L, et al. Complement is a central mediator of radiotherapy-induced tumor-specific immunity and clinical response. Immunity. 2015;42:767–777. doi: 10.1016/j.immuni.2015.03.009. [DOI] [PubMed] [Google Scholar]
- 53.Elvington M, et al. Complement-dependent modulation of antitumor immunity following radiation therapy. Cell Rep. 2014;8:818–830. doi: 10.1016/j.celrep.2014.06.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Becker JC, Andersen MH, Schrama D, Thor Straten P. Immune-suppressive properties of the tumor microenvironment. Cancer Immunol Immunother. 2013;62:1137–1148. doi: 10.1007/s00262-013-1434-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Morgan BP, Gasque P. Extrahepatic complement biosynthesis: where, when and why? Clin Exp Immunol. 1997;107:1–7. doi: 10.1046/j.1365-2249.1997.d01-890.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lubbers R, van Essen MF, van Kooten C, Trouw LA. Production of complement components by cells of the immune system. Clin Exp Immunol. 2017;188:183–194. doi: 10.1111/cei.12952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cho MS, et al. Autocrine effects of tumor-derived complement. Cell Rep. 2014;6:1085–1095. doi: 10.1016/j.celrep.2014.02.014. This article shows that tumour cells secrete complement proteins that act in an autocrine fashion to induce tumorigenesis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Piao C, et al. Complement 5a enhances hepatic metastases of colon cancer via monocyte chemoattractant protein-1-mediated inflammatory cell infiltration. J Biol Chem. 2015;290:10667–10676. doi: 10.1074/jbc.M114.612622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kohl J. Self, non-self, and danger: a complementary view. Adv Exp Med Biol. 2006;586:71–94. doi: 10.1007/0-387-34134-X_6. [DOI] [PubMed] [Google Scholar]
- 60.Ajona D, et al. Investigation of complement activation product c4d as a diagnostic and prognostic biomarker for lung cancer. J Natl Cancer Inst. 2013;105:1385–1393. doi: 10.1093/jnci/djt205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ishida Y, et al. Activation of complement system in adult T-cell leukemia (ATL) occurs mainly through lectin pathway: a serum proteomic approach using mass spectrometry. Cancer Lett. 2008;271:167–177. doi: 10.1016/j.canlet.2008.06.004. [DOI] [PubMed] [Google Scholar]
- 62.Hsieh CC, et al. The role of complement component 3 (C3) in differentiation of myeloid-derived suppressor cells. Blood. 2013;121:1760–1768. doi: 10.1182/blood-2012-06-440214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Shalapour S, Karin M. Immunity, inflammation, and cancer: an eternal fight between good and evil. J Clin Invest. 2015;125:3347–3355. doi: 10.1172/JCI80007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 65.Parkin DM. The global health burden of infection-associated cancers in the year 2002. Int J Cancer. 2006;118:3030–3044. doi: 10.1002/ijc.21731. [DOI] [PubMed] [Google Scholar]
- 66.Umansky V, Blattner C, Gebhardt C, Utikal J. The role of myeloid-derived suppressor cells (MDSC) in cancer progression. Vaccines (Basel) 2016;4:e36. doi: 10.3390/vaccines4040036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Corrales L, et al. Anaphylatoxin C5a creates a favorable microenvironment for lung cancer progression. J Immunol. 2012;189:4674–4683. doi: 10.4049/jimmunol.1201654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Nitta H, et al. Cancer cells release anaphylatoxin C5a from C5 by serine protease to enhance invasiveness. Oncol Rep. 2014;32:1715–1719. doi: 10.3892/or.2014.3341. [DOI] [PubMed] [Google Scholar]
