

Abstract
Plasma membrane Ca2+ ATPases (PMCAs) are a major system for calcium extrusion from all cells. Different PMCA isoforms and splice variants are involved in the precise temporal and spatial handling of Ca2+ signals and the re-establishment of resting Ca2+ levels in the nervous system. Lack or inappropriate expression of specific PMCAs leads to characteristic neuronal phenotypes, which may be reciprocally exacerbated by genetic predisposition through alleles in other genes that modify PMCA interactions, regulation, and function. PMCA dysfunction is often poorly compensated in neurons and may lead to changes in synaptic transmission, altered excitability and, with long-term calcium overload, eventual cell death. Decrease and functional decline of PMCAs are hallmarks of neurodegeneration during aging, and mutations in specific PMCAs are responsible for neuronal dysfunction and accelerated neurodegeneration in many sensory and cognitive diseases.
Keywords: Calcium homeostasis, cerebellar ataxia, excitotoxicity, neurodegenerative disease, plasma membrane calcium ATPase
Graphical Abstract
Introduction
Communication in the nervous system is dependent on the generation, propagation, and decoding of signals. Signal transmission at pre- and post-synaptic sites generally involves calcium in its ionized form (Ca2+) as a universal and versatile signaling agent. Ca2+ is crucial not only for neuronal development, differentiation, synaptic strengthening and disassembly, but also plays a central role in neuronal cell death [4]. Consequently, the intracellular Ca2+ concentration [Ca2+]i must be precisely controlled in space and time to enable and sustain proper neuronal function [4, 72]. In turn, aberrant Ca2+ regulation results in neuronal dysfunction and is an early event in the development and progression of major neurodegenerative disorders including Alzheimer’s disease and multiple sclerosis [3, 60, 69].
Because Ca2+ cannot be synthetized or destroyed, changes in its cellular concentration require specific uptake and extrusion mechanisms in the cell membrane. While Ca2+ influx is mediated by a large number of voltage-, ligand- or mechanically-gated channels and transporters, Ca2+ expulsion is dependent on Ca2+ pumps and Na+/Ca2+ (or Na+/Ca2+-K+) exchangers [5, 50]. Among these, the plasma membrane Ca2+ pumps (also known as plasma membrane Ca2+ ATPases or PMCAs) are the major high-affinity Ca2+ extrusion system capable of exporting Ca2+ ions out of the cell against a large concentration gradient. These pumps are thus important gatekeepers of overall Ca2+ homeostasis and are also involved in spatially and temporally defined Ca2+ signaling events [14, 98].
In this review we will start with a brief overview of the PMCA isoforms, their basic functional and regulatory properties, and their tissue- and cell-specific expression in the nervous system. In the following sections, the emerging role of the PMCAs in neuropathology will be discussed with an emphasis on recent findings combining in vitro studies with animal model-derived data and genetic studies obtained in human populations. The sections are divided according to major neuronal functions, systems or diseases, however, we note that these distinctions are somewhat arbitrary as the PMCAs affect Ca2+ regulation in all neuronal tissues, and PMCA dysfunction may be a common trait in all of these conditions.
Overview of the PMCAs in the nervous system
PMCAs are members of the large family of ion-transport ATPases responsible for vectorial movement of ions across biological membranes [79]. They are P-type ATPases characterized by the formation of a phosphorylated aspartate intermediate during the ion-transport reaction cycle [81]. PMCAs are membrane proteins of about 1200 amino acids with calculated molecular masses of 125–140 kDa. Based on biochemical and structural studies the bulk of their mass, including their N- and C-terminal tail, faces the cytosol. PMCAs have ten membrane-spanning segments, with major cytosolic loops between membrane-spanning segments 2–3 and 4–5 (Figure 1). Based on their homology to the SERCA (sarco/endoplasmic reticulum Ca2+ ATPase) pumps, for which detailed structural information in different functional states is available [77, 102], the PMCAs are thought to undergo similar conformational changes and operate by a similar mechanism during the ion transport cycle. Structural and mechanistic aspects of the PMCAs have been covered in several recent reviews [13, 14, 78] and will not be discussed further in this contribution.
Figure 1. Scheme of the PMCA and major alternative splice pattern of human ATP2B genes.

Top: Schematic representation of the PMCA with major domains indicated M, membrane; A, actuator, P, phosphorylation; N, nucleotide-binding; and R, regulatory domain. The ten membrane-spanning regions are numbered, the N- and C-terminal ends are indicated, and the conserved aspartate (Asp) residue phosphorylated during the reaction cycle, the catalytic ATP-binding site (ATP), and the CaM-binding region (CBD) are labeled. The direction of Ca2+ transport is indicated by an arrow. Arrows also mark the positions where alternative splicing leads to isoform diversity at sites A and C. Bottom: Splicing options of human PMCA genes ATP2B1-ATP2B4. The exon structure in the region of alternative splicing at sites A and C is shown for each ATB2B (PMCA) gene. Constitutively spliced exons are represented by gray boxes, alternatively spliced exons are colored and their size is shown in nucleotides. Splice options are indicated by connecting lines, and the resulting splice variants are labeled by their lowercase symbol. Note that only splice option “x” has been found in PMCA1 at site A, and that variant “e” in PMCA3 and PMCA4 results from a read-through of the last alternatively spliced exon into the following intron (shown as thin white box).
Four genes encode mammalian PMCAs, giving rise to PMCA isoforms 1–4. Each gene contains alternatively spliced exons whose differential usage results in PMCA variants differing in two specific regions of the protein: the first intracellular loop (splice site A) and the C-terminal tail (splice site C). Over 20 different PMCA splice variants have been identified at the transcript and/or protein level [99]. A schematic overview of the PMCA structure and the major human splice variants is presented in Figure 1. Alternative splicing at site A results in the inclusion or exclusion of a short peptide sequence in the first cytosolic loop, which may affect the membrane targeting of the PMCA isoform [1, 27, 53]. Alternative splicing at site C affects the length and sequence of the C-tail, which thus varies significantly among different PMCA isoforms. Importantly, the C-tail contains the binding site(s) for calmodulin (CaM), the major regulatory protein affecting PMCA activity [99]. Some C-terminal alternative splice variants also differ in their ability to interact with PDZ (PSD95/Dlg/ZO-1) domain-containing signaling and scaffolding proteins, affecting the type of multi-protein complex to which these PMCA variants can contribute [97].
As essential regulators of cellular Ca2+ homeostasis, PMCAs are found in all animal cells. PMCA expression in the mammalian brain has been studied in multiple species, showing that all four PMCA isoforms and virtually all known splice variants are expressed in the nervous system, albeit at highly variable levels and with distinct tissue- and cell-specificity [11, 16, 94, 95, 109]. PMCA1, often referred to as “housekeeping” PMCA isoform, is expressed early in (mouse) embryonic development [111] and is present in all tissues, including the nervous system, throughout adulthood. PMCA4, which is also expressed in most tissues, shows differential expression patterns in different regions of the brain. PMCA2 and PMCA3 are predominantly found in excitable tissues and highly enriched in specific cell types such as cerebellar Purkinje cells. In the adult rat brain, PMCA1 is enriched in the cortex and hippocampus and present at lower levels in the cerebellum. PMCA2 and PMCA3 are abundant in the cerebellum and forebrain, and PMCA4 is found at higher levels in the superficial layers of the cortex than in the hippocampus and cerebellum [16].
Using isoform- and splice variant-specific antibodies in high-resolution immunohistochemistry and immunogold electron microsopy, several PMCA isoform variants have been localized more precisely to specific neuron types and to subcellular areas such as dendrites, spines and pre- or post-synaptic membranes within single cells. For example, PMCA1x/a – the major PMCA1 splice variant expressed in the mature brain – is localized in a punctate pattern in the soma and dendrite membranes of pyramidal neurons in the somatosensory cortex and hippocampus [59]. In the rat cerebellar cortex PMCA2 and PMCA3 are prominent in the molecular layer, but whereas PMCA2 is concentrated in postsynaptic spine membranes of Purkinje cells, PMCA3 is present in somata of basket/stellate cells and co-localized with VGLUT1 in what appear to be presynaptic axon terminals of parallel fibers [18]. PMCA2w (likely PMCA2w/b) has been shown to concentrate at the postsynaptic density in hippocampal spines [17]; in contrast, PMCA2a was specifically localized in parvalbumin-positive GABA-ergic presynaptic terminals of inhibitory neurons in areas including the cortex and hippocampus [19].
The distinct expression and cellular localization of PMCA isoforms in different regions and cell types of the brain suggests that these pumps perform specific functions uniquely adapted to the physiological requirements of the particular neurons. PMCA isoforms and splice variants vary greatly in their basal calcium pumping activity and in the kinetics of activation by CaM as well as their regulation by phosphorylation, phospholipid interaction, and proteolysis [14, 78, 97]. Depending on the cellular context and the dynamic interactions between cells, different PMCAs may be needed to control bulk cytosolic [Ca2+] and to regulate spatially and temporally limited Ca2+ signals. Considering the importance of precise Ca2+ regulation in the proper function of the nervous system, it is not surprising that physiological, genetic, and biochemical studies have suggested an involvement of the PMCAs in neuronal dysfunction including several types of neurodegenerative disease [15].
