

Abstract
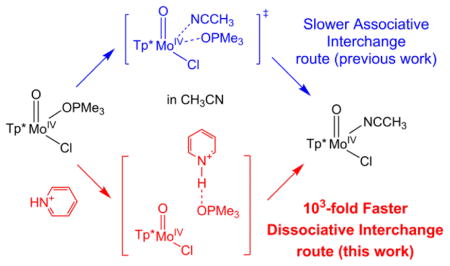
We report that pyridinium ions (HPyr+) accelerate the conversion of [Tp*MoIVOCl(OPMe3)] (1) to [Tp*MoIVOCl(NCCH3)] (2) by 103 fold, affording 2 in near-quatitative yield; Tp* = hydrotris(3,5-dimethyl-1-pyrazolyl)borate. This novel reactivity and the mechanism of this reaction were investigated in details. The formation of 2 followed pseudo-first-order kinetics, with the observed pseudo first-order rate constant (kobs) linearly correlated with [HPyr+]. An Eyring plot revealed that this HPyr+-facilitated reaction has a small positive value of ΔS‡ indicative of a dissociative interchange mechanism (Id), different from the slower associative interchange mechanism in the absence of HPyr+ marked with a negative ΔS‡. Interestingly, log(kobs) was found to be linearly correlated to the acidity of substituted pyridinium ions. This novel reactivity is further investigated using combined DFT and ab initio coupled cluster methods. Different reaction pathways, including Id, associative interchange (Ia), and possible alternative routes in the absence or presence of HPyr+, were considered, and enthalpy and free energies were calculated for each pathway. Our computational results further underscored that the Id route is energetically favored in the presence of HPyr+, in contrast with the preferred Ia-NNO pathway in the absence of HPyr+. Our computational results also revealed molecular-level details for the HPyr+-facilitated Id route. Specifically, HPyr+ initially becomes hydrogen-bonded to the oxygen atom of the Mo(IV)-OPMe3 moiety, which lowers the activation barrier for the Mo–OPMe3 bond cleavage in a rate-limiting step to dissociate the OPMe3 product. The implications of our results were discussed in the context of molybdoenzymes, particularly the reductive half-reaction of sulfite oxidase.
Keywords: Molybdenum, Molybdoenzymes, Coordination complexes, Oxygen atom transfer reactions, Reaction mechanism
Graphic Abstract
1. Introduction
Molybdoenzymes are found in all forms of life. Most of these important enzymes catalyze a wide range of oxygen atom transfer (OAT) reactions of key importance to human health [1, 2], and contain a mononuclear molybdenum (Mo) center in their active sites. Among these molybdoenzymes, the widely distributed sulfite oxidase (SO) catalyzes the conversion of toxic sulfite to sulfate, which is the last step in the catabolism of sulfur-containing amino acids cysteine and methionine, and several xenobiotic compounds [3–6]. The lack of an effective SO in humans leads to severe neurological abnormalities, including mental retardation and death in infancy, with the lethality due partially to intracellular sulfite build-up [7].
To advance our understanding of SO and other molybdenum/tungsten-containing enzymes and design better bio-inspired catalysts for OAT reactions, it is imperative to advance our understandings of how these enzymes operate on the molecular level. For this purpose, small molecule molybdenum model compounds have been developed, with several reviews recently published on this topic [8–10]. Representative examples of cis-MoVIO2 complexes exhibiting OAT reactivities are supported by different ligands including bis(dithiocarbamate) [11, 12], bis(dithiolene) [13, 14], bis(dithiolatopyridine) [15], trispyrazolylborate [16, 17], thiosemicarbazone [18, 19], or iminophenolate [20]. OAT reaction mechanisms of model cis-MoVIO2 compounds [21–25] lent credence to the proposal that the sulfite lone pair attacks the equatorial MoVI=O group to afford a Mo(IV)-sulfate species, followed by product liberation to complete the reductive half-reaction of SO [26–28]. However, OAT rates for small molecule molybdenum model complexes, even in the best systems, are still orders of magnitude slower compared to those of molybdoenzymes [29]. This mismatch suggests that there is still a lack of understanding into different molecular characteristics and other factors that contribute to the very fast individual steps in catalytic cycles of molybdoenzymes.
One potentially important factor is the possible interaction with Brønsted and/or Lewis acids, which have been shown to modulate other non-redox [30] and redox reactions [31] of transition metal compounds. For molybdoenzymes, reduction of {MoVIO2} is often accompanied by proton transfer to form {MoV(O)(OH)} and/or {MoIV(O)(OH2)} species, and vice versa [32]. Such coupled electron and proton transfer (CEPT) processes are modelled in the [Tp*MoVIO2(SPh)] system (Tp* = hydrotris(3,5-dimethyl-1-pyrazolyl)borate; see Scheme 1A): after transferring an oxygen atom to PPh3, the original {MoVIO2} complex can be regenerated in two CEPT processes via a putative {MoIV(O)(OH2)} species and a {MoV(O)(OH)} intermediate [33]. In other model systems, the (de)protonation of ligands does not necessitate molybdenum oxidation state changes. For example, Sarkar et al. synthesized dimeric and monomeric desoxo Mo(IV) and W(IV) bis(dithiolene) complexes from acidification of [MIVO(mnt)2]2− (M = Mo or W) in the presence of thiols (mnt2− = 1,2-dicyanoethylenedithiolate) [34, 35]. Kim et al. later reported that the addition of tosylic acid to [MIVOS2C2Ph2)2]2− (M = Mo or W) yielded the deoxygenated [MIV(MeCN)2(S2C2Ph2)2] product [36]. More recently, Itoh, Kirk, and coworkers showed that dioxido-molybdenum(VI) complexes reacted with a strong organic acid in the presence of an alcohol or a thiol to produce oxido-alcoholato or oxido-thiolato molybdenum(VI) complexes with bis-dithiolene coordination [37].
Scheme 1.

(A) Chemical structure of the Tp* ligand. (B) HPyr+-facilitated conversion of [Tp*MoIVOCl(OPMe3)] (1) to [Tp*MoIVOCl(NCCH3)] (2).
In addition to involvement of protons in CEPT and removals of tightly bound terminal oxo ligands, contributions of hydrogen-bonding interactions to structures and properties of molybdenum-oxo and analogous complexes were also demonstrated in model systems [38–50]. In a few cases, such interactions were capable of directly facilitating OAT reactions mediated by molybdenum compounds [40, 42, 44, 47, 50]. For example, pyridinium (HPyr+) cation was previously found to be hydrogen-bonded to the Mo=O moiety of [HPyr]2[MoIVO(mnt)2] complex [42], which lengthens the Mo=O bond by 0.04 Ǻ. Interestingly, [HPyr]2[MoIVO(mnt)2] complex was more reactive towards oxidation by trimethylamine N-oxide, compared to [NEt4]2[MoIVO(mnt)2] complex [42]. Unfortunately, most of these examples exhibited a meager rate enhancement of less than 10-fold [40, 44, 47, 50]. In addition, detailed mechanistic evaluations of these systems have not been carried out to pinpoint which elementary step(s) of OAT reactions is/are accelerated by acids or hydrogen-bonding interactions.
In the current work, we report a novel reactivity that HPyr+ leads to 103-fold increase in the rate of phosphine oxide product liberation from [Tp*MoIVOCl(OPMe3)] (1; see Scheme 1B), a synthetic analog of [E′P] complex (product-bound molybdenum complex). This was previously shown to be the rate-limiting second step of the OAT reaction from [Tp*MoVIO2Cl] to PMe3 [51]. Using detailed experimental kinetic studies in tandem with combined DFT/coupled cluster computational studies, we further reveal molecular-level details of how the reaction route is altered by HPyr+ to account for the significantly accelerated product dissociation.
2. Results
2.1 Pyridinium (HPyr+) facilitates the conversion of 1 to 2
Previously, [Tp*MoIVOCl(OPR3)], an intermediate in the oxygen transfer reaction from cis-MoVIO2 to tertiary phosphines, was reported to cleanly decay into [Tp*MoIVOCl(NCCH3)] (2) in CH3CN [16, 17, 51–59]. Decreasing the steric hindrance of the phosphine stabilizes the MoIV–OPR3 species so that complex 1 was isolated. Therefore, this system represents a good synthetic analog to study the effects of acids in OAT reactions of molybdenum-oxo complexes relevant to the reductive half-reaction of SO.
In this work, we report that interestingly the reaction of 1 to 2 was greatly facilitated by the addition of HPyr+ as a weak acid (Scheme 1B). As shown in Figure 1, complex 1 was fairly stable and exhibited no sign of decay within the first 300 seconds in CH3CN at 10 °C. However, the addition of 10 equiv. HPyr+ caused a rapid near-quantitative conversion of 1 (λmax: 820 nm) to 2 (λmax: 720 nm) that was completed within the next 300 seconds. Isosbestic points were observed at 760, 365, and 338 nm. As a control experiment, the addition of 10 equiv. HPyr+ to 2 did not produce detectable spectral changes within the same period of time (Figure S1).