- 69.Han X, Zha H, Yang F, Guo B, Zhu B. Tumor-derived tissue factor aberrantly activates complement and facilitates lung tumor progression via recruitment of myeloid-derived suppressor cells. Int J Mol Sci. 2017;18:e22. doi: 10.3390/ijms18010022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.An LL, et al. Complement C5a induces PD-L1 expression and acts in synergy with LPS through Erk1/2 and JNK signaling pathways. Sci Rep. 2016;6:33346. doi: 10.1038/srep33346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wang Y, et al. Autocrine complement inhibits IL10-dependent T-cell-mediated antitumor immunity to promote tumor progression. Cancer Discov. 2016;6:1022–1035. doi: 10.1158/2159-8290.CD-15-1412. This article suggests a novel strategy to enhance the effect of cancer immunotherapy by inhibiting complement receptors. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ajona D, et al. A combined PD-1/C5a blockade synergistically protects against lung cancer growth and metastasis. Cancer Discov. 2017;7:694–703. doi: 10.1158/2159-8290.CD-16-1184. This article shows evidence for a beneficial role of combined therapeutic inhibition of complement and immune checkpoint molecules. [DOI] [PubMed] [Google Scholar]
- 73.Sharma P, Allison JP. The future of immune checkpoint therapy. Science. 2015;348:56–61. doi: 10.1126/science.aaa8172. [DOI] [PubMed] [Google Scholar]
- 74.Tsuji S, et al. Network-based analysis for identification of candidate genes for colorectal cancer progression. Biochem Biophys Res Commun. 2016;476:534–540. doi: 10.1016/j.bbrc.2016.05.158. [DOI] [PubMed] [Google Scholar]
- 75.Weaver DJ, Jr, et al. C5a receptor-deficient dendritic cells promote induction of Treg and Th17 cells. Eur J Immunol. 2010;40:710–721. doi: 10.1002/eji.200939333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gunn L, et al. Opposing roles for complement component C5a in tumor progression and the tumor microenvironment. J Immunol. 2012;189:2985–2994. doi: 10.4049/jimmunol.1200846. This article offers insights into the dual role of complement in cancer development. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Nabizadeh JA, et al. The complement C3a receptor contributes to melanoma tumorigenesis by inhibiting neutrophil and CD4+ T cell responses. J Immunol. 2016;196:4783–4792. doi: 10.4049/jimmunol.1600210. [DOI] [PubMed] [Google Scholar]
- 78.Guglietta S, et al. Coagulation induced by C3aR-dependent NETosis drives protumorigenic neutrophils during small intestinal tumorigenesis. Nat Commun. 2016;7:11037. doi: 10.1038/ncomms11037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ning C, et al. Complement activation promotes colitis-associated carcinogenesis through activating intestinal IL-1beta/IL-17A axis. Mucosal Immunol. 2015;8:1275–1284. doi: 10.1038/mi.2015.18. [DOI] [PubMed] [Google Scholar]
- 80.Phieler J, et al. The complement anaphylatoxin C5a receptor contributes to obese adipose tissue inflammation and insulin resistance. J Immunol. 2013;191:4367–4374. doi: 10.4049/jimmunol.1300038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Phieler J, Garcia-Martin R, Lambris JD, Chavakis T. The role of the complement system in metabolic organs and metabolic diseases. Semin Immunol. 2013;25:47–53. doi: 10.1016/j.smim.2013.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Doerner SK, et al. High-fat diet-induced complement activation mediates intestinal inflammation and neoplasia, independent of obesity. Mol Cancer Res. 2016;14:953–965. doi: 10.1158/1541-7786.MCR-16-0153. This is the first study showing evidence of activation of complement by a high-fat diet and promotion of tumorigenesis independent of obesity. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Vlaicu SI, et al. Role of C5b-9 complement complex and response gene to complement-32 (RGC-32) in cancer. Immunol Res. 2013;56:109–121. doi: 10.1007/s12026-012-8381-8. [DOI] [PubMed] [Google Scholar]
- 84.Towner LD, Wheat RA, Hughes TR, Morgan BP. Complement membrane attack and tumorigenesis: a systems biology approach. J Biol Chem. 2016;291:14927–14938. doi: 10.1074/jbc.M115.708446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Nunez-Cruz S, et al. Genetic and pharmacologic inhibition of complement impairs endothelial cell function and ablates ovarian cancer neovascularization. Neoplasia. 2012;14:994–1004. doi: 10.1593/neo.121262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Bulla R, et al. C1q acts in the tumour microenvironment as a cancer-promoting factor independently of complement activation. Nat Commun. 2016;7:10346. doi: 10.1038/ncomms10346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.DeAngelis RA, et al. A complement-IL-4 regulatory circuit controls liver regeneration. J Immunol. 