The role of PMCAs in excitotoxicity
Excitotoxicity, the excessive activation of receptors for the neurotransmitter glutamate, damages neurons as a result of excessive Ca2+ influx. Most neurodegenerative disorders have an excitotoxic component and in some, like stroke and epilepsy, glutamate plays a major role. PMCAs are important in this process because they help neurons cope with the increased [Ca2+]i and because they are vulnerable to Ca2+-mediated toxicity. Changes in PMCA expression affect sensitivity to glutamate in several excitotoxicity models. Increased expression of message for PMCA2b and c was found in cerebellar granule cells that survived a glutamate insult, suggesting that increased expression enhanced survival [106]. Kainate-induced seizures decreased expression of PMCA isoforms 1 and 2 in hippocampal principal neurons, possibly contributing to the reduced survival of these cells [42]. The importance of PMCA2 in the protection against (excitotoxic) cell death was also demonstrated in spinal cord neurons: siRNA-mediated reduction of PMCA2 by about 50% caused significant cell death within 48 to 72 h in vitro, whereas administration of an AMPA receptor antagonist rescued PMCA2 expression and promoted cell survival in a mouse model of experimental autoimmune encephalopathy [64]. Acute excitotoxic increases in Ca2+ activate proteases that alter PMCA structure and function. Activation of caspases following excitotoxic insult leads to PMCA2 degradation, Ca2+ overload and death [91]. The loss of PMCA2 accelerated the cell death process to “seal the cell’s fate” [91]. Thus, caspase-mediated degradation of PMCAs may confer Ca2+ sensitivity to cell death processes not initially triggered by increases in [Ca2+]i, linking apoptotic to necrotic cell death pathways. In contrast, caspase 3 increases the activity of PMCA4b, indicating that functional changes are specific to the PMCA isoform [80]. Glutamate induced increases in [Ca2+]i activate calpain with a subsequent decrease in PMCA-mediated Ca2+ efflux rate [83]. This might seem at odds with studies showing that calpain constitutively activates PMCA4 by cleaving the autoinhibitory domain on the C-terminal of the pump [56]. However, an initial proteolytic increase in PMCA function followed by degradation, either by continued digestion or increased susceptibility to other proteases, is a plausible sequence of events. Ca2+ influx via NMDA receptors is more detrimental to PMCA function than comparable [Ca2+]i increases mediated by voltage-gated Ca2+ channels [38]. The source specificity of glutamate neurotoxicity is thought to result from the selective localization of Ca2+-sensitive signaling cascades near the mouths of certain NMDA receptor subtypes [88]. Of note, reciprocal immunoprecipitation experiments showed that PMCA2b associates via the postsynaptic density protein PSD95 with the NR1 and NR2a subunits of the NMDA receptor in the rat brain [43]. The close proximity of PMCA2 and NMDA receptors allows tight spatio-temporal control of postsynaptic Ca2+ signaling and its modulation. Cross-talk between the calcium extrusion (PMCA) and calcium influx (glutamate receptor) systems is important for physiological synapse plasticity; for example, PMCA-mediated Ca2+ extrusion is reduced in an activity-dependent manner in rat hippocampal spines via Ca2+-mediated inactivation of PMCA function [89]. While important for normal synapse function and plasticity, the intimate relationship between PMCAs and glutamate receptors at excitatory synapses carries significant risk for a catastrophic outcome if one or both of the systems are faulty. Hence even relatively minor disturbances of PMCA function or prolonged stimulation of Ca2+ influx may result in excitotoxic insults leading to neuronal death. In general, increased PMCA expression or activation attenuates glutamate toxicity. However, cellular changes in PMCA function in response to an excitotoxic challenge are unique to the specific insult and the particular PMCA isoforms involved.
Cerebral ischemia evokes an excitotoxic response that is further compounded by metabolic changes. PMCAs are exquisitely sensitive to changes in cellular ATP and extracellular pH. PMCAs in cerebellar and sensory neurons preferentially consume ATP generated through glycolysis [48, 55]. This reliance on anaerobic metabolism confers upon PMCAs a resistance to anoxia relative to other ATP-dependent ion pumps [52]. During cerebral ischemia PMCA1 immunoreactivity decreases [66] and following an initial recovery in function a subsequent decline in PMCA expression contributes to the delayed neuronal death of those neurons that survive the initial insult [75]. Overall, the reliance on ATP generated by glycolysis underlies an acute reduction in PMCA mediated Ca2+ extrusion during hypoxia/hypoglycemia followed by changes in PMCA expression that contribute to delayed neurotoxicity. Exposure to a brief non-lethal period of ischemia generates tolerance to a subsequent lethal ischemic insult. An increase in PMCA activity following preconditioning contributes in part to this protection [76]. Perhaps pharmacological treatments that increase PMCA expression might form the basis for neuroprotective strategies [29].
The PMCAs have been studied in animal models of epilepsy both after acute induction of seizure activity and in chronic epilepsy. A reduction in PMCA expression in the hippocampus within 1–2 days after seizure induction likely reflects cell loss in the areas affected by acute excitotoxicity. In the chronic stages of epilepsy long-term changes in calcium regulatory mechanisms are observed. Basal [Ca2+]i levels were increased in hippocampal CA1 neurons, and the recovery time to baseline after an induced Ca2+ spike was longer than in control neurons [86]. In agreement with an altered Ca2+ homeostasis in surviving neurons in the epileptic brain, Bravo-Martinez et al. [12] showed that PMCA1 transcripts were increased whereas PMCA3 expression was decreased one month after epileptogenic stimulation. This altered expression pattern of the PMCA isoforms likely contributes to the different excitable properties of the epileptic neurons.
The role of PMCAs in altered sensory function
PMCA function is critically important to hearing, vision and nociception. The prominent role of PMCAs in sensory function is exemplified by defects in the ATP2B2 gene encoding PMCA2 that produce dramatic defects in hearing and vision due to the lack of redundancy for the highly specialized roles of this PMCA isoform in cells of the inner ear and retina [47, 115]. A prominent role for PMCAs in primary sensory neurons of the dorsal root ganglion (DRG) is revealed by marked changes in pump function and expression following nerve injury [36].
PMCA2 is localized to the stereocilia of cochlea and vestibular hair cells [26, 53]. The pump transports Ca2+ to the endolymph following entry via Ca2+ permeable mechanotransducer (MT) channels activated by sound-, acceleration- or gravity-induced deflection of the stereocilia [47]. Elevated [Ca2+]i within the stereocilia desensitizes the MT channels and is responsible for the adaptation process [8]. Maintenance of endolymph Ca2+ levels is critical for regulating Ca2+ binding proteins in the tip-links that couple hair cells to each other and enable bending of the stereocilia to gate MT channels [41]. Depletion of endolymph [Ca2+] leads to degeneration of the tip-links, consistent with the hearing loss and impaired balance observed in PMCA2 knockout mice [63]. PMCA isoform 2 splice variant w/a is the only PMCA isoform expressed in the stereocilia of hair cells [49]. This lack of redundancy makes hearing and balance highly susceptible to mutations in PMCA2 that reduce Ca2+ transport [39]. Mutations that impair catalytic activity [10, 25] or reduce delivery of functional pump to the stereocilia [96, 101] impair hearing and balance. A reduced rate of Ca2+ clearance from the stereocilia would be expected to prolong adaptation and reduce endolymph [Ca2+] altering mechanotransduction [67, 107]. PMCA2 mutations that produce a functional null display haploinsufficiency [70]. Interestingly, heterozygotes expressing a Gly283 to Ser mutation exhibit approximately 30% of wild type PMCA2 function [82] and display hearing and balance comparable to control animals. This may explain why PMCA2 mutations without obvious phenotype when expressed alone, potentiate the deafness phenotype of co-existing mutations of other hearing related genes and sensitize individuals to noise-induced hearing loss [62, 90]. Thus, PMCA hypomorphs impair normal hearing and balance and predispose to inner ear toxicity induced by genetic and sensory insults. We speculate that PMCA mutations in general may potentiate the severity of neurodegenerative diseases mediated by Ca2+ overload.
PMCAs play a prominent role in synaptic transmission within the retina. They are the dominant mechanism for removal of Ca2+ from the presynaptic terminals of bipolar cell ribbon synapses [117]. PMCAs regulate intraterminal [Ca2+]i at rest and following stimulation, modulating luminance coding and adaptation to background illumination [105]. PMCA2 is expressed both presynaptically in rod terminals and postsynaptically in bipolar cells [30]. Rod spherules employ high affinity PMCAs to maintain low [Ca2+]i in darkness, which increases their sensitivity and signal-to-noise ratio [58]. The sensitivity of transmission at the rod output synapse is reduced by approximately 50% in deafwaddler mice [30], consistent with a presynaptic site of action. Thus, PMCAs serve as key modulators of scotopic visual signaling.