Figure 1.

(Top) 1.0 mM [Tp*MoIVOCl(OPMe3)] (1, monitored at 820 nm) decays to [Tp*MoIVOCl(NCCH3)] (2, monitored at 720 nm) in CH3CN; the reaction is facilitated by the addition of 10 equiv. pyridinium (HPy+) triflate at 330 s. (Bottom) UV-visible spectra for the HPy+-facilitated conversion of 1 to 2. Temperature: 10 °C.
2.2 Kinetic Studies
To elucidate the role of HPyr+ in this conversion, we carried out kinetic studies using time-resolved UV-visible spectroscopy. As shown in the top panel of Figure 2, the decay of 1 was accompanied by the concurrent formation of 2. No intermediates were observed for the entire process either in the presence or absence of HPyr+. Kinetic studies with 5 – 45 mM (5 – 45 equiv.) pyridinium triflate suggest that the decay of 1 (monitored at 820nm) or the formation of 2 (monitored at 720 nm) can be nicely fit with a single exponential function to afford the pseudo first-order rate constant (kobs). Plotting kobs against the concentration of HPyr+ shows a linear correlation with a second-order rate constant (k2) of 6.4(7) × 10−1 M−1s−1 at 10 °C (Figure 2, bottom).
Figure 2.

(Top) Decay of 1 (monitored at 820 nm; initial concentration 1.0 mM) and formation of 2 (monitored at 720 nm) in the presence of 10 equiv. HPyr+ in CH3CN at 10 °C. (Bottom) Plots of kobs(720) vs. added [HPyr+] for the conversion of 1.0 mM 1 to 2 in CH3CN at 10 °C. Error bars show one standard deviation of kobs from at least three independent measurements.
2.3 Eyring Plot
To provide further experimental evidence to probe mechanisms of the HPyr+-facilitated conversion, kinetic data over a temperature range of +15 °C to −15 °C were collected. The Eyring plot analysis of the temperature dependence of the kobs affords the activation parameters. ΔH‡ and ΔS‡ were determined to be +84(3) kJ/mol and +16(11) J/(mol • K), respectively (Figure 3 top panel). The ΔH‡ value is essentially identical to that of the solvolysis of [Tp*MoIVOCl(OPMe3)] in CH3CN in the absence of HPyr+ [+87(1) kJ/mol; see Table 1] [59]. However, the ΔS‡ values with vs. without HPyr+ are quite different (see Table 1). The negative ΔS‡ value of −31(4) J/(mol • K) for product liberation of 1 in the absence of HPyr+ is consistent with an associative interchange mechanism (Ia) [59]. This negative entropy of activation can be attributed to a loss of rotational and translational freedom of the incoming acetonitrile molecule in the transition state. Conversely, the HPyr+-facilitated conversion of 1 to 2 has a small positive value of ΔS‡, indicative of a dissociative interchange mechanism (Id).
Figure 3.

(Top) The Eyring plot for the conversion of 1.0 mM 1 to 2 in CH3CN in the presence of 10 equiv. HPyr+. Temperature range: −15 to +15 °C. Error bars show one standard deviation from at least three independent measurements. (Bottom) Correlation of kobs (monitored at 720 nm) with the acidities of various substituted pyridinium tosylates. Conditions: 1.0 mM 1 in CH3CN, 10 °C, 10 equiv. pyridinium, 2,6-lutidinium, or 2,4,6-collidinium tosylate. kobs of 3-bromopyridinium at 10 °C was extrapolated from an Eyring plot collected at a temperature range of -30 to 0 °C (Figure S2).
Table 1.
Comparison of activation parameters for the conversion of 1 to 2 in CH3CN.
| Conditions | ΔH‡ (kJ/mol) | ΔS‡ (J/(mol • K)) | References |
|---|---|---|---|
| With 10 mM HPyr+ | 84(3) | 16(11) | This work |
| No HPyr+ | 87(1) | −31(4) | [59] |
Based on the determined ΔH‡ and ΔS‡ values, ΔG‡ was estimated to be ~79 kJ/mol for HPyr+-facilitated 1-to-2 conversion at 298 K, compared to ~96 kJ/mol without HPyr+. The reduced ΔG‡ value translates to ~103 fold increase in the reaction rates, as 5.69 kJ/mol in ΔG‡ corresponds to a factor of ten in the rate constant [60]. The change of ΔG‡ is consistent with the significant increase in the kobs of 1-to-2 conversion from 1.1 × 10−5 s−1 in the absence of any acids [59] to 1.8 × 10−2 s−1 in the presence of 10 mM HPyr+ at 10 °C.
2.4 Substituted pyridinium ions with various acidities
Our next piece of experimental mechanistic evidence comes from kinetic studies using substituted pyridinium ions with different acidities to replace HPyr+. We synthesized and purified the tosylate salts of a series of substituted pyridinium ions with various acidities, including 3-bromopyridinium (pKa 9.64 in CH3CN), pyridinium (pKa 12.53 in CH3CN), 2,6-lutidinium (pKa 14.13 in CH3CN), and 2,4,6-collidinium (pKa 14.98 in CH3CN) tosylate [61]. Interestingly, a plot of log(kobs) versus the pKa values of these substituted pyridinium tosylate salts demonstrated a proportional decrease (Figure 3, bottom). The negative slope is consistent with an acid-facilitated reaction, with the most acidic 3-bromopyridinium ion in this series facilitating the 1-to-2 conversion at a reaction rate of ~104-fold faster than the least acidic 2,4,6-collidinium ion. Although 2,6-lutidinium and 2,4,6-collidinium are more sterically demanding than pyridinium, the linear correlation of log(kobs) versus pKa suggests that the electronic factor of pyridinium ions, rather than the steric factor, plays a major role in this pyridinium-facilitated conversion of 1 to 2.
2.5 Computational Studies
In order to gain deeper molecular-level understanding of this HPyr+-facilitated ligand exchange at Mo(IV) center, computational studies were carried out to shed further light on this fascinating transformation. To demonstrate the validity of the computational model, we first compared the calculated equilibrium geometry of [Tp*MoOCl(OPMe3)] with its available X-ray structure [52]. As shown in Figure 4, the structure calculated at the M06L/def2-SV(P)+PCM(MeCN) level of theory showed a similar conformation with the experimental structure, and the relative errors in bond lengths did not exceed 2%. The use of two other common density functionals B3LYP* and OPBE led to larger disagreement with the experimental structure. In order to achieve higher accuracy, we further refined the electronic energy by the single-point calculations at the LDPNO-CCSD(T)/def2-TZVP level of theory (see section 5.3 Computational Methods for further details). Next, we utilized the combined coupled cluster/DFT computational protocol to obtain the mechanistic insights into the product release step, as follows. ΔH‡ and ΔG‡ values of all pathways that were investigated computationally are summarized in Table 2.
Figure 4.

Comparison of the equilibrium geometry of [Tp*MoOCl(OPMe3)], obtained at the M06L/def2-SV(P)+PCM(MeCN) level of theory (A), and the corresponding X-ray structure (B) [52]. Hydrogen atoms are hidden for clarity.
Table 2.
Summary of computed ΔH‡ (ΔG‡) values of various reaction pathways in kJ/mol.
2.5.1 Id pathway in the absence of HPyr+
We first considered a possible dissociative interchange (Id) mechanism in the absence of HPyr+. The dissociation of the Mo–OPMe3 bond of complex 1 in the absence of HPyr+ led to a gradual increase in energy and did not show a transition state to afford the five coordinate [Tp*MoOCl] intermediate (Figure 5A). The reaction enthalpy and free energy of this step were calculated to be 104 and 93 kJ/mol (Figure 5C), respectively. The subsequent event of binding an acetonitrile molecule is exothermic. Therefore it could be concluded that the overall activation energy of this dissociative reaction route is associated with the formation of the [Tp*MoOCl] intermediate.
Figure 5.

Energetic diagrams for the dissociative interchange mechanism (Id) in absence (panel C) or presence (panel D) of HPyr+ cation, and structures of key intermediates including the five-coordinate [Tp*MoOCl] (panel A; appear in Id pathways shown in figures 5C and 5D) and [1•••HPyr+] (panel B; appear in Id pathway shown in figure 5D). The energy diagrams were constructed using a combination of coupled cluster and DFT approaches (see section 5.3 Computational Methods for details). The numbers denote the enthalpies of compounds relative to the reactant, and numbers in parentheses denote the corresponding Gibbs free energy changes (kJ/mol).
2.5.2 Id pathway in the presence of HPyr+
In the presence of HPyr+, our computational results reveal that HPyr+ initially binds to the oxygen atom of the Me3PO ligand of complex 1. The equilibrium geometry of the resulting complex (denoted as [1•••HPyr+] complex) has a close-to-linear arrangement of the O•••H•••N atoms (169.1°) and an O•••H distances of 180.1 pm (Figure 5B), which are characteristic of hydrogen bonding interaction. An additional relaxed scan calculation, in which the O•••H distance was gradually increased while other structural parameters were fully optimized, demonstrated lack of a maximum (i.e. transition state) between the conversion of 1 and [1•••HPyr+] species.