2012;188:641–648. doi: 10.4049/jimmunol.1101925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Haynes T, et al. Complement anaphylatoxin C3a is a potent inducer of embryonic chick retina regeneration. Nat Commun. 2013;4:2312. doi: 10.1038/ncomms3312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Contractor T, et al. Sexual dimorphism of liver metastasis by murine pancreatic neuroendocrine tumors is affected by expression of complement C5. Oncotarget. 2016;7:30585–30596. doi: 10.18632/oncotarget.8874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Riihila P, et al. Complement component C3 and complement factor B promote growth of cutaneous squamous cell carcinoma. Am J Pathol. 2017;187:1186–1197. doi: 10.1016/j.ajpath.2017.01.006. [DOI] [PubMed] [Google Scholar]
- 91.Boire A, et al. Complement component 3 adapts the cerebrospinal fluid for leptomeningeal metastasis. Cell. 2017;168:1101–1113. doi: 10.1016/j.cell.2017.02.025. This article shows for the first time the invement of complement in metastasis to the nervous system. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kaida T, et al. C5a receptor (CD88) promotes motility and invasiveness of gastric cancer by activating RhoA. Oncotarget. 2016;7:84798–84809. doi: 10.18632/oncotarget.12656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Nitta H, et al. Enhancement of human cancer cell motility and invasiveness by anaphylatoxin C5a via aberrantly expressed C5a receptor (CD88) Clin Cancer Res. 2013;19:2004–2013. doi: 10.1158/1078-0432.CCR-12-1204. [DOI] [PubMed] [Google Scholar]
- 94.Cho MS, et al. Complement component 3 is regulated by TWIST1 and mediates epithelial-mesenchymal transition. J Immunol. 2016;196:1412–1418. doi: 10.4049/jimmunol.1501886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Abdelbaset-Ismail A, et al. Activation of the complement cascade enhances motility of leukemic cells by downregulating expression of HO-1. Leukemia. 2017;31:446–458. doi: 10.1038/leu.2016.198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Bandini S, et al. Early onset and enhanced growth of autochthonous mammary carcinomas in C3-deficient Her2/neu transgenic mice. Oncoimmunology. 2013;2:e26137. doi: 10.4161/onci.26137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Diebolder CA, et al. Complement is activated by IgG hexamers assembled at the cell surface. Science. 2014;343:1260–1263. doi: 10.1126/science.1248943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Cook EM, et al. Antibodies that efficiently form hexamers upon antigen binding can induce complement-dependent cytotoxicity under complement-limiting conditions. J Immunol. 2016;197:1762–1775. doi: 10.4049/jimmunol.1600648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Baig NA, et al. Induced resistance to ofatumumab-mediated cell clearance mechanisms, including complement-dependent cytotoxicity, in chronic lymphocytic leukemia. J Immunol. 2014;192:1620–1629. doi: 10.4049/jimmunol.1302954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Beurskens FJ, et al. Exhaustion of cytotoxic effector systems may limit monoclonal antibody-based immunotherapy in cancer patients. J Immunol. 2012;188:3532–3541. doi: 10.4049/jimmunol.1103693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Golay J. Direct targeting of cancer cells with antibodies: what can we learn from the successes and failure of unconjugated antibodies for lymphoid neoplasias? J Autoimmun. 2017 doi: 10.1016/j.jaut.2017.06.002. http://dx.doi.org/10.1016/j.jaut.2017.06.002. [DOI] [PubMed]
- 102.Wang SY, Racila E, Taylor RP, Weiner GJ. NK-cell activation and antibody-dependent cellular cytotoxicity induced by rituximab-coated target cells is inhibited by the C3b component of complement. Blood. 2008;111:1456–1463. doi: 10.1182/blood-2007-02-074716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Wang SY, et al. Depletion of the C3 component of complement enhances the ability of rituximab-coated target cells to activate human NK cells and improves the efficacy of monoclonal antibody therapy in an in vivo model. Blood. 2009;114:5322–5330. doi: 10.1182/blood-2009-01-200469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Janelle V, et al. Transient complement inhibition promotes a tumor-specific immune response through the implication of natural killer cells. Cancer Immunol Res. 2014;2:200–206. doi: 10.1158/2326-6066.CIR-13-0173. [DOI] [PubMed] [Google Scholar]
