

Abstract
Background
Hyperalphalipoproteinemia (HALP) is inversely correlated with coronary heart disease (CHD) although genetic variants associated with high serum levels of HDL-C have not been shown to be cardioprotective.
Objective
To uncover novel genetic variants associated with HALP and possibly with reduced risk of CHD.
Methods
Exome sequencing data, HDL-C and triglyceride (TG) levels were analyzed in 1645 subjects. They included the University of Maryland outpatients with high HDL-C (n=12), Cardiovascular Health Study (CHS) (n=210), Jackson Heart Study (JHS) (n=402), Multi-Ethnic Study of Atherosclerosis (MESA) (n=404), Framingham Heart Study (FHS) (n=463) and old Order Amish (n=154).
Results
Novel nonsynonymous SNPs (nsSNPs) were identified in men and women with primary HALP (mean HDL-C, 145 +/− 30 mg/dL). Using PolyPhen-2 and Combined Annotation Dependent Depletion (CADD) to estimate the predictive effect of each nsSNP on the gene product, rare, deleterious polymorphisms in UGT1A3, PLLP, PLEKHH1, ANK2, DIS3L, ACACB and LRP4 were identified in 16 subjects with HALP but not in any tested subject with low HDL-C (< 40 mg/dL). In addition, a single novel polymorphism, rs376849274, was found in OSBPL1A. The majority of these candidate genes have been implicated in fat and lipid metabolism and none of these subjects has a history of CHD despite 75% of subjects having risk factors for CHD. Overall, the probability of finding these nsSNPs in a non-high HDL-C population ranges from 1 × 10−17 to 1 × 10−25.
Conclusion
Novel functional polymorphisms in 8 candidate genes are associated with HALP in the absence of CHD. Future study is required to examine the extent to which these genes may affect HDL function and serve as potential therapeutic targets for CHD risk reduction.
Introduction
For the past several decades, an inverse association between high-density lipoprotein cholesterol (HDL-C) and coronary heart disease (CHD) has been well recognized (1–3). In addition to its role in reverse cholesterol transport, HDL possesses antioxidant and anti-inflammatory properties that are believed to contribute to its cardioprotective role (4). However, while family-based and cross-sectional studies have suggested that primary hyperalphalipoproteinemia (HALP) is associated with longevity (5–6), HDL-C-raising variants in the established gene candidates linked to HALP, cholesteryl ester transfer protein (CETP), hepatic lipase (HL), and endothelial lipase (LIPG), have not resulted in cardioprotection or longevity (7–10). Moreover, a rare variant in scavenger receptor class B type 1 (SRB1) that raises HDL-C has been associated with elevated CHD risk (11), and randomized human outcome trials have failed to demonstrate clinical benefit following pharmacologically-mediated CETP inhibition (12–15). Therefore, the goal of the present study was to identify novel gene candidates associated with HALP and reduced CHD risk.
Methods
We identified 62 subjects with HALP (defined as 80 mg/dL or greater) (16–17) from 5 different sources with whole exome sequencing to identify novel nonsynonymous SNPs (nsSNPs) associated with the high HDL-C phenotype. The five population HALP sources were the University of Maryland Preventive Cardiology clinic (Baltimore, MD) (n = 12), the Cardiovascular Health Study (CHS) (n = 8), Jackson Heart Study (JHS) (n = 9), Multi-Ethnic Study of Atherosclerosis (MESA) (n = 22), the Framingham Heart Study (FHS) (n = 15), and the Old Order Amish population of Lancaster, PA (n =6) (18–22).
HALP subjects were initially selected from the University of Maryland Preventive Cardiology clinic and derived from 2 families (pedigree #1, pedigree #2) and 6 biologically unrelated subjects with HALP. Whole exome sequencing was performed using the Illumina Genome Analyzer platform methodology as previously described (23). Mutation analysis included all coding, intron-exon regions and promoter regions; questionable readings were verified using the Broad Institute’s Integrative Genomics Viewer (IGV). Genetic variants were confirmed within the NHLBI GO Exome Sequencing Project’s exome variant server (EVS) and the University of Michigan’s BRAVO server. Allele frequencies were identified using the Broad Institute’s Exome Aggregation Consortium browser using sequencing data from over 60,000 subjects. lntronic or synonymous mutations were excluded from analysis. All study procedures were approved by the Institutional Review Board of the University of Maryland School of Medicine.