Our CCSD(T)//M06L calculations further show that, as a result of HPyr+ bound to Me3PO, the dissociation of the Mo–OPMe3 bond requires less energy to afford the [Tp*MoOCl] intermediate (i.e., ΔH = 74 kJ/mol, ΔG = 58 kJ/mol; see Figure 5D), compared with the Id pathway without HPyr+. In other words, the interaction with HPyr+ makes Me3PO ligand to be a better leaving group.
To shed light on whether the HPyr+-facilitated OPMe3 product dissociation in the Id pathway entails a complete proton transfer or a hydrogen-bonding interaction, we also explored the reaction mechanism in which complex 1 becomes protonated at the oxygen atom of the Mo(IV)-bound OPMe3 group, with the proton donated from pyridinium cation. The complete proton transfer step was found to be highly endothermic (ΔG = 112.2 kJ/mol, ΔH = 167.3 kJ/mol), requiring more energy (ΔΔG = 32 kJ/mol and ΔΔH = 68 kJ/mol) than that needed for the hydrogen-bond facilitated cleavage of the Mo—OPMe3 bond via the Id-pathway shown in Figure 5D. This result is consistent with the very low basicity of the Mo(IV)-bound OPMe3 moiety in 1, with its computed pKa value to be 20.4 and 16.2 units lower than that of pyridine and free Me3PO, respectively (calculations were based on the equation ; see Scheme S12). These findings indicate that a complete protonation of the Mo–OPMe3 moiety of complex 1 is unfavorable compared to the hydrogen-bond facilitated Id pathway presented in Figure 5D.
2.5.3 Ia pathways in the absence of HPyr+
For the associative interchange Ia mechanism, several pathways could be envisioned depending on the orientation of the acetonitrile molecule attacking the Mo center. In one possible scenario (denoted as Ia-NNO pathway), an acetonitrile molecule approaches the Mo atom from the octahedral face formed by two Tp* nitrogen atoms and the oxygen atom of the phosphine oxide ligand (Figure 6A). Our computational results suggest that the attack of the acetonitrile molecule leads to the dissociation of the Mo–OPMe3 bond and formation of Mo–NCCH3 bond in one elementary reaction step via a hepta-coordinated transition state. The activation enthalpy and free energy for this reaction were equal to 89 and 90 kJ/mol, respectively (Figure 6B), in excellent agreement with the experimental kinetic studies (i.e. 87 and 96 kJ/mol; T = 298 K).
Figure 6.

Depiction of the attacking orientation for acetonitrile molecule (A), and the corresponding free energy diagram (B) in the Ia-NNO mechanism in the absence of HPyr+. The numbers denote the enthalpies of compounds relative to the reactant, and numbers in parentheses denote the corresponding Gibbs free energy changes (kJ/mol).
Our DFT calculations also revealed another possible Ia pathway (denoted as Ia-ClOO pathway), in which an acetonitrile molecule approaches the Mo atom from the octahedral face formed by the chlorine and two oxygen donors (Figure 7A). An unusual intermediate with hepta-coordinated molybdenum center was formed as a result (ΔH = 97 kJ/mol, ΔG = 98 kJ/mol, see Figures 7B and 7C). Subsequent cleavage of Mo–OPMe3 bond requires additional 22 kJ/mol of free energy, thus constituting an overall free energy barrier of 120 kJ/mol. The barrier is 30 kJ/mol higher than that of the Ia-NNO pathway in the absence of HPyr+ (Figure 6B). Our finding that Ia-NNO is favoured over Ia-ClOO reaction route is consistent with previous calculations by Basu, Hall, and co-workers that reported the preferred direction of CH3CN attacking [Tp iPrMoIVO(OPMe3)(OPh)] via the Ia mechanism [55].
Figure 7.

Depiction of the attacking orientation for acetonitrile molecule (A), the structure of the resulting seven-coordinated molybdenum intermediate (B), and the corresponding free energy diagram (C) of the Ia-ClOO mechanism in the absence of HPyr+. The numbers denote the enthalpies of compounds relative to the reactant, and numbers in parentheses denote the corresponding Gibbs free energy changes (kJ/mol).
In addition to Ia-NNO and as Ia-ClOO pathways, we also considered additional Ia pathways with different directions by which the acetonitrile molecule approaches the molybdenum center. Our computational results suggest that other pathways have significantly higher activation barriers than the Ia-NNO pathway (see the Supporting Information for details), and in some cases side products are produced. We therefore conclude that these pathways are non-competitive with the dominant Ia-NNO reaction mechanism for reaction of 1 to 2 in the absence of HPyr+.
2.5.4 Ia pathways in the presence of HPyr+
As HPyr+ binds to the oxygen atom of the Me3PO ligand of 1 (Figure 5B), the original binding site for the attacking molecule of MeCN via the Ia-NNO pathway becomes sterically more congested. We localized the corresponding transition state and found that reaction enthalpy and free energy to be 95 and 96 kJ/mol, respectively (Figure 8C). Compared with the Ia-NNO pathway without HPyr+ (Figure 6B), the significantly increased activation barrier here is attributed to steric hindrance between HPyr+ and incoming CH3CN molecule.
Figure 8.

Depiction of the attacking orientation for acetonitrile molecule (A), the structure of the resulting seven-coordinated molybdenum transition state (B), and the corresponding free energy diagram (C) in the Ia-NNO mechanism in the presence of HPyr+. The numbers denote the enthalpies of compounds relative to the reactant, and numbers in parentheses denote the corresponding Gibbs free energy changes (kJ/mol).
Conversely in the Ia-ClOO pathway, the incoming acetonitrile molecule attacks the Mo center from a second octahedral face different from the one that is blocked by the bound HPyr+ (Figure 9A). We therefore hypothesize that this pathway might be facilitated by the presence of HPyr+. Indeed, our CCSD(T)//M06L calculations showed that the presence of HPyr+ caused a significant drop in the activation enthalpy from 104 kJ/mol (Figure 7C) to 60 kJ/mol (Figure 9C) for the Ia-ClOO pathway. In fact, Ia-ClOO pathway has lower free energy barrier (64 kJ/mol; see Figure 9C) than Ia-NNO route (96 kJ/mol; see Figure 8C) in the presence of HPyr+. However, it should be noted that Ia-ClOO mechanism has an intrinsic penalty relative to the Id mechanism, as shown in the comparison of overall free energy barrier in Figure 7C (120 kJ/mol for Ia-ClOO pathway) vs. Figure 5C (93 kJ/mol for Id), which somewhat counterpoises the stabilization exerted by HPyr+ cation. Due to this effect, the Ia-ClOO pathway is unfavourable in comparison with the Id mechanism (Figure 5D) both in the absence and presence of HPyr+.
Figure 9.

Depiction of the attacking orientation for acetonitrile molecule (A), the structure of the resulting seven-coordinated molybdenum intermediate (B), and the corresponding free energy diagram (C) in the Ia-ClOO mechanism in the presence of HPyr+. The numbers denote the enthalpies of compounds relative to the reactant, and numbers in parentheses denote the corresponding Gibbs free energy changes (kJ/mol).
2.5.5. Alternative pathways
Finally, we explored alternative reaction routes in which HPyr+ binds initially to the oxo ligand of 1, rather than the phosphine oxide ligand. We first considered such an alternative Id pathway. For binding step of HPyr+ to the oxo ligand of 1, enthalpy and free energy were 2 and 11 kJ/mol more favourable, respectively, than those for coordination of HPyr+ to the oxygen atom in trimethylphosphine oxide ligand. However, lack of stabilization of the leaving phosphine oxide leads to large activation enthalpy (84 kJ/mol, see Figure S6) and free energy (74 kJ/mol) to produce 2 as product, thus making this reactive pathway non-competitive with the favoured Id mechanism in which HPyr+ binds to the oxygen of the trimethylphosphine oxide moiety.
We also inspected if the coordination of HPyr+ to the oxygen of the Mo=O moiety could accelerate the solvolysis via an Ia reaction route, and observed a moderate stabilization of the transition state, which was not sufficient for making this reaction more favourable than the Id mechanism discussed previously (i.e., compare activation energies in Figure 5B vs Figure S7). These findings indicate that coordination of HPyr+ to the oxygen atom of the Mo=O moiety does not facilitate the solvolysis of 1 to afford 2.