- 105.Werlenius O, et al. Reactive oxygen species induced by therapeutic CD20 antibodies inhibit natural killer cell-mediated antibody-dependent cellular cytotoxicity against primary CLL cells. Oncotarget. 2016;7:32046–32053. doi: 10.18632/oncotarget.8769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Spiller OB, Criado-Garcia O, Rodriguez De Cordoba S, Morgan BP. Cytokine-mediated up-regulation of CD55 and CD59 protects human hepatoma cells from complement attack. Clin Exp Immunol. 2000;121:234–241. doi: 10.1046/j.1365-2249.2000.01305.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Ohta R, et al. Mouse complement receptor-related gene y/p65-neutralized tumor vaccine induces antitumor activity in vivo. J Immunol. 2004;173:205–213. doi: 10.4049/jimmunol.173.1.205. [DOI] [PubMed] [Google Scholar]
- 108.Bjorge L, et al. Ascitic complement system in ovarian cancer. Br J Cancer. 2005;92:895–905. doi: 10.1038/sj.bjc.6602334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Okroj M, Hsu YF, Ajona D, Pio R, Blom AM. Non-small cell lung cancer cells produce a functional set of complement factor I and its soluble cofactors. Mol Immunol. 2008;45:169–179. doi: 10.1016/j.molimm.2007.04.025. [DOI] [PubMed] [Google Scholar]
- 110.Kapka-Skrzypczak L, et al. CD55, CD59, factor H and factor H-like 1 gene expression analysis in tumors of the ovary and corpus uteri origin. Immunol Lett. 2015;167:67–71. doi: 10.1016/j.imlet.2015.06.017. [DOI] [PubMed] [Google Scholar]
- 111.Ajona D, Hsu YF, Corrales L, Montuenga LM, Pio R. Down-regulation of human complement factor H sensitizes non-small cell lung cancer cells to complement attack and reduces in vivo tumor growth. J Immunol. 2007;178:5991–5998. doi: 10.4049/jimmunol.178.9.5991. [DOI] [PubMed] [Google Scholar]
- 112.Manches O, et al. In vitro mechanisms of action of rituximab on primary non-Hodgkin lymphomas. Blood. 2003;101:949–954. doi: 10.1182/blood-2002-02-0469. [DOI] [PubMed] [Google Scholar]
- 113.Golay J, et al. CD20 levels determine the in vitro susceptibility to rituximab and complement of B-cell chronic lymphocytic leukemia: further regulation by CD55 and CD59. Blood. 2001;98:3383–3389. doi: 10.1182/blood.v98.12.3383. [DOI] [PubMed] [Google Scholar]
- 114.Hu W, et al. Human CD59 inhibitor sensitizes rituximab-resistant lymphoma cells to complement-mediated cytolysis. Cancer Res. 2011;71:2298–2307. doi: 10.1158/0008-5472.CAN-10-3016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Zell S, et al. Down-regulation of CD55 and CD46 expression by anti-sense phosphorothioate oligonucleotides (S-ODNs) sensitizes tumour cells to complement attack. Clin Exp Immunol. 2007;150:576–584. doi: 10.1111/j.1365-2249.2007.03507.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Geis N, et al. Inhibition of membrane complement inhibitor expression (CD46, CD55, CD59) by siRNA sensitizes tumor cells to complement attack in vitro. Curr Cancer Drug Targets. 2010;10:922–931. doi: 10.2174/156800910793357952. [DOI] [PubMed] [Google Scholar]
- 117.Mamidi S, Cinci M, Hasmann M, Fehring V, Kirschfink M. Lipoplex mediated silencing of membrane regulators (CD46, CD55 and CD59) enhances complement-dependent anti-tumor activity of trastuzumab and pertuzumab. Mol Oncol. 2013;7:580–594. doi: 10.1016/j.molonc.2013.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Sherbenou DW, et al. Antibody-drug conjugate targeting CD46 eliminates multiple myeloma cells. J Clin Invest. 2016;126:4640–4653. doi: 10.1172/JCI85856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Imai M, Ohta R, Varela JC, Song H, Tomlinson S. Enhancement of antibody-dependent mechanisms of tumor cell lysis by a targeted activator of complement. Cancer Res. 2007;67:9535–954. doi: 10.1158/0008-5472.CAN-07-1690. [DOI] [PubMed] [Google Scholar]
