

Abstract
Autophagy is a lysosomal degradation pathway that is critical to maintaining neuronal homeostasis and viability. Autophagy sequesters damaged and aged cellular components from the intracellular environment, and shuttles these diverse macromolecules to lysosomes for destruction. This active surveillance of the quality of the cytoplasm and organelles is essential in neurons to sustain their long-term functionality and viability. Indeed, defective autophagy is linked to neurodevelopmental abnormalities and neurodegeneration in mammals. Here, we review the mechanisms of autophagy in neurons and functional roles for autophagy in neuronal homeostasis. We focus on the compartment-specific dynamics of autophagy in neurons, and how autophagy might perform non-canonical functions critical for neurons. We suggest the existence of multiple populations of autophagosomes with compartment-specific functions important for neural activity and function.
Keywords: Autophagy, neurons, axonal transport, neurodegeneration, homeostasis
Introduction: the challenges of being a neuron
Autophagy is an evolutionarily conserved catabolic process that maintains cellular homeostasis by degrading and recycling proteins and organelles (Ariosa and Klionsky, 2016, Weidberg et al., 2011, Mizushima et al., 2011, Feng et al., 2014). In this process, cellular components targeted for destruction are engulfed and packaged within an organelle termed an autophagosome (Fig. 1). Autophagosomes transport enveloped cargo toward proteolytically-active lysosomes, and fusion between these organelles results in degradation of autophagosome contents (Fig. 1). The amino acids and lipids generated through the process of degradation can then be recycled to fuel new biosynthetic reactions. Thus, autophagy provides a mechanism to constitutively regulate protein and organelle integrity, thereby balancing the synthesis of new cellular components with the degradation of old. Further, levels of autophagy are dynamically modulated in response to various modalities of stress including nutrient deprivation, protein aggregation, and disease (Yang and Klionsky, 2010, Schneider and Cuervo, 2014).
Figure 1. Diagram of the progression of autophagy.
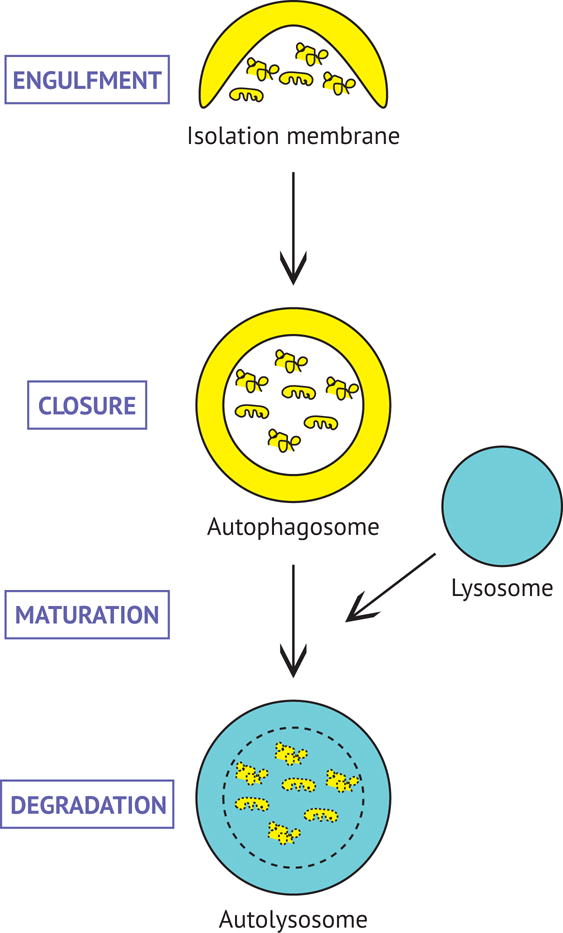
Cargoes are sequestered within double-membraned autophagosomes which then fuse with lysosomes for degradation. Autophagic cargo, as well as the inner membrane of the autophagosome, are degraded by resident lysosomal hydrolases. Degradation products (e.g. amino acids and lipids) can be re-used for new biosynthetic reactions.
Neurons are particularly dependent on autophagy to maintain cellular homeostasis (Maday, 2016, Yamamoto and Yue, 2014, Ariosa and Klionsky, 2016). Being post-mitotic, neurons are simply unable to dilute out dysfunctional proteins and organelles via cell division. Further, neurons need to live for the lifetime of the individual, which in the case of humans is ~90 years or longer! Most neurons in the brain are generated during embryogenesis, and unlike other cell types that are replaced frequently (e.g. intestinal cells are replaced every 2–3 days, red blood cells every 4 months, and hepatocytes every 5 months), neurons are typically the age of the individual (Spalding et al., 2005). As a consequence, neuronal proteins and organelles are exceptionally vulnerable to overuse and damage. Thus, neurons require robust quality control pathways to sustain functionality over this extended lifetime (Wang et al., 2017, Cajigas et al., 2010).
Neurons also face unique spatial challenges due to their extreme morphology and polarized architecture. The soma, central headquarters of the neuron, is considered the main site of protein synthesis and degradation, and can be located up to meters away from sites of action for many neuronal proteins and organelles. The axon is another specialized compartment that communicates information across large distances reaching up to meters in length, posing a logistical challenge of how homeostatic pathways such as autophagy are adapted to regulate protein and organelle quality across this extended landscape.
Here, we will briefly review the roles for autophagy in neuronal development, homeostasis, and survival. While several lines of evidence support the essential role of autophagy in maintaining neuronal health and functionality, the mechanisms and regulation driving autophagy in neurons are only beginning to emerge. Burgeoning evidence has demonstrated that neurons have evolved mechanisms to regulate autophagy in compartment-specific manners to facilitate neuron-specific functions. We will review in detail the mechanisms of autophagy in each compartment of the neuron, which enable the development and functionality of a mature nervous system. Uncovering the diverse roles for autophagy in the neuron will provide insights into how autophagy can be modulated for therapeutic purposes to treat disorders characterized by neuronal dysfunction and degeneration.
Autophagy is essential for neuronal survival
Autophagy plays a critical role in regulating neuronal homeostasis and viability (Hara et al., 2006, Komatsu et al., 2006, Komatsu et al., 2007, Nishiyama et al., 2007, Liang et al., 2010). Evidence supporting this role stems from various mouse models deficient for key genes in the autophagy pathway. Genetic inactivation of Atg5 or Atg7, core proteins required for autophagosome formation (Feng et al., 2014, Mizushima et al., 2011, Weidberg et al., 2011), in the nervous system is sufficient to cause axon swelling and neuron death in mice (Hara et al., 2006, Komatsu et al., 2006). Neuronal loss is observed in pyramidal cells in the cerebral cortex and hippocampus, and most prominently in the Purkinje cell layer of the cerebellum (Hara et al., 2006, Komatsu et al., 2006). Loss of autophagy is also associated with a progressive increase in ubiquitin-positive aggregates specifically within neurons and not within surrounding glia (Hara et al., 2006, Komatsu et al., 2006), indicating the importance of autophagy in the constitutive surveillance of proteome quality. Interestingly, different regions of the brain exhibit a differential response to the loss of autophagy, indicating neuron-specific vulnerabilities to autophagy deficiency. While Purkinje cells appear most vulnerable to autophagy deficiency, they do not present aggregate pathology (Hara et al., 2006), suggesting that autophagy may play different roles in different neuronal subtypes. Autophagy-deficient animals display progressive deficits in motor function (Hara et al., 2006, Komatsu et al., 2006) and lethality at 28 weeks (Komatsu et al., 2006). Similarly, neural-specific deletion of FIP200, a protein required for the initiation of autophagosome formation (Feng et al., 2014, Hara et al., 2008), also results in Purkinje cell degeneration and death, cerebellar ataxia, and animal lethality (Liang et al., 2010). Thus, a basal level of autophagy is critical to maintain neuronal homeostasis and protect against neurodegeneration.
In these mouse models, however, knockout of Atg5 or Atg7 was effective in neural progenitor cells, including neuron and glial cell precursors. Therefore, non-cell autonomous contributions from glia cannot be excluded. As a result, neuron-specific knockout mice were generated to assess cell autonomous effects of autophagy loss. Genetic inactivation of Atg5 or Atg7 specifically in Purkinje cells results in similar phenotypes of progressive axon degeneration followed by neuron death in mice (Komatsu et al., 2007, Nishiyama et al., 2007). Animals also display a progressive decline in motor coordination (Komatsu et al., 2007, Nishiyama et al., 2007). Taken together, these neuron-specific models demonstrate that autophagy is an essential and constitutively active process in neurons that plays a critical role in neuroprotection.
Recent work has demonstrated that loss of neuronal function is the primary cause of neonatal lethality in mice with a global deficiency in autophagy (Yoshii et al., 2016). While Atg5-null mice die within one day of birth (Kuma et al., 2004), restoration of autophagy specifically within the brain is sufficient to rescue neonatal lethality, extending lifetime into adulthood (Yoshii et al., 2016). Thus, loss of autophagy causes a decline in neuronal function leading to neonatal lethality. Neuronal dysfunction appears to manifest as a suckling defect, rendering neonates incapable of surviving the initial starvation period after birth (Yoshii et al., 2016, Kuma et al., 2004).