3. Discussion
3.1 Reaction Mechanism of HPyr+-facilitated 1-to-2 conversion
As shown in the Result section, the decay of 1 (monitored at 820 nm) occurred simultaneously with the formation of 2 (monitored at 720 nm), and isosbestic points were found at 760, 365, and 338 nm. No other intermediates were observed during the reaction. The time courses of A(820) and A(720) followed pseudo first-order kinetic behaviour in the presence of 5–45 equiv. HPyr+. The formation rate of 2 is first-order with respect to [HPyr+], and saturation kinetics is not observed for the range of [HPyr+] studied. Activation parameters are consistent with a dissociative interchange Id mechanism. While the 1-to-2 conversion operates via an associative interchange Ia reaction route in the absence of HPyr+, the presence of HPyr+ switches it to a dissociative interchange Id pathway, as evidenced by the change in ΔS‡ (Table 1). On the basis of our accumulated experimental data, we suggest a reaction mechanism for the HPyr+-facilitated conversion from 1 to 2, where the OPMe3 ligand of complex 1 dissociates via Id mechanism with the help of HPyr+ in a rate-limiting bimolecular step, followed by rapid coordination of an incoming CH3CN molecule to produce 2 (Scheme 2).#
Scheme 2.

Proposed reaction mechanism for HPyr+-facilitated conversion from 1 to 2.
The experimental results are corroborated by our combined coupled cluster/DFT computations, which provide further insights to validate the roles played by HPyr+ and supplement key mechanistic understandings. In the absence of HPyr+, our computational results confirm the Ia-NNO (Figure 6) pathway has the lowest activation energy among all pathways that were considered, consistent with published experimental data [59]. Our computational results further show that acidic HPyr+ facilitates the Mo–OPMe3 bond scission by making OPMe3 to be a better leaving group (Figure 5). This beneficial effect of HPyr+ to lower activation energy for OPMe3 dissociation could potentially be manifested in both Ia and Id pathways. However, in the Ia-NNO pathway, the original attacking site of CH3CN is now occupied by the HPyr+ molecule, raising the activation energy in this route due to the unfavourable steric hindrance (Figure 8). Therefore, Id becomes the energetically preferred reaction route for the conversion of 1 to 2 in the presence of HPyr+ (Figure 5D). In addition to confirming Id mechanism in the most favourable reaction route, our computational results also reveal molecular-level details for this route in the presence of HPyr+. Specifically, HPyr+ initially becomes hydrogen-bonded to the oxygen atom of the Mo(IV)-OPMe3 moiety (Figure 5B) after the co-solvated encounter complex is formed. The binding of HPyr+ promotes the rate-limiting Mo–OPMe3 bond scission that releases of the phosphine oxide product from the Mo(IV) center and affords a five-coordinate [Tp*MoIVOCl] intermediate. This species rapidly reacts with an incoming CH3CN molecule to generate 2 as product.
Interestingly, our computational results reveal another possible Ia reaction route that we denote as the Ia-ClOO pathway, with the CH3CN approaching the Mo center from a direction different from that in the Ia-NNO pathway. While this Ia-ClOO pathway has a higher activation energy than Ia-NNO reaction route in the absence of HPyr+ (Figure 7), the presence of HPyr+ makes the Ia-ClOO pathway energetically more favourable than the Ia-NNO reaction route (Figures 8 and 9). However, Id mechanism still has the lowest activation energy compared to both Ia-ClOO and Ia-NNO pathways in the presence of HPyr+, consistent with our experimental data.
Alternative reaction routes involve initial binding of HPyr+ to the oxygen atom of the Mo(IV)=O moiety to afford a Mo(IV)=O•••H+•••Py species. Our computational studies suggest that while this binding could be facile, these alternative reaction routes have sizably higher activation energies to afford the product. Therefore these alternative routes are considered to be non-competitive with the Id pathway that has the lowest activation energy.
3.2 Potential relevance to OAT mechanisms of molybdoenzymes and related model complexes
In OAT mediated by molybdoenzymes, an enzyme-substrate [ES] complex (Michaelis-Menten complex) is formed from a bimolecular reaction of enzyme (E) and substrate (S) [62, 63]. The newly formed [ES] complex then undergoes a unimolecular reaction to afford enzyme-product [E′P] complex, which in turn produce the product P along with E′. For the reductive half-reaction of SO, the catalysis is initiated by the attack of sulfite substrate on the oxidized form of enzymes (MoVI) to generate a MoIV–O–SO3 intermediate. Subsequent cleavage of Mo–O bond releases the sulfate product [32, 64]. Using phosphines as proxy substrates, MoIV–O–PR3 intermediates (a synthetic counterpart of the [E′P] complex) were previously trapped from OAT reactions mediated by [MoVIO2] model complexes supported by Tp* ligand framework [51, 53–55, 57, 59]. Such intermediates then underwent Mo–O bond cleavage to release the OPR3 product and generate a MoIV-solvent complex, analogous to the following process in the reductive half-reaction of SO:
| Eq (1) |
Our results in this work demonstrate that the reaction rate of the product release from molybdenum(IV) centers can be dramatically increased by a weak acid like HPyr+, as the mechanisms of the product release step is now switched to:
| Eq (2) |
Our computational studies provide additional insights that it is the initial hydrogen bonding interaction between the HPyr+ and the oxygen atom of phosphine oxide ligand that makes phosphine oxide ligand to be a better leaving group and facilitates the scission of the Mo–OPMe3 bond in a rate-limiting step. As a hydrogen bonding interaction between a nearby acidic residue (or a water molecule) and the nascent SO42− product bound to molybdenum center could also be present in SO, we cannot rule out the intriguing possibility that a mechanism conceptually similar to that of our system is operative to facilitate product liberation in the reductive half-reaction of SO and potentially other molybdoenzymes that transfer an oxygen atom to their substrates. For human SO, Rajagopalan et al. has concluded the sulfate product release and/or the first -electron transfer step from MoIV to heme is likely the rate-limiting step of , as the formation rate of the enzyme-substrate complex was shown to be faster (~1900 s−1 at 10 °C) than (~70 s−1 at 25 °C) at pH of 7.0 [64, 65]. Y343F variant exhibited a much impaired (1.75 s−1 at pH 7.0) that was dependent on pH, unlike the wild type protein. As the rate of the individual reaction step typically cannot be easily determined in enzymes including SO, it remains to be conclusively established whether the weakly acidic Y343 residue (possibly in coordination with the nearby Arg138 residue), or a likely water molecule in the binding pocket of molybdenum cofactor (e.g., in the case of Y343F mutant [65]) was utilized to facilitate product release, analogous to the fundamental chemistry that was demonstrated to be very chemically feasible by our model system.
Finally, we want to caution our readers some potential concerns to extend our results from a model system onto SO and other molybdoenzymes. The proxy substrate used in our system was different from the biological substrate in SO. Admittedly, phosphine proxy substrates will not accurately mimic the full electronic structures of biological substrates. Considering the difference in frontier orbital interactions of proxy vs. biological substrates with a {MoVIO2} center [23], this is a bigger concern when studying the attack of substrate on {MoVIO2} center (i.e., E + S → [EP] in the reductive half-reaction of SO), which is not the focus of the present work. On the other hand, we propose that a similar acid-facilitated product release could be invoked in the reductive half-reaction of SO, because a hydrogen bonding interaction that primes the product to be a better leaving group is also likely to be present in active sites of SO. Computational studies are currently underway to elucidate whether such an acid-facilitated product release is indeed operative in the reductive half-reaction of SO.
4. Conclusions
In this work, we report that a weak acid HPyr+ causes a 103-fold increase in the conversion rate of 1 to 2 by altering the operative mechanism of this step. Accumulated pieces of experimental mechanistic evidence include: 1) the decay of 1 was concurrent with the formation of 2, without other intermediates observed in the conversion; 2) the reaction was first order with regard to both [1] and [HPyr+]; 3) The Eyring plot provided a positive ΔS‡, suggesting that the operative mechanism was switched from associative interchange Ia mechanism in the absence of an acid, to dissociative interchange Id mechanism in the current scenario; 4) kobs was dependent upon the acidity of substituted pyridinium ions, with up to 104-fold rate enhancement for the most acidic 3-bromopyridinium ion in this series. Therefore we propose a mechanism to account for the novel reactivity (Scheme 2), with HPyr+ facilitating the release of the OPMe3 ligand of complex 1 via Id mechanism in a rate-limiting bimolecular step, followed by rapid attack of a CH3CN molecule to afford 2. Comprehensive computational studies were then carried out to investigate molecular-level details of reaction trajectories and compare activation energies of different pathways, including Id, Ia, and possible alternative routes in the absence or presence of HPyr+. The computational results further corroborate experimental conclusions that the dominant route of the 1-to-2 conversion is switched from a slower Ia pathway when HPyr+ is not in the vicinity, to a faster Id reaction route with the help of HPyr+. This is because HPyr+ lowers the activation barrier of OPMe3 ligand dissociation by making OPMe3 into a better leaving group.
