

Abstract
Cannabinoid Receptor Interacting Protein isoform 1b (CRIP1b) is known to interact with the CB1 receptor. Alternative splicing of the CNRIP1 gene produces CRIP1a and CRIP1b with a difference in the third exon only. Exons 1 and 2 encode for a functional domain in both proteins. CRIP1a is involved in regulating CB1 receptor internalization, but the function of CRIP1b is not very well characterized. Since there are significant identities in functional domains of these proteins, CRIP1b is a potential target for drug discovery. We report here predicted structure of CRIP1b followed by its interaction analysis with CB1 receptor by in-silico methods. A number of complementary computational techniques, including, homology modeling, ab-initio and protein threading, were applied to generate three-dimensional molecular models for CRIP1b. The computed model of CRIP1b was refined, followed by docking with C terminus of CB1 receptor to generate a model for the CRIP1b- CB1 receptor interaction. The structure of CRIP1b obtained by homology modelling using RHO_GDI-2 as template is a sandwich fold structure having beta sheets connected by loops, similar to predicted CRIP1a structure. The best scoring refined model of CRIP1b in complex with the CB1 receptor C terminus peptide showed favourable polar interactions. The overall binding pocket of CRIP1b was found to be overlapping to that of CRIP1a. The Arg82 and Cys126 of CRIP1b are involved in the majority of hydrogen bond interactions with the CB1 receptor and are possible key residues required for interactions between the CB1 receptor and CRIP1b.
Keywords: CRIP1b, CRIP1a, CB1 receptor, molecular modeling, G Protein-coupled receptor
Graphical abstract
The CRIP1b (cyan) in complex with the CB1 receptor C-terminal peptide (blue) showing the hydrogen bond interactions between CRIP1b and C-terminal tail of the CB1 receptor (S464TDTSAEAL472). Here, the computational models of CRIP1b and CB1 receptor C-terminal peptide are used to depict the relation between CRIP1b and CB1 receptor.
1. Background
Cannabinoid receptors are a family of G protein-coupled receptors that are involved in a wide variety of physiological and homeostatic processes [1–5]. The two cannabinoid receptors, the CB1 receptor and the CB2 receptor, have been identified including multiple splice variants of CB1in humans [1, 2]. The CB1 receptor is predominantly expressed in brain where it seems to play an important role in regulating analgesia, appetite, obesity, learning and memory, synaptic plasticity, substance abuse disorders, and neuro-protection [1, 3–5].TheCB2 receptor, in contrast is found primarily in peripheral tissue, including tissue associated with immune system like spleen and tonsils, and is involved in cannabinoid mediated immune response[1,2]. Cannabinoid receptors bind and are activated by the lipid derived endocannabinoid ligands, anandamide and 2-arachidonoylglycerol[3,4]. CB1 receptor signaling and regulation have assumed increased biomedical significance because of their involvement in variety of physiological disorders and diseases [3–5].
The CB1 receptor signals mainly through the activation of G proteins of Gi/o family, and regulates Gi/o-mediated inhibition of adenylyl cyclase, inhibition of Ca2+ channels, interaction with receptor tyrosine kinases and non-receptor Src kinases to activate the Extracellular Signal-Regulated Kinase (ERK1/2), and other cellular signaling via interactions with Gi/o proteins[1,2]. The CB1 receptor also interacts with associated proteins that serve as regulators of cellular signaling (FAN, β-arrestins) or receptor trafficking (β-arrestins, SGIP1, GASP) [6–8]. In addition, the CB1receptoralso interacts with CRIP1a and CRIP1b, the cannabinoid receptor interacting proteins (CRIPs) that function to regulate cellular signaling and CB1 receptor trafficking [9–16].
CRIP1a and CRIP1b are cytosolic proteins that were first discovered when the Lewis laboratory determined that deletion of the CB1 receptor C-terminal released a tonic inhibition of N-type Ca2+ channels in neurons, concluding that the C-terminal performed an auto-inhibition function [5,6]. A yeast two-hybrid screen identified CRIP1a as a key associated protein [11]. The cannabinoid receptor interacting protein isoforms 1a and 1b (CRIP1a and CRIP1b) are generated by alternative splicing of CNRIP1 gene located on chromosome number 2 of the human genome [11]. CRIP1a is a 164-amino acid protein, whereas CRIP1b is a 128-amino acid protein, and both interact with the CB1 cannabinoid receptor. Although CRIP1a is conserved among mammals and fish, the presence of CRIP1b is limited to primates [11]. CRIP1 proteins have been has been identified immunologically in neurons in the superior cervical ganglion [11], striatum [17], glutamatergic CA3 pyramidal neurons in the hippocampus [18, 19] and retina [20].
The function of CRIP1a has been clearly established in CB1 receptor selection of Gi/o protein preference and signaling, and competition for β-arrestin to attenuate CB1 receptor internalization[14], whereas the function of CRIP1b is not known [11]. Exogenous CRIP1a reversed CB1 receptor-mediated tonic inhibition of Ca2+ currents, whereas CRIP1b could not[11]. CRIP1a-knockdown clones in the model neuroblastoma N18TG2 cells exhibited enhanced ERK1/2 phosphorylation efficacy in response to CP55940 and displayed a leftward shift in CP55940-mediated inhibition of forskolin-stimulated cAMP accumulation [12,13]. CB1 receptor-mediated Gi3 and Go activation by CP55940 was attenuated by CRIP1a over-expression, but robustly enhanced in CRIP1a-knockdown clones. Conversely, CP55940-mediated Gi1 and Gi2 activation was significant enhanced in cells over-expressing CRIP1a, but not inCRIP1a-knockdown clones [14]. These studies suggest that endogenous levels of CRIP1a modulate CB1 receptor-mediated signal transduction by facilitating a Gi/o subtype preference for Gi1 and Gi2, accompanied by an overall suppression of G protein-mediated signaling in neuronal cells [12,13].
CRIP1a plays a role in the control of agonist-driven CB1R cell surface regulation by competing with β-arrestins and thereby attenuating agonist-mediated internalization[14,15]. CRIP1a over-expression attenuated the agonist-induced redistribution of endogenous β-arrestin to punctae aggregates and subsequent loss of cell surface CB1receptors. Conversely, CRIP1a knock-down augmented agonist-mediated β-arrestin redistribution to punctae[14]. Co-immunoprecipitation studies indicated thatCRIP1a competes with β-arrestin for binding to the CB1 receptor, which attenuates the action of β-arrestin to mediate CB1R internalization[14].Peptides mimicking the CB1 receptor C-terminus could pull-down CRIP1a from cell extracts and recombinant CRIP1a. Co-immunoprecipitation of CB1 receptor protein complexes demonstrated that distal C-terminal peptides competed for the CB1 receptor association with CRIP1a [14], demonstrating the importance of this domain of the CB1 receptor for CRIP binding to the CB1 receptor. A similar distal C-terminal sequence found in the mGlu8a receptor has also been found to interact with CRIP1a, and may sub serve the same functions as for regulation of the CB1 receptor [16].
Efforts to establish a cellular function for CRIP1b have not yielded definitive conclusions. Both CRIP1a and CRIP1b proteins interact with the last nine amino acids (S464TDTSAEAL472) of the distal CB1-C terminus tail of the human CB1 cannabinoid receptor (Figure 1). The CB1 C-terminus binding is dependent upon a domain (amino acids 34 to 100) common to both CRIP1a and CRIP1b, as determined by studies of deletion mutations [11]. CRIP1a and CRIP1b possess distinctly divergent C-terminal domains, such that CRIP1b has the potential to exhibit functional differences from CRIP1a. CRIP1 proteins share no sequence homology to other proteins, so they cannot be categorized into a particular functional family based upon primary structure. Although recent modeling studies of CRIP1a by Ahmad et al., [21] revealed a structure containing beta sheets connected by loops, there is no report on the structure of CRIP1b so far.
Figure 1.