Alterations in autophagy are associated with neurodegeneration in humans (Maday, 2016, Yamamoto and Yue, 2014, Menzies et al., 2017, Ramesh and Pandey, 2017). Recently, a mutation in the core autophagy protein Atg5 was identified to cause childhood ataxia, a movement disorder associated with degeneration of Purkinje cells in the cerebellum (Kim et al., 2016). This mutation decreases autophagic flux, directly linking defective autophagy with human disease (Kim et al., 2016). Genetic evidence also links mutations in autophagy receptors and regulatory proteins with human neurodegenerative disease (Maday, 2016, Frake et al., 2015), although whether autophagic defects arise early in autophagosome biogenesis or later in autophagosome clearance is under investigation.
Autophagosomes accumulate in vulnerable neurons in various neurodegenerative diseases ranging from Amyotrophic Lateral Sclerosis, Alzheimer’s, Parkinson’s, and Huntington’s Diseases (Maday, 2016, Yamamoto and Yue, 2014, Menzies et al., 2017, Nixon et al., 2005, Son et al., 2012), but the underlying cause for this apparent increase is unclear. Autophagosomes accumulate if the balance between autophagosome formation and degradation is disrupted. For example, autophagy may be upregulated during neurodegeneration in effort to clear away the excess of misfolded protein that is prevalent in many of these diseases. Alternatively, defects downstream in autophagic clearance would prevent the turnover of autophagosomes and result in a buildup of autophagosome organelles. Emerging evidence provides support for the latter hypothesis. Defects in axonal transport (Maday et al., 2014, Millecamps and Julien, 2013) combined with inefficient lysosome function (Gowrishankar et al., 2015, Lee et al., 2010, Xie et al., 2015, Lee et al., 2011) have been observed in models of neurodegeneration, which may render the autophagic pathway ineffective at clearing away “cellular trash”, contributing to neuronal death.
Autophagy in development of the nervous system
In addition to protecting the viability of mature neurons, autophagy may also play an important role in the development of the nervous system (Fimia et al., 2007, Dragich et al., 2016, Stavoe et al., 2016, Tang et al., 2014). Autophagy has been implicated early in neurodevelopment during formation of the neural tube (Fimia et al., 2007). Mice deficient for AMBRA1, a regulator of autophagy enriched in the neuroepithelium, have defects in closure of the neural tube, associated with an imbalance between cell proliferation and apoptosis of the neuroepithelium (Fimia et al., 2007).
Autophagy may regulate the development of axon tracts in the central nervous system (CNS) (Dragich et al., 2016). In mice with a systemic deficiency for Alfy, a CNS-enriched adaptor for selective autophagy (Filimonenko et al., 2010), interhemispheric axon tracts such as the corpus callosum, anterior commissure, and hippocampal commissure do not develop normally and fail to cross the midline (Dragich et al., 2016). Some axonal projections in the CNS follow unusual trajectories, suggesting a defect in axon guidance (Dragich et al., 2016). In fact, while Alfy-deficient neurons retain the ability to extend axons in vitro, they fail to grow in response to Netrin-1-based guidance cues (Dragich et al., 2016). Since these animals are specifically deficient in the degradation of ubiquitinated substrates, and not bulk autophagy, selective forms of autophagy may play a key role in axon guidance and establishing connectivity in the developing brain.
Recently, other autophagy genes have been implicated in axon outgrowth during brain development (Kannan et al., 2017, Yamaguchi et al., 2017). Mice with loss of function of both copies of WDR47, a modulator of autophagy, or a single copy of Atg16L1, a core autophagy protein, display abnormalities in the corpus callosum (Kannan et al., 2017). Neural-specific depletion of Atg9, a core autophagy protein that directs membrane to the growing autophagosome (Feng et al., 2014, Mizushima et al., 2011, Weidberg et al., 2011), also results in abnormal development of axon tracts including the corpus callosum and anterior commissure (Yamaguchi et al., 2017). Curiously, while Atg9-deficient neurons show defects in neurite outgrowth in vitro, Atg7-deficient neurons do not exhibit defects in neurite outgrowth, (Yamaguchi et al., 2017), raising the possibility of autophagy-independent roles for autophagy genes, such as Atg9, during brain development. In some conditions, autophagy may function as a negative regulator of axon outgrowth with reduced autophagy levels resulting in increased axon length (Ban et al., 2013, Stavoe et al., 2016). While the precise mechanisms underlying the contributions of autophagy in axonal growth and guidance are unknown, autophagy in the leading edge of the axon may facilitate membrane recycling or turnover of signaling information in the migrating growth cone.
Autophagy may also play a critical role in the development of synapses (Stavoe et al., 2016, Shen and Ganetzky, 2009, Rudnick et al., 2017). The autophagy pathway is required for assembly of presynaptic compartments in C. elegans by regulating synaptic vesicle clustering (Stavoe et al., 2016). Autophagy may also promote formation of the presynaptic axon terminal at the neuromuscular junction (Shen and Ganetzky, 2009, Rudnick et al., 2017), and inhibition of autophagy results in alterations in neurotransmission (Rudnick et al., 2017). Taken together, these observations indicate that autophagy may impact synaptogenesis via regulation of synaptic structure and function.
Lastly, autophagy may function later in neurodevelopment and mediate the elimination of excess dendritic spines (Tang et al., 2014). Tang et al. demonstrated that deficits in spine pruning exhibited in mouse models of autism with hyperactive mTOR, are attributed to low levels of autophagy (Tang et al., 2014). Mice with selective loss of autophagy in pyramidal neurons exhibit an increase in dendritic spines in the cortex and social deficits akin to autism-like behaviors (Tang et al., 2014). Taken together, autophagy is implicated in various stages of neurodevelopment to ensure the stereotyped connectivity of the CNS, and alterations in autophagy may be associated with neurodevelopmental disorders.
In total, the loss of autophagy manifests differently depending on the gene targeted and neuron-type examined. Loss of core autophagy proteins can result in neurodevelopmental defects (e.g. Atg9 and Atg16L1), while others (e.g. Atg5, Atg7, FIP200) display a neurodegenerative phenotype. Since neural-specific loss of Atg5 and Atg7 protein expression is significantly reduced by embryonic day 15.5 (Hara et al., 2006, Komatsu et al., 2006), developmental defects may still contribute to the degeneration observed postnatally in these mouse models. In other words, these neurons may have reduced longevities as a result of not being established properly in the first place. Roles for autophagy, independent of neurodevelopment, could be examined directly with a conditional knockout of autophagy in adult neurons.
The diversity of phenotypes generated by the reduction of core autophagy and modulatory proteins may be further confounded by the possibility that perturbing autophagy proteins may have pleiotropic effects. Indeed, autophagy proteins may “moonlight” as key players in other cellular processes. The fact that mouse models deficient for Atg9, FIP200, Alfy, and AMBRA1 die at an earlier developmental stage than neural-specific loss of Atg5 or Atg7 hints at this possibility. Future studies are necessary to define additional functions for these autophagy-related proteins.
Further, loss of the same gene can yield different phenotypes depending on the type of neuron examined. In the case of C. elegans, Atg9 loss-of-function mutants exhibit synaptogenesis defects in a population of interneurons, and axonal growth defects in a population of sensory neurons (Stavoe et al., 2016). In summary, the role of autophagy may evolve with time and depend on neuron-specific demands.
For the remainder of the review, we will focus on the compartment-specific mechanisms and regulation of autophagy that contribute to neuronal homeostasis and function. While our knowledge of what is contained within neuronal autophagosomes is limited, several identified cargoes have illuminated possible functions for autophagy in the neuron. Further, deciphering the signals that regulate autophagy levels in neurons has provided key insights into neuron-specific functions for autophagy. We will discuss roles for autophagy at the synapse, and explore emerging, non-canonical roles for autophagosomes as conduits for neurotrophin signaling across the axon.
Compartment-specific dynamics of autophagosomes in neurons
Given the extreme spatial challenges facing neurons, how is autophagy coordinated in each compartment of the neuron, i.e. the axon, dendrites, and the soma? Further, how are the dynamics of autophagy in neurons uniquely suited to facilitate compartment-specific demands and functions? For example, the axon is dependent on supply from the soma, but how is protein and organelle quality managed far away from sites of synthesis and degradation that are concentrated in the soma? Insights into the features of autophagosome motility in neurons have emerged from live-cell imaging studies of GFP-LC3, the canonical marker for the autophagosome (Kabeya et al., 2000, Mizushima et al., 2004).