To the best of our knowledge, this is the first detailed demonstration of such a process in molybdenum-oxo chemistry. Our studies show that the fundamental ligand exchange chemistry, which is the rate-limiting step of OAT reactions in some systems [51], can become very feasible and greatly accelerated with the help of a weak acid rendering hydrogen bonding interaction with the oxygen atom of the leaving group. Our finding of speeding up product release step in OAT reactions could be important for designing better-performing metal catalysts for these reactions. Such a process could also be invoked in molybdenum oxotransferases, potentially expanding our knowledge on how acids could be used to facilitate the transportation of small molecule substrates or products into or away from the molybdenum active sites. The acid-facilitated product release demonstrated in the current model system prompts us to computationally investigate whether a similar mechanism is indeed invoked to efficiently expel the product molecule from the molybdenum center in the reductive half-reaction of SO as an on-going project. We will also experimentally study how other previously unrecognized factors modulate OAT reactivities using small molecule Mo model complexes.
5. Materials and Methods
5.1 Materials and Syntheses
All reagents, including anhydrous grade solvents (acetonitrile, pentane, and toluene), were purchased from commercial sources such as Sigma-Aldrich Co., and used as received unless stated otherwise. Unless otherwise specified, the synthesis of air-sensitive complexes utilized standard anaerobic and anhydrous Schlenk line techniques or an Mbraun glovebox with a high-purity research grade N2 atmosphere. The KTp* ligand [KTp* = potassium tris(3,5-dimethyl-1-pyrazolyl)borohydride] and Tp*MoVIO2Cl complex were prepared by literature methods [66][17]. Pyridinium triflate was purchased from TCI America, and used without further purification.
[Tp*MoIVOCl(OPMe3)] was prepared by following a literature method [54]. Specifically, 20 mg Tp*MoVIO2Cl (0.043 mmol) was dissolved in 2 mL cold anhydrous CH3CN at −20 °C (or in 2 mL anhydrous toluene at room temperatures) inside a glovebox. If CH3CN is used as the solvent, the armor beads-filled cold well was used to keep the reactions at −20 °C. 1.5 equiv. PMe3 was added to the solution, and the reaction mixture was stirred until all solids were dissolved and a dark brown solution was resulted. After evaporating all the acetonitrile or toluene solvent, 1 mL cold toluene and 10 mL cold pentane were added to precipitate the [Tp*MoIVOCl(OPMe3)] product. The product was collected by filtration at low temperatures, washed with cold pentane, and dried in vacuum. Yield: 9 mg (~40%).
p-Toluenesulfonate (tosylate) salts of pyridinium, 2,6-lutidinium, 2,4,6-collidinium, and 3-bromopyridinium were synthesized by slight modifications of published procedures [67]. The general procedure is as follows: to 1 mL CH3CN solution of 2.15 mmol (substituted) pyridine, 0.37 g (2.15 mmol) tosylic acid in 2 mL CH3CN solution was added drop by drop. The solution was allowed to stir at room temperatures for 5 minutes until the heat generated by this reaction dissipated. ~5 mL Et2O was added, and the resulting white precipitate was collected by vacuum filtration and washed with Et2O three times. The yield was 0.31 g (58%) for pyridinium tosylate, 0.36 g (60%) for 2,6-lutidinium tosylate, 0.36 g (57%) for 2,4,6-collidinium tosylate, and 0.51 g (71%) for 3-bromopyridinium tosylate, respectively.
Pyridinium tosylate. 1H NMR (CDCl3): δ 9.02 (d, 2H, HPyr+ o-CH), 8.43 (t, 1H, HPyr+ p-CH), 7.97 (t, 2H, HPyr+ m-CH), 7.84 (d, 2H, tosylate CH), 7.20 (d, 2H, tosylate CH), 2.36 (s, 3H, tosylate CH3).
2,6-Lutidinium tosylate. 1H NMR (CDCl3): δ 8.09 (t, 1H, 2,6-lutidinium CH), 7.84 (d, 2H, tosylate CH), 7.44 (d, 2H, 2,6-lutidinium CH), 7.20 (d, 2H, tosylate CH), 2,90 (s, 6H, 2,6-lutidinium CH3), 2.37 (s, 3H, tosylate CH3).
2,4,6-Collidinium tosylate. 1H NMR (CDCl3): δ 7.84 (d, 2H, tosylate CH), 7.20 (s, 2H, 2,4,6-collidinium CH), 7.19 (d, 2H, tosylate CH), 2.52 (s, 6H, 2,4,6-collidinium o-CH3), 2.52 (s, 3H, 2,4,6-collidinium p-CH3), 2.37 (s, 3H, tosylate CH3).
3-Bromopyridinium tosylate. 1H NMR (CDCl3): δ 9.03 (s, 1H, 3-bromopyridnium 2-CH), 8.98 (d, 1H, 3-bromopyridinium 6-CH), 8.48 (d, 1H, 3-bromopyridnium 4-CH) 7.82 (m, 3H, tosylate CH and 3-bromo-pyridnium 5-CH), 7.21 (d, 2H, tosylate CH), 2.37 (s, 3H, tosylate CH3).
5.2 Physical Methods
UV-visible spectra were collected on a HP8453 diode-array spectrometer equipped with a cryostat from Unisoku Scientific Instruments (Osaka, Japan) for low temperature control. Progress of the conversion from [Tp*MoIVOCl(OPMe3)] to [Tp*MoIVOCl(NCCH3)] in CH3CN was followed by monitoring the changes in the absorbance at 820 nm for the decay of 1 and the absorbance at 720 nm for the formation of 2, with an isobestic point at 760 nm (Figure 1b). Plots of reaction progress [A(820)-A(760) and (A720)-A(760)] against time were fit to pseudo-first-order rate equations that provided good fits for determination of observed rate constants (kobs). Errors associated with reaction rates came from at least three independent trials. Second order rate constants (k2) were determined from the slope of the linear plots of kobs vs. [HPyr+] (Origin 2017 b9.4.0220). Activation parameters were derived from a standard Eyring plot. pKa values of pyridinium, 2,6-lutidinium, and 2,4,6-collidinium in acetonitrile used in Figure 3 bottom panel were taken from the absolute pKa values summarized in Table 1 of Ref 61; pKa value of 3-bromopyridinium was converted from its pKa in water by following the equation: pKa(MeCN) = 6.04 + 1.269 pKa(H2O) [61].
5.3 Computational Methods
Electronic structure calculations were performed with the Gaussian 09 package, revision D01 or E01 [68]. For the density functional theory (DFT) calculations we used M06L functional [69] and def2-SV(P) basis set [70] (for Mo, the 28 core electrons were treated using the effective core potential approach) [71]. Solvent effects were included using the implicit integral equation formalism polarizable continuum model (IEF-PCM, also referred as PCM) [72–76] with the acetonitrile solvent parameters. In all DFT calculations, ultrafine Lebedev’s grid was used with 99 radial shells per atom and 590 angular points in each shell. The wave function stability tests [77] were performed to ensure that the closed-shell singlet states were the solutions with the lowest energy. Tight cutoffs on forces and atomic displacement were used to determine convergence in geometry optimization procedure. Hessians were calculated for the optimized structures to confirm absence of imaginary frequencies or presence of a single imaginary frequency for minima and transition states, respectively. Additional single-point DFT calculations were undertaken for the optimized equilibrium structures to confirm that triplet electronic state is sufficiently high in energy (i.e. ~100 kJ/mol) as compared with the ground electronic state thus ruling out the possibility of the spin crossover for the reaction mechanism.
The obtained enthalpy and free energy of binding of HPyr+ to 1 were further refined by the single-point calculations at the DLPNO-CCSD(T)/def2-TZVP level [78] using Orca v4.0 program [79] according to the following scheme (eqs 3 and 4) [80]:
| (Eq. 3) |
| (Eq. 4) |
where Egas and Esolv are the electronic energies in gas- and solvent phase from the DFT calculations, and enthalpic/entropic corrections Hcorr and Gcorr were calculated by DFT using the standard approximations of quantum harmonic oscillator (for the vibrational component of the partition functions), rigid rotor (for the rotational component), and particle-in-the box (for the translational component) for T = 298.15 K and P = 1 atm.
The absence of transition states in the binding of HPyr+ to 1 (Figure 5B), in the release of Me3PO, and in subsequent binding of acetonitrile (for the Id mechanism in Figure 5A) was confirmed by the relaxed scan procedure, see Figure S5.
The reaction complex approach was used for the evaluation of the enthalpy/free energy changes along reaction coordinate in order to minimize the error in the calculated entropy contribution [80–84].
Supplementary Material
Figure S1. UV-visible spectra monitored at 720 nm after the addition of 10 equiv. pyridinium triflate to 1.0 mM [Tp*MoIVOCl(NCCH3)] (2) in acetonitrile at 10 °C as a control experiment.
Figure S2. The Eyring plot for the conversion of 1.0 mM [Tp*MoIVOCl(OPMe3)] (1) to 2 in CH3CN in the presence of 10 equiv. 3-bromopyridinium tosylate. Temperature range: −30 to 0°C. ΔH‡ = 68(2) kJ/mol. ΔS‡ = −14(7) J/mol. Error bars show one standard deviation from at least three independent measurements.
Figure S3. The Ia-NNCl mechanism that leads to the side products.
Figure S4. An illustration of the direction of CH3CN attack in the Ia-NNOxo mechanism.