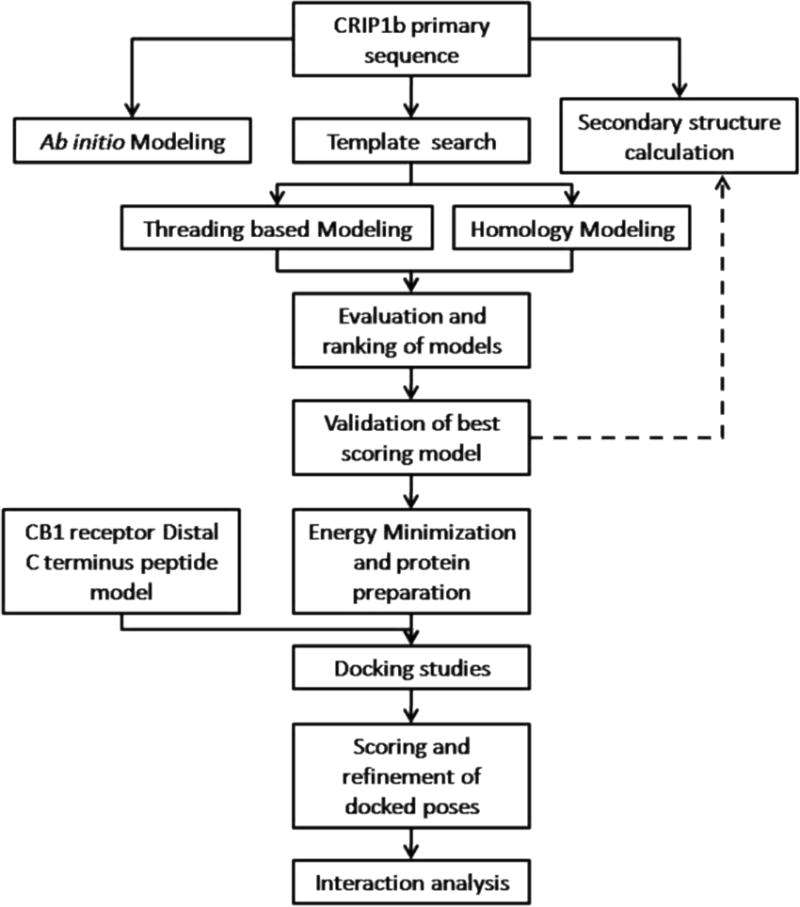
Schematic outline of the general workflow for CRIP1b modeling and its interaction analysis withCB1 receptor.
The present study deals with structure prediction of CRIP1b followed by analysis of its interaction with aCB1 receptor peptide by in-silico methods. A number of complementary computational techniques, including, homology modeling, ab-initio and protein threading, were applied to generate three dimensional molecular models for CRIP1b. The computed model of CRIP1b was refined, followed by docking with the C-terminus of CB1 receptor to generate a model for the CRIP1b- CB1 receptor interaction. The steps followed are described in a flow chart given in Figure 1.The aim of this study was to finding the key residues involved in CRIP1b- CB1 receptor interaction in order to provide clues to CRIP1b function.
2. Methods
2.1 CB1 receptor C terminus peptide modeling
The terminal 9-mer sequence from the distal CB1 receptor C-terminus (S464TDTSAEAL472) was modeled using PEP-Fold [22]. This method, based on structural alphabet SA letters to describe the conformations of four consecutive residues, couples the predicted series of SA letters to a greedy algorithm and a coarse-grained force field. The most-native clusters of all peptides are ranked in the top 10 generated models [23].
2.2 CRIP1b modeling and Validation
The secondary structure for CRIP1b was predicted by various algorithms like HMMSTR, SSPRO4, SSPAL, JNET, SPARROW, SABLE, NNSSP, NETSURFP, SSP, PSS FINDER, SPINE, and PSIPRED[24–34]. A consensus of secondary structure results was generated by using GeneSilico Metaserver [35].
The suitable template for the CRIP1b model was selected by a homology search against Protein Data Bank using SwissModel server, NCBI-Blastp, PDBBLAST, CSBLAST, COMA, Hhblits, HHSearch, FFAS and Phyre [36–45]. The three dimensional model was built using MODELLER in Accelrys Discovery Studio 2.5 [46]. Loop refinement protocol was incorporated to increase the accuracy of the model [47]. The energy minimization was performed by Conjugate gradient minimization protocol using CHARMM force field in Discovery studio [48–50].
Apart from homology modeling, ab-initio and threading methods using Robetta, Muster and iTasser, were used to calculate a model for CRIP1b [51–55]. The Robetta algorithm first attempts homology modeling by searching for confident homologs from BLAST, PSI-BLAST, FFAS03 or 3D-Jury. If a homolog is not available, then the de novo Rosetta fragment insertion method is used for prediction. Muster and iTasser following a threading algorithm, based on fold recognition. The iTasser (Iterative Threading ASSEmbly Refinement) follows a hierarchical approach, starting with LOMETS to identify structural templates from the PDB followed by full-length atomic model construction using iterative template fragment assembly simulations [53–55]. MUSTER (MUlti-Sources ThreadER) combines sequence profile alignment with multiple structural information data to generate sequence-template alignments for model prediction [52].
The quality of the models generated by different methods was analyzed using Discrete Optimized Protein Energy, or DOPE score. DOPE is an atomic distance-dependent statistical potential from a sample of native structures, and is extracted from a non-redundant set of crystallographic structures [56]. The model geometry was validated by using Ramachandran plot [57] in Discovery Studio. The PyMol was employed to aid visualizations, analysis and image preparation of all the obtained models [58].
2.3 Protein-peptide docking
The HADDOCK (High Ambiguity Driven DOCKing) server was used for docking CRIP1b- CB1 receptor nine-mer peptide [59–61]. As the site where C-terminus of CB1 receptor binds exactly on CRIP1b is not known, the 9-mer peptide was docked with all possible binding sites with no restriction for unbiased evaluation. The HADDOCK performs docking in three stages, starting with randomization of orientations and rigid-body minimization, and then semi-flexible simulated annealing in torsion angle space is performed, followed by refinement in Cartesian space with explicit solvent. During the final step, the complex is immersed in a suitable solvent environment, using TIP3P water model [62]. Then, the models are subjected to molecular dynamics simulation at 300K [63]. Initially position restraints are applied on the non-interface heavy atoms, which are relaxed in later stages to facilitate side chain optimization [59–61]. Docking results obtained as ligand poses on different binding site clusters based on positional interface ligand RMSD were individually inspected, after which high scoring models were passed into the refinement step.
2.4 Refinement of docked complexes
The structures of CRIP1b in complex with the CB1-receptor C-terminus 9-mer peptide (S464TDTSAEAL472) obtained from docking results were subjected to FireDock (Fast Interaction REfinement in molecular DOCKing) for rescoring and refinement [64]. The rearrangement of the interface side-chains followed by adjustment of the relative orientation of the molecules, are incorporated in the refinement process. The refined structures are ranked according to their binding energy, which is represented by global energy. The atomic contact energy (ACE), van der Waals forces (Attractive and Repulsive VdW) and hydrogen bonds (HB) contribute to the calculation of global energy, leading to final ranking [64,65]. The best scoring complex was further analyzed with PyMOL, Discovery Studio and Protein Interactions Calculator (PIC)[66]. PIC computes various interactions such as disulphide bonds, interactions between hydrophobic residues, ionic interactions, hydrogen bonds, aromatic-aromatic interactions, aromatic–sulphur interactions and cation–π interactions within a protein or between proteins in a complex [66].
3. Results and Discussion
3.1 Model of 9-mer peptide(S464TDTSAEAL472) of CB1 receptor C terminus tail
A de novo approach using PEP-Fold server was used to calculate the structure of the 9-mer peptide (S464TDTSAEAL472) (Figure 2). The PEP-Fold ranks generated models according to their sOPEP, the coarse grained energy. The best-ranked model was selected for docking studies. The energy minimization of the selected peptide was performed by Conjugate gradient protocol under CHARMM force field.
Figure 2.
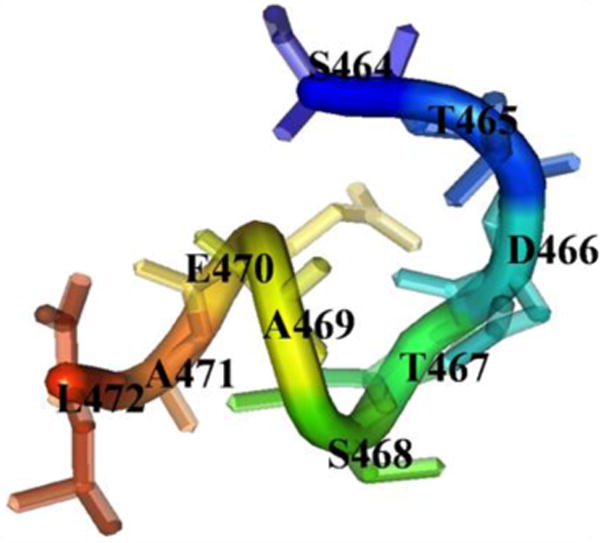
The de novo model of peptide from distal tail of CB1 receptor C-terminus
3.2 The CRIP1b models
The human CRIP1b sequence was retrieved from NCBI with accession NP_001104571. The CRIP1b sequence was subjected to secondary structure calculation. The consensus result of various algorithms showed beta sheets as the major secondary structure component (Figure 3).