Under basal conditions of growth, autophagosomes in the axon are preferentially generated in the distal end of the axon (Fig. 2A) (Maday et al., 2012, Maday and Holzbaur, 2014, Hollenbeck, 1993). GFP-LC3-positive autophagosomes initially appear as punctate structures that progressively increase in size, reaching up to 800 nm in diameter (Maday et al., 2012, Maday and Holzbaur, 2014). Autophagy core proteins Atg13 and Atg5 are recruited, in an ordered fashion, to subdomains of the endoplasmic reticulum (ER), indicating that autophagosomes are generated from ER membranes present within the distal axon (Maday and Holzbaur, 2014).
Figure 2. Overview of compartment-specific autophagy in neurons.
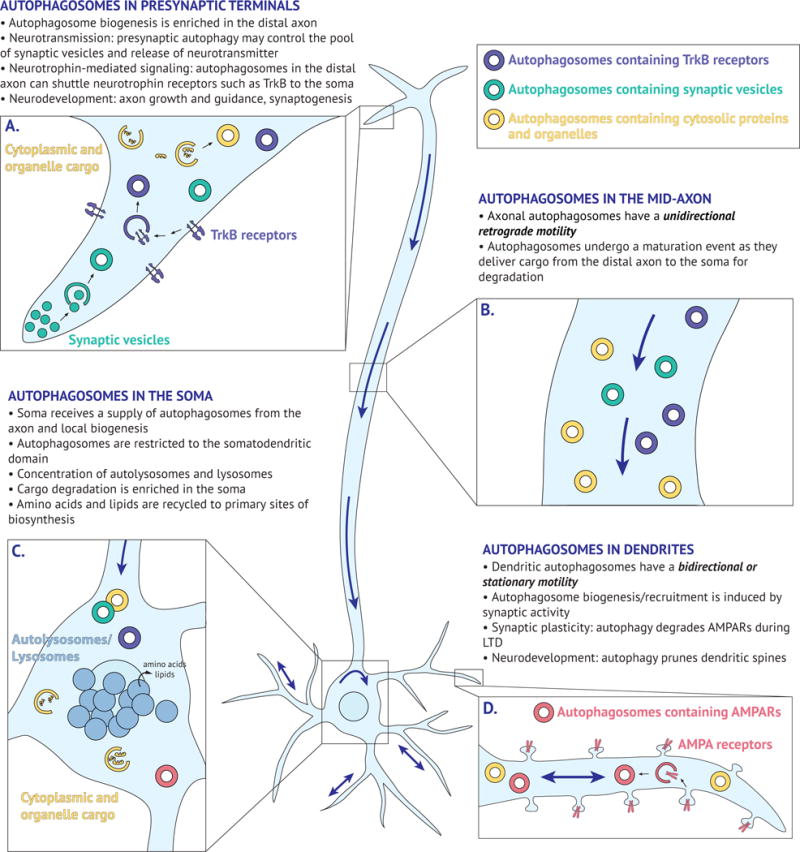
Key characteristics of autophagosome dynamics and function in the pre-synaptic terminal (A), mid-axon (B), soma (C), and dendrites (D). Blue arrows denote directionality of autophagosome movement.
Following formation, autophagosomes then undergo robust retrograde transport toward the soma (Fig. 2B) (Hollenbeck, 1993, Maday et al., 2012, Maday and Holzbaur, 2014, Lee et al., 2011, Yue, 2007). This movement requires the microtubule-based motor, dynein, and is correlated with a transition in maturation state (Maday et al., 2012, Lee et al., 2011). Newly formed autophagosomes in the distal axon are initially LAMP1-negative, but acquire markers of late endosomes/lysosomes as they journey toward the soma (Maday et al., 2012, Lee et al., 2011). Thus, nascent autophagosomes fuse with late endosomes and/or lysosomes upon exit from the distal axon, and this initial advance in maturation may trigger retrograde transport to the soma (Cheng et al., 2015, Fu et al., 2014). Proximal to the cell body, autophagosomes have fully acidified and matured into degradative autolysosomes (Maday et al., 2012, Lee et al., 2011). Therefore, at steady state, axonal autophagy is a vectorial pathway that requires long-distance transport to deliver cargo from the distal axon to the soma. This distal autophagy may function to maintain axonal homeostasis and regulate the quality of the neuronal proteome at sites far removed from the soma.
The soma, however, contains multiple populations of autophagosomes at different maturation states, including input received from the axon combined with locally generated autophagosomes (Fig. 2C) (Maday and Holzbaur, 2016). Evidence for autophagosome biogenesis in dendrites has been limited under basal conditions, but dendritic autophagosomes are elevated under conditions of enhanced synaptic activity (Shehata et al., 2012), either by increased local biogenesis or the recruitment of pre-existing autophagosomes to sites of synaptic function. Once in the soma, autophagosomes are confined within the somatodendritic domain, unable to enter into the axon (Maday and Holzbaur, 2016). Autophagosomes freely travel into dendrites where they exhibit either stationary or bidirectional motility (Maday and Holzbaur, 2014, Maday and Holzbaur, 2016), likely due to the mixed polarity of the underlying microtubule cytoskeleton (Baas et al., 1988, Kleele et al., 2014). The majority of autophagosomes that enter into dendrites, however, return to the soma (Maday and Holzbaur, 2016). In effect, this restriction to the somatodendritic compartment likely facilitates cargo degradation by promoting fusion with proteolytically-active lysosomes that are enriched in this region of the neuron (Fig. 2C) (Gowrishankar et al., 2015, Lee et al., 2011). Concentrating degradative activity in the soma may in turn facilitate efficient recycling of biosynthetic building blocks to primary sites of protein synthesis. In sum, autophagosomes exhibit compartment-specific dynamics within the neuron that may facilitate autophagosome function.
Degradation of autophagic cargo
Lysosomes with high proteolytic activity are concentrated in the cell body (Lee et al., 2011, Gowrishankar et al., 2015), making the soma enriched for degradative activity (Fig. 2C). Inhibition of lysosome function results in an accumulation of autophagosomes specifically within the soma, and not in the axon or dendrites (Maday and Holzbaur, 2016). Therefore, the final destination for autophagosome clearance in the neuron is predominantly the cell body, and autophagosomes originating in the distal axon travel the entire distance of the axon to deliver their cargo to the soma for degradation.
While the contents of autophagosomes are largely unknown, retrograde axonal autophagosomes contain engulfed cytosolic as well as organelle cargoes (Maday et al., 2012). A small percentage (~10%) of autophagosomes contain fragments of mitochondria (Maday et al., 2012). A key question is whether this retrograde axonal pathway for autophagy represents constitutive or selective autophagy, or both? Constitutive or basal autophagy recycles the cytoplasm and its constituents, in a non-selective and continuous manner, to balance synthesis with degradation. Selective autophagy is induced by stress, such as organelle damage, and specific substrates are removed through recognition by adaptors linking cargo to the autophagic machinery (Stolz et al., 2014, Zaffagnini and Martens, 2016, Xu et al., 2015). Recent work has suggested that selective autophagy may be compartmentalized within the neuron (Ashrafi et al., 2014, Cai et al., 2012, Sung et al., 2016, Dragich et al., 2016).
Selective removal of damaged mitochondria by autophagy (mitophagy), has been extensively studied in HeLa cells (Narendra et al., 2008, Narendra et al., 2010), and involves the serine/threonine kinase PINK1 and E3 ubiquitin ligase Parkin. The contributions of PINK1 and Parkin to mitophagy in neurons, however, is less clear (Martinez-Vicente, 2017). While under basal conditions, few autophagosomes are generated in the mid-axon, acute and focally-induced mitochondrial damage can activate autophagy locally within the mid-axon of primary hippocampal neurons (Ashrafi et al., 2014). This axonal mitophagy requires recruitment of PINK1 and Parkin (Ashrafi et al., 2014). In contrast, globally-induced mitochondrial damage stimulates retrograde transport of mitochondria for autophagic engulfment within the soma (Cai et al., 2012). Lastly, recent reports have shown that mitophagy is rare in motor neurons in Drosophila, and Parkin functions exclusively in the soma to regulate mitochondrial quality control by balancing mitochondrial fission and fusion (Sung et al., 2016). Future work will be important to resolve the compartment selectivity to mitophagy and whether the type of mitochondrial damage or insult affects these dynamics.
Ubiquitinated protein aggregates are targeted for autophagic degradation by the adaptor Alfy (Filimonenko et al., 2010). Alfy is enriched in the axon (Dragich et al., 2016), suggesting that the axon may have its own machinery for executing selective autophagy. This finding also suggests the existence of compartment-specific populations of autophagosomes that are essential for proper axonal guidance and development.
In addition to intra-neuronal autophagy, garbage can be shuttled to neighboring glia for degradation. Mitochondria can be extruded from retinal ganglion cell axons and internalized and degraded by neighboring astrocytes (Davis et al., 2014). Adult neurons in C. elegans discard cellular garbage through the secretion of vesicles, called exophers, containing protein aggregates and organelles (Melentijevic et al., 2017). Production of exophers increases when endogenous quality control pathways, including autophagy, are compromised (Melentijevic et al., 2017). Taken together, these findings indicate that neurons have alternative mechanisms for eliminating cellular garbage, involving non-canonical intercellular pathways for degradation.