Figure S5. Electronic energy profiles for the dissociation of the MeCN···Mo bond in [Tp*MoOCl(MeCN)] (A), Me3PO···Mo bond in [Tp*MoOCl(Me3PO)] (B), and HPyr+···O(PMe3) bond in [Tp*MoOCl(Me3PO)(HPyr+)] (C), calculated using the relaxing scan technique at the M06L/def2-SV(P)+PCM(MeCN) level of theory.
Figure S6. Alternative Id mechanism in which HPyr+ is coordinated to the oxygen atom of the oxo ligand.
Figure S7. Alternative Ia mechanism in which HPyr+ is coordinated to the oxygen atom of the Mo=O moiety.
Acknowledgments
This work was supported by the U.S. National Institute of Health (project number: 1SC2GM121183-01), the Extreme Science and Engineering Discovery Environment (XSEDE; project number TG-CHE170004), and New Mexico State University.
Footnotes
The overall conversion of 1-to-2 was observed and treated as pseudo first-order kinetics, rather than saturation kinetics, with -d[1]/dt equal to d[2]/dt at any time of this reaction. No other intermediates, including [1•••HPyr+], accumulated to detectable amount. Therefore, the acid-base pre-equilibrium step of HPyr+ binding to the oxygen atom of the Mo-OPMe3 moiety after the co-solvated encounter complex is formed must be rapid, with [1•••HPyr+]/[1] ≪ 1 under the current experimental conditions.
Notes and references
- 1.Pushie MJ, Cotelesage JJ, George GN. Molybdenum and tungsten oxygen transferases--and functional diversity within a common active site motif. Metallomics. 2014;6:15–24. doi: 10.1039/c3mt00177f. [DOI] [PubMed] [Google Scholar]
- 2.Schwarz G, Mendel RR, Ribbe MW. Molybdenum cofactors, enzymes and pathways. Nature. 2009;460:839–847. doi: 10.1038/nature08302. [DOI] [PubMed] [Google Scholar]
- 3.Feng C, Tollin G, Enemark JH. Sulfite oxidizing enzymes. Biochim Biophys Acta - Proteins Proteomics. 2007;1774:527–539. doi: 10.1016/j.bbapap.2007.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hille R. The molybdenum oxotransferases and related enzymes. Dalt Trans. 2013;42:3029–3042. doi: 10.1039/c2dt32376a. [DOI] [PubMed] [Google Scholar]
- 5.Kappler U, Enemark JH. Sulfite-oxidizing enzymes. J Biol Inorg Chem. 2015;20:253–264. doi: 10.1007/s00775-014-1197-3. [DOI] [PubMed] [Google Scholar]
- 6.Maia LB, Moura JJG. Nitrite reduction by xanthine oxidase family enzymes: A new class of nitrite reductases. J Biol Inorg Chem. 2015;16:443–460. doi: 10.1007/s00775-010-0741-z. [DOI] [PubMed] [Google Scholar]
- 7.Rothery RA, Weiner JH. Shifting the metallocentric molybdoenzyme paradigm: the importance of pyranopterin coordination. J Biol Inorg Chem. 2015;20:349–372. doi: 10.1007/s00775-014-1194-6. [DOI] [PubMed] [Google Scholar]
- 8.Majumdar A. Structural and functional models in molybdenum and tungsten bioinorganic chemistry: description of selected model complexes, present scenario and possible future scopes. Dalton Trans. 2014;43:8990–9003. doi: 10.1039/c4dt00631c. [DOI] [PubMed] [Google Scholar]
- 9.Schulzke C, Ghosh AC. Molybdenum and tungsten oxidoreductase models. Eur J Inorg Chem. 2011:1189–1199. [Google Scholar]
- 10.Heinze K. Bioinspired functional analogs of the active site of molybdenum enzymes: Intermediates and mechanisms. Coord Chem Rev. 2015;300:121–141. [Google Scholar]
- 11.Chen GJ, Mcdonald JW, Newton WE. Synthesis of Mo(IV) and Mo(V) Complexes Using Oxo Abstraction by Phosphines. Mechanistic Implications. Inorg Chem. 1976;15:2612–2615. [Google Scholar]
- 12.Reynolds MS, Berg JM, Holm RH. Kinetics of Oxygen Atom Transfer Reactions Involving Oxomolybdenum Complexes. General Treatment for Reaction with Intermediates oxo-bridged molybdenum(V) dimer formation. Inorg Chem. 1984;23:3057–3062. [Google Scholar]
- 13.Sugimoto H, Tatemoto S, Suyama K, et al. Dioxomolybdenum(VI) complexes with ene-1,2-dithiolate ligands: Synthesis, Spectroscopy, and oxygen atom transfer reactivity. Inorg Chem. 2009;48:10581–10590. doi: 10.1021/ic901112s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Das SK, Chaudhury PK, Biswas D, Sarkar S. Modeling for the Active Site of Sulfite Oxidase: Synthesis, Characterization, and Reactivity of [MoVIO2(mnt)2]2− (mnt2− = 1,2-Dicyanoethylenedithiolate) J Am Chem Soc. 1994;116:9061–9070. [Google Scholar]
- 15.Berg JM, Holm RH. Synthetic Approach to the Mononuclear Active Sites of Molybdoenzymes: Catalytic Oxygen Atom Transfer Reactions by Oxomolybdenum(IV, VI) Complexes with Saturation Kinetics and without Molybdenum(V) Dimer Formation. J Am Chem Soc. 1984;106:3035–3036. [Google Scholar]
- 16.Smith PD, Millar AJ, Young CG, et al. Detection, Isolation, and Characterization of Intermediates in Oxygen Atom Transfer Reactions in Molybdoenzyme Model Systems. J Am Chem Soc. 2000;122:9298–9299. [Google Scholar]
- 17.Roberts SA, Young CG, Kipke CA, et al. Dioxomolybdenum(VI) Complexes of the Hydrotris(3,5-dimethyl-1-pyrazolyl) borate Ligand. Synthesis and Oxygen Atom Transfer Reactions. Inorg Chem. 1990;29:3650–3656. [Google Scholar]
- 18.Stelzig L, Kotte S, Krebs B. Molybdenum complexes with tridentate NS2 ligands. Synthesis, Crystal Structures and Spectroscopic Properties. Dalt Trans. 1998:2921–2926. [Google Scholar]
- 19.Ducrot A, Scattergood B, Coulson B, et al. Electronic Fine-Tuning of Oxygen Atom Transfer Reactivity of cis-Dioxomolybdenum(VI) Complexes with Thiosemicarbazone Ligands. Eur J Inorg Chem. 2015:3562–3571. [Google Scholar]
- 20.Zwettler N, Judmaier ME, Strohmeier L, et al. Oxygen activation and catalytic aerobic oxidation by Mo(IV)/(VI) complexes with functionalized iminophenolate ligands. Dalton Trans. 2016;45:14549–60. doi: 10.1039/c6dt01692h. [DOI] [PubMed] [Google Scholar]
- 21.Thapper A, Deeth RJ, Nordlander E. Computer modeling of the oxygen-atom transfer reaction between hydrogen sulfite and a molybdenum(VI) dioxo complex. Inorg Chem. 1999;38:1015–1018. doi: 10.1021/ic980646g. [DOI] [PubMed] [Google Scholar]
- 22.Thomson LM, Hall MB. A Theoretical Study of the Primary Oxo Transfer Reaction of a Dioxo Molybdenum(VI) Compound with Imine Thiolate Chelating Ligands: A Molybdenum Oxotransferase Analogue. J Am Chem Soc. 2001;123:3995–4002. doi: 10.1021/ja003258y. [DOI] [PubMed] [Google Scholar]
- 23.Pal K, Chaudhury PK, Sarkar S. Structure of the Michaelis complex and function of the catalytic center in the reductive half-reaction of computational and synthetic models of sulfite oxidase. Chem - An Asian J. 2007;2:956–964. doi: 10.1002/asia.200700020. [DOI] [PubMed] [Google Scholar]
- 24.Hernandez-Marin E, Ziegler T. Theoretical study of the oxidation reaction and electron spin resonance parameters involving sulfite oxidase. Inorg Chem. 2009;48:1323–1333. doi: 10.1021/ic801158t. [DOI] [PubMed] [Google Scholar]
- 25.Metz S, Thiel W. Theoretical studies on the reactivity of molybdenum enzymes. Coord Chem Rev. 2011;255:1085–1103. [Google Scholar]
- 26.Hille R. The reaction mechanism of oxomolybdenum enzymes. BBA - Bioenerg. 1994;1184:143–169. doi: 10.1016/0005-2728(94)90220-8. [DOI] [PubMed] [Google Scholar]
- 27.Brody M, Hille R. The reaction of chicken liver sulfite oxidase with dimethylsulfite. Biochim Biophys Acta. 1995;1253:133. doi: 10.1016/0167-4838(95)00194-4. [DOI] [PubMed] [Google Scholar]