Increased PMCA activity contributes to the increased excitability of sensory neurons in response to injury. Bradykinin and ATP, mediators of local inflammation, activate PMCA4 in DRG neurons via protein kinase C-mediated phosphorylation [104]. The increased Ca2+ clearance rate decreases the activation of Ca2+ activated K+ channels to reduce the amplitude of the action potential after hyperpolarization. Similarly, nerve injury alters Ca2+ signaling in primary sensory neurons by increasing PMCA function in axotomized mice [44]. This increase in Ca2+ clearance rate results from an increase in the extracellular matrix protein thrombospondin-4 which activates protein kinase C [51]. Short oligosaccharides derived from another element of the extracellular matrix, hyaluronic acid, are also produced by injury. Short oligosaccharides were found to inhibit a CD44-activated kinase cascade in sensory neurons, accelerating PMCA4-mediated Ca2+ clearance with a subsequent decrease in the activity of Ca2+-activated K+ channels [46]. Thus, degradation of the extracellular matrix provides an indirect mechanism via PMCA stimulation to increase the excitability of sensory neurons following injury. Brief intense increases in [Ca2+]i, such as those evoked by injury, stimulate PMCA function priming the pump for the next Ca2+ challenge [84], and possibly protecting the cell from accumulating toxic levels of Ca2+. This Ca2+ memory phenomenon may result from a slow dissociation of calmodulin from the PMCA [23, 24]. Overall, the acute response to injury appears to be an increase in PMCA-mediated Ca2+ clearance that increases excitability and improves tolerance to potentially toxic Ca2+ loads. However, the sustained response to injury is less clear. PMCA2 gene expression is down regulated by more the 50% following contusive spinal cord injury [100]. Axotomy decreases expression of PMCA isoforms 1–3 in large DRG neurons but increases isoform 4 in small sensory neurons [74]. DRG neurons of different size convey information from different sensory modalities, suggesting that changes in PMCA expression may contribute to injury–induced changes in sensory perception. Finally, PMCAs may affect sensory perception in a sex-specific manner: in a recent study, the 50% reduction of PMCA2 in the dorsal horn neurons of Atp2b2+/− heterozygote mice was shown to affect the mechanical and heat-induced pain sensitivity of female, but not male, mice in a modality-specific manner [61]. Remarkably, this was accompanied by significant changes in the expression of several glutamate and GABA receptor subunits and astroglial glutamate transporters in the dorsal horn of the female (but not the male) mice, although PMCA2 was similarly reduced in male and female Atp2b2 heterozygotes. How the reduction in PMCA2 and consequent change of Ca2+ signaling affects the expression of the excitatory toolkit in a sex-specific manner is unknown. However, PMCA2 is an integral component of signaling complexes that include glutamate receptors, and the composition, abundance and function of these complexes may be regulated in an estrogen-dependent manner resulting in sex-specific differences in nociception.
PMCAs in cerebellar function: Synaptic function and role in ataxias
PMCA isoforms 2 and 3 are abundant in cerebellum. Their heterogeneous distribution underlies isoform-specific roles in synaptic transmission that when disturbed produce motor phenotypes. PMCA2 is highly expressed in Purkinje neuron soma, dendrites and spines and PMCA3 highly expressed in parallel fiber, but not climbing fiber, terminals [18]. PMCA2 is also expressed in parallel fiber terminals and both the 2 and 3 isoforms are expressed in the basket and stellate cells of the molecular layer [16, 31].
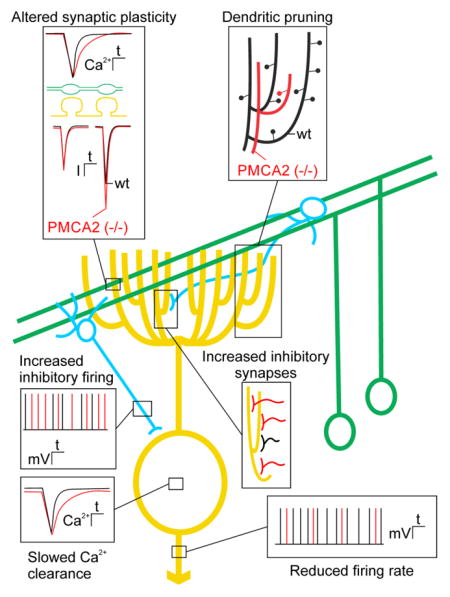
PMCA2 knockout mice exhibit overt cerebellar ataxia resulting from impaired Purkinje neuron Ca2+ homeostasis, as well as altered excitatory and inhibitory input to Purkinje neurons (Figure 2). In these mice, Purkinje neuron Ca2+ recovery kinetics are slowed and basal [Ca2+]i is elevated, resulting in a reduced firing rate due to increased activity of Ca2+-activated K+ channels [32] (Figure 2, bottom insets). Inhibition of PMCA2 results in pruning of the elaborate Purkinje neuron dendritic tree, suggesting that PMCA2-mediated [Ca2+]i homeostasis is required for maintenance of dendritic structure [92] (Figure 2, upper right). This dramatic simplification of Purkinje neuron structure alters dendritic integration [33]. The parallel fiber to Purkinje neuron synapse displays marked paired-pulse facilitation, a form of short-term synaptic plasticity that results from enhanced neurotransmitter release due to the accumulation of residual Ca2+ in the presynaptic terminal [2]. PMCA2 plays a major role in regulating resting [Ca2+]i and in returning presynaptic [Ca2+]i to basal levels following weak stimulation of parallel fibers [87]. Thus, in PMCA2 knockout animals the kinetics of Ca2+ removal from parallel fiber nerve terminals is slowed resulting in enhanced paired-pulse facilitation [33] (Figure 2, upper left). Altered short-term plasticity likely contributes to the motor impairment in these animals. Finally, PMCA2−/− mice exhibit increased synaptic inhibition onto Purkinje neurons, likely resulting from an adaptive response that includes an increased number of stellate and basket cells and increased firing rate of these molecular layer interneurons [34] (Figure 2, middle insets). The loss of the “fast” PMCA2a splice variant expressed in basket cell terminals may also contribute to enhanced GABA release onto Purkinje neurons [19]. Thus, several factors contribute to changes in Purkinje neuron spike timing that are responsible for ataxia in PMCA2−/− mice. These include direct effects on Purkinje neuron function and structure as well as altered plasticity of the parallel fiber input and increased inhibitory input.
Figure 2. Impaired Purkinje neuron function in PMCA2 knockout mice highlights the role of PMCAs in synaptic transmission.

Scheme shows major neuronal connections in a simplified cerebellar circuit showing Purkinje neuron (yellow), parallel fibers (green) and molecular layer interneurons (blue). Insets show pathophysiological changes in PMCA2−/− mice (red) overlaid on wild type responses (wt; black). Loss of PMCA2 alters short-term synaptic plasticity at parallel fiber Purkinje neuron synapses. Slowed Ca2+ clearance from presynaptic terminals results in enhanced paired-pulse facilitation of excitatory postsynaptic currents (inset upper left). Loss of PMCA2 results in dendritic pruning. Dendritic purning produces a dramatic simplification of the Purkinje neuron dentritic tree (inset upper right). PMCA2−/− animals exhibit maladaptive changes in inhibitory basket and stellate cells (blue). There are increases in the number of inhibitory synaptic inputs and the rate of inhibitory firing (insets in middle right and left). Ca2+ transient recovery in the Purkinje neuron cell body is prolonged and spontaneous action potential frequency is slowed in PMCA2−/− mice (insets on lower left and lower right).
Subtle changes in [Ca2+]i regulation observed in PMCA2 heterozygous mice also result in impaired function and reduced survival of Purkinje neurons. Purkinje neurons from PMCA2+/− mice exhibit slowed Ca2+ recovery kinetics and reduced frequency of action potential firing, disrupting the accuracy of cerebellar processing and motor coordination [35]. A number of adaptive changes contribute to altered cerebellar function in PMCA2+/− mice. The expression of AMPA receptors and metabotropic glutamate receptors is decreased and Ca2+ influx via voltage-gated Ca2+ channels increased possibly underlying an age-dependent loss of Purkinje neurons [37]. In contrast, in mice homozygous for the PMCA2 point mutant wriggle mouse sagami, Purkinje neurons also have slow Ca2+ recovery kinetics, but exhibit reduced Ca2+ influx presumably due to down-regulation of voltage-gated Ca2+ channels [103]. Thus, the nature of the compensatory/consequential changes that contribute to loss of motor control appear unique to the specific PMCA2 defect.