Figure 3.
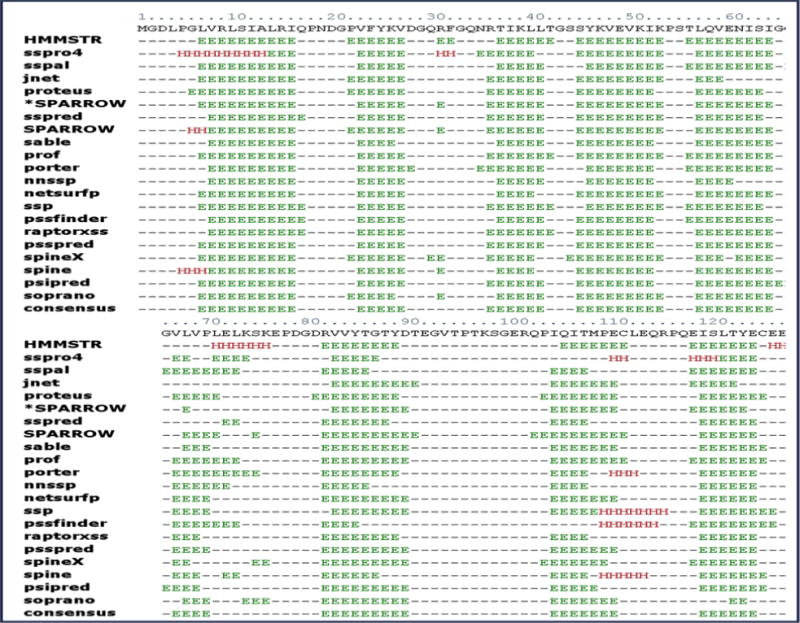
The secondary structure of CRIP1b predicted by various protocols, the consensus of results was taken into account to estimate secondary structural content in CRIP1b. The beta sheets are represented by ‘E’ and alpha helices by ‘H’.
The Blastp results (Supporting file: Table S1) did not show any suitable sequence homolog for the purposes of modeling structures. Other tools were utilized for more sensitive data mining in search of CRIP1b structural homologs. The results produced by PDBBLAST, CSBLAST, COMA, Hhblits, HHSearch, FFAS and Phyre were low scoring (Supporting file: Table S1 A-G). The SwissModel suggested RHO GDP-DISSOCIATION INHIBITOR 2 (1DS6_B)[63] as a suitable template for modeling with 27.5% identity with CRIP1b (Supporting file: Table S2).This template was also recently suggested for CRIP1a homolog by Ahmed and colleagues [21]. Because our goal was to seek commonality or distinct differences from CRIP1a, we chose to use this template. The RHO-GDI2 structure-CRIP1b sequence alignment (figure 4) was used as input for computing CRIP1b models, using Modeller (Discover studio), followed by loop refinement and energy minimization (Figure 1A). Apart from homology modeling, other approaches like ab-initio and threading were also employed to obtain CRIP1b models. For comparison, we also calculated CRIP1a model, using the same template RHO-GDI 2.
Figure 4. Sequence alignment of Rho-GDI 2/1DS6_B (template) and CRIP1b sequence.
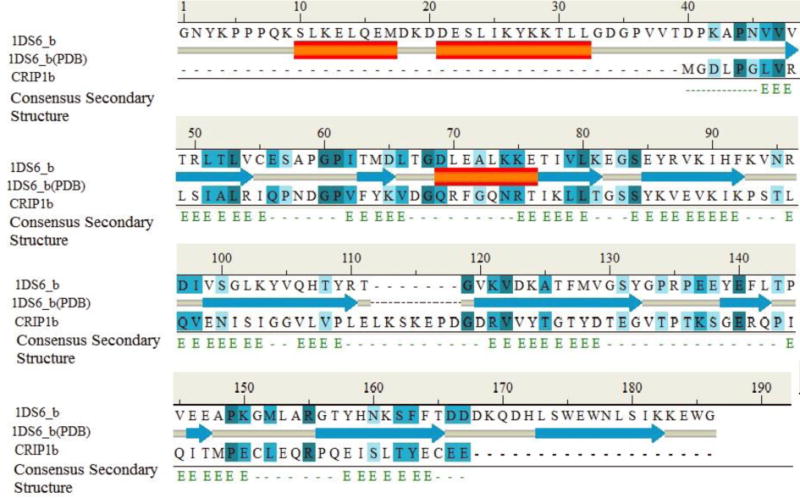
The ‘1DS6_b(PDB)’ represents secondary structure of Rho-GDI2 in PDB, and consensus secondary structure represents predicted secondary structure of CRIP1b.
3.3 Selection of a Valid Model
Because multiple CRIP1b models were generated by different approaches, it was important to choose a valid model with the best quality for further studies. For this purpose DOPE scores of all the models were calculated to facilitate their ranking. One of the CRIP1b models generated by homology modeling (with RHO_GDI 2 as template) showed the lowest DOPE value (Figure 5). The ab initio models generated by Robetta were closest to, but not better than, the models generated by Discovery studio in terms of their high DOPE score, (Table 1). The best ranking CRIP1b structure was validated with Ramachandran plot having all the residues in favorable region(Supporting file: Figure S1). The secondary structure data was also in agreement with the CRIP1b model, showing a sandwich fold model with beta sheets.
Figure 5.
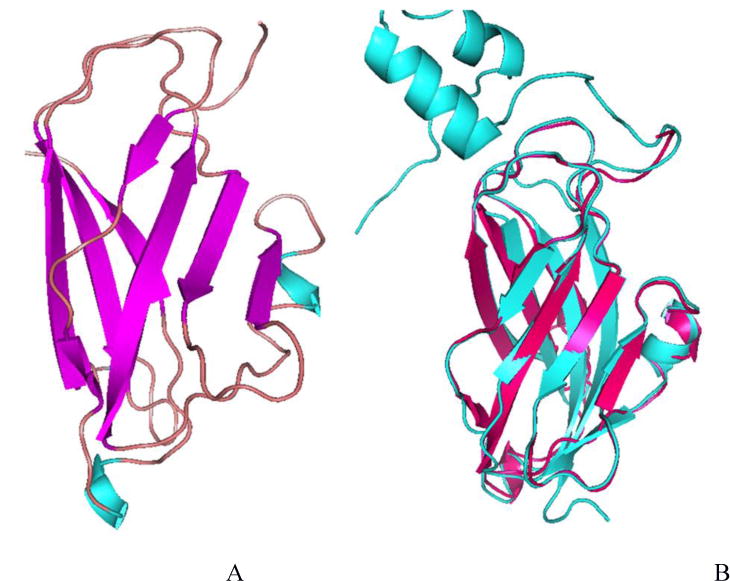
The CRIP1b model (A) and its alignment with the template Rho-GDI 2, cyan colored (B)
Table 1. The DOPE score of CRIP1b models.
The models are arranged according to their DOPE score.
| CRIP1b Model | DOPE Score |
|---|---|
| DS_model4 | −13222.31836 |
| DS_model1 | −12861.45606 |
| Robetta_model1 | −12259.22266 |
| Robetta_model5 | −12102.96289 |
| Robetta_model2 | −12075.74316 |
| Robetta_model3 | −11994.18164 |
| Robetta_model4 | −11134.72266 |
| DS_model3 | −10764.875 |
| DS_model2 | −10442.48145 |
| DS_model5 | −10083.26953 |
| Muster_model2 | −7846.150391 |
| Muster_model9 | −7585.559082 |
| Itasser_model1 | −7320.54834 |
| Muster_model4 | −7285.194336 |
| Itasser_model2 | −7151.129883 |
| Muster_model3 | −7069.747559 |
| Itasser_model4 | −6522.483887 |
| Muster_model7 | −6313.573242 |
| Muster_model8 | −6313.573242 |
| Muster_model5 | −5812.94873 |
| Muster_model6 | −5812.632324 |
| Muster_model1 | −5803.634277 |
| MultiCom_model | −5381.104004 |
| Itasser_model5 | −5299.068359 |
| Muster_model10 | −4933.700684 |
| Itasser_model3 | −1822.536377 |
Because CRIP1a and CRIP1b sequences are identical within the initial 110 residues, comparison between their models was performed. Although the model of CRIP1a is already reported by Ahmed et al.[21], the PDB coordinates of model are not available, so we derived the CRIP1a model in the same manner as CRIP1b to draw comparisons between the two. The residues lying in the C-terminal region (amino acids 155–164) were not covered by the template; hence they were modeled by an ab initio approach using Robetta and incorporated to form a full length model of CRIP1a. The homology model (with RHO-GDI 2 as template) showed the best DOPE scores; secondary structure data and Ramachandran Plot validated it. CRIP1a also showed a sandwich fold model with beta sheets (Supporting file: Figure S2A). The CRIP1a and CRIP1b models were aligned (Supporting file: Figure S2B), the majority of their secondary structure is not only similar to each other, but also agrees with the CRIP1a model reported by Ahmed et al. [21].The best ranked models of CRIP1b as well as CRIP1a were chosen for further interaction studies with the CB1 receptor C-terminus 9-mer peptide.