- 28.Hille R. The Mononuclear Molybdenum Enzymes. Chem Rev. 1996;96:2757–2816. doi: 10.1021/cr950061t. [DOI] [PubMed] [Google Scholar]
- 29.Basu P, Burgmayer SJN. Recent developments in the study of molybdoenzyme models. J Biol Inorg Chem. 2015;20:373–383. doi: 10.1007/s00775-014-1228-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Langford CH, Gray HB. Ligand substitution processes. W.A. Benjamin, Inc; New York, New York: 1966. [Google Scholar]
- 31.Fukuzumi S, Ohkubo K, Lee YM, Nam W. Lewis Acid Coupled Electron Transfer of Metal-Oxygen Intermediates. Chem - A Eur J. 2015;21:17548–17559. doi: 10.1002/chem.201502693. [DOI] [PubMed] [Google Scholar]
- 32.Maia LB, Moura I, Moura JJG. Molybdenum and Tungsten-Containing Enzymes. In: Hille R, Schulzke C, Kirk ML, editors. Molybdenum Tungsten Enzym Biochem. The Royal Society of Chemistry; Cambridge, UK: 2016. pp. 1–80. [Google Scholar]
- 33.Xiao ZG, Bruck MA, Enemark JH, et al. A catalytic cycle related to molybdenum enzymes containing [(MoVIO2)]2+ oxidized active sites. Inorg Chem. 1996;35:7508–7515. [Google Scholar]
- 34.Das SK, Biswas D, Maiti R, Sarkar S. Modeling the tungsten sites of inactive and active forms of hyperthermophilic Pyrococcus furiosus aldehyde ferredoxin oxidoreductase. J Am Chem Soc. 1996;118:1387–1397. [Google Scholar]
- 35.Majumdar A, Pal K, Nagarajan K, Sarkar S. Desoxo molybdenum(IV) and tungsten(IV) bis(dithiolene) complexes: Monomer-dimer interconversion involving reversible thiol bridge formation. Inorg Chem. 2007;46:6136–6147. doi: 10.1021/ic7006777. [DOI] [PubMed] [Google Scholar]
- 36.Seo J, Williard PG, Kim E. Deoxygenation of mono-oxo bis(dithiolene) Mo and W complexes by protonation. Inorg Chem. 2013;52:8706–12. doi: 10.1021/ic4008747. [DOI] [PubMed] [Google Scholar]
- 37.Sugimoto H, Sato M, Asano K, et al. A Model for the Active-Site Formation Process in DMSO Reductase Family Molybdenum Enzymes Involving Oxido-Alcoholato and Oxido-Thiolato Molybdenum(VI) Core Structures. Inorg Chem. 2016;55:1542–1550. doi: 10.1021/acs.inorgchem.5b02395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ueyama N, Okamura TA, Nakamura A. Structure and Properties of Molybdenum(IV, V) Arenethiolates with a Neighboring Amide Group. Significant Contribution of NH•••S Hydrogen Bond to the Positive Shift of Redox Potential of Mo(V)/Mo(IV) J Am Chem Soc. 1992;114:8129–8137. [Google Scholar]
- 39.Davies ES, Beddoes RL, Collison D, et al. Synthesis of oxomolybdenum bis(dithiolene) complexes related to the cofactor of the oxomolybdoenzymes †. J Chem Soc Dalt Trans. 1997;2:3985–3996. [Google Scholar]
- 40.Oku H, Ueyama N, Nakamura A. Association of Oxo-Molybdenum Dithiolene Complexes with a Multiamide Additive and Its Influence on the Ease of O-Atom Transfer. Inorg Chem. 1997;36:1504–1516. doi: 10.1021/ic951277g. [DOI] [PubMed] [Google Scholar]
- 41.Cooney JJA, Carducci MD, McElhaney AE, et al. New oxovanadium bis(1,2-dithiolate) compounds that mimic the hydrogen-bonding interactions at the active sites of mononuclear molybdenum enzymes. Inorg Chem. 2002;41:7086–7093. doi: 10.1021/ic025856e. [DOI] [PubMed] [Google Scholar]
- 42.Maiti R, Nagarajan K, Sarkar S. Synthesis and structure of [L][MoIVO(mnt)2] {L = [(C2H5)4N]+, [C5H5NH]+, [(C2H5)3NH]+, [lysinium]2+ and (mnt2− = 1,2-diacyanoethylenedithiolate)} in relevance to molybdenum cofactor of diverse class of molybdoenzymes. J Mol Struct. 2003;656:169–176. [Google Scholar]
- 43.Sugimoto H, Tarumizu M, Tanaka K, et al. A new series of molybdenum-(IV), -(V), and -(VI) dithiolate compounds as active site models of molybdoenzymes: preparation, crystal structures, spectroscopic/electrochemical properties and reactivity in oxygen atom transfer. Dalt Trans. 2005:3558. doi: 10.1039/b503828f. [DOI] [PubMed] [Google Scholar]
- 44.Baba K, Okamura T, Suzuki C, et al. O-Atom-Transfer Oxidation of [Molybdenum(IV) Oxo{3,6-(acylamino)2-1,2-benzenedithiolato}2]2− Promoted by Intramolecular NH•••S Hydrogen Bonds. Inorg Chem. 2006;45:894–901. doi: 10.1021/ic051493h. [DOI] [PubMed] [Google Scholar]
- 45.Okamura TA, Taniuchi K, Lee K, et al. Crystal structures and 77Se NMR spectra of molybdenum(IV) areneselenolates having intramolecular NH•••Se hydrogen bonds. Inorg Chem. 2006;45:9374–9380. doi: 10.1021/ic061140y. [DOI] [PubMed] [Google Scholar]
- 46.Ng VWL, Taylor MK, Hill LMR, et al. Novel O,O′-donor oxo-MoIV hydrotris(3-isopropylpyrazolyl)borate complexes formed by chelation of potentially hydrogen-bonding phenolate ligands on reduction of dioxo-MoVI complexes. Eur J Inorg Chem. 2010;2:3261–3269. [Google Scholar]
- 47.Okamura TA, Tatsumi M, Omi Y, et al. Selective and effective stabilization of MoVI=O Bonds by NH•••S hydrogen bonds via trans influence. Inorg Chem. 2012;51:11688–11697. doi: 10.1021/ic301597d. [DOI] [PubMed] [Google Scholar]
- 48.Okamura AT, Omi Y, Fujii M, et al. Significant differences of monooxotungsten(IV) and dioxotungsten(VI) benzenedithiolates containing two intramolecular NH•••S hydrogen bonds from molybdenum analogues. Dalt Trans. 2015;44:18090–18100. doi: 10.1039/c5dt03278d. [DOI] [PubMed] [Google Scholar]
- 49.Okamura T, Omi Y, Hirano Y, Onitsuka K. Comparative studies on the contribution of NH•••S hydrogen bonds in tungsten and molybdenum benzenedithiolate complexes. Dalt Trans. 2016;45:15651–15659. doi: 10.1039/c6dt02250b. [DOI] [PubMed] [Google Scholar]
- 50.Okamura T, Yamada T, Hasenaka Y, et al. Unexpected reaction promoted by NH+•••O=Mo hydrogen bonds in nonpolar solvents. Eur J Inorg Chem. 2016;2016:2952–2961. [Google Scholar]
- 51.Basu P, Kail BW, Young CG. Influence of the Oxygen Atom Acceptor on the Reaction Coordinate and Mechanism of Oxygen Atom Transfer from the Dioxo-Mo(VI) Complex, TpiPrMoO2(OPh), to Tertiary Phosphines. Inorg Chem. 2010;49:4895–4900. doi: 10.1021/ic902500h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nemykin VN, Laskin J, Basu P. Isolation, characterization of an intermediate in an oxygen atom-transfer reaction, and the determination of the bond dissociation energy. J Am Chem Soc. 2004;126:8604–8605. doi: 10.1021/ja049121f. [DOI] [PubMed] [Google Scholar]
- 53.Millar AJ, Doonan CJ, Smith PD, et al. Oxygen atom transfer in models for molybdenum enzymes: isolation and structural, spectroscopic, and computational studies of intermediates in oxygen atom transfer from molybdenum(VI) to phosphorus(III) Chem - A Eur J. 2005;11:3255–3267. doi: 10.1002/chem.200401101. [DOI] [PubMed] [Google Scholar]
- 54.Nemykin VN, Basu P. Oxygen atom transfer reactivity from a dioxo-Mo(VI) complex to tertiary phosphines: Synthesis, characterization, and structure of phosphoryl intermediate complexes. Inorg Chem. 2005;44:7494–7502. doi: 10.1021/ic0508324. [DOI] [PubMed] [Google Scholar]
- 55.Kail BW, Pérez LM, Zarić SD, et al. Mechanistic investigation of the oxygen-atom-transfer reactivity of dioxo-molybdenum(VI) complexes. Chem - A Eur J. 2006;12:7501–7509. doi: 10.1002/chem.200600269. [DOI] [PubMed] [Google Scholar]