Defects in PMCA3 are also causally linked to cerebellar ataxia as first demonstrated in an X-exome sequencing study in a family suffering from X-linked congenital cerebellar ataxia [116]. In affected individuals the ATP2B3 gene (located on the human X-chromosome) carries a point mutation resulting in a G1107D amino acid change in the region encoding the calmodulin-binding domain of the PMCA3 [116]. This mutation impairs pump activation and autoinhibition reducing Ca2+ clearance rate [20]. More recently, a different PMCA3 missense mutation (R482H) was identified in another patient afflicted by cerebellar ataxia. This mutation impairs Ca2+ clearance rate and elevates basal [Ca2+]i but, is innocuous alone. However, when combined with missense mutations in the 1α subunit of laminin it results in developmental delay, generalized hypotonia and cerebellar ataxia [21]. This appears to be another case of PMCA mutations acting as genetic modifiers, similar to PMCA2 mutants contributing to hearing loss discussed above. Even PMCA mutations that do not result in an overt functional deficit when analyzed in vitro may contribute to ataxia, as illustrated by the R35C missense mutation in PMCA3 that co-segregates with the X-linked tremor-ataxia phenotype in the shaker rat [40]. PMCAs are integrated in signaling complexes in vivo, and even subtle differences in their protein interactions or membrane targeting may suffice to result in changes in Ca2+ regulation with pronounced physiological consequences. The R35C mutant of PMCA3 may exemplify this concept although proof of its causative involvement in the shaker rat phenotype is still elusive.
The specialized and non-redundant roles for PMCA gene products in cerebellum and the dramatic phenotypes that result from impaired cerebellar synaptic transmission highlight the diverse roles played by PMCAs in the CNS. Ca2+ pumps regulate synaptic plasticity, dendritic structure, and cellular excitability. In the absence of the fine control of [Ca2+]i provided by PMCAs compensatory changes in inhibitory neurons, expression of neurotransmitter receptors and Ca2+ channel function are important consequences that contribute to impaired motor function.
PMCAs in schizophrenia and autism spectrum disorder
Altered and aberrant communication between neurons is a hallmark of complex neurological diseases such as schizophrenia. Because of the essential role of Ca2+ in synaptic plasticity and signal transmission it is not surprising that the PMCA is among the proteins showing highly significant differences in abundance in postmortem brain tissue (from the anterior temporal lobe) in normal vs schizophrenic patients [68]. Unexpectedly, however, the affected isoform is PMCA4, and it is upregulated in the schizophrenic brain compared to the normal controls. Unfortunately, information on the specific role of PMCA4 in neuronal function and survival is limited, hampering interpretation of these data. Since the above proteomic study was performed on whole tissue, it is also likely that cells other than neurons (e.g. glia) contributed to the observed protein changes. PMCA2 may also play a role in schizophrenia: in a pharmacogenomics study, single nucleotide polymorphism (SNP) markers in the ATP2B2 (PMCA2) gene were highly predictive of treatment response to the schizophrenia drug risperidone [54]. In mice, PMCA2 gene expression in the prefrontal cortex is altered with risperidone exposure [54]. However, in the absence of functional data on PMCA2-mediated Ca2+ regulation in the schizophrenic vs normal brain the above data remain strictly correlative.
Aberrant neuronal calcium homeostasis is also implicated in autism-spectrum disorders. These complex disorders show a high degree of heritability, with multiple genetic loci contributing to disease susceptibility. Among these, the ATP2B2 gene showed linkage with autism susceptibility in a family-based association study [22]. Association of specific ATP2B2 alleles with autism was further reported in studies on Italian and Chinese populations [85, 108]. The functional consequences of these alleles on the PMCA2 are not yet known. However, because most of the allele-specific single-nucleotide polymorphisms reside in introns of the ATP2B2 gene, they most likely affect PMCA2 mRNA transcription or splicing, resulting in an altered level of PMCA2 or a change in PMCA2 localization.
PMCAs in neuroinflammation, the aging brain and in Alzheimer’s disease
An inflammatory response often accompanies neurodegeneration. In multiple sclerosis and its animal model experimental autoimmune encephalomyelitis (EAE), an autoimmune reaction impairs axonal function. PMCA2 expression is markedly depressed in spinal neurons early in the course of EAE and it recovers during remission [73]. Inhibition of PMCA function or knockout of PMCA2 expression in spinal cord neurons in vitro mimics much of the neuropathology observed in EAE [65]. Furthermore, PMCA2 heterozygous mice display an age-dependent loss of motor neurons, consistent with a critical role for PMCA2 in the survival of these cells [93]. The mechanism by which this demyelinating disease alters PMCA expression is unclear. The inflammatory cytokine inerleukin-1β modulates PMCA function via a tyrosine kinase pathway [45]; perhaps cytokines also modulate PMCA gene expression.
PMCA function in brain synaptic membranes decreases significantly with aging, and most of this decrease is due to loss of activity rather than to decreased PMCA abundance [114]. A likely reason for the decrease in PMCA function during aging is oxidative stress and the ensuing damage resulting in structural modification and conformational changes of the PMCA as well as its surrounding membrane lipids [112]. The PMCA specific activity is higher in membrane lipid rafts than in non-raft domains, and the raft PMCA appears to be particularly vulnerable to age-dependent decrease in activity [57]. Cholesterol, by increasing lipid order, may partially protect PMCA activity in aging membranes but cannot overcome the decline in PMCA function with aging. The decrease of PMCA in the aging brain could also be due to chronic stress, since high doses of the stress hormone corticosterone reduce PMCA1 expression in the rat hippocampus [9].
Impairment of Ca2+ handling is exacerbated in the Alzheimer’s disease (AD) compared to the normally aging brain. The Ca2+ sensitivity and activity of the PMCA are reduced in the cortex of AD compared to age-matched control brains, and biochemical studies have shown that the amyloid β-peptide characteristic for AD inhibits the activity of the PMCA [6]. The strongest inhibitory effect of Aβ was seen with PMCA4 [6], which is abundantly expressed in the dentate gyrus and CA2 of the human hippocampus [110]. Recent studies have shown that PMCA inhibition by Aβ is effectively antagonized by CaM either by competing for access to the PMCA [7] or by directly binding to Aβ [28]. CaM levels are reduced by over 50% in the frontal and temporal cortex in AD compared to normal brains [71]. This decrease of CaM in the AD brain thus further compounds the “normal” age-dependent decline of PMCA function because CaM not only acts as major activator of the PMCA but may also shield the pump from Aβ inhibition as well as proteolytic and oxidative damage [113].
Conclusion
PMCAs are causally implicated in an ever-growing number of neurodegenerative disorders and complex syndromes, emphasizing the specialized roles that PMCA isoforms and splice variants fulfill in the regulation and fine-tuning of neuronal Ca2+ signaling and communication. Altered expression and function of specific PMCA isoforms due to mutation, targeting defects, changing binding partners, or oxidative damage are a characteristic feature of many neuronal diseases. Reduced PMCA function results in altered Ca2+ homeostasis and may lead to a sustained increase in [Ca2+]i, which can become toxic to the cell. Future work will continue to show the tight integration of different PMCAs in dynamic processes of neuronal adaptation, function, and repair. Concomitantly, our understanding of the role of PMCA isoforms in diverse neurodegenerative disorders will be enhanced. Ultimately, this may help in the identification of potential strategies for selective PMCA-centered therapeutic interventions.
Highlights.
PMCA dysfunction impairs precise Ca2+ signaling in neurons producing neurotoxicity
Loss of PMCA function accompanies excitotoxicity and contributes to ischemia and spinal cord pathology
PMCA2 and PMCA3 mutations are linked to cerebellar ataxias and sensory neuron diseases
PMCA mutations are often pathogenic when combined with modifier gene mutations
Decreases in PMCA function during aging and in AD may accelerate neurodegeneration
Acknowledgments
This work was funded by the National Institutes of Health [grant number DA07304 to SAT].