Molecular docking of CRIP1b with the CB1 receptor C-terminus peptide
To study interactions between CRIP1b and CB1 receptor C-terminus distal tail, the CRIP1b was docked with the 9-mer peptide (S464TDTSAEAL472) model generated earlier. Here, we have used HADDOCK 2.2, which implements solvated docking approach along with conventional Haddock docking protocol [67]. Each molecule and its associated solvation shell are considered as rigid body, due to which an encounter complex is formed with a water layer between the two protein chains (in our case CRIP1b and CB1R peptide). The non-interfacial waters are removed and during subsequent energy minimization stage the remaining waters and protein chains are treated are separate rigid bodies. Afterwards a biased Monte Carlo procedure is applied to remove water molecules, based on their probabilities for water-mediated contacts for specific amino acids. This procedure is repeated until only 25% of the initial interfacial water molecules remain. During the final steps, water molecules are removed if their interaction energy is unfavorable. The interaction energy (sum of van der Waals and electrostatic water–protein energies) should be less than 0.0 kcal/mol.
As the site where the C-terminus of CB1 binds exactly on CRIP1b is not known, the 9-mer peptide was docked with all possible sites on CRIP1b. The Haddock analysis returned 36 poses, grouped in nine clusters based on positional interface ligand RMSD. Twelve poses from the top three clusters were scattered at different positions on the CRIP1b surface (Figure 6). The binding pockets of these three clusters revealed the residues involved (Figure6). The CRIP1a model was also docked with CB1 receptor C-terminus peptide (Supporting file: Figure S3).
Figure 6. Docked poses of CB1 receptor C terminus peptide on CRIP1b model.
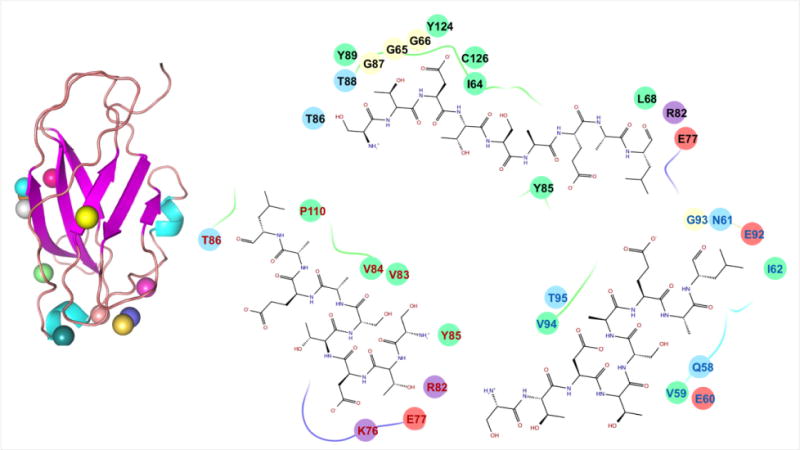
Docking studies yielded various docked complexes with peptide on distinct positions. All the poses were refined and scored. On the left side of figure, the colored spheres show the positions of peptide on CRIP1b in different poses. While on the right side, binding pockets of top scoring poses from three different clusters are written with differently colored residue fonts. Also, the chemical properties of residues are represented by oval background of different colors i.e., negative charged by pink, positive charged by violet, hydrophobic by green, polar by light blue and glycine by cream.
Refined poses of CRIP1b in complex with the CB1receptor C-terminus distal tail
All of the docked poses of CRIP1b were subjected to refinement, followed by binding energy (referred to as global energy in Kcal/mol) calculation (Table 2). The best scoring refined pose of CRIP1b in complex with CB1 receptor C-terminus peptide was taken into account for interaction analyses. The best scoring pose for CRIP1a in complex with CB1 receptor C-terminus peptide was retrieved under same set of parameters (Supporting file: Table S3). The interactions for both complexes were calculated by PIC.
Table 2. FireDock scores.
The docked poses of CRIP1b in Complex with the CB1 9-mer peptide were refined and ranked by their binding energy (Global energy), which were contributed by attractive and repulsive Van der Waals (VdW) forces, atomic contact energy(ACE) and hydrogen bonds(HB).
| Rank | Solution Number | Global Energy | Attractive VdW | Repulsive VdW | ACE | HB |
|---|---|---|---|---|---|---|
| 1 | 11 | −38.77 | −17.43 | 3.72 | −8.89 | 0.00 |
| 2 | 27 | −38.19 | −14.59 | 1.42 | −10.07 | 0.00 |
| 3 | 12 | −35.05 | −17.69 | 4.42 | −4.54 | −1.16 |
| 4 | 34 | −33.46 | −20.68 | 6.30 | −6.97 | −1.47 |
| 5 | 19 | −31.99 | −19.96 | 8.32 | −8.86 | −1.61 |
| 6 | 26 | −31.76 | −20.19 | 3.95 | −5.47 | −2.04 |
| 7 | 8 | −31.36 | −18.81 | 7.90 | −5.35 | −3.84 |
| 8 | 2 | −30.78 | −19.25 | 8.78 | −10.22 | −3.06 |
| 9 | 35 | −28.31 | −17.35 | 5.69 | −9.30 | −0.50 |
| 10 | 10 | −28.17 | −18.47 | 4.22 | −4.06 | −2.22 |
| 11 | 7 | −27.43 | −14.61 | 1.10 | −3.12 | −2.64 |
| 12 | 17 | −26.42 | −18.67 | 5.56 | −4.24 | −1.82 |
| 13 | 29 | −23.96 | −12.25 | 4.14 | −6.86 | −1.10 |
| 14 | 6 | −23.81 | −19.43 | 4.94 | −7.83 | −2.44 |
| 15 | 23 | −23.73 | −20.79 | 6.42 | −0.73 | −4.38 |
| 16 | 30 | −22.75 | −15.15 | 3.61 | −5.71 | −1.67 |
| 17 | 36 | −22.71 | −14.68 | 7.75 | −6.77 | −1.41 |
| 18 | 1 | −22.47 | −12.56 | 6.79 | −7.48 | −1.82 |
| 19 | 37 | −22.47 | −12.56 | 6.79 | −7.48 | −1.82 |
| 20 | 21 | −22.36 | −16.79 | 6.70 | −6.95 | −1.72 |
| 21 | 15 | −20.78 | −13.24 | 3.74 | −3.67 | −0.36 |
| 22 | 24 | −20.38 | −12.79 | 2.14 | −2.77 | 0.00 |
| 23 | 31 | −18.24 | −17.65 | 4.80 | −2.24 | −1.98 |
| 24 | 4 | −16.62 | −20.56 | 7.33 | −3.08 | −1.02 |
| 25 | 20 | −16.41 | −17.54 | 6.70 | −5.84 | 0.00 |
| 26 | 9 | −15.62 | −14.81 | 0.66 | −0.40 | −1.69 |
| 27 | 32 | −15.45 | −11.91 | 0.75 | −4.81 | 0.00 |
| 28 | 28 | −14.71 | −13.76 | 4.69 | −3.96 | −1.16 |
| 29 | 16 | −14.07 | −17.12 | 12.77 | −2.34 | −1.52 |
| 30 | 33 | −12.81 | −13.58 | 4.74 | −2.54 | −1.58 |
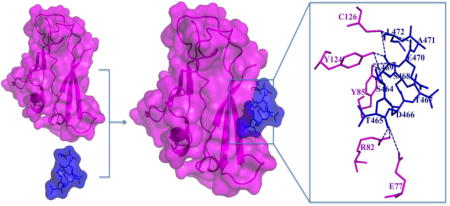
The last nine amino acids of the C-terminus of the CB1 receptor (Figure 2) are mostly polar in nature. The polar interactions are expected to be the major interaction between CRIP1b and CB1 receptor. Our model suggests that CRIP1b forms hydrophobic and hydrogen bond interactions with the CB1 receptor C-terminus peptide (Table 3). The hydrogen bonds involve Glu77, Arg82, Tyr85, Tyr124, Cys126 from CRIP1b and Ser464, Thr465, Ser468, Ala469, Glu470, Leu472 from the CB1 receptor peptide (Figure 7). Similarly interactions for CRIP1a with CB1receptor peptide were calculated and hydrophobic and hydrogen bond interactions were found (Supporting file: Table S4). The residues, namely Ser63, Asp81, Val83, Tyr 126, Tyr128, from CRIP1a were involved in the majority of interactions (Supporting file: Figure S4). The binding pockets of CRIP1b and CRIP1a lies in similar region between residues 61 and 128, mainly encompassing residues at position 82, 85 and 126. If we compare the binding pockets within a 4Å radius of the CB1 receptor peptide in CRIP1a reported by Ahmed and colleagues [21] with our data, a large number of common residues are observed. The residues at position 64, 68, 82, 85, 124 and 126 lie in binding pockets of CRIP1b as well as CRIP1a. This is in agreement with the model of CRIP1a obtained by Ahmed et al.[21].