We then analyzed the whole exome sequencing data from our subjects in conjunction with complete exome sequencing of HALP subjects from the CHS, JHS, MESA, FHS and the Old Order Amish population of Lancaster, PA. As part of the NHLBI GO Exome Sequencing Project, subjects from JHS, MESA, FHS and FHS had undergone whole exome sequencing completed using Illumina Genome Analyzer IIX or Illumina HiSeq 2000. Similarly, the Old Order Amish population of Lancaster, PA had undergone whole genome sequencing using Illumina HiSeq X Ten as part of the Amish Complex Disease Research Program at the University of Maryland. This available data were readily used to identify rare nsSNPs that were shared by members of the four families as well as individuals from the large population based studies.
The nsSNPs identified in the 62 HALP subjects were then compared with complete exome sequencing data from 1573 subjects without HALP from CHS (n=202), JHS (n=393), MESA (n=382), FHS (n=448) and Old Order Amish (n=148). Of these, 324 subjects exhibited low HDL-C (< 40 mg/dL); nsSNPs shared with HALP and low HDL-C subjects were excluded from further analysis.
We then used two widely utilized in silico functional and ensemble algorithms to determine the predicted functional significance of an amino acid change on each gene product and select for deleterious nsSNPs (24). PolyPhen-2 is a bioinformatics system that integrates features from eight protein sequences and three protein structures to generate a numerical score (0–1) of nsSNP deleteriousness (25–26). PolyPhen-2 defines ranges of nsSNP gene product as “possibly-damaging” (>0.45 and </= 0.95) or “probably-damaging” (>0.95) compared to those viewed as “benign” (</= 0.45). Combined Annotation-Dependent Depletion (CADD) is a framework that integrates 63 distinct measures of nsSNP deleteriousness into scaled C-scores ranging from 1 to 99 based expected outcome of each variant (27). A CADD score >15 considers the nsSNP to be deleterious to the gene product.
Results
Complete exome sequencing was available in 1645 subjects with 62 (3.8%) exhibiting the HALP phenotype (Table 1). We identified 8 novel candidate genes not previously known to affect HDL-C metabolism (Table 2). Each of these genes, UGT1A3, PLLP, PLEKHH1, ANK2, DIS3L, ACACB and LRP4, displayed nsSNPs with very rare allele frequencies (< 0.0009) that were identified as deleterious by PolyPhen-2 and the CADD score. A novel polymorphism, rs376849274, was also identified in OSBPL1A, a gene previously shown to affect HDL-C metabolism (28). Importantly, these nsSNPs were observed only in subjects with HALP and not among any of the 324 low HDL-C subjects with complete exome sequencing.
Table 1.
Prevalence of HALP in Subjects with Whole Exome Sequencing
| Study | Total Subjects | Subjects with HALP* | Prevalence of HALP |
|---|---|---|---|
| Cardiovascular Health Study | 210 | 8 | 3.8% |
| Jackson Heart Study | 402 | 9 | 2.2% |
| Multi - Ethnic Study of Atherosclerosis | 404 | 22 | 5.5% |
| Framingham Heart Study | 463 | 15 | 3.2% |
| UMD Preventive Cardiology Clinic | 12 | 12 | N/A |
| Old Order Amish | 154 | 6 | 3.9% |
| Total | 1645 | 62 | 3.8% |
Subjects with HDL-C >/= 80 mg/dL
Table 2.