Abbreviations
- AD
Alzheimer’s disease
- CaM
calmodulin
- CNS
central nervous system
- PMCA
plasma membrane calcium pump
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Antalffy G, Mauer AS, Pászty K, Hegedus L, Padányi R, Enyedi A, Strehler EE. Plasma membrane calcium pump (PMCA) isoform 4 is targeted to the apical membrane by the w-splice insert from PMCA2. Cell Calcium. 2012;51:171–178. doi: 10.1016/j.ceca.2011.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Atluri PP, Regehr WG. Determinants of the time course of facilitation at the granule cell to Purkinje cell synapse. J Neurosci. 1996;16:5661–5671. doi: 10.1523/JNEUROSCI.16-18-05661.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Berridge MJ. Calcium hypothesis of Alzheimer’s disease. Pflügers Arch. 2010;459 doi: 10.1007/s00424-009-0736-1. [DOI] [PubMed] [Google Scholar]
- 4.Berridge MJ. Calcium signaling remodeling and disease. Biochem Soc Trans. 2012;40:297–309. doi: 10.1042/BST20110766. [DOI] [PubMed] [Google Scholar]
- 5.Berridge MJ, Bootman MD, Roderick HL. Calcium signalling: dynamics, homeostasis and remodelling. Nature Rev Mol Cell Biol. 2003;4:517–529. doi: 10.1038/nrm1155. [DOI] [PubMed] [Google Scholar]
- 6.Berrocal M, Marcos D, Sepúlveda MR, Perez M, Avila J, Mata AM. Altered Ca2+ dependence of synaptosomoal plasma membrane Ca2+-ATPase in human brain affected by Alzheimer’s disease. FASEB J. 2009;23:1826–1834. doi: 10.1096/fj.08-121459. [DOI] [PubMed] [Google Scholar]
- 7.Berrocal M, Sepúlveda MR, Vazquez-Hernandez M, Mata AM. Calmodulin antagonizes amyloid-b peptides-mediated inhibition of brain plasma membrane Ca2+-ATPase. Biochim Biophys Acta. 2012;1822:961–969. doi: 10.1016/j.bbadis.2012.02.013. [DOI] [PubMed] [Google Scholar]
- 8.Beurg M, Nam JH, Chen Q, Fettiplace R. Calcium balance and mechanotransduction in rat cochlear hair cells. J Neurophysiol. 2010;104:18–34. doi: 10.1152/jn.00019.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bhargava A, Meijer OC, Dallman MF, Pearce D. Plasma membrane calcium pump isoform 1 gene expression is repressed by corticosterone and stress in rat hippocampus. J Neurosci. 2000;20:3129–3138. doi: 10.1523/JNEUROSCI.20-09-03129.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bortolozzi M, Brini M, Parkinson N, Crispino G, Scimemi P, De Siati RD, Di Leva F, Parker A, Ortolano S, Arslan E, Brown SD, Carafoli E, Mammano F. The novel PMCA2 pump mutation Tommy impairs cytosolic calcium clearance in hair cells and links to deafness in mice. J Biol Chem. 2010;285:37693–37703. doi: 10.1074/jbc.M110.170092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brandt P, Neve RL. Expression of plasma membrane calcium-pumping ATPase mRNAs in developing rat brain and adult brain subregions: Evidence for stage-specific expression. J Neurochem. 1992;59:1566–1569. doi: 10.1111/j.1471-4159.1992.tb08476.x. [DOI] [PubMed] [Google Scholar]
- 12.Bravo-Martinez J, Delgado-Coello B, García DE, Mas-Oliva J. Analysis of plasma membrane Ca2+-ATPase gene expression during epileptogenesis employing single hippocampal CA1 neurons. Exp Biol Med. 2011;236:409–417. doi: 10.1258/ebm.2011.010342. [DOI] [PubMed] [Google Scholar]
- 13.Brini M, Calì T, Ottolini D, Carafoli E. The plasma membrane calcium pump in health and disease. FEBS J. 2013;280:5385–5397. doi: 10.1111/febs.12193. [DOI] [PubMed] [Google Scholar]
- 14.Brini M, Carafoli E. Calcium pumps in health and disease. Physiol Rev. 2009;89:1341–1378. doi: 10.1152/physrev.00032.2008. [DOI] [PubMed] [Google Scholar]
- 15.Brini M, Carafoli E. The plasma membrane calcium pumps: focus on the role in (neuro) pathology. Biochem Biophys Res Commun. 2017;483:1116–1124. doi: 10.1016/j.bbrc.2016.07.117. [DOI] [PubMed] [Google Scholar]
- 16.Burette A, Rockwood JM, Strehler EE, Weinberg RJ. Isoform-specific distribution of the plasma membrane Ca2+ ATPase in the rat brain. J Comp Neurol. 2003;467:464–476. doi: 10.1002/cne.10933. [DOI] [PubMed] [Google Scholar]
- 17.Burette A, Strehler EE, Weinberg RJ. A plasma membrane Ca2+ ATPase isoform at the postsynaptic density. Neuroscience. 2010;169:987–993. doi: 10.1016/j.neuroscience.2010.05.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Burette A, Weinberg RJ. Perisynaptic organization of plasma membrane calcium pumps in cerebellar cortex. J Comp Neurol. 2007;500:1127–1135. doi: 10.1002/cne.21237. [DOI] [PubMed] [Google Scholar]
- 19.Burette AC, Strehler EE, Weinberg RJ. “Fast” plasma membrane calcium pump PMCA2a concentrates in GABAergic terminals in the adult rat brain. J Comp Neurol. 2009;512:500–513. doi: 10.1002/cne.21909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cali T, Frizzarin M, Luoni L, Zonta F, Pantano S, Cruz C, Bonza MC, Bertipaglia I, Ruzzene M, De Michelis MI, Damiano N, Marin O, Zanni G, Zanotti G, Brini M, Lopreiato R, Carafoli E. The ataxia related G1107D mutation of the plasma membrane Ca2+ ATPase isoform 3 affects its interplay with calmodulin and the autoinhibition process. Biochim Biophys Acta. 2017;1863:165–173. doi: 10.1016/j.bbadis.2016.09.007. [DOI] [PubMed] [Google Scholar]
- 21.Cali T, Lopreiato R, Shimony J, Vineyard M, Frizzarin M, Zanni G, Zanotti G, Brini M, Shinawi M, Carafoli E. A novel mutation in isoform 3 of the plasma membrane Ca2+ pump impairs cellular Ca2+ homeostasis in a patient with cerebellar ataxia and laminin subunit 1alpha mutations. J Biol Chem. 2015;290:16132–16141. doi: 10.1074/jbc.M115.656496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Carayol J, Sacco R, Tores F, Rousseau F, Lewin P, Hager J, Persico AM. Converging evidence for an association of ATP2B2 allelic variants with autism in male subjects. Biol Psychiatry. 2011;70:880–887. doi: 10.1016/j.biopsych.2011.05.020. [DOI] [PubMed] [Google Scholar]
- 23.Caride AJ, Filoteo AG, Penheiter AR, Pászty K, Enyedi Á, Penniston JT. Delayed activation of the plasma membrane calcium pump by a sudden increase in Ca2+: fast pumps reside in fast cells. Cell Calcium. 2001;30:49–57. doi: 10.1054/ceca.2001.0212. [DOI] [PubMed] [Google Scholar]
- 24.Caride AJ, Penheiter AR, Filoteo AG, Bajzer Z, Enyedi A, Penniston JT. The plasma membrane calcium pump displays memory of past calcium spikes. Differences between isoforms 2b and 4b. J Biol Chem. 2001;276:39797–39804. doi: 10.1074/jbc.M104380200. [DOI] [PubMed] [Google Scholar]
- 25.Carpinelli MR, Manning MG, Kile BT, Burt RA. Two ENU-induced alleles of Atp2b2 cause deafness in mice. PLoS One. 2013;8:e67479. doi: 10.1371/journal.pone.0067479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen Q, Mahendrasingam S, Tickle JA, Hackney CM, Furness DN, Fettiplace R. The development, distribution and density of the plasma membrane calcium ATPase 2 calcium pump in rat cochlear hair cells. Eur J Neurosci. 2012;36:2302–2310. doi: 10.1111/j.1460-9568.2012.08159.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chicka MC, Strehler EE. Alternative splicing of the first intracellular loop of plasma membrane Ca2+-ATPase isoform 2 alters its membrane targeting. J Biol Chem. 2003;278:18464–18470. doi: 10.1074/jbc.M301482200. [DOI] [PubMed] [Google Scholar]
- 28.Corbacho I, Berrocal M, Török K, Mata AM, Gutierrez-Merino C. High affinity binding of amyloid b--peptide to calmodulin: structural and functional implications. Biochem Biophys Res Commun. 2017;486:992–997. doi: 10.1016/j.bbrc.2017.03.151. [DOI] [PubMed] [Google Scholar]