Table 3. Interactions between CRIP1b and the CB1 receptor C-terminus 9-merpeptide.
The Chain A represents CRIP1b while chain B represents CB1-R peptide. The distances are represented as Dd-a (between Donor and Acceptor) and Dh-a(between Hydrogen and Acceptor) while angles are represented as A(d-H-N)(between Donor-H-N) and A(a-O=C)(between Acceptor-O=C). The MO stands for Multiple Occupancy. The undefined values are indicated as “-“.
| A. Binding pocket residues of CRIP1b within 4 Angstroms of CB1 R -peptide | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Position | 64 | 68 | 77 | 79 | 82 | 85 | 86 | 124 | 126 |
| Residue | ILE | LEU | GLU | ASP | ARG | TYR | TYR | TYR | CYS |
| B. Hydrophobic Interactions within 5 Angstroms | |||||
|---|---|---|---|---|---|
| Position | Residue | Chain | Position | Residue | Chain |
| 64 | ILE | A | 472 | LEU | B |
| 68 | LEU | A | 469 | ALA | B |
| 85 | TYR | A | 469 | ALA | B |
| 89 | TYR | A | 472 | LEU | B |
| 124 | TYR | A | 469 | ALA | B |
| C. Ionic Interactions within 6 Angstroms | |||||
|---|---|---|---|---|---|
| Position | Residue | Chain | Position | Residue | Chain |
| 82 | ARG | A | 466 | ASP | B |
| D. Protein-Protein Main Chain-Side Chain Hydrogen Bonds | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Donor | Acceptor | Parameters | ||||||||||
| POS | CHAIN | RES | ATOM | POS | CHAIN | RES | ATOM | MO | Dd-a | Dh-a | A(d-N) | A(a-O=C) |
| 77 | A | Glu | OE1 | 465 | B | Thr | O | 1 | 3.35 | 2.70 | 119.29 | 172.19 |
| 77 | A | Glu | OE1 | 465 | B | Thr | O | 2 | 3.35 | 3.43 | 76.62 | 172.19 |
| 82 | A | Arg | NH1 | 465 | B | Thr | O | 1 | 2.50 | 3.13 | 45.15 | 133.79 |
| 82 | A | Arg | NH1 | 465 | B | Thr | O | 2 | 2.50 | 2.33 | 87.44 | 133.79 |
| 82 | A | Arg | NH2 | 465 | B | Thr | O | 1 | 2.82 | 3.58 | 37.22 | 122.79 |
| 82 | A | Arg | NH2 | 465 | B | Thr | O | 2 | 2.82 | 2.65 | 88.58 | 122.79 |
| 85 | A | Tyr | OH | 468 | B | Ser | O | – | 3.05 | — | — | 87.81 |
| 124 | A | Tyr | OH | 469 | B | Ala | O | – | 2.38 | — | — | 135.51 |
| 126 | A | Cys | SG | 469 | B | Ala | O | – | 2.98 | — | — | 147.60 |
| 126 | A | Cys | SG | 470 | B | Glu | O | – | 3.92 | — | — | 101.32 |
| 126 | A | Cys | SG | 472 | B | Leu | O | – | 2.30 | — | — | 125.72 |
| 126 | A | Cys | SG | 472 | B | Leu | OXT | – | 3.51 | — | — | 64.40 |
| 464 | B | Ser | N | 124 | A | Tyr | OH | – | 3.16 | — | 179.42 | — |
| 469 | B | Ala | N | 85 | A | Tyr | OH | – | 3.27 | 3.97 | 39.03 | — |
Figure 7. Interaction between CRIP1b (wheat colored) and the CB1-receptor 9-mer peptide (magenta stick model).
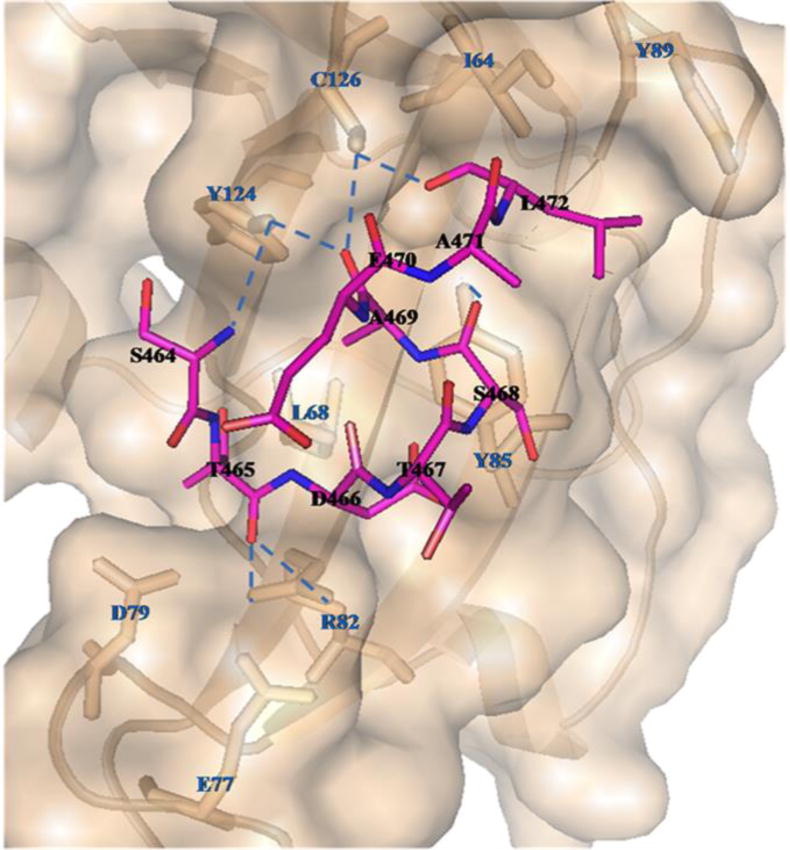
The CRIP1b residues involved in the binding domain are represented by wheat colored sticks; those involved in hydrogen bonds are connected to the CB1 receptor peptide by blue dashed lines.
Conclusion
There are previous experimental data indicating CRIP1-CB1 receptor interactions [11]; however, due to the absence of crystal/solution structures of the complex or CRIP1b protein, these interactions have never been studied at the atomic level. For CRIP1 proteins, the majority of studies have been performed on CRIP1a and the data on CRIP1b are very limited. Our study is the first attempt to study structure and interactions of CRIP1b with the CB1 receptor. To predict and ensure accurate three-dimensional models of CRIP1b structure; a number of different modeling techniques (Ab initio, fold recognition and homology modeling using RHO-GDI 2 as a template) were incorporated. RhoGDI 2 is known to regulate the GDP/GTP exchange reaction of Rho proteins by inhibiting the dissociation of GDP and the subsequent binding of GTP to them [68,69]. The CB₁ cannabinoid receptors promote focal adhesion kinase (FAK) activation, and data suggest that CB₁-stimulated FAK phosphorylation and activation can be mediated by G12/13 regulation of Rac1 and RhoA [70]. Although we have data indicating a role for CRIP1a in the Gi/o family regulation, we have not explored the interactions between CRIP proteins, the G12/13 proteins, and the Rho family proteins. Rho-GDI 2 is characterized by a sandwich fold beta structure, and exhibits the best identity with CRIP1b. The template (Rho-GDI 2) used for homology modeling by us is same as the one used by Ahmad et al., for CRIP1a studies (obtained by threading), but we obtained it from SwissModel template search [38]. The sequence alignment of Rho-GDI2 and CRIP1b encompassed all 128 residues of CRIP1b. Apart from homology models, other models obtained from different methods were also individually evaluated and ranked accordingly. The best scoring CRIP1b model (with Rho-GDI2 as a template) having a beta sandwich fold validated by Ramachandran Plot was selected for interaction studies.