- 120.Verma MK, et al. A novel hemolytic complement-sufficient NSG mouse model supports studies of complement-mediated antitumor activity in vivo. J Immunol Methods. 2017;446:47–53. doi: 10.1016/j.jim.2017.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Diaz-Zaragoza M, Hernandez-Avila R, Viedma-Rodriguez R, Arenas-Aranda D, Ostoa-Saloma P. Natural and adaptive IgM antibodies in the recognition of tumor-associated antigens of breast cancer (Review) Oncol Rep. 2015;34:1106–1114. doi: 10.3892/or.2015.4095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Ricklin D, Lambris JD. Complement therapeutics. Semin Immunol. 2016;28:205–207. doi: 10.1016/j.smim.2016.07.001. [DOI] [PubMed] [Google Scholar]
- 123.Morgan BP, Harris CL. Complement, a target for therapy in inflammatory and degenerative diseases. Nat Rev Drug Discov. 2015;14:857–877. doi: 10.1038/nrd4657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Buckanovich RJ, et al. Tumor vascular proteins as biomarkers in ovarian cancer. J Clin Oncol. 2007;25:852–861. doi: 10.1200/JCO.2006.08.8583. [DOI] [PubMed] [Google Scholar]
- 125.Manning ML, Williams SA, Jelinek CA, Kostova MB, Denmeade SR. Proteolysis of complement factors iC3b and C5 by the serine protease prostate-specific antigen in prostatic fluid and seminal plasma. J Immunol. 2013;190:2567–2574. doi: 10.4049/jimmunol.1200856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Ricklin D, Lambris JD. Complement in immune and inflammatory disorders: pathophysiological mechanisms. J Immunol. 2013;190:3831–3838. doi: 10.4049/jimmunol.1203487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Gros P, Milder FJ, Janssen BJ. Complement driven by conformational changes. Nat Rev Immunol. 2008;8:48–58. doi: 10.1038/nri2231. [DOI] [PubMed] [Google Scholar]
- 128.Nilsson B, Nilsson Ekdahl K. The tick-over theory revisited: is C3 a contact-activated protein? Immunobiology. 2012;217:1106–1110. doi: 10.1016/j.imbio.2012.07.008. [DOI] [PubMed] [Google Scholar]
- 129.Mastellos DC, Reis ES, Ricklin D, Smith RJ, Lambris JD. Complement C3-targeted therapy: replacing long-held assertions with evidence-based discovery. Trends Immunol. 2017;38:383–394. doi: 10.1016/j.it.2017.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Amara U, et al. Molecular intercommunication between the complement and coagulation systems. J Immunol. 2010;185:5628–5636. doi: 10.4049/jimmunol.0903678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Elvington M, Liszewski MK, Atkinson JP. Evolution of the complement system: from defense of the single cell to guardian of the intravascular space. Immunol Rev. 2016;274:9–15. doi: 10.1111/imr.12474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Le Friec G, et al. The CD46-Jagged1 interaction is critical for human TH1 immunity. Nat Immunol. 2012;13:1213–1221. doi: 10.1038/ni.2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Schmidt CQ, Lambris JD, Ricklin D. Protection of host cells by complement regulators. Immunol Rev. 2016;274:152–171. doi: 10.1111/imr.12475. This review summarizes the function of complement inhibitors and their invement in pathologic conditions. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Longhurst H, et al. Prevention of hereditary angioedema attacks with a subcutaneous C1 inhibitor. N Engl J Med. 2017;376:1131–1140. doi: 10.1056/NEJMoa1613627. [DOI] [PubMed] [Google Scholar]
- 135.Degn SE, et al. MAp19, the alternative splice product of the MASP2 gene. J Immunol Methods. 2011;373:89–101. doi: 10.1016/j.jim.2011.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Degn SE, et al. MAp44, a human protein associated with pattern recognition molecules of the complement system and regulating the lectin pathway of complement activation. J Immunol. 2009;183:7371–7378. doi: 10.4049/jimmunol.0902388. [DOI] [PubMed] [Google Scholar]
- 137.Rooryck C, et al. Mutations in lectin complement pathway genes COLEC11 and MASP1 cause 3MC syndrome. Nat Genet. 2011;43:197–203. doi: 10.1038/ng.757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Blom AM, et al. A novel non-synonymous polymorphism (pArg240His) in C4b-binding protein is associated with atypical hemolytic uremic syndrome and leads to impaired alternative pathway cofactor activity. J Immunol. 2008;180:6385–6391. doi: 10.4049/jimmunol.180.9.6385. [DOI] [PubMed] [Google Scholar]