Neuron-specific functions for autophagy at the synapse
Neuronal synapses are sites of high demand for cellular degradation pathways (Wang et al., 2017, Cajigas et al., 2010). Neurons fire action potentials at rates of up to ~50 impulses per second (LeDoux and Lorden, 2002, Harris and Attwell, 2012, Bean, 2007, Hausser et al., 2004), rendering synaptic proteins and organelles susceptible to being overworked and damaged. Further, synapses are dynamic, being remodeled in response to developmental and sensory cues to refine connections and circuits in the brain (Hering and Sheng, 2001, Nimchinsky et al., 2002). Therefore, synapses require efficient degradation systems to maintain integrity of the local proteome and sustain function (Maday, 2016, Vijayan and Verstreken, 2017, Wang et al., 2017, Cajigas et al., 2010). In fact, synaptic activity regulates levels of autophagy in neurons (Shehata et al., 2012, Wang et al., 2015, Soukup et al., 2016). In turn, autophagy impacts synaptic function with emerging roles at both pre- and post-synaptic domains (Vijayan and Verstreken, 2017, Shen et al., 2015). A significant role for autophagy at synapses may help explain why Purkinje cells are the most vulnerable to the loss of autophagy because they have the highest basal firing rates in the brain (LeDoux and Lorden, 2002, Harris and Attwell, 2012).
Neuronal stimulation induces autophagosome biogenesis in presynaptic regions (Wang et al., 2015, Soukup et al., 2016, Vanhauwaert et al., 2017). While formation of these autophagosomes utilizes core machinery conserved from yeast (Maday and Holzbaur, 2014), several neural-specific regulatory factors modulate rates of autophagosome biogenesis at the synapse (Soukup et al., 2016, Vanhauwaert et al., 2017, Murdoch et al., 2016, Okerlund et al., 2017). Endophillin A, an endocytic adaptor enriched at the presynaptic terminal, promotes autophagy by generating highly curved membranes, which serve as a platform to recruit core autophagy proteins that form autophagosomes (Soukup et al., 2016, Murdoch et al., 2016). Synaptojanin 1, a lipid phosphatase important for synaptic vesicle trafficking, is also required for autophagosome biogenesis at presynaptic terminals (Vanhauwaert et al., 2017). Bassoon, a scaffolding protein localized to the presynaptic nerve terminal, inhibits presynaptic autophagy by sequestering the core autophagy protein Atg5, making it unavailable for autophagosome biogenesis (Okerlund et al., 2017). How these opposing regulatory mechanisms are coordinated to control presynaptic autophagy levels is unknown. In addition, how synaptic activity is sensed by the aforementioned mechanisms and how calcium influx may be involved remains uncertain.
While the precise function of autophagy in the presynaptic domain is largely unknown, identification of engulfed cargoes has provided key insights into this process. Interestingly, presynaptic autophagosomes appear to engulf structures resembling synaptic vesicles (Fig. 2A) (Hernandez et al., 2012, Okerlund et al., 2017). Pharmacological activation of autophagy reduces the number of synaptic vesicles at axon terminals and loss of autophagy specifically within dopaminergic neurons increases evoked dopamine release in mice (Hernandez et al., 2012). Thus, presynaptic autophagy negatively regulates neurotransmission by controlling the size of the synaptic vesicle pool and neurotransmitter release (Hernandez et al., 2012). Autophagy could impact neurotransmission either by sequestering vesicles containing neurotransmitter, or by regulating the pool of empty vesicles available for neurotransmitter loading.
Synaptic activity also regulates autophagy in the postsynaptic domain (Shehata et al., 2012). Synaptic stimulation in the presence of Bafilomycin A1, to inhibit lysosome-mediated degradation, results in a moderate but significant increase in GFP-LC3-positive puncta within the dendritic shaft, above levels observed in cells treated with Bafilomycin A1-alone, suggesting an activity-dependent induction of autophagosome formation or recruitment (Shehata et al., 2012). Postsynaptic autophagy plays a role in synaptic plasticity, activity-dependent changes in synaptic strength (Shehata et al., 2012). Persistent increases in synaptic strength, Long-Term Potentiation (LTP), or weakening of synaptic strength, Long-Term Depression (LTD), require structural and functional changes at the synapse (Vitureira and Goda, 2013). In LTP, persistent activation of NMDA receptors leads to an increase in AMPA receptor insertion into the postsynaptic membrane (Vitureira and Goda, 2013). Conversely, in LTD, moderate activation of NMDA receptors results in downregulation of AMPA receptors in the postsynaptic membrane (Vitureira and Goda, 2013). Autophagy plays a role in LTD by mediating the trafficking and elimination of AMPA receptors (Fig. 2D). Low-dose activation of NMDA receptors with NMDA, to model chemical-LTD, activates autophagy-dependent degradation of AMPA receptors (Shehata et al., 2012). Similarly, lysosomes are recruited to synapses in an activity-dependent manner, internalize AMPA receptors, and regulate synaptic activity and density (Goo et al., 2017). Thus, autophagy plays a role in the plasticity, or changes in strength, of a particular synapse, which are foundational principles underlying learning and memory (Nicholls et al., 2008, Takeuchi et al., 2014, Luscher and Malenka, 2012).
Further supporting a role for autophagy in synaptic plasticity, BDNF suppresses autophagy in hippocampal neurons, and this BDNF-induced decrease in autophagy facilitates LTP and the persistence of memories in mice (Nikoletopoulou et al., 2017). Since autophagosomes contained PICK1, PSD-95 and SHANK3, autophagy may regulate synaptic plasticity in this model by degrading postsynaptic scaffolds (Nikoletopoulou et al., 2017). Lastly, CNS-specific deletion of WDR45, a gene that encodes a protein involved in autophagosome formation, is linked to deficits in learning and memory in mice (Zhao et al., 2015).
As a product of the degradation process, autophagy generates biosynthetic building blocks that fuel the synthesis of new proteins. It is well known that local translation within dendrites is an essential process for synaptic function and plasticity (Rangaraju et al., 2017). Thus, it is possible that recruitment of autophagosomes to synapses promotes synaptic formation and function via new protein synthesis, a way of creation by destruction. Consistent with this idea, the proteasome is recruited to synapses in an activity-dependent manner (Bingol and Schuman, 2006), and fuels the formation of new spine outgrowth (Hamilton et al., 2012). Under certain conditions, autophagy may function in a similar capacity. Therefore, autophagy may have dual functions at the synapse; destroying ‘unwanted’ synapses and sustaining and creating ‘wanted’ synapses.
Non-canonical roles for autophagosomes as vehicles for neurotrophin-mediated signaling?
Are autophagosomes more than just garbage trucks? Axonal autophagosomes exhibit a striking motility pattern by traveling the length of the axon from tip to soma, making these organelles uniquely suited to deliver specific signals to the soma. This predominantly unidirectional, retrograde transport is mirrored by one other organelle, the signaling endosome (Maday et al., 2014, Cui et al., 2007). Signaling endosomes regulate neuronal survival by delivering target-derived neurotrophin-mediated signaling information from the distal axon to the soma to effect changes in gene expression (Chowdary et al., 2012, Harrington and Ginty, 2013). The precise nature of these carriers is unclear, but evidence supports an early endosomal lineage (positive for EEA1 and Rab5) (Cui et al., 2007, Deinhardt et al., 2006, Delcroix et al., 2003), and they may mature to Rab7-positive compartments (Deinhardt et al., 2006, Sandow et al., 2000), raising the possibility that signaling endosomes and autophagosomes may merge. In fact, a population of axonal autophagosomes are positive for the brain-derived neurotrophic factor (BDNF) receptor, TrkB (Fig. 2A) (Kononenko et al., 2017), and addition of BDNF stimulates retrograde motility of organelles co-positive for LC3 and TrkB (Kononenko et al., 2017). By contrast, BDNF has also been shown to decrease autophagy levels in cortical and hippocampal neurons (Nikoletopoulou et al., 2017, Wu et al., 2017). In the brain, this suppression of autophagy is important for BDNF-induced synaptic plasticity (Nikoletopoulou et al., 2017), although the underlying mechanisms remain to be investigated.
For transported signals to be sustained to effect changes in transcription, carrier autophagosomes would need to avoid destruction by the lysosome. Thus, multiple populations of autophagosomes may exist within the neuron, including a subset that may not fully mature to degradative autolysosomes, but rather function as a vehicle for transmitting signaling information. Mechanistically, these “signaling autophagosomes” could potentially avoid their canonical lysosomal fate by being selectively deficient in the SNARE protein Syntaxin 17 (Itakura et al., 2012), which is an essential component of the membrane fusion machinery required for the fusion of autophagosomes with lysosomes. Alternatively, these autophagosomes may associate with Coronin-1, an effector protein that prevents signaling endosome fusion with lysosomes (Suo et al., 2014). Collectively, these findings begin to suggest a new paradigm for non-canonical functions of autophagosomes distinct from their canonical role in degradative pathways.