The 9-mer peptide (464STDTSAEAL472) from the CB1 receptor distal C-terminus, was modeled using PEPFOLD, and docked as a ligand to the best-scored model of CRIP1b. The HADDOCK protocol generates multiple poses based on energy and scoring functions. These poses were further refined and the best scoring pose in terms of its interaction energy was chosen for interaction analysis. The best scoring refined model of CRIP1b in complex with the CB1 receptor C-terminus peptide showed favorable polar interactions. Overall, the binding pocket of CRIP1b was found to be similar to that of CRIP1a (Figure 8). The Arg82 and Cys126 of CRIP1b, involved in the majority of hydrogen bond interactions with the 9-mer peptide, are possible key residues required for interactions between the CB1 receptor and CRIP1b. Though we didn’t see bonds with Lys130 as described by Ahmed et al., but Lys130 exists in the binding pocket of CRIP1a in our results(Supporting file: Table S4A). The Presence of Lys130 within the 4 angstroms radius of CB1R peptide indicates the high possibility of contacts between Lys130 of CRIP1a and CB1R peptide. Apart from Lys130, the other residues (Asn61, Ser63, Asp81, Arg82, Tyr126 and Asn127) of CRIP1a overlap with binding pocket defined by Ahmad et al., within the 4 angstroms radius of CB1R peptide. The CRIP1a binding pocket defined by us not only overlaps with data generated by Ahmad et al., but also with CRIP1b binding sites residues lying in region 64 to 126.
Figure 8. Binding pockets of CRIP1a (green) and CRIP1b (cyan) for CB1 receptor peptide (magenta).
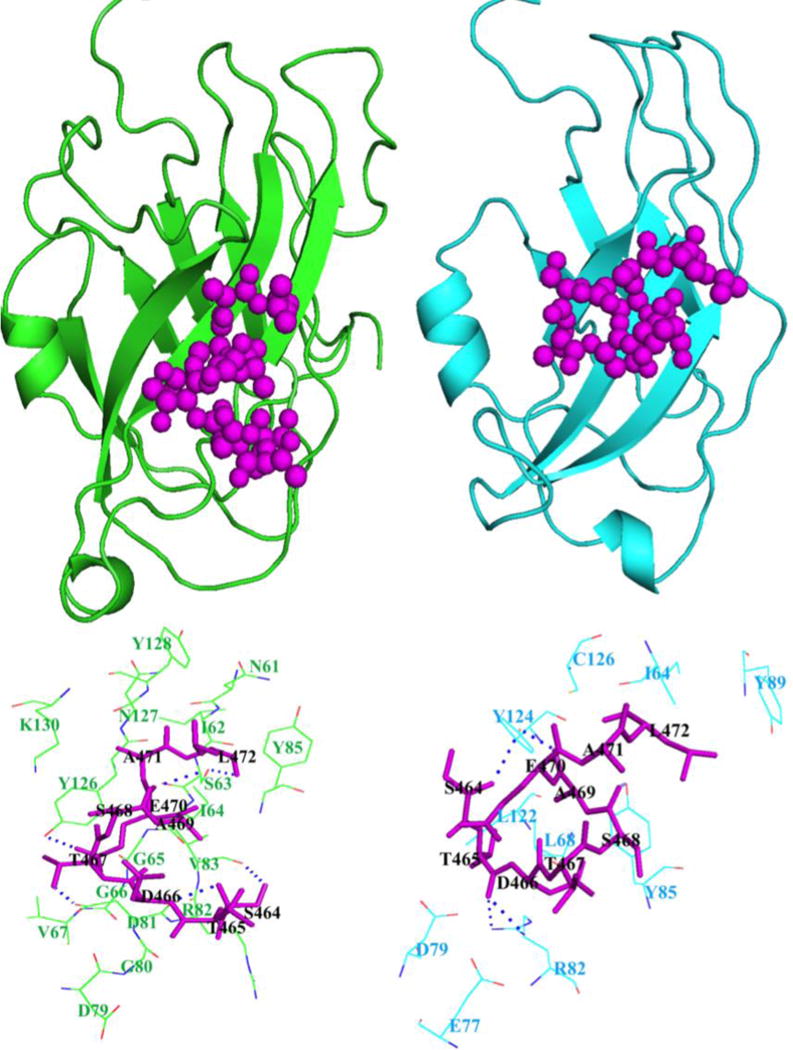
The above portion of image represents positions of CB1 receptor peptide on CRIP1a and 1b, while lower portion shows their respective binding pockets.
Our computational studies indicate that CRIP1b shares similar CB1 receptor binding pocket residues with CRIP1a, but there are functional differences between the two proteins. As it is known that CRIP1a (164 residues) and CRIP1b (128 residues) amino acid sequences are identical for the initial 110 residues, it can be postulated that the remaining 54 residues of CRIP1a may contribute to differences in their function. CRIP1a significantly attenuates tonic inhibition of voltage-gated Ca2+ channels by the CB1receptor but CRIP1b does not. CRIP1a but not CRIP1b has a predicted palmitoyalation site that may contribute to its localization at the plasma membranes [11]. Also, CRIP1a but not CRIP1b contains a PDZ class I ligand in its C-terminal tail, which could indicate a potential for interactions with proteins containing PDZ domains [11]. There is no such ligand in the CRIP1b C-terminal tail, which could account for the functional differences between CRIP1a and CRIP1b, in spite of having similar CB1R interaction binding pocket.
As there were previously no experimental data available for CRIP1b structure, information obtained from our modeling studies may be used to guide design of future site directed mutagenesis experiments. Understanding structure and interactions of CRIP1 proteins will lead to understanding underlying mechanisms of regulation of the CB1 receptor.
Supplementary Material
Highlights.
The three dimensional molecular model of CRIP1b is derived.
The CRIP1b is a sandwich fold structure.
Molecular docking studies establish Arg82 and Cys126 as the key residues of CRIP1b, involved in interaction with CB1 receptor
The binding pocket of CRIP1b has a overlap to that of CRIP1a.
Acknowledgments
One of the authors (Sudha M Cowsik) thanks UPE II grant and DST Purse grant, JNU, Delhi for partial support. Pratishtha Singh is grateful to DST-INSPIRE for providing fellowship. Dr. Anjali Ganjiwale is thankful to UGC-FRP for support. The CRIP1b studies were supported in part by NIH grant R01-DA03690 to Allyn C Howlett. We acknowledge help from Prof Yathindra, Director, Institute of Bioinformatics and Applied Biotechnology (IBAB). The work was partly completed using Bioinformatics facility at IBAB, Bangalore, INDIA.
FUNDING SOURCES
UPE –II grant and DST Purse, Jawaharlal Nehru University, Delhi
CRIP1b studies were supported in part by NIH grant R01-DA03690.
ABBREVIATIONS
- CRIP
Cannabinoid receptor interacting protein
- FAN
Factor Associated with Neutral sphingomyelinase
- GASP
GPCR-associated sorting protein
- TIP3P
transferable intermolecular potential with 3 points
- THC
Tetra-hydro cannabinol
- DOPE
Discrete optimized protein energy
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
AUTHOR CONTRIBUTIONS
Allyn C. Howlett and Sudha M Cowsik developed the concept map for the investigation of CRIP1b. Pratishtha Singh, Anjali Ganjiwale and Sudha M Cowsik performed the computational studies of CRIP1b. Pratishtha Singh and Anjali Ganjiwale drafted the manuscript, and Sudha M Cowsik and Allyn C. Howlett revised and edited the manuscript.
COMPETING INTERESTS
The authors declare no competing interests.