- 139.Mohlin FC, et al. Analysis of genes coding for CD46, CD55, and C4b-binding protein in patients with idiopathic, recurrent, spontaneous pregnancy loss. Eur J Immunol. 2013;43:1617–1629. doi: 10.1002/eji.201243196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Ross GD, et al. Disease-associated loss of erythrocyte complement receptors (CR1, C3b receptors) in patients with systemic lupus erythematosus and other diseases involving autoantibodies and/or complement activation. J Immunol. 1985;135:2005–2014. [PubMed] [Google Scholar]
- 141.Rondelli T, et al. Polymorphism of the complement receptor 1 gene correlates with the hematologic response to eculizumab in patients with paroxysmal nocturnal hemoglobinuria. Haematologica. 2014;99:262–266. doi: 10.3324/haematol.2013.090001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Cockburn IA, et al. A human complement receptor 1 polymorphism that reduces Plasmodium falciparum rosetting confers protection against severe malaria. Proc Natl Acad Sci USA. 2004;101:272–277. doi: 10.1073/pnas.0305306101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Ohi H, et al. Two cases of mesangiocapillary glomerulonephritis with CR1 deficiency. Nephron. 1986;43:307. doi: 10.1159/000183861. [DOI] [PubMed] [Google Scholar]
- 144.Klein RJ, et al. Complement factor H polymorphism in age-related macular degeneration. Science. 2005;308:385–389. doi: 10.1126/science.1109557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Edwards AO, et al. Complement factor H polymorphism and age-related macular degeneration. Science. 2005;308:421–424. doi: 10.1126/science.1110189. [DOI] [PubMed] [Google Scholar]
- 146.Haines JL, et al. Complement factor H variant increases the risk of age-related macular degeneration. Science. 2005;308:419–421. doi: 10.1126/science.1110359. [DOI] [PubMed] [Google Scholar]
- 147.Abrera-Abeleda MA, et al. Variations in the complement regulatory genes factor H (CFH) and factor H related 5 (CFHR5) are associated with membranoproliferative glomerulonephritis type II (dense deposit disease) J Med Genet. 2006;43:582–589. doi: 10.1136/jmg.2005.038315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Bernabeu-Herrero ME, et al. Complement factor H, FHR-3 and FHR-1 variants associate in an extended haplotype conferring increased risk of atypical hemolytic uremic syndrome. Mol Immunol. 2015;67:276–286. doi: 10.1016/j.molimm.2015.06.021. [DOI] [PubMed] [Google Scholar]
- 149.Vernon KA, et al. Partial complement factor H deficiency associates with C3 glomerulopathy and thrombotic microangiopathy. J Am Soc Nephrol. 2016;27:1334–1342. doi: 10.1681/ASN.2015030295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Clark SJ, et al. Identification of factor H-like protein 1 as the predominant complement regulator in Bruch’s membrane: implications for age-related macular degeneration. J Immunol. 2014;193:4962–4970. doi: 10.4049/jimmunol.1401613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Hakobyan S, Tortajada A, Harris CL, de Cordoba SR, Morgan BP. Variant-specific quantification of factor H in plasma identifies null alleles associated with atypical hemolytic uremic syndrome. Kidney Int. 2010;78:782–788. doi: 10.1038/ki.2010.275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Bresin E, et al. Combined complement gene mutations in atypical hemolytic uremic syndrome influence clinical phenotype. J Am Soc Nephrol. 2013;24:475–486. doi: 10.1681/ASN.2012090884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.van de Ven JP, et al. A functional variant in the CFI gene confers a high risk of age-related macular degeneration. Nat Genet. 2013;45:813–817. doi: 10.1038/ng.2640. [DOI] [PubMed] [Google Scholar]
- 154.Reis ES, Falcao DA, Isaac L. Clinical aspects and molecular basis of primary deficiencies of complement component C3 and its regulatory proteins factor I and factor H. Scand J Immunol. 2006;63:155–168. doi: 10.1111/j.1365-3083.2006.01729.x. [DOI] [PubMed] [Google Scholar]