- 29.Cuomo O, Vinciguerra A, Cerullo P, Anzilotti S, Brancaccio P, Bilo L, Scorziello A, Molinaro P, Di Renzo G, Pignataro G. Ionic homeostasis in brain conditioning. Front Neurosci. 2015;9:277. doi: 10.3389/fnins.2015.00277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Duncan JL, Yang H, Doan T, Silverstein RS, Murphy GJ, Nune G, Liu X, Copenhagen D, Tempel BL, Rieke F, Krizaj D. Scotopic visual signaling in the mouse retina is modulated by high-affinity plasma membrane calcium extrusion. J Neurosci. 2006;26:7201–7211. doi: 10.1523/JNEUROSCI.5230-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Eakin TJ, Antonelli MC, Malchiodi EL, Baskin DG, Stahl W. Localization of the plasma membrane Ca2+-ATPase isoform PMCA3 in rat cerebellum, choroid plexus and hippocampus. Molec Brain Res. 1995;29:71–80. doi: 10.1016/0169-328x(94)00231-3. [DOI] [PubMed] [Google Scholar]
- 32.Empson RM, Akemann W, Knöpfel T. The role of the calcium transporter protein plasma membrane calcium atpase pmca2 in cerebellar Purkinje neuron function. Funct Neurol. 2010;25:153–158. [PubMed] [Google Scholar]
- 33.Empson RM, Garside ML, Knöpfel T. Plasma membrane Ca2+ ATPase 2 contributes to short-term synapse plasticity at the parallel fibre to Purkinje neurone synapse. J Neurosci. 2007;27:3753–3758. doi: 10.1523/JNEUROSCI.0069-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Empson RM, Huang H, Nagaraja RY, Roome CJ, Knöpfel T. Enhanced synaptic inhibition in the cerebellar cortex of the ataxic PMCA2(−/−) knockout mouse. Cerebellum. 2013;12:667–675. doi: 10.1007/s12311-013-0472-0. [DOI] [PubMed] [Google Scholar]
- 35.Empson RM, Turner PR, Nagaraja RY, Beesley PW, Knöpfel T. Reduced expression of the Ca2+ transporter protein PMCA2 slows Ca2+ dynamics in mouse cerebellar Purkinje neurones and alters the precision of motor coordination. J Physiol. 2010;588:907–922. doi: 10.1113/jphysiol.2009.182196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fakira AK, Elkabes S. Role of plasma membrane calcium ATPase 2 in spinal cord pathology. World J Biol Chem. 2010;26:103–108. doi: 10.4331/wjbc.v1.i5.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fakira AK, Gaspers LD, Thomas AP, Li H, Jain MR, Elkabes S. Purkinje cell dysfunction and delayed death in plasma membrane calcium ATPase 2-heterozygous mice. Mol Cell Neurosci. 2012;51:22–31. doi: 10.1016/j.mcn.2012.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ferragamo MJ, Reinardy JL, Thayer SA. Ca2+-dependent, stimulus-specific modulation of the plasma membrane Ca2+ pump in hippocampal neurons. J Neurophysiol. 2009;101:2563–2571. doi: 10.1152/jn.90774.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ficarella R, Di Leva F, Bortolozzi M, Ortolano S, Donaudy F, Petrillo M, Melchionda S, Lelli A, Domi T, Fedrizzi L, Lim D, Shull GE, Gasparini P, Brini M, Mammano F, Carafoli E. A functional study of plasma-membrane calcium-pump isoform 2 mutants causing digenic deafness. Proc Nat Acad Sci USA. 2007;104:1516–1521. doi: 10.1073/pnas.0609775104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Figueroa KP, Paul S, Cali T, Lopreiato R, Karan S, Frizzarin M, Ames D, Zanni G, Brini M, Dansithong W, Milash B, Scoles DR, Carafoli E, Pulst SM. Spontaneous shaker rat mutant – a new model for X-linked tremor/ataxia. Dis Mod Mech. 2016;9:553–562. doi: 10.1242/dmm.022848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Furness DN, Katori Y, Nirmal Kumar B, Hackney CM. The dimensions and structural attachments of tip links in mammalian cochlear hair cells and the effects of exposure to different levels of extracellular calcium. Neuroscience. 2008;154:10–21. doi: 10.1016/j.neuroscience.2008.02.010. [DOI] [PubMed] [Google Scholar]
- 42.Garcia ML, Murray KD, Garcia VB, Strehler EE, Isackson PJ. Seizure induced alterations of plasma membrane calcium ATPase isoforms 1, 2, and 3 mRNA and protein in rat hippocampus. Molec Brain Res. 1997;45:230–238. doi: 10.1016/s0169-328x(96)00253-7. [DOI] [PubMed] [Google Scholar]
- 43.Garside ML, Turner PR, Austen B, Strehler EE, Beesley PW, Empson RM. Molecular interactions of the plasma membrane calcium ATPase 2 at pre-and post-synaptic sites in rat cerebellum. Neuroscience. 2009;162:383–395. doi: 10.1016/j.neuroscience.2009.04.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gemes G, Oyster KD, Pan B, Wu H-E, Bangaru MLY, Tang Q, Hogan QH. Painful nerve injury increases plasma membrane Ca2+-ATPase activity in axotomized sensory neurons. Mol Pain. 2012;8:46. doi: 10.1186/1744-8069-8-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ghosh B, Green MV, Krogh KA, Thayer SA. Interleukin-1beta activates an Src family kinase to stimulate the plasma membrane Ca2+ pump in hippocampal neurons. J Neurophysiol. 2016;115:1875–1885. doi: 10.1152/jn.00541.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ghosh B, Li Y, Thayer SA. Inhibition of the plasma membrane Ca2+ pump by CD44 receptor activation of tyrosine kinases increases the action potential afterhyperpolarization in sensory neurons. J Neurosci. 2011;31:2361–2370. doi: 10.1523/JNEUROSCI.5764-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Giacomello M, De Mario A, Primerano S, Brini M, Carafoli E. Hair cells, plasma membrane Ca2+ ATPase and deafness. Int J Biochem Cell Biol. 2012;44:679–683. doi: 10.1016/j.biocel.2012.02.006. [DOI] [PubMed] [Google Scholar]
- 48.Gover TD, Moreira TH, Kao JP, Weinreich D. Calcium regulation in individual peripheral sensory nerve terminals of the rat. J Physiol. 2007;578:481–490. doi: 10.1113/jphysiol.2006.119008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Grati M, Aggarwal N, Strehler EE, Wenthold RJ. Molecular determinants for differential membrane trafficking of PMCA1 and PMCA2 in mammalian hair cells. J Cell Sci. 2006;119:2995–3007. doi: 10.1242/jcs.03030. [DOI] [PubMed] [Google Scholar]
- 50.Guerini D, Coletto L, Carafoli E. Exporting calcium from cells. Cell Calcium. 2005;38:281–289. doi: 10.1016/j.ceca.2005.06.032. [DOI] [PubMed] [Google Scholar]
- 51.Guo Y, Zhang Z, Wu H-E, Luo ZD, Hogan QH, Pan B. Increased thrombospondin-4 after nerve injury mediates disruption of intracellular calcium signaling in primary sensory neurons. Neuropharmacol. 2017;117:292–304. doi: 10.1016/j.neuropharm.2017.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Henrich M, Buckler KJ. Cytosolic calcium regulation in rat afferent vagal neurons during anoxia. Cell Calcium. 2013;54:416–427. doi: 10.1016/j.ceca.2013.10.001. [DOI] [PubMed] [Google Scholar]
- 53.Hill JK, Williams DE, LeMasurier DE, Dumont RA, Strehler EE, Gillespie PG. Splice-site A choice targets plasma-membrane Ca2+-ATPase isoform 2 to hair bundles. J Neurosci. 2006;26:6172–6180. doi: 10.1523/JNEUROSCI.0447-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ikeda M, Tomita Y, Mouri A, Koga M, Okochi T, Yoshimura R, Yamanouchi Y, Kinoshita Y, Hashimoto R, Williams HJ, Takeda M, Nakamura J, Nabeshima T, Owen MJ, O’Donovan MC, Honda H, Arinami T, Ozaki N, Iwata N. Identification of novel candidate genes for treatment response to risperidone and susceptibility for schizophrenia: integrated analysis among pharmacogenomics, mouse expression, and genetic case-control association approaches. Biol Psychiatry. 2010;67:263–269. doi: 10.1016/j.biopsych.2009.08.030. [DOI] [PubMed] [Google Scholar]
- 55.Ivannikov MV, Sugimori M, Llinas RR. Calcium clearance and its energy requirements in cerebellar neurons. Cell Calcium. 2010;47:507–513. doi: 10.1016/j.ceca.2010.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.James P, Vorherr T, Krebs J, Morelli A, Catello G, McCormick DJ, Penniston JT, DeFlora A, Carafoli E. Modulation of erythrocyte Ca2+ ATPase by selective calpain cleavage of the calmodulin-binding domain. J Biol Chem. 1989;264:8289–8296. [PubMed] [Google Scholar]
- 57.Jiang L, Bechtel MD, Galeva NA, Williams TD, Michaelis EK, Michaelis ML. Decreases in plasma membrane Ca2+-ATPase in brain synaptic membrane rafts from aged rats. J Neurochem. 2012;123:689–699. doi: 10.1111/j.1471-4159.2012.07918.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Johnson JJE, Perkins GA, Giddabasappa A, Chaney S, Xiao W, White AD, Brown JM, Waggoner J, Ellisman MH, Fox DA. Spatiotemporal regulation of ATP and Ca2+ dynamics in vertebrate rod and cone ribbon synapses. Mol Vis. 2007;13:887–919. [PMC free article] [PubMed] [Google Scholar]