Outstanding questions
Autophagy plays a critical role in maintaining neuronal homeostasis and viability. Autophagosomes exhibit compartment-specific dynamics that may facilitate their range of functions (Fig. 2). In addition to its role in degradative processes, non-canonical roles for autophagy as signaling organelles are also emerging, although this is a nascent field of investigation. Emerging evidence also supports roles for autophagy in regulating neuronal function by impacting synaptic transmission and plasticity. These functions for autophagy may manifest in different neuronal subtypes and at different developmental stages. Accordingly, the primary role for autophagy in neurons may evolve over time to meet the needs of the cell. Evidence also indicates that different brain regions modulate autophagy differentially in response to stress such as nutrient deprivation (Nikoletopoulou et al., 2017). Further, functions for autophagy shift within a neuron population during the progression of neurodegenerative disease (Rudnick et al., 2017). Perhaps, these regional and temporal differences in autophagy may contribute to the neuron-specific vulnerabilities observed in neurodegenerative disease.
Future work to define the cargo packaged within autophagosomes will be critical to further our understanding of the diverse roles of autophagy in neuronal function. Moreover, how the balance shifts between selective versus non-selective autophagy in stress and disease will empower insights into the mechanisms underlying disease pathogenesis. Since modulation of autophagy has therapeutic value (Barmada et al., 2014), harnessing selective forms of autophagy might enable us to clear deleterious cargo associated with neurodegenerative disease, while simultaneously preserving healthy proteins.
How is autophagy coordinated in the brain between neurons and glia? Do glia supplement the neuronal capacity for degradation? And is this intersection of glia and neurons a point of vulnerability in neurodegenerative disease? Glia are key regulators of neuronal homeostasis and alterations in neuron-glia communication are linked to the progression of neurodegenerative disease (Boillee et al., 2006, Lee et al., 2012). In fact, astrocytes generated from ALS patient iPSCs decrease autophagy levels in neurons (Madill et al., 2017), raising the possibility that quality control pathways can be regulated in a non-cell autonomous fashion, and contribute to disease progression. Additionally, microglia in the brain also have extended lifespans with ~50% of the population surviving an entire mouse lifetime (Fuger et al., 2017). Therefore, how are quality control pathways in glia adapted to sustain functionality over an extended lifetime? A comprehensive understanding of the mechanisms and regulation of homeostatic pathways in the brain will be critical to determining how alterations in these processes contribute to neuronal dysfunction and disease.
Acknowledgments
We thank James Shorter, Aditi Kulkarni, Jessica Chen, and Amy Ghiretti for helpful feedback on the manuscript. This work was funded by NIH grants K99NS082619 and R00NS082619 to S.M. The authors declare no competing financial interests.
References
- ARIOSA AR, KLIONSKY DJ. Autophagy core machinery: overcoming spatial barriers in neurons. J Mol Med (Berl) 2016;94:1217–1227. doi: 10.1007/s00109-016-1461-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ASHRAFI G, SCHLEHE JS, LAVOIE MJ, SCHWARZ TL. Mitophagy of damaged mitochondria occurs locally in distal neuronal axons and requires PINK1 and Parkin. J Cell Biol. 2014;206:655–70. doi: 10.1083/jcb.201401070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BAAS PW, DEITCH JS, BLACK MM, BANKER GA. Polarity orientation of microtubules in hippocampal neurons: uniformity in the axon and nonuniformity in the dendrite. Proc Natl Acad Sci U S A. 1988;85:8335–9. doi: 10.1073/pnas.85.21.8335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BAN BK, JUN MH, RYU HH, JANG DJ, AHMAD ST, LEE JA. Autophagy negatively regulates early axon growth in cortical neurons. Mol Cell Biol. 2013;33:3907–19. doi: 10.1128/MCB.00627-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BARMADA SJ, SERIO A, ARJUN A, BILICAN B, DAUB A, ANDO DM, TSVETKOV A, PLEISS M, LI X, PEISACH D, SHAW C, CHANDRAN S, FINKBEINER S. Autophagy induction enhances TDP43 turnover and survival in neuronal ALS models. Nat Chem Biol. 2014;10:677–85. doi: 10.1038/nchembio.1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BEAN BP. The action potential in mammalian central neurons. Nat Rev Neurosci. 2007;8:451–65. doi: 10.1038/nrn2148. [DOI] [PubMed] [Google Scholar]
- BINGOL B, SCHUMAN EM. Activity-dependent dynamics and sequestration of proteasomes in dendritic spines. Nature. 2006;441:1144–8. doi: 10.1038/nature04769. [DOI] [PubMed] [Google Scholar]
- BOILLEE S, YAMANAKA K, LOBSIGER CS, COPELAND NG, JENKINS NA, KASSIOTIS G, KOLLIAS G, CLEVELAND DW. Onset and progression in inherited ALS determined by motor neurons and microglia. Science. 2006;312:1389–92. doi: 10.1126/science.1123511. [DOI] [PubMed] [Google Scholar]
- CAI Q, ZAKARIA HM, SIMONE A, SHENG ZH. Spatial parkin translocation and degradation of damaged mitochondria via mitophagy in live cortical neurons. Curr Biol. 2012;22:545–52. doi: 10.1016/j.cub.2012.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CAJIGAS IJ, WILL T, SCHUMAN EM. Protein homeostasis and synaptic plasticity. EMBO J. 2010;29:2746–52. doi: 10.1038/emboj.2010.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHENG XT, ZHOU B, LIN MY, CAI Q, SHENG ZH. Axonal autophagosomes recruit dynein for retrograde transport through fusion with late endosomes. J Cell Biol. 2015;209:377–86. doi: 10.1083/jcb.201412046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHOWDARY PD, CHE DL, CUI B. Neurotrophin signaling via long-distance axonal transport. Annu Rev Phys Chem. 2012;63:571–94. doi: 10.1146/annurev-physchem-032511-143704. [DOI] [PubMed] [Google Scholar]
- CUI B, WU C, CHEN L, RAMIREZ A, BEARER EL, LI WP, MOBLEY WC, CHU S. One at a time, live tracking of NGF axonal transport using quantum dots. Proc Natl Acad Sci U S A. 2007;104:13666–71. doi: 10.1073/pnas.0706192104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DAVIS CH, KIM KY, BUSHONG EA, MILLS EA, BOASSA D, SHIH T, KINEBUCHI M, PHAN S, ZHOU Y, BIHLMEYER NA, NGUYEN JV, JIN Y, ELLISMAN MH, MARSH-ARMSTRONG N. Transcellular degradation of axonal mitochondria. Proc Natl Acad Sci U S A. 2014;111:9633–8. doi: 10.1073/pnas.1404651111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DEINHARDT K, SALINAS S, VERASTEGUI C, WATSON R, WORTH D, HANRAHAN S, BUCCI C, SCHIAVO G. Rab5 and Rab7 control endocytic sorting along the axonal retrograde transport pathway. Neuron. 2006;52:293–305. doi: 10.1016/j.neuron.2006.08.018. [DOI] [PubMed] [Google Scholar]
- DELCROIX JD, VALLETTA JS, WU C, HUNT SJ, KOWAL AS, MOBLEY WC. NGF signaling in sensory neurons: evidence that early endosomes carry NGF retrograde signals. Neuron. 2003;39:69–84. doi: 10.1016/s0896-6273(03)00397-0. [DOI] [PubMed] [Google Scholar]