- 155.Nomura M, et al. Genomic analysis of idiopathic infertile patients with sperm-specific depletion of CD46. Exp Clin Immunogenet. 2001;18:42–50. doi: 10.1159/000049086. [DOI] [PubMed] [Google Scholar]
- 156.Medof ME, et al. Relationship between decay accelerating factor deficiency, diminished acetylcholinesterase activity, and defective terminal complement pathway restriction in paroxysmal nocturnal hemoglobinuria erythrocytes. J Clin Invest. 1987;80:165–174. doi: 10.1172/JCI113043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Wilcox LA, Ezzell JL, Bernshaw NJ, Parker CJ. Molecular basis of the enhanced susceptibility of the erythrocytes of paroxysmal nocturnal hemoglobinuria to hemolysis in acidified serum. Blood. 1991;78:820–829. [PubMed] [Google Scholar]
- 158.Nevo Y, et al. CD59 deficiency is associated with chronic hemolysis and childhood relapsing immune-mediated polyneuropathy. Blood. 2013;121:129–135. doi: 10.1182/blood-2012-07-441857. [DOI] [PubMed] [Google Scholar]
- 159.Milis L, Morris CA, Sheehan MC, Charlesworth JA, Pussell BA. Vitronectin-mediated inhibition of complement: evidence for different binding sites for C5b-7 and C9. Clin Exp Immunol. 1993;92:114–119. doi: 10.1111/j.1365-2249.1993.tb05956.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.McDonald JF, Nelsestuen GL. Potent inhibition of terminal complement assembly by clusterin: characterization of its impact on C9 polymerization. Biochemistry. 1997;36:7464–7473. doi: 10.1021/bi962895r. [DOI] [PubMed] [Google Scholar]
- 161.Sigler C, Annis K, Cooper K, Haber H, Van deCarr S. Examination of baseline levels of carboxypeptidase N and complement components as potential predictors of angioedema associated with the use of an angiotensin-converting enzyme inhibitor. Arch Dermatol. 1997;133:972–975. [PubMed] [Google Scholar]
- 162.Mathews KP, Pan PM, Amendola MA, Lewis FH. Plasma protease inhibitor and anaphylatoxin inactivator levels in chronic urticaria/angioedema and in patients experiencing anaphylactoid reactions to radiographic contrast media. Int Arch Allergy Appl Immunol. 1986;79:220–223. doi: 10.1159/000233975. [DOI] [PubMed] [Google Scholar]
- 163.Downs-Canner S, et al. Complement inhibition: a novel form of immunotherapy for colon cancer. Ann Surg Oncol. 2016;23:655–662. doi: 10.1245/s10434-015-4778-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Liang T, et al. Identification of complement C3f-desArg and its derivative for acute leukemia diagnosis and minimal residual disease assessment. Proteomics. 2010;10:90–98. doi: 10.1002/pmic.200900513. [DOI] [PubMed] [Google Scholar]
- 165.Miguet L, et al. Discovery and identification of potential biomarkers in a prospective study of chronic lymphoid malignancies using SELDI-TOF-MS. J Proteome Res. 2006;5:2258–2269. doi: 10.1021/pr060058y. [DOI] [PubMed] [Google Scholar]
- 166.Villanueva J, et al. Differential exoprotease activities confer tumor-specific serum peptidome patterns. J Clin Invest. 2006;116:271–284. doi: 10.1172/JCI26022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Gast MC, et al. Serum protein profiling for diagnosis of breast cancer using SELDI-TOF MS. Oncol Rep. 2009;22:205–213. doi: 10.3892/or_00000426. [DOI] [PubMed] [Google Scholar]
- 168.Michlmayr A, et al. Modulation of plasma complement by the initial dose of epirubicin/docetaxel therapy in breast cancer and its predictive value. Br J Cancer. 2010;103:1201–1208. doi: 10.1038/sj.bjc.6605909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Chung L, et al. Novel serum protein biomarker panel revealed by mass spectrometry and its prognostic value in breast cancer. Breast Cancer Res. 2014;16:R63. doi: 10.1186/bcr3676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Imamura T, et al. Influence of the C5a-C5a receptor system on breast cancer progression and patient prognosis. Breast Cancer. 2016;23:876–885. doi: 10.1007/s12282-015-0654-3. [DOI] [PubMed] [Google Scholar]