- 59.Kenyon KA, Bushong EA, Mauer AS, Strehler EE, Weinberg RJ, Burette AC. Cellular and subcellular localizatin of the neuron-specific plasma membrane calcium ATPase PMCA1a in the rat brain. J Comp Neurol. 2010;518:3169–3183. doi: 10.1002/cne.22409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Khachaturian ZS. Calcium hypothesis of Alzheimer’s disease and brain aging. Ann NY Acad Sci. 1994;747:1–11. doi: 10.1111/j.1749-6632.1994.tb44398.x. [DOI] [PubMed] [Google Scholar]
- 61.Khariv V, Ni L, Ratnayake A, Sampath S, Lutz BM, Tao X-X, Heary RF, Elkabes S. Impaired sensitivity to pain stimuli in plasma membrane calcium ATPase 2 (PMCA2) heterozygous mice: a possible modality- and sex-specific role for PMCA2 in nociception. FASEB J. 2017;31:224–237. doi: 10.1096/fj.201600541R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kozel PJ, Davis RR, Krieg EF, Shull GE, Erway LC. Deficiency in plasma membrane calcium ATPase isoform 2 increases susceptibility to noise-induced hearing loss in mice. Hearing Res. 2002;164:231–239. doi: 10.1016/s0378-5955(01)00420-8. [DOI] [PubMed] [Google Scholar]
- 63.Kozel PJ, Friedman RA, Erway LC, Yamoah EN, Liu LH, Riddle T, Duffy JJ, Doetschman T, Miller ML, Cardell EL, Shull GE. Balance and hearing deficits in mice with a null mutation in the gene encoding plasma membrane Ca2+-ATPase isoform 2. J Biol Chem. 1998;273:18693–18696. doi: 10.1074/jbc.273.30.18693. [DOI] [PubMed] [Google Scholar]
- 64.Kurnellas MP, Li H, Jain MR, Giraud SN, Nicot AB, Ratnayake A, Heary RF, Elkabes S. Reduced expression of plasma membrane calcium ATPase 2 and collapsin response mediator protein 1 promotes death of spinal cord neurons. Cell Death Diff. 2010;17:1501–1510. doi: 10.1038/cdd.2010.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kurnellas MP, Nicot A, Shull GE, Elkabes S. Plasma membrane calcium ATPase deficiency causes neuronal pathology in the spinal cord: a potential mechanism for neurodegeneration in multiple sclerosis and spinal cord injury. FASEB J. 2005;19:298–300. doi: 10.1096/fj.04-2549fje. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lehotsky J, Kaplan P, Racay P, Mezesova V, Raeymaekers L. Distribution of plasma membrane Ca2+ pump (PMCA) isoforms in the gerbil brain: effect of ischemia-reperfusion injury. Neurochem Int. 1999;35:221–227. doi: 10.1016/s0197-0186(99)00062-5. [DOI] [PubMed] [Google Scholar]
- 67.Mammano F. Ca2+ homeostasis defects and hereditary hearing loss. Biofactors. 2011;37:182–188. doi: 10.1002/biof.150. [DOI] [PubMed] [Google Scholar]
- 68.Martins-de-Souza D, Gattaz WF, Schmitt A, Rewerts C, Marangoni S, Novello JC, Maccarrone G, Turck CW, Dias-Neto E. Alterations in oligodendrocyte proteins, calcium homeostasis and new potential markers in schizophrenia anterior temporal lobe are revealed by shotgun protome analysis. J Neural Transm. 2009;116:275–289. doi: 10.1007/s00702-008-0156-y. [DOI] [PubMed] [Google Scholar]
- 69.Mattson MP. Calcium and neurodegeneration. Aging Cell. 2007;6:337–350. doi: 10.1111/j.1474-9726.2007.00275.x. [DOI] [PubMed] [Google Scholar]
- 70.McCullough BJ, Tempel BL. Haplo-insufficiency revealed in deafwaddler mice when tested for hearing loss and ataxia. Hearing Res. 2004;195:90–102. doi: 10.1016/j.heares.2004.05.003. [DOI] [PubMed] [Google Scholar]
- 71.McLachlan DRC, Wong L, Bergeron C, Baimbridge KG. Calmodulin and calbindin D28K in Alzheimer disease. Alzheimer Dis Assoc Disord. 1987;1:171–179. doi: 10.1097/00002093-198701030-00009. [DOI] [PubMed] [Google Scholar]
- 72.Miller RJ. The control of neuronal Ca2+ homeostasis. Prog Neurobiol. 1991;37:255–285. doi: 10.1016/0301-0082(91)90028-y. [DOI] [PubMed] [Google Scholar]
- 73.Nicot A, Kurnellas M, Elkabes S. Temporal pattern of plasma membrane calcium ATPase 2 expression in the spinal cord correlates with the course of clinical symptoms in two rodent models of autoimmune encephalomyelitis. Eur J Neurosci. 2005;21:2660–2670. doi: 10.1111/j.1460-9568.2005.04086.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ogura H, Tachibana T, Yamanaka H, Kobayashi K, Obata K, Dai Y, Yoshiya S, Noguchi K. Axotomy increases plasma membrane Ca2+ pump isoform4 in primary afferent neurons. Neuroreport. 2007;18:17–22. doi: 10.1097/WNR.0b013e328011e6e3. [DOI] [PubMed] [Google Scholar]
- 75.Oguro K, Nakamura M, Masuzawa T. Histochemical study of Ca(2+)-ATPase activity in ischemic CA1 pyramidal neurons in the gerbil hippocampus. Acta Neuropathologica. 1995;90:448–453. doi: 10.1007/BF00294804. [DOI] [PubMed] [Google Scholar]
- 76.Ohta S, Furuta S, Matsubara I, Kohno K, Kumon Y, Sakaki S. Calcium movement in ischemia-tolerant hippocampal CA1 neurons after transient forebrain ischemia in gerbils. J Cereb Blood Flow Metab. 1996;16:915–926. doi: 10.1097/00004647-199609000-00015. [DOI] [PubMed] [Google Scholar]
- 77.Olesen C, Picard M, Winther AM, Gyrup C, Morth JP, Oxvig C, Møller JV, Nissen P. the structural basis of calcium transport by the calcium pump. Nature. 2007;450:1036–1042. doi: 10.1038/nature06418. [DOI] [PubMed] [Google Scholar]
- 78.Padányi R, Pászty K, Hegedüs L, Varga K, Papp B, Penniston JT, Enyedi Á. Multifaceted plasma membrane Ca2+ pumps: from structure to intracellular Ca2+ handling and cancer. Biochim Biophys Acta. 2016;1863:1351–1363. doi: 10.1016/j.bbamcr.2015.12.011. [DOI] [PubMed] [Google Scholar]
- 79.Palmgren MG, Nissen P. P-type ATPases. Ann Rev Biophys. 2011;40:243–266. doi: 10.1146/annurev.biophys.093008.131331. [DOI] [PubMed] [Google Scholar]
- 80.Pászty K, Verma AK, Padányi R, Filoteo AG, Penniston JT, Enyedi Á. Plasma membrane Ca2+ ATPase isoform 4b is cleaved and activated by caspase-3 during the early phase of apoptosis. J Biol Chem. 2002;277:6822–6829. doi: 10.1074/jbc.M109548200. [DOI] [PubMed] [Google Scholar]
- 81.Pedersen PL, Carafoli E. Ion motive ATPases. I. Ubiquity, properties, and significance to cell function. Trends Biochem Sci. 1987;12:146–150. [Google Scholar]
- 82.Penheiter AR, Filoteo AG, Croy CL, Penniston JT. Characterization of the deafwaddler mutant of the rat plasma membrane calcium-ATPase 2. Hearing Res. 2001;162:19–28. doi: 10.1016/s0378-5955(01)00356-2. [DOI] [PubMed] [Google Scholar]
- 83.Pottorf WJ, II, Johanns TM, Derrington SM, Strehler EE, Enyedi Á, Thayer SA. Glutamate-induced protease-mediated loss of plasma membrane Ca2+ pump activity in rat hippocampal neurons. J Neurochem. 2006;98:1646–1656. doi: 10.1111/j.1471-4159.2006.04063.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Pottorf WJ, II, Thayer SA. Transient rise in intracellular calcium produces a long-lasting increase in plasma membrane calcium pump activity in rat sensory neurons. J Neurochem. 2002;83:1002–1008. doi: 10.1046/j.1471-4159.2002.01221.x. [DOI] [PubMed] [Google Scholar]
- 85.Prandini P, Pasquali A, Malerba G, Marostica A, Zusi C, Xumerle L, Muglia P, Da Ros L, Ratti E, Trabetti E, Pignatti PF (ITAN), T.I.A.N. The association of rs4307059 and rs35678 markers with autism spectrum disorders is replicated in Italian families. Psychiatr Genet. 2012;22:177–181. doi: 10.1097/YPG.0b013e32835185c9. [DOI] [PubMed] [Google Scholar]
- 86.Raza M, Pal S, Rafiq A, DeLorenzo RJ. Long-term alteration of calcium homeostatic mechanisms in the pilocarpine model of temporal lobe epilepsy. Brain Res. 2001;903:1–12. doi: 10.1016/s0006-8993(01)02127-8. [DOI] [PubMed] [Google Scholar]
- 87.Roome CJ, Knöpfel T, Empson RM. Functional contributions of the plasma membrane calcium ATPase and the sodium-calcium exchanger at mouse parallel fibre to Purkinje neuron synapses. Pflügers Arch. 2013;465:319–331. doi: 10.1007/s00424-012-1172-1. [DOI] [PubMed] [Google Scholar]