References
- 1.Howlett AC, Barth F, Bonner TI, Cabral G, Casellas P, Devane WA, et al. International Union of Pharmacology. XXVII. Classification of Cannabinoid Receptors. Pharmacol Rev. 2002;54:161–202. doi: 10.1124/pr.54.2.161. [DOI] [PubMed] [Google Scholar]
- 2.Howlett AC, Abood ME. CB1 and CB2 Receptor Pharmacology, Advances in Pharmacology. In: Kendall D, Alexander S, editors. Cannabinoid Pharmacology. Vol. 80. Elsevier Publications; 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hillard CJ. Circulating Endocannabinoids: From Whence Do They Come and Where are They Going? Neuropsychopharmacology: official publication of the American College of Neuropsychopharmacology. 2017 doi: 10.1038/npp.2017.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kendall DA, Yudowski GA. Cannabinoid receptors in the central nervous system: their signaling and roles in disease. Frontiers in cellular neuroscience. 2016:10. doi: 10.3389/fncel.2016.00294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ligresti A, De Petrocellis L, Di Marzo V. From phytocannabinoids to cannabinoid receptors and endocannabinoids: pleiotropic physiological and pathological roles through complex pharmacology. Physiological reviews. 2016;96(4):1593–1659. doi: 10.1152/physrev.00002.2016. [DOI] [PubMed] [Google Scholar]
- 6.Howlett AC, Blume LC, Dalton GD. CB1 cannabinoid receptors and their associated proteins. Curr Med Chem. 2010;17:1382–1393. doi: 10.2174/092986710790980023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Smith TH, Sim-Selley LJ, Selley DE. Cannabinoid CB1 receptor-interacting proteins: novel targets for central nervous system drug discovery? Br J Pharmacol. 2010;160:454–466, i. doi: 10.1111/j.1476-5381.2010.00777.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hajkova A, Techlovska S, Dvorakova M, Chambers JN, Kumpost J, Hubalkova P, et al. SGIP1 alters internalization and modulates signaling of activated cannabinoid receptor 1 in a biased manner. Neuropharmacology. 2016;107:201–214. doi: 10.1016/j.neuropharm.2016.03.008. [DOI] [PubMed] [Google Scholar]
- 9.Nie J, Lewis DL. Structural domains of the CB1 cannabinoid receptor that contribute to constitutive activity and G-protein sequestration. J Neurosci. 2001a;21:8758–8764. doi: 10.1523/JNEUROSCI.21-22-08758.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nie J, Lewis DL. The proximal and distal C-terminal tail domains of the CB1 cannabinoid receptor mediate G protein coupling. Neuroscience. 2001b;107:161–167. doi: 10.1016/s0306-4522(01)00335-9. [DOI] [PubMed] [Google Scholar]
- 11.Niehaus JL, Liu Y, Wallis KT, Egertova M, Bhartur SG, Mukhopadhyay S, et al. CB1 cannabinoid receptor activity is modulated by the cannabinoid receptor interacting protein CRIP 1a. Mol Pharmacol. 2007;72:1557–1566. doi: 10.1124/mol.107.039263. [DOI] [PubMed] [Google Scholar]
- 12.Blume LC, Eldeeb K, Bass CE, Selley DE, Howlett AC. Cannabinoid receptor interacting protein (CRIP1a) attenuates CB1R signaling in neuronal cells. Cell Signal. 2015;27:716–726. doi: 10.1016/j.cellsig.2014.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Smith TH, Blume LC, Straiker A, Cox JO, David BG, McVoy JR, Sayers KW, Poklis JL, Abdullah RA, Egertová M, Chen CK, Mackie K, Elphick MR, Howlett AC, Selley DE. Cannabinoid receptor-interacting protein 1a modulates CB1 receptor signaling and regulation. Mol Pharmacol. 2015;87:747–65. doi: 10.1124/mol.114.096495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Blume LC, Patten T, Eldeeb K, Leone-Kabler S, Ilyasov AA, Keegan BM, et al. Cannabinoid Receptor Interacting Protein 1a Competition with β-Arrestin for CB1 Receptor Binding Sites. Molecular Pharmacology. 2017;91(2):75–86. doi: 10.1124/mol.116.104638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Blume LC, Leone-Kabler S, Luessen DJ, Marrs GS, Lyons E, Bass CE, et al. Cannabinoid receptor interacting protein suppresses agonist-driven CB1 receptor internalization and regulates receptor replenishment in an agonist-biased manner. J Neurochem. 2016;139:396–407. doi: 10.1111/jnc.13767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mascia F, Klotz L, Lerch J, Ahmed MH, Zhang Y, Enz R. CRIP1a inhibits endocytosis of G‐ protein coupled receptors activated by endocannabinoids and glutamate by a common molecular mechanism. Journal of Neurochemistry. 2017;141(4):577–591. doi: 10.1111/jnc.14021. [DOI] [PubMed] [Google Scholar]
- 17.Blume LC, Bass CE, Childers SR, Dalton GD, Roberts D, Richardson JM, Howlett AC. Striatal CB1 and D2 receptors regulate expression of each other, CRIP1A and delta opioid systems. Journal of neurochemistry. 2013;124(6):808–820. doi: 10.1111/jnc.12139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Guggenhuber S, Alpar A, Chen R, Schmitz N, Wickert M, Mattheus T, et al. Cannabinoid receptor-interacting protein Crip1a modulates CB1 receptor signaling in mouse hippocampus. Brain Structure and Function. 2016;221(4):2061–2074. doi: 10.1007/s00429-015-1027-6. [DOI] [PubMed] [Google Scholar]
- 19.Ludányi A, Erőss L, Czirják S, Vajda J, Halász P, Watanabe M, et al. Downregulation of the CB1 cannabinoid receptor and related molecular elements of the endocannabinoid system in epileptic human hippocampus. Journal of Neuroscience. 2008;28(12):2976–2990. doi: 10.1523/JNEUROSCI.4465-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shu-Jung Hu S, Arnold A, Hutchens JM, Radicke J, Cravatt BF, Wager-Miller J, et al. Architecture of cannabinoid signaling in mouse retina. Journal of Comparative Neurology. 2010;518(18):3848–3866. doi: 10.1002/cne.22429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ahmed MH, Kellogg GE, Selley DE, Safo MK, Zhang Y. Predicting the molecular interactions of CRIP1a–cannabinoid 1 receptor with integrated molecular modeling approaches. Bioorganic & medicinal chemistry letters. 2014;24(4):1158–1165. doi: 10.1016/j.bmcl.2013.12.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shen Y, Maupetit J, Derreumaux P, Tufféry P. Improved PEP-FOLD approach for peptide and miniprotein structure prediction. Journal of chemical theory and computation. 2014;10(10):4745–4758. doi: 10.1021/ct500592m. [DOI] [PubMed] [Google Scholar]
- 23.Thévenet P, Shen Y, Maupetit J, Guyon F, Derreumaux P, Tufféry P. PEP-FOLD: an updated de novo structure prediction server for both linear and disulfide bonded cyclic peptides. Nucleic acids research. 2012;40(W1):W288–W293. doi: 10.1093/nar/gks419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bystroff C, Thorsson V, Baker D. HMMSTR: a hidden Markov model for local sequence-structure correlations in proteins. Journal of molecular biology. 2000;301(1):173–190. doi: 10.1006/jmbi.2000.3837. [DOI] [PubMed] [Google Scholar]
- 25.Cheng J, Randall AZ, Sweredoski MJ, Baldi P. SCRATCH: a protein structure and structural feature prediction server. Nucleic acids research. 2005;33(suppl 2):W72–W76. doi: 10.1093/nar/gki396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Salamov AA, Solovyev VV. Protein secondary structure prediction using local alignments. Journal of molecular biology. 1997;268(1):31–36. doi: 10.1006/jmbi.1997.0958. [DOI] [PubMed] [Google Scholar]
- 27.Cuff JA, Barton GJ. Application of multiple sequence alignment profiles to improve protein secondary structure prediction. Proteins: Structure, Function, and Bioinformatics. 2000;40(3):502–511. doi: 10.1002/1097-0134(20000815)40:3<502::aid-prot170>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 28.Bettella F, Rasinski D, Knapp EW. Protein secondary structure prediction with SPARROW. Journal of chemical information and modeling. 2012;52(2):545–556. doi: 10.1021/ci200321u. [DOI] [PubMed] [Google Scholar]
- 29.Adamczak R, Porollo A, Meller J. Accurate prediction of solvent accessibility using neural networks–based regression. Proteins: Structure, Function, and Bioinformatics. 2004;56(4):753–767. doi: 10.1002/prot.20176. [DOI] [PubMed] [Google Scholar]
- 30.Adamczak R, Porollo A, Meller J. Combining prediction of secondary structure and solvent accessibility in proteins. Proteins: Structure, Function, and Bioinformatics. 2005;59(3):467–475. doi: 10.1002/prot.20441. [DOI] [PubMed] [Google Scholar]
- 31.Wagner M, Adamczak R, Porollo A, Meller J. Linear regression models for solvent accessibility prediction in proteins. Journal of Computational Biology. 2005;12(3):355–369. doi: 10.1089/cmb.2005.12.355. [DOI] [PubMed] [Google Scholar]
- 32.Salamov AA, Solovyev VV. Prediction of protein secondary structure by combining nearest-neighbor algorithms and multiple sequence alignments. Journal of molecular biology. 1995;247(1):11–15. doi: 10.1006/jmbi.1994.0116. [DOI] [PubMed] [Google Scholar]