- 171.Habermann JK, et al. Increased serum levels of complement C3a anaphylatoxin indicate the presence of colorectal tumors. Gastroenterology. 2006;131:1020–1029. doi: 10.1053/j.gastro.2006.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Storm L, et al. Evaluation of complement proteins as screening markers for colorectal cancer. Cancer Immunol Immunother. 2015;64:41–50. doi: 10.1007/s00262-014-1615-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Xi W, et al. Enrichment of C5a-C5aR axis predicts poor postoperative prognosis of patients with clear cell renal cell carcinoma. Oncotarget. 2016;7:80925–80934. doi: 10.18632/oncotarget.13108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Xi W, et al. High level of anaphylatoxin C5a predicts poor clinical outcome in patients with clear cell renal cell carcinoma. Sci Rep. 2016;6:29177. doi: 10.1038/srep29177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Maher SG, et al. Serum proteomic profiling reveals that pretreatment complement protein levels are predictive of esophageal cancer patient response to neoadjuvant chemoradiation. Ann Surg. 2011;254:809–816. doi: 10.1097/SLA.0b013e31823699f2. [DOI] [PubMed] [Google Scholar]
- 176.Agatea L, et al. Peptide patterns as discriminating biomarkers in plasma of patients with familial adenomatous polyposis. Clin Colorectal Cancer. 2016;15:e75–e92. doi: 10.1016/j.clcc.2015.12.002. [DOI] [PubMed] [Google Scholar]
- 177.Song Q, Zhang Z, Liu Y, Han S, Zhang X. The tag SNP rs10746463 in decay-accelerating factor is associated with the susceptibility to gastric cancer. Mol Immunol. 2015;63:473–478. doi: 10.1016/j.molimm.2014.10.006. [DOI] [PubMed] [Google Scholar]
- 178.Bouwens TA, et al. Complement activation in Glioblastoma multiforme pathophysiology: evidence from serum levels and presence of complement activation products in tumor tissue. J Neuroimmunol. 2015;278:271–276. doi: 10.1016/j.jneuroim.2014.11.016. [DOI] [PubMed] [Google Scholar]
- 179.Bassig BA, et al. Polymorphisms in complement system genes and risk of non-Hodgkin lymphoma. Environ Mol Mutag. 2012;53:145–151. doi: 10.1002/em.21675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Lin K, et al. Complement component 3 is a prognostic factor of nonsmall cell lung cancer. Mol Med Rep. 2014;10:811–817. doi: 10.3892/mmr.2014.2230. [DOI] [PubMed] [Google Scholar]
- 181.Zhang Y, et al. A common CD55 rs2564978 variant is associated with the susceptibility of non-small cell lung cancer. Oncotarget. 2017;8:6216–6221. doi: 10.18632/oncotarget.14053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Swierzko AS, et al. Mannose-binding lectin (MBL) and MBL-associated serine protease-2 (MASP-2) in women with malignant and benign ovarian tumours. Cancer Immunol Immunother. 2014;63:1129–1140. doi: 10.1007/s00262-014-1579-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Mikami M, et al. Fully-sialylated alpha-chain of complement 4-binding protein: diagnostic utility for ovarian clear cell carcinoma. Gynecol Oncol. 2015;139:520–528. doi: 10.1016/j.ygyno.2015.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Hanas JS, et al. Biomarker identification in human pancreatic cancer sera. Pancreas. 2008;36:61–69. doi: 10.1097/mpa.0b013e3180d0a738. [DOI] [PubMed] [Google Scholar]
- 185.Chen J, et al. Profiling the potential tumor markers of pancreatic ductal adenocarcinoma using 2D-DIGE and MALDI-TOF-MS: up-regulation of complement C3 and alpha-2-HS-glycoprotein. Pancreatology. 2013;13:290–297. doi: 10.1016/j.pan.2013.03.010. [DOI] [PubMed] [Google Scholar]
- 186.Lee MJ, et al. Identification of human complement factor B as a novel biomarker candidate for pancreatic ductal adenocarcinoma. J Proteome Res. 2014;13:4878–4888. doi: 10.1021/pr5002719. [DOI] [PubMed] [Google Scholar]
- 187.Karczmarski J, et al. Pre-analytical-related variability influencing serum peptide profiles demonstrated in a mass spectrometry-based search for colorectal and prostate cancer biomarkers. Acta Biochim Pol. 2013;60:417–425. [PubMed] [Google Scholar]
- 188.Villanueva J, et al. Serum peptidome patterns that distinguish metastatic thyroid carcinoma from cancer-free controls are unbiased by gender and age. Mol Cell Proteomics. 2006;5:1840–1852. doi: 10.1074/mcp.M600229-MCP200. [DOI] [PubMed] [Google Scholar]