- DRAGICH JM, KUWAJIMA T, HIROSE-IKEDA M, YOON MS, EENJES E, BOSCO JR, FOX LM, LYSTAD AH, OO TF, YARYGINA O, MITA T, WAGURI S, ICHIMURA Y, KOMATSU M, SIMONSEN A, BURKE RE, MASON CA, YAMAMOTO A. Autophagy linked FYVE (Alfy/WDFY3) is required for establishing neuronal connectivity in the mammalian brain. Elife. 2016;5 doi: 10.7554/eLife.14810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FENG Y, HE D, YAO Z, KLIONSKY DJ. The machinery of macroautophagy. Cell Res. 2014;24:24–41. doi: 10.1038/cr.2013.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FILIMONENKO M, ISAKSON P, FINLEY KD, ANDERSON M, JEONG H, MELIA TJ, BARTLETT BJ, MYERS KM, BIRKELAND HC, LAMARK T, KRAINC D, BRECH A, STENMARK H, SIMONSEN A, YAMAMOTO A. The selective macroautophagic degradation of aggregated proteins requires the PI3P-binding protein Alfy. Mol Cell. 2010;38:265–79. doi: 10.1016/j.molcel.2010.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FIMIA GM, STOYKOVA A, ROMAGNOLI A, GIUNTA L, DI BARTOLOMEO S, NARDACCI R, CORAZZARI M, FUOCO C, UCAR A, SCHWARTZ P, GRUSS P, PIACENTINI M, CHOWDHURY K, CECCONI F. Ambra1 regulates autophagy and development of the nervous system. Nature. 2007;447:1121–5. doi: 10.1038/nature05925. [DOI] [PubMed] [Google Scholar]
- FRAKE RA, RICKETTS T, MENZIES FM, RUBINSZTEIN DC. Autophagy and neurodegeneration. J Clin Invest. 2015;125:65–74. doi: 10.1172/JCI73944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FU MM, NIRSCHL JJ, HOLZBAUR EL. LC3 binding to the scaffolding protein JIP1 regulates processive dynein-driven transport of autophagosomes. Dev Cell. 2014;29:577–90. doi: 10.1016/j.devcel.2014.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FUGER P, HEFENDEHL JK, VEERARAGHAVALU K, WENDELN AC, SCHLOSSER C, OBERMULLER U, WEGENAST-BRAUN BM, NEHER JJ, MARTUS P, KOHSAKA S, THUNEMANN M, FEIL R, SISODIA SS, SKODRAS A, JUCKER M. Microglia turnover with aging and in an Alzheimer’s model via long-term in vivo single-cell imaging. Nat Neurosci. 2017 doi: 10.1038/nn.4631. [DOI] [PubMed] [Google Scholar]
- GOO MS, SANCHO L, SLEPAK N, BOASSA D, DEERINCK TJ, ELLISMAN MH, BLOODGOOD BL, PATRICK GN. Activity-dependent trafficking of lysosomes in dendrites and dendritic spines. J Cell Biol. 2017;216:2499–2513. doi: 10.1083/jcb.201704068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GOWRISHANKAR S, YUAN P, WU Y, SCHRAG M, PARADISE S, GRUTZENDLER J, DE CAMILLI P, FERGUSON SM. Massive accumulation of luminal protease-deficient axonal lysosomes at Alzheimer’s disease amyloid plaques. Proc Natl Acad Sci U S A. 2015;112:E3699–708. doi: 10.1073/pnas.1510329112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HAMILTON AM, OH WC, VEGA-RAMIREZ H, STEIN IS, HELL JW, PATRICK GN, ZITO K. Activity-dependent growth of new dendritic spines is regulated by the proteasome. Neuron. 2012;74:1023–30. doi: 10.1016/j.neuron.2012.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HARA T, NAKAMURA K, MATSUI M, YAMAMOTO A, NAKAHARA Y, SUZUKI-MIGISHIMA R, YOKOYAMA M, MISHIMA K, SAITO I, OKANO H, MIZUSHIMA N. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature. 2006;441:885–9. doi: 10.1038/nature04724. [DOI] [PubMed] [Google Scholar]
- HARA T, TAKAMURA A, KISHI C, IEMURA S, NATSUME T, GUAN JL, MIZUSHIMA N. FIP200, a ULK-interacting protein, is required for autophagosome formation in mammalian cells. J Cell Biol. 2008;181:497–510. doi: 10.1083/jcb.200712064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HARRINGTON AW, GINTY DD. Long-distance retrograde neurotrophic factor signalling in neurons. Nat Rev Neurosci. 2013;14:177–87. doi: 10.1038/nrn3253. [DOI] [PubMed] [Google Scholar]
- HARRIS JJ, ATTWELL D. The energetics of CNS white matter. J Neurosci. 2012;32:356–71. doi: 10.1523/JNEUROSCI.3430-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HAUSSER M, RAMAN IM, OTIS T, SMITH SL, NELSON A, DU LAC S, LOEWENSTEIN Y, MAHON S, PENNARTZ C, COHEN I, YAROM Y. The beat goes on: spontaneous firing in mammalian neuronal microcircuits. J Neurosci. 2004;24:9215–9. doi: 10.1523/JNEUROSCI.3375-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HERING H, SHENG M. Dendritic spines: structure, dynamics and regulation. Nat Rev Neurosci. 2001;2:880–8. doi: 10.1038/35104061. [DOI] [PubMed] [Google Scholar]
- HERNANDEZ D, TORRES CA, SETLIK W, CEBRIAN C, MOSHAROV EV, TANG G, CHENG HC, KHOLODILOV N, YARYGINA O, BURKE RE, GERSHON M, SULZER D. Regulation of presynaptic neurotransmission by macroautophagy. Neuron. 2012;74:277–84. doi: 10.1016/j.neuron.2012.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HOLLENBECK PJ. Products of endocytosis and autophagy are retrieved from axons by regulated retrograde organelle transport. J Cell Biol. 1993;121:305–15. doi: 10.1083/jcb.121.2.305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ITAKURA E, KISHI-ITAKURA C, MIZUSHIMA N. The hairpin-type tail-anchored SNARE syntaxin 17 targets to autophagosomes for fusion with endosomes/lysosomes. Cell. 2012;151:1256–69. doi: 10.1016/j.cell.2012.11.001. [DOI] [PubMed] [Google Scholar]
- KABEYA Y, MIZUSHIMA N, UENO T, YAMAMOTO A, KIRISAKO T, NODA T, KOMINAMI E, OHSUMI Y, YOSHIMORI T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000;19:5720–8. doi: 10.1093/emboj/19.21.5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KANNAN M, BAYAM E, WAGNER C, RINALDI B, KRETZ PF, TILLY P, ROOS M, MCGILLEWIE L, BAR S, MINOCHA S, CHEVALIER C, PO C, SANGER MOUSE GENETICS, P. CHELLY J, MANDEL JL, BORGATTI R, PITON A, KINNEAR C, LOOS B, ADAMS DJ, HERAULT Y, COLLINS SC, FRIANT S, GODIN JD, YALCIN B. WD40-repeat 47, a microtubule-associated protein, is essential for brain development and autophagy. Proc Natl Acad Sci U S A. 2017;114:E9308–E9317. doi: 10.1073/pnas.1713625114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KIM M, SANDFORD E, GATICA D, QIU Y, LIU X, ZHENG Y, SCHULMAN BA, XU J, SEMPLE I, RO SH, KIM B, MAVIOGLU RN, TOLUN A, JIPA A, TAKATS S, KARPATI M, LI JZ, YAPICI Z, JUHASZ G, LEE JH, KLIONSKY DJ, BURMEISTER M. Mutation in ATG5 reduces autophagy and leads to ataxia with developmental delay. Elife. 2016;5 doi: 10.7554/eLife.12245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KLEELE T, MARINKOVIC P, WILLIAMS PR, STERN S, WEIGAND EE, ENGERER P, NAUMANN R, HARTMANN J, KARL RM, BRADKE F, BISHOP D, HERMS J, KONNERTH A, KERSCHENSTEINER M, GODINHO L, MISGELD T. An assay to image neuronal microtubule dynamics in mice. Nat Commun. 2014;5:4827. doi: 10.1038/ncomms5827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KOMATSU M, WAGURI S, CHIBA T, MURATA S, IWATA J, TANIDA I, UENO T, KOIKE M, UCHIYAMA Y, KOMINAMI E, TANAKA K. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature. 2006;441:880–4. doi: 10.1038/nature04723. [DOI] [PubMed] [Google Scholar]
- KOMATSU M, WANG QJ, HOLSTEIN GR, FRIEDRICH VL, JR, IWATA J, KOMINAMI E, CHAIT BT, TANAKA K, YUE Z. Essential role for autophagy protein Atg7 in the maintenance of axonal homeostasis and the prevention of axonal degeneration. Proc Natl Acad Sci U S A. 2007;104:14489–94. doi: 10.1073/pnas.0701311104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KONONENKO NL, CLASSEN GA, KUIJPERS M, PUCHKOV D, MARITZEN T, TEMPES A, MALIK AR, SKALECKA A, BERA S, JAWORSKI J, HAUCKE V. Retrograde transport of TrkB-containing autophagosomes via the adaptor AP-2 mediates neuronal complexity and prevents neurodegeneration. Nat Commun. 2017;8:14819. doi: 10.1038/ncomms14819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KUMA A, HATANO M, MATSUI M, YAMAMOTO A, NAKAYA H, YOSHIMORI T, OHSUMI Y, TOKUHISA T, MIZUSHIMA N. The role of autophagy during the early neonatal starvation period. Nature. 2004;432:1032–6. doi: 10.1038/nature03029. [DOI] [PubMed] [Google Scholar]
- LEDOUX MS, LORDEN JF. Abnormal spontaneous and harmaline-stimulated Purkinje cell activity in the awake genetically dystonic rat. Exp Brain Res. 2002;145:457–67. doi: 10.1007/s00221-002-1127-4. [DOI] [PubMed] [Google Scholar]