- 33.Petersen B, Petersen TN, Andersen P, Nielsen M, Lundegaard C. A generic method for assignment of reliability scores applied to solvent accessibility predictions. BMC structural biology. 2009;9(1):51. doi: 10.1186/1472-6807-9-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Solov’ev VV, Salamov VV. A method of calculating the discrete secondary structures of globular proteins. Molekuliarnaiabiologiia. 1990;25(3):810–824. [PubMed] [Google Scholar]
- 35.Kurowski MA, Bujnicki JM. GeneSilico protein structure prediction meta-server. Nucleic Acids Research. 2003;31(13):3305–3307. doi: 10.1093/nar/gkg557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Arnold K, Bordoli L, Kopp J, Schwede T. The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics. 2006;22(2):195–201. doi: 10.1093/bioinformatics/bti770. [DOI] [PubMed] [Google Scholar]
- 37.Bordoli L, Kiefer F, Arnold K, Benkert P, Battey J, Schwede T. Protein structure homology modeling using SWISS-MODEL workspace. Nature protocols. 2009;4(1):1–13. doi: 10.1038/nprot.2008.197. [DOI] [PubMed] [Google Scholar]
- 38.Arnold K, Bordoli L, Kopp J, Schwede T. The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics. 2006;22(2):195–201. doi: 10.1093/bioinformatics/bti770. [DOI] [PubMed] [Google Scholar]
- 39.Angermüller C, Biegert A, Söding J. Discriminative modelling of context-specific amino acid substitution probabilities. Bioinformatics. 2012;28(24):3240–3247. doi: 10.1093/bioinformatics/bts622. [DOI] [PubMed] [Google Scholar]
- 40.Biegert A, Söding J. Sequence context-specific profiles for homology searching. Proceedings of the National Academy of Sciences. 2009;106(10):3770–3775. doi: 10.1073/pnas.0810767106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Margelevičius M, Laganeckas M, Venclovas Č. COMA server for protein distant homology search. Bioinformatics. 2010;26(15):1905–1906. doi: 10.1093/bioinformatics/btq306. [DOI] [PubMed] [Google Scholar]
- 42.Remmert M, Biegert A, Hauser A, Söding J. HHblits: lightning-fast iterative protein sequence searching by HMM-HMM alignment. Nature methods. 2012;9(2):173–175. doi: 10.1038/nmeth.1818. [DOI] [PubMed] [Google Scholar]
- 43.Söding J. Protein homology detection by HMM–HMM comparison. Bioinformatics. 2005;21(7):951–960. doi: 10.1093/bioinformatics/bti125. [DOI] [PubMed] [Google Scholar]
- 44.Xu D, Jaroszewski L, Li Z, Godzik A. FFAS-3D: improving fold recognition by including optimized structural features and template re-ranking. Bioinformatics. 2013 doi: 10.1093/bioinformatics/btt578. btt578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kelley LA, Sternberg MJ. Protein structure prediction on the Web: a case study using the Phyre server. Nature protocols. 2009;4(3):363–371. doi: 10.1038/nprot.2009.2. [DOI] [PubMed] [Google Scholar]
- 46.Eswar N, Marti-Renom MA, Webb B, Madhusudhan MS, Eramian D, Shen M, Pieper U, Sali A, Comparative Protein Structure Modeling With MODELLER . Current Protocols in Bioinformatics. Vol. 15. John Wiley & Sons, Inc; 2006. pp. 5.6.1–5.6.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fiser A, Do RKG, Šali A. Modeling of loops in protein structures. Protein science. 2000;9(9):1753–1773. doi: 10.1110/ps.9.9.1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hestenes MR, Stiefel E. Methods of conjugate gradients for solving linear systems. Vol. 49. NBS; 1952. p. 1. [Google Scholar]
- 49.Anisimov VM, Lamoureux G, Vorobyov IV, Huang N, Roux B, MacKerell AD., Jr Determination of Electrostatic Parameters for a Polarizable Force Field Based on the Classical Drude Oscillator. J Chem Theory Comput. 2005;1:153–168. doi: 10.1021/ct049930p. [DOI] [PubMed] [Google Scholar]
- 50.Yu H, Whitfield TW, Harder E, Lamoureux G, Vorobyov I, Anisimov VM, MacKerell AD, Jr, Roux B. Simulating Monovalent and Divalent Ions in Aqueous Solution Using a Drude Polarizable Force Field. J Chem Theory Comput. 2010;6:774–786. doi: 10.1021/ct900576a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kim DE, Chivian D, Baker D. Protein structure prediction and analysis using the Robetta server. Nucleic acids research. 2004;32(suppl 2):W526–W531. doi: 10.1093/nar/gkh468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wu S, Zhang Y. MUSTER: improving protein sequence profile–profile alignments by using multiple sources of structure information. Proteins: Structure, Function, and Bioinformatics. 2008;72(2):547–556. doi: 10.1002/prot.21945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhang Y. I-TASSER server for protein 3D structure prediction. BMC bioinformatics. 2008;9(1):40. doi: 10.1186/1471-2105-9-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Roy A, Kucukural A, Zhang Y. I-TASSER: a unified platform for automated protein structure and function prediction. Nature protocols. 2010;5(4):725–738. doi: 10.1038/nprot.2010.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yang J, Yan R, Roy A, Xu D, Poisson J, Zhang Y. The I-TASSER Suite: protein structure and function prediction. Nature methods. 2015;12(1):7–8. doi: 10.1038/nmeth.3213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shen MY, Sali A. Statistical potential for assessment and prediction of protein structures. Protein science. 2006;15(11):2507–2524. doi: 10.1110/ps.062416606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ramachandran GN, Ramakrishnan C, Sasisekharan V. Stereochemistry of polypeptide chain configurations. Journal of molecular biology. 1963;7(1):95–99. doi: 10.1016/s0022-2836(63)80023-6. [DOI] [PubMed] [Google Scholar]
- 58.DeLano WL. The PyMOL molecular graphics system 2002 [Google Scholar]
- 59.Dominguez C, Boelens R, Bonvin AM. HADDOCK: A protein– protein docking approach based on biochemical or biophysical information. Journal of the American Chemical Society. 2003;125(7):1731–1737. doi: 10.1021/ja026939x. [DOI] [PubMed] [Google Scholar]
- 60.De Vries SJ, Van Dijk M, Bonvin AM. The HADDOCK web server for data-driven biomolecular docking. Nature protocols. 2010;5(5):883–897. doi: 10.1038/nprot.2010.32. [DOI] [PubMed] [Google Scholar]
- 61.Van Zundert GCP, Rodrigues JPGLM, Trellet M, Schmitz C, Kastritis PL, Karaca E, et al. The HADDOCK2. 2 web server: user-friendly integrative modeling of biomolecular complexes. Journal of molecular biology. 2016;428(4):720–725. doi: 10.1016/j.jmb.2015.09.014. [DOI] [PubMed] [Google Scholar]
- 62.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of simple potential functions for simulating liquid water. The Journal of chemical physics. 1983;79(2):926–935. [Google Scholar]
- 63.Karplus M, Kuriyan J. Molecular dynamics and protein function. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(19):6679–6685. doi: 10.1073/pnas.0408930102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Andrusier N, Nussinov R, Wolfson HJ. FireDock: fast interaction refinement in molecular docking. Proteins: Structure, Function, and Bioinformatics. 2007;69(1):139–159. doi: 10.1002/prot.21495. [DOI] [PubMed] [Google Scholar]
- 65.Mashiach E, Schneidman-Duhovny D, Andrusier N, Nussinov R, Wolfson HJ. FireDock: a web server for fast interaction refinement in molecular docking. Nucleic acids research. 2008;36(suppl 2):W229–W232. doi: 10.1093/nar/gkn186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tina KG, Bhadra R, Srinivasan N. PIC: protein interactions calculator. Nucleic acids research. 2007;35(suppl 2):W473–W476. doi: 10.1093/nar/gkm423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Van Dijk AD, Bonvin AM. Solvated docking: introducing water into the modelling of biomolecular complexes. Bioinformatics. 2006;22(19):2340–2347. doi: 10.1093/bioinformatics/btl395. [DOI] [PubMed] [Google Scholar]
- 68.Scheffzek K, Stephan I, Jensen ON, Illenberger D, Gierschik P. The Rac–RhoGDI complex and the structural basis for the regulation of Rho proteins by RhoGDI. Nature structural & molecular biology. 2000;7(2):122–126. doi: 10.1038/72392. [DOI] [PubMed] [Google Scholar]
- 69.Scherle P, Behrens T, Staudt LM. Ly-GDI, a GDP-dissociation inhibitor of the RhoA GTP-binding protein, is expressed preferentially in lymphocytes. Proceedings of the National Academy of Sciences. 1993;90(16):7568–7572. doi: 10.1073/pnas.90.16.7568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Dalton GD, Peterson LJ, Howlett AC. CB 1 cannabinoid receptors promote maximal FAK catalytic activity by stimulating cooperative signaling between receptor tyrosine kinases and integrins in neuronal cells. Cellular signalling. 2013;25(8):1665–1677. doi: 10.1016/j.cellsig.2013.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.