- 56.Sengar RS, Nemykin VN, Basu P. Synthesis, electrochemistry, geometric and electronic structure of oxo-molybdenum compounds involved in an oxygen atom transferring system. J Inorg Biochem. 2008;102:748–756. doi: 10.1016/j.jinorgbio.2007.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Basu P, Nemykin VN, Sengar RS. Substituent effect on oxygen atom transfer reactivity from oxomolybdenum centers: Synthesis, structure, electrochemistry, and mechanism. Inorg Chem. 2009;48:6303–6313. doi: 10.1021/ic900579s. [DOI] [PubMed] [Google Scholar]
- 58.Keith JM, Tomić ZD, Zarić SD, Hall MB. Oxygen atom transfer catalysis:Ligand effects on the key reaction barrier in molybdenum (VI) dioxo systems. J Mol Catal A Chem. 2010;324:15–23. [Google Scholar]
- 59.Basu P, Kail BW, Adams AK, Nemykin VN. Quantitation of the ligand effect in oxo-transfer reactions of dioxo-Mo(VI) trispyrazolyl borate complexes. Dalton Trans. 2013;42:3071–81. doi: 10.1039/c2dt32349d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Anslyn E, Dougherty DA. Modern Physical Organic Chemistry. University Science Books; Sausalito, California: 2004. [Google Scholar]
- 61.Kaljurand I, Kütt A, Sooväli L, et al. Extension of the self-consistent spectrophotometric basicity scale in acetonitrile to a full span of 28 pKa units: Unification of different basicity scales. J Org Chem. 2005;70:1019–1028. doi: 10.1021/jo048252w. [DOI] [PubMed] [Google Scholar]
- 62.Fischer C, Fischer L. Comparative Kinetics of Enzymes and Models. In: Hille R, Schulzke C, Kirk ML, editors. Molybdenum Tungsten Enzym Bioinorg Chem. 1. The Royal Society of Chemistry; Cambridge, UK: 2016. pp. 94–129. [Google Scholar]
- 63.Bholanath P, Sarkar R, Sarkar S. Electron transfer mechanisms in molybdenum and tungsten model compounds. In: Hille R, Schulzke C, Kirk MK, editors. Molybdenum Tungsten Enzym Bioinorg Chem. The Royal Society of Chemistry; Cambridge, UK: 2016. pp. 68–94. [Google Scholar]
- 64.Kappler U, Schwarz G. The sulfite oxidase family of molybdenum enzymes. In: Hille R, Schulzke C, Kirk ML, editors. Molybdenum Tungsten Enzym Biochem. Cambridge, UK: 2016. pp. 240–273. [Google Scholar]
- 65.Wilson HL, Rajagopalan KV. The Role of Tyrosine 343 in Substrate Binding and Catalysis by Human Sulfite Oxidase. J Biol Chem. 2004;279:15105–15113. doi: 10.1074/jbc.M314288200. [DOI] [PubMed] [Google Scholar]
- 66.Trofimenko S. Boron-pyrazole chemistry. IV. Carbon- and Boron-substituted Poloy(1-pyrazolyl)borate. J Am Chem Soc. 1967;89:6288–6294. [Google Scholar]
- 67.Galan AA. Pyridinium p-toluenesulfonate. E-EROS Encycl reagents Org Synth 2001 [Google Scholar]
- 68.Frisch MJ, Trucks GW, Schlegel HB, et al. Gaussian 09, Revision E.01. 2010. [Google Scholar]
- 69.Zhao Y, Truhlar DG. A new local density functional for main-group thermochemistry, transition metal bonding, thermochemical kinetics, and noncovalent interactions. J Chem Phys. 2006;125:194101. doi: 10.1063/1.2370993. [DOI] [PubMed] [Google Scholar]
- 70.Weigend F, Ahlrichs R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys Chem Chem Phys. 2005;7:3297. doi: 10.1039/b508541a. [DOI] [PubMed] [Google Scholar]
- 71.Andrae D, Haeussermann U, Dolg M, et al. Energy-adjustedab initio pseudopotentials for the second and third row transition elements. Theor Chim Acta. 1990;77:123–141. [Google Scholar]
- 72.Miertuš S, Scrocco E, Tomasi J. Electrostatic interaction of a solute with a continuum. A direct utilizaion of AB initio molecular potentials for the prevision of solvent effects. Chem Phys. 1981;55:117–129. [Google Scholar]
- 73.Cancès E, Mennucci B, Tomasi J. A new integral equation formalism for the polarizable continuum model: Theoretical background and applications to isotropic and anisotropic dielectrics. J Chem Phys. 1997;107:3032–3041. [Google Scholar]
- 74.Tomasi J, Mennucci B, Cammi R. Quantum Mechanical Continuum Solvation Models. Chem Rev. 2005;105:2999–3094. doi: 10.1021/cr9904009. [DOI] [PubMed] [Google Scholar]
- 75.Ribeiro RF, Marenich AV, Cramer CJ, Truhlar DG. Use of Solution-Phase Vibrational Frequencies in Continuum Models for the Free Energy of Solvation. J Phys Chem B. 2011;115:14556–14562. doi: 10.1021/jp205508z. [DOI] [PubMed] [Google Scholar]
- 76.Cossi M, Barone V, Mennucci B, Tomasi J. Ab initio study of ionic solutions by a polarizable continuum dielectric model. Chem Phys Lett. 1998;286:253–260. [Google Scholar]
- 77.Seeger R, Pople JA. Self-consistent molecular orbital methods. XVIII. Constraints and stability in Hartree–Fock theory. J Chem Phys. 1977;66:3045–3050. [Google Scholar]
- 78.Riplinger C, Neese F. An efficient and near linear scaling pair natural orbital based local coupled cluster method. J Chem Phys. 2013;138:34106. doi: 10.1063/1.4773581. [DOI] [PubMed] [Google Scholar]
- 79.Neese F. The ORCA program system. Wiley Interdiscip Rev Comput Mol Sci. 2012;2:73–78. [Google Scholar]
- 80.Mondal B, Neese F, Ye S. Control in the Rate-Determining Step Provides a Promising Strategy To Develop New Catalysts for CO2 Hydrogenation: A Local Pair Natural Orbital Coupled Cluster Theory Study. Inorg Chem. 2015;54:7192–7198. doi: 10.1021/acs.inorgchem.5b00469. [DOI] [PubMed] [Google Scholar]
- 81.Ye S, Riplinger C, Hansen A, et al. Electronic Structure Analysis of the Oxygen-Activation Mechanism by FeII- and α-Ketoglutarate (αKG)-Dependent Dioxygenases. Chem - A Eur J. 2012;18:6555–6567. doi: 10.1002/chem.201102829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Xue G, Geng C, Ye S, et al. Hydrogen-Bonding Effects on the Reactivity of [X–FeIII–O–FeIV=O] (X = OH, F) Complexes toward C–H Bond Cleavage. Inorg Chem. 2013;52:3976–3984. doi: 10.1021/ic3027896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Song J, Klein EL, Neese F, Ye S. The Mechanism of Homogeneous CO2 Reduction by Ni(cyclam): Product Selectivity, Concerted Proton–Electron Transfer and C–O Bond Cleavage. Inorg Chem. 2014;53:7500–7507. doi: 10.1021/ic500829p. [DOI] [PubMed] [Google Scholar]
- 84.Cao S-W, Liu X-F, Yuan Y-P, et al. Artificial photosynthetic hydrogen evolution over g-C3N4 nanosheets coupled with cobaloxime. Phys Chem Chem Phys. 2013;15:18363–6. doi: 10.1039/c3cp53350f. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. UV-visible spectra monitored at 720 nm after the addition of 10 equiv. pyridinium triflate to 1.0 mM [Tp*MoIVOCl(NCCH3)] (2) in acetonitrile at 10 °C as a control experiment.
Figure S2. The Eyring plot for the conversion of 1.0 mM [Tp*MoIVOCl(OPMe3)] (1) to 2 in CH3CN in the presence of 10 equiv. 3-bromopyridinium tosylate. Temperature range: −30 to 0°C. ΔH‡ = 68(2) kJ/mol. ΔS‡ = −14(7) J/mol. Error bars show one standard deviation from at least three independent measurements.
Figure S3. The Ia-NNCl mechanism that leads to the side products.
Figure S4. An illustration of the direction of CH3CN attack in the Ia-NNOxo mechanism.
Figure S5. Electronic energy profiles for the dissociation of the MeCN···Mo bond in [Tp*MoOCl(MeCN)] (A), Me3PO···Mo bond in [Tp*MoOCl(Me3PO)] (B), and HPyr+···O(PMe3) bond in [Tp*MoOCl(Me3PO)(HPyr+)] (C), calculated using the relaxing scan technique at the M06L/def2-SV(P)+PCM(MeCN) level of theory.
Figure S6. Alternative Id mechanism in which HPyr+ is coordinated to the oxygen atom of the oxo ligand.
Figure S7. Alternative Ia mechanism in which HPyr+ is coordinated to the oxygen atom of the Mo=O moiety.