- 88.Sattler R, Xiong Z, Lu W-Y, Hafner M, MacDonald JF, Tymianski M. Specific coupling of NMDA receptor activation to nitric oxide neurotoxicity by PSD-95 protein. Science. 1999;284:1845–1848. doi: 10.1126/science.284.5421.1845. [DOI] [PubMed] [Google Scholar]
- 89.Scheuss V, Yasuda R, Sobczyk A, Svoboda K. Nonlinear [Ca2+] signaling in dendrites and spines caused by activity-dependent depression of Ca2+ extrusion. J Neurosci. 2006;26:8183–8194. doi: 10.1523/JNEUROSCI.1962-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Schultz JM, Yang Y, Caride AJ, Filoteo AG, Penheiter AR, Lagziel A, Morell RJ, Mohiddin SA, Fananapazir L, Madeo AC, Penniston JT, Griffith AJ. Modification of human hearing loss by plasma-membrane calcium pump PMCA2. New England J of Med. 2005;352:1557–1564. doi: 10.1056/NEJMoa043899. [DOI] [PubMed] [Google Scholar]
- 91.Schwab BL, Guerini D, Didszun C, Bano D, Ferrando-May E, Fava E, Tam J, Xu D, Xanthoudakis S, Nicholson DW, Carafoli E, Nicotera P. Cleavage of plasma membrane calcium pumps by caspases: a link between apoptosis and necrosis. Cell Death Diff. 2002;9:818–831. doi: 10.1038/sj.cdd.4401042. [DOI] [PubMed] [Google Scholar]
- 92.Sherkhane P, Kapfhammer JP. The plasma membrane Ca2+-ATPase2 (PMCA2) is involved in the regulation of Purkinje cell dendritic growth in cerebellar organotypic slice cultures. Neural Plast. 2013;2013:321685. doi: 10.1155/2013/321685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Souayah N, Sharovetskaya A, Kurnellas MP, Myerson M, Deitch JS, Elkabes S. Reductions in motor unit number estimates (MUNE) precede motor neuron loss in the plasma membrane calcium ATPase 2 (PMCA2)-heterozygous mice. Exp Neurol. 2008;214:341–346. doi: 10.1016/j.expneurol.2008.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Stahl WL, Eakin TJ, Owens JWM, Breininger JF, Filuk PE, Anderson WR. Plasma membrane Ca2+-ATPase isoforms: distribution of mRNAs in rat brain by in situ hybridization. Molec Brain Res. 1992;16:223–231. doi: 10.1016/0169-328x(92)90229-5. [DOI] [PubMed] [Google Scholar]
- 95.Stauffer TP, Guerini D, Celio MR, Carafoli E. Immunolocalization of the plasma membrane Ca2+ pump isoforms in the rat brain. Brain Res. 1997;748:21–29. doi: 10.1016/s0006-8993(96)01282-6. [DOI] [PubMed] [Google Scholar]
- 96.Street VA, McKee-Johnson JW, Fonseca RC, Tempel BL, Noben-Trauth K. Mutations in a plasma membrane Ca2+-ATPase gene cause deafness in deafwaddler mice. Nature Genet. 1998;19:390–394. doi: 10.1038/1284. [DOI] [PubMed] [Google Scholar]
- 97.Strehler EE. Plasma membrane calcium ATPases: From generic Ca2+ sump pumps to versatile systems for fine-tuning cellular Ca2+ Biochem Biophys Res Commun. 2015;460:26–33. doi: 10.1016/j.bbrc.2015.01.121. [DOI] [PubMed] [Google Scholar]
- 98.Strehler EE, Caride AJ, Filoteo AG, Xiong Y, Penniston JT, Enyedi Á. Plasma membrane Ca2+ ATPases as dynamic regulators of cellular calcium handling. Ann N Y Acad Sci. 2007;1099:226–236. doi: 10.1196/annals.1387.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Strehler EE, Zacharias DA. Role of alternative splicing in generating isoform diversity among plasma membrane calcium pumps. Physiol Rev. 2001;81:21–50. doi: 10.1152/physrev.2001.81.1.21. [DOI] [PubMed] [Google Scholar]
- 100.Tachibana T, Noguchi K, Ruda MA. Analysis of gene expression following spinal cord injury in rat using complementary DNA microarray. Neurosci Lett. 2002;327:133–137. doi: 10.1016/s0304-3940(02)00375-0. [DOI] [PubMed] [Google Scholar]
- 101.Takahashi K, Kitamura K. A point mutation in a plasma membrane Ca2+-ATPase gene causes deafness in Wriggle Mouse Sagami. Biochem Biophys Res Commun. 1999;261:773–778. doi: 10.1006/bbrc.1999.1102. [DOI] [PubMed] [Google Scholar]
- 102.Toyoshima C. How Ca2+-ATPase pumps ions across the sarcoplasmic reticulum membrane. Biochim Biophys Acta. 2009;1793:941–946. doi: 10.1016/j.bbamcr.2008.10.008. [DOI] [PubMed] [Google Scholar]
- 103.Ueno T, Kameyama K, Hirata M, Ogawa M, Hatsuse H, Takagaki Y, Ohmura M, Osawa N, Kudo Y. A mouse with a point mutation in plasma membrane Ca2+-ATPase isoform 2 gene showed the reduced Ca2+ influx in cerebellar neurons. Neurosci Res. 2002;42:287–297. doi: 10.1016/s0168-0102(02)00008-1. [DOI] [PubMed] [Google Scholar]
- 104.Usachev YM, DeMarco SJ, Campbell C, Strehler EE, Thayer SA. Bradykinin and ATP accelerate Ca2+ efflux from rat sensory neurons via protein kinase C and the plasma membrane Ca2+ pump isoform 4. Neuron. 2002;33:113–122. doi: 10.1016/s0896-6273(01)00557-8. [DOI] [PubMed] [Google Scholar]
- 105.Wan QF, Nixon E, Heidelberger R. Regulation of presynaptic calcium in a mammalian synaptic terminal. J Neurophysiol. 2012;108:3059–3067. doi: 10.1152/jn.00213.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Wanigasekara Y, Armati PJ, Roufogalis BD. Ca2+-ATPase isoforms are expressed in neuroprotection in rat, but not human, neurons. Neuroreport. 2003;14:2421–2424. doi: 10.1097/00001756-200312190-00026. [DOI] [PubMed] [Google Scholar]
- 107.Wood JD, Muchinsky SJ, Filoteo AG, Penniston JT, Tempel BL. Low endolymph calcium concentrations in deafwaddler2J mice suggest that PMCA2 contributes to endolymph calcium maintenance. J Assoc Res Otolaryngol. 2004;5:99–110. doi: 10.1007/s10162-003-4022-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Yang W, Liu J, Zheng F, Jia M, Zhao L, Lu T, Ruan Y, Zhang J, Yue W, Zhang D, Wang L. The evidence for association of ATP2B2 polymorphisms with autism in Chinese Han population. PLoS One. 2013;8:e61021. doi: 10.1371/journal.pone.0061021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Zacharias DA, Dalrymple SJ, Strehler EE. Transcript distribution of plasma membrane Ca2+ pump isoforms and splice variants in the human brain. Molec Brain Res. 1995;28:263–272. doi: 10.1016/0169-328x(94)00215-z. [DOI] [PubMed] [Google Scholar]
- 110.Zacharias DA, DeMarco SJ, Strehler EE. mRNA expression of the four isoforms of the human plasma membrane Ca2+-ATPase in human hippocampus. Molecular Brain Research. 1997;45:173–176. doi: 10.1016/s0169-328x(97)00009-0. [DOI] [PubMed] [Google Scholar]
- 111.Zacharias DA, Kappen C. Developmental expression of the four plasma membrane calcium ATPase (Pmca) genes in the mouse. Biochim Biophys Acta. 1999;1428:397–405. doi: 10.1016/s0304-4165(99)00058-6. [DOI] [PubMed] [Google Scholar]
- 112.Zaidi A. Plasma membrane Ca2+-ATPases: targets of oxidative stress in brain aging and neurodegeneration. World J Biol Chem. 2010;1:271–280. doi: 10.4331/wjbc.v1.i9.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Zaidi A, Barton L, Sharov VS, Schöneich C, Michaelis EK, Michaelis ML. Oxidative inactivation of purified plasma membrane Ca2+-ATPase by hydrogen peroxide and protection by calmodulin. Biochmistry. 2003;42:12001–12010. doi: 10.1021/bi034565u. [DOI] [PubMed] [Google Scholar]
- 114.Zaidi A, Squier TC, Michaelis ML. Age-related decrease in brain synaptic membrane Ca2+-ATPase in F344/BNF1 rats. Neurobiol Aging. 1998;19:487–495. doi: 10.1016/s0197-4580(98)00078-5. [DOI] [PubMed] [Google Scholar]
- 115.Zanazzi G, Matthews G. The molecular architecture of ribbon presynaptic terminals. Mol Neurobiol. 2009;39:130–148. doi: 10.1007/s12035-009-8058-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Zanni G, Cali T, Kalscheuer VM, Ottolini D, Barresi S, Lebrun N, Montecchi-Palazzi L, Hu H, Chelly J, Bertini E, Brini M, Carafoli E. Mutation of plasma membrane Ca2+ ATPase isoform 3 in a family with X-linked congenital cerebellar ataxia impairs Ca2+ homeostasis. Proc Nat Acad Sci USA. 2012;109:14514–14519. doi: 10.1073/pnas.1207488109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Zenisek D, Matthews G. The role of mitochondria in presynaptic calcium handling at a ribbon synapse. Neuron. 2000;25:229–237. doi: 10.1016/s0896-6273(00)80885-5. [DOI] [PubMed] [Google Scholar]