- LEE JH, YU WH, KUMAR A, LEE S, MOHAN PS, PETERHOFF CM, WOLFE DM, MARTINEZ-VICENTE M, MASSEY AC, SOVAK G, UCHIYAMA Y, WESTAWAY D, CUERVO AM, NIXON RA. Lysosomal proteolysis and autophagy require presenilin 1 and are disrupted by Alzheimer-related PS1 mutations. Cell. 2010;141:1146–58. doi: 10.1016/j.cell.2010.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LEE S, SATO Y, NIXON RA. Lysosomal proteolysis inhibition selectively disrupts axonal transport of degradative organelles and causes an Alzheimer’s-like axonal dystrophy. J Neurosci. 2011;31:7817–30. doi: 10.1523/JNEUROSCI.6412-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LEE Y, MORRISON BM, LI Y, LENGACHER S, FARAH MH, HOFFMAN PN, LIU Y, TSINGALIA A, JIN L, ZHANG PW, PELLERIN L, MAGISTRETTI PJ, ROTHSTEIN JD. Oligodendroglia metabolically support axons and contribute to neurodegeneration. Nature. 2012;487:443–8. doi: 10.1038/nature11314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LIANG CC, WANG C, PENG X, GAN B, GUAN JL. Neural-specific deletion of FIP200 leads to cerebellar degeneration caused by increased neuronal death and axon degeneration. J Biol Chem. 2010;285:3499–509. doi: 10.1074/jbc.M109.072389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LUSCHER C, MALENKA RC. NMDA receptor-dependent long-term potentiation and long-term depression (LTP/LTD) Cold Spring Harb Perspect Biol. 2012;4 doi: 10.1101/cshperspect.a005710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MADAY S. Mechanisms of neuronal homeostasis: Autophagy in the axon. Brain Res. 2016;1649:143–150. doi: 10.1016/j.brainres.2016.03.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MADAY S, HOLZBAUR EL. Autophagosome biogenesis in primary neurons follows an ordered and spatially regulated pathway. Dev Cell. 2014;30:71–85. doi: 10.1016/j.devcel.2014.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MADAY S, HOLZBAUR EL. Compartment-Specific Regulation of Autophagy in Primary Neurons. J Neurosci. 2016;36:5933–45. doi: 10.1523/JNEUROSCI.4401-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MADAY S, TWELVETREES AE, MOUGHAMIAN AJ, HOLZBAUR EL. Axonal transport: cargo-specific mechanisms of motility and regulation. Neuron. 2014;84:292–309. doi: 10.1016/j.neuron.2014.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MADAY S, WALLACE KE, HOLZBAUR EL. Autophagosomes initiate distally and mature during transport toward the cell soma in primary neurons. J Cell Biol. 2012;196:407–17. doi: 10.1083/jcb.201106120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MADILL M, MCDONAGH K, MA J, VAJDA A, MCLOUGHLIN P, O’BRIEN T, HARDIMAN O, SHEN S. Amyotrophic lateral sclerosis patient iPSC-derived astrocytes impair autophagy via non-cell autonomous mechanisms. Mol Brain. 2017;10:22. doi: 10.1186/s13041-017-0300-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MARTINEZ-VICENTE M. Neuronal Mitophagy in Neurodegenerative Diseases. Front Mol Neurosci. 2017;10:64. doi: 10.3389/fnmol.2017.00064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MELENTIJEVIC I, TOTH ML, ARNOLD ML, GUASP RJ, HARINATH G, NGUYEN KC, TAUB D, PARKER JA, NERI C, GABEL CV, HALL DH, DRISCOLL M. C. elegans neurons jettison protein aggregates and mitochondria under neurotoxic stress. Nature. 2017;542:367–371. doi: 10.1038/nature21362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MENZIES FM, FLEMING A, CARICASOLE A, BENTO CF, ANDREWS SP, ASHKENAZI A, FULLGRABE J, JACKSON A, JIMENEZ SANCHEZ M, KARABIYIK C, LICITRA F, LOPEZ RAMIREZ A, PAVEL M, PURI C, RENNA M, RICKETTS T, SCHLOTAWA L, VICINANZA M, WON H, ZHU Y, SKIDMORE J, RUBINSZTEIN DC. Autophagy and Neurodegeneration: Pathogenic Mechanisms and Therapeutic Opportunities. Neuron. 2017;93:1015–1034. doi: 10.1016/j.neuron.2017.01.022. [DOI] [PubMed] [Google Scholar]
- MILLECAMPS S, JULIEN JP. Axonal transport deficits and neurodegenerative diseases. Nat Rev Neurosci. 2013;14:161–76. doi: 10.1038/nrn3380. [DOI] [PubMed] [Google Scholar]
- MIZUSHIMA N, YAMAMOTO A, MATSUI M, YOSHIMORI T, OHSUMI Y. In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol Biol Cell. 2004;15:1101–11. doi: 10.1091/mbc.E03-09-0704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MIZUSHIMA N, YOSHIMORI T, OHSUMI Y. The role of Atg proteins in autophagosome formation. Annu Rev Cell Dev Biol. 2011;27:107–32. doi: 10.1146/annurev-cellbio-092910-154005. [DOI] [PubMed] [Google Scholar]
- MURDOCH JD, ROSTOSKY CM, GOWRISANKARAN S, ARORA AS, SOUKUP SF, VIDAL R, CAPECE V, FREYTAG S, FISCHER A, VERSTREKEN P, BONN S, RAIMUNDO N, MILOSEVIC I. Endophilin-A Deficiency Induces the Foxo3a-Fbxo32 Network in the Brain and Causes Dysregulation of Autophagy and the Ubiquitin-Proteasome System. Cell Rep. 2016;17:1071–1086. doi: 10.1016/j.celrep.2016.09.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NARENDRA D, TANAKA A, SUEN DF, YOULE RJ. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol. 2008;183:795–803. doi: 10.1083/jcb.200809125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NARENDRA DP, JIN SM, TANAKA A, SUEN DF, GAUTIER CA, SHEN J, COOKSON MR, YOULE RJ. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 2010;8:e1000298. doi: 10.1371/journal.pbio.1000298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NICHOLLS RE, ALARCON JM, MALLERET G, CARROLL RC, GRODY M, VRONSKAYA S, KANDEL ER. Transgenic mice lacking NMDAR-dependent LTD exhibit deficits in behavioral flexibility. Neuron. 2008;58:104–17. doi: 10.1016/j.neuron.2008.01.039. [DOI] [PubMed] [Google Scholar]
- NIKOLETOPOULOU V, SIDIROPOULOU K, KALLERGI E, DALEZIOS Y, TAVERNARAKIS N. Modulation of Autophagy by BDNF Underlies Synaptic Plasticity. Cell Metab. 2017;26:230–242 e5. doi: 10.1016/j.cmet.2017.06.005. [DOI] [PubMed] [Google Scholar]
- NIMCHINSKY EA, SABATINI BL, SVOBODA K. Structure and function of dendritic spines. Annu Rev Physiol. 2002;64:313–53. doi: 10.1146/annurev.physiol.64.081501.160008. [DOI] [PubMed] [Google Scholar]
- NISHIYAMA J, MIURA E, MIZUSHIMA N, WATANABE M, YUZAKI M. Aberrant membranes and double-membrane structures accumulate in the axons of Atg5-null Purkinje cells before neuronal death. Autophagy. 2007;3:591–6. doi: 10.4161/auto.4964. [DOI] [PubMed] [Google Scholar]
- NIXON RA, WEGIEL J, KUMAR A, YU WH, PETERHOFF C, CATALDO A, CUERVO AM. Extensive involvement of autophagy in Alzheimer disease: an immuno-electron microscopy study. J Neuropathol Exp Neurol. 2005;64:113–22. doi: 10.1093/jnen/64.2.113. [DOI] [PubMed] [Google Scholar]
- OKERLUND ND, SCHNEIDER K, LEAL-ORTIZ S, MONTENEGRO-VENEGAS C, KIM SA, GARNER LC, GUNDELFINGER ED, REIMER RJ, GARNER CC. Bassoon Controls Presynaptic Autophagy through Atg5. Neuron. 2017;93:897–913 e7. doi: 10.1016/j.neuron.2017.01.026. [DOI] [PubMed] [Google Scholar]
- RAMESH N, PANDEY UB. Autophagy Dysregulation in ALS: When Protein Aggregates Get Out of Hand. Front Mol Neurosci. 2017;10:263. doi: 10.3389/fnmol.2017.00263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RANGARAJU V, TOM DIECK S, SCHUMAN EM. Local translation in neuronal compartments: how local is local? EMBO Rep. 2017;18:693–711. doi: 10.15252/embr.201744045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RUDNICK ND, GRIFFEY CJ, GUARNIERI P, GERBINO V, WANG X, PIERSAINT JA, TAPIA JC, RICH MM, MANIATIS T. Distinct roles for motor neuron autophagy early and late in the SOD1G93A mouse model of ALS. Proc Natl Acad Sci U S A. 2017;114:E8294–E8303. doi: 10.1073/pnas.1704294114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SANDOW SL, HEYDON K, WEIBLE MW, 2ND, REYNOLDS AJ, BARTLETT SE, HENDRY IA. Signalling organelle for retrograde axonal transport of internalized neurotrophins from the nerve terminal. Immunol Cell Biol. 2000;78:430–5. doi: 10.1046/j.1440-1711.2000.00924.x. [DOI] [PubMed] [Google Scholar]
- SCHNEIDER JL, CUERVO AM. Autophagy and human disease: emerging themes. Curr Opin Genet Dev. 2014;26:16–23. doi: 10.1016/j.gde.2014.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SHEHATA M, MATSUMURA H, OKUBO-SUZUKI R, OHKAWA N, INOKUCHI K. Neuronal stimulation induces autophagy in hippocampal neurons that is involved in AMPA receptor degradation after chemical long-term depression. J Neurosci. 2012;32:10413–22. doi: 10.1523/JNEUROSCI.4533-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SHEN DN, ZHANG LH, WEI EQ, YANG Y. Autophagy in synaptic development, function, and pathology. Neurosci Bull. 2015;31:416–26. doi: 10.1007/s12264-015-1536-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SHEN W, GANETZKY B. Autophagy promotes synapse development in Drosophila. J Cell Biol. 2009;187:71–9. doi: 10.1083/jcb.200907109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SON JH, SHIM JH, KIM KH, HA JY, HAN JY. Neuronal autophagy and neurodegenerative diseases. Exp Mol Med. 2012;44:89–98. doi: 10.3858/emm.2012.44.2.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SOUKUP SF, KUENEN S, VANHAUWAERT R, MANETSBERGER J, HERNANDEZ-DIAZ S, SWERTS J, SCHOOVAERTS N, VILAIN S, GOUNKO NV, VINTS K, GEENS A, DE STROOPER B, VERSTREKEN P. A LRRK2-Dependent EndophilinA Phosphoswitch Is Critical for Macroautophagy at Presynaptic Terminals. Neuron. 2016;92:829–844. doi: 10.1016/j.neuron.2016.09.037. [DOI] [PubMed] [Google Scholar]
- SPALDING KL, BHARDWAJ RD, BUCHHOLZ BA, DRUID H, FRISEN J. Retrospective birth dating of cells in humans. Cell. 2005;122:133–43. doi: 10.1016/j.cell.2005.04.028. [DOI] [PubMed] [Google Scholar]
- STAVOE AK, HILL SE, HALL DH, COLON-RAMOS DA. KIF1A/UNC-104 Transports ATG-9 to Regulate Neurodevelopment and Autophagy at Synapses. Dev Cell. 2016;38:171–85. doi: 10.1016/j.devcel.2016.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- STOLZ A, ERNST A, DIKIC I. Cargo recognition and trafficking in selective autophagy. Nat Cell Biol. 2014;16:495–501. doi: 10.1038/ncb2979. [DOI] [PubMed] [Google Scholar]
- SUNG H, TANDARICH LC, NGUYEN K, HOLLENBECK PJ. Compartmentalized Regulation of Parkin-Mediated Mitochondrial Quality Control in the Drosophila Nervous System In Vivo. J Neurosci. 2016;36:7375–91. doi: 10.1523/JNEUROSCI.0633-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SUO D, PARK J, HARRINGTON AW, ZWEIFEL LS, MIHALAS S, DEPPMANN CD. Coronin-1 is a neurotrophin endosomal effector that is required for developmental competition for survival. Nat Neurosci. 2014;17:36–45. doi: 10.1038/nn.3593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TAKEUCHI T, DUSZKIEWICZ AJ, MORRIS RG. The synaptic plasticity and memory hypothesis: encoding, storage and persistence. Philos Trans R Soc Lond B Biol Sci. 2014;369:20130288. doi: 10.1098/rstb.2013.0288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TANG G, GUDSNUK K, KUO SH, COTRINA ML, ROSOKLIJA G, SOSUNOV A, SONDERS MS, KANTER E, CASTAGNA C, YAMAMOTO A, YUE Z, ARANCIO O, PETERSON BS, CHAMPAGNE F, DWORK AJ, GOLDMAN J, SULZER D. Loss of mTOR-dependent macroautophagy causes autistic-like synaptic pruning deficits. Neuron. 2014;83:1131–43. doi: 10.1016/j.neuron.2014.07.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VANHAUWAERT R, KUENEN S, MASIUS R, BADEMOSI A, MANETSBERGER J, SCHOOVAERTS N, BOUNTI L, GONTCHARENKO S, SWERTS J, VILAIN S, PICILLO M, BARONE P, MUNSHI ST, DE VRIJ FM, KUSHNER SA, GOUNKO NV, MANDEMAKERS W, BONIFATI V, MEUNIER FA, SOUKUP SF, VERSTREKEN P. The SAC1 domain in synaptojanin is required for autophagosome maturation at presynaptic terminals. EMBO J. 2017;36:1392–1411. doi: 10.15252/embj.201695773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VIJAYAN V, VERSTREKEN P. Autophagy in the presynaptic compartment in health and disease. J Cell Biol. 2017;216:1895–1906. doi: 10.1083/jcb.201611113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VITUREIRA N, GODA Y. Cell biology in neuroscience: the interplay between Hebbian and homeostatic synaptic plasticity. J Cell Biol. 2013;203:175–86. doi: 10.1083/jcb.201306030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WANG T, MARTIN S, PAPADOPULOS A, HARPER CB, MAVLYUTOV TA, NIRANJAN D, GLASS NR, COOPER-WHITE JJ, SIBARITA JB, CHOQUET D, DAVLETOV B, MEUNIER FA. Control of autophagosome axonal retrograde flux by presynaptic activity unveiled using botulinum neurotoxin type a. J Neurosci. 2015;35:6179–94. doi: 10.1523/JNEUROSCI.3757-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WANG YC, LAUWERS E, VERSTREKEN P. Presynaptic protein homeostasis and neuronal function. Curr Opin Genet Dev. 2017;44:38–46. doi: 10.1016/j.gde.2017.01.015. [DOI] [PubMed] [Google Scholar]
- WEIDBERG H, SHVETS E, ELAZAR Z. Biogenesis and cargo selectivity of autophagosomes. Annu Rev Biochem. 2011;80:125–56. doi: 10.1146/annurev-biochem-052709-094552. [DOI] [PubMed] [Google Scholar]
- WU CL, CHEN CH, HWANG CS, CHEN SD, HWANG WC, YANG DI. Roles of p62 in BDNF-dependent autophagy suppression and neuroprotection against mitochondrial dysfunction in rat cortical neurons. J Neurochem. 2017;140:845–861. doi: 10.1111/jnc.13937. [DOI] [PubMed] [Google Scholar]
- XIE Y, ZHOU B, LIN MY, WANG S, FOUST KD, SHENG ZH. Endolysosomal Deficits Augment Mitochondria Pathology in Spinal Motor Neurons of Asymptomatic fALS Mice. Neuron. 2015;87:355–70. doi: 10.1016/j.neuron.2015.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- XU Z, YANG L, XU S, ZHANG Z, CAO Y. The receptor proteins: pivotal roles in selective autophagy. Acta Biochim Biophys Sin (Shanghai) 2015;47:571–80. doi: 10.1093/abbs/gmv055. [DOI] [PubMed] [Google Scholar]
- YAMAGUCHI J, SUZUKI C, NANAO T, KAKUTA S, OZAWA K, TANIDA I, SAITOH T, SUNABORI T, KOMATSU M, TANAKA K, AOKI S, SAKIMURA K, UCHIYAMA Y. Atg9a deficiency causes axon-specific lesions including neuronal circuit dysgenesis. Autophagy. 2017:0. doi: 10.1080/15548627.2017.1314897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YAMAMOTO A, YUE Z. Autophagy and its normal and pathogenic states in the brain. Annu Rev Neurosci. 2014;37:55–78. doi: 10.1146/annurev-neuro-071013-014149. [DOI] [PubMed] [Google Scholar]
- YANG Z, KLIONSKY DJ. Eaten alive: a history of macroautophagy. Nat Cell Biol. 2010;12:814–22. doi: 10.1038/ncb0910-814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YOSHII SR, KUMA A, AKASHI T, HARA T, YAMAMOTO A, KURIKAWA Y, ITAKURA E, TSUKAMOTO S, SHITARA H, EISHI Y, MIZUSHIMA N. Systemic Analysis of Atg5-Null Mice Rescued from Neonatal Lethality by Transgenic ATG5 Expression in Neurons. Dev Cell. 2016;39:116–130. doi: 10.1016/j.devcel.2016.09.001. [DOI] [PubMed] [Google Scholar]
- YUE Z. Regulation of neuronal autophagy in axon: implication of autophagy in axonal function and dysfunction/degeneration. Autophagy. 2007;3:139–41. doi: 10.4161/auto.3602. [DOI] [PubMed] [Google Scholar]
- ZAFFAGNINI G, MARTENS S. Mechanisms of Selective Autophagy. J Mol Biol. 2016;428:1714–24. doi: 10.1016/j.jmb.2016.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZHAO YG, SUN L, MIAO G, JI C, ZHAO H, SUN H, MIAO L, YOSHII SR, MIZUSHIMA N, WANG X, ZHANG H. The autophagy gene Wdr45/Wipi4 regulates learning and memory function and axonal homeostasis. Autophagy. 2015;11:881–90. doi: 10.1080/15548627.2015.1047127. [DOI] [PMC free article] [PubMed] [Google Scholar]