Novel Candidate Genes Associated with the HALP Phenotype
| Chromosome | Gene name | Number of novel nsSNPs | Number of subjects | Average Allele Frequency | Average HDL (mg/dL) | Average Triglycerides (mg/dL) |
|---|---|---|---|---|---|---|
| 2 | UGT1A3 | 1 | 3* | 0.004267 | 117 | 59 |
| 14 | PLEKHH1 | 2 | 3* | 0.000231263 | 120 | 66 |
| 16 | PLLP | 3 | 4ˆ | 0.000097737 | 147 | 63 |
| 4 | ANK2 | 3 | 3 | 0.00016578 | 111 | 84 |
| 15 | DIS3L | 2 | 2 | 0.00004945 | 96 | 56 |
| 12 | ACACB | 3 | 3 | 0.00024454 | 92 | 76 |
| 11 | LRP4 | 2 | 2 | 0.00007419 | 86 | 341 |
| 1 | OSBPL1A | 1 | 1 | 0.000008269 | 92 | 35 |
includes siblings from pedigree 1
includes siblings from pedigree 2
Cardiovascular and metabolic health data (Table 3) demonstrated several paradoxical findings in the very high HDL-C cohort, inasmuch as 25% were obese, 64% were hypertensive and 56% smoked cigarettes. Yet, despite the considerable risk imposed by the high prevalence of factors known to promote atherothrombosis, none of the HALP subjects had a history of CHD and there was a history of familial longevity (2 or more family members living to at least age 90).
Table 3.
nsSNPs Associated with the HALP Phenotype
| Gene | Amino Acid | rsID | Study | Allele Frequency | HDL-C | Sex | Race | PolyPhen-2 Score | CADDS Score |
|---|---|---|---|---|---|---|---|---|---|
| UGT1A3 | p.(R45W) | rs45625338 | MESA | 0.004267 | 84 | F | EA | probably-damaging: 0.998 | 19.2 |
| p.(R45W) | rs45625338 | UMD | 0.004267 | 116 | M* | EA | probably-damaging:0.998 | 19.2 | |
| p.(R45W) | rs45625338 | UMD | 0.004267 | 158 | F* | EA | probably-damaging: 0.998 | 19.2 | |
| PLEKHH1 | p.(K1311Q) | rs371360639 | MESA | 0.00005799 | 93 | F | AA | probably-damaging: 1.0 | 28.4 |
| p.(R1179C) | rs200119528 | UMD | 0.0003179 | 116 | M* | EA | probably-damaging:1.0 | 20.8 | |
| p.(R1179C) | rs200119528 | UMD | 0.0003179 | 158 | F* | EA | probably-damaging: 1.0 | 20.8 | |
| PLLP | p.(A53V) | rs149486153 | MESA | 0.0001155 | 101 | F | EA | probably-damaging: 1.0 | 23.6 |
| p.(L114R) | MESA | 0.00003305 | 92 | M | EA | probably-damaging: 1.0 | n/a | ||
| p.(A141T) | rs143221173 | UMD | 0.0001212 | 192 | Fˆ | EA | probably-damaging: 0.958 | 24.8 | |
| p.(A141T) | rs143221173 | UMD | 0.0001212 | 201 | Fˆ | EA | probably-damaging: 0.958 | 24.8 | |
| ANK2 | p.(T527A) | MESA | 0.0000247 | 97 | F | EA | possibly-damaging: 0.804 | n/a | |
| p.(E1458G) | rs72544141 | CHS | 0.0004222 | 115 | M | EA | possibly-damaging: 0.812 | 23.7 | |
| p.(T2242M) | rs37648404 | JHS | 0.00005044 | 121 | F | AA | probably-damaging: 1.0 | 30 | |
| DIS3L | p.(E130A) | rs3677091 | JHS | 0.00008241 | 90 | F | AA | probably-damaging: 0.981 | 19.91 |
| p.(R928Q) | MESA | 0.00001649 | 102 | F | AA | probably-damaging: 0.993 | 34 | ||
| ACACB | p.(G646D) | rs372168822 | FHS | 0.000008251 | 92 | F | EA | probably-damaging: 1.0 | 29.2 |
| p.(R1119C) | rs150478780 | CHS | 0.00001647 | 83 | F | EA | probably-damaging: 1.0 | 35 | |
| p.(R1586H) | rs142393083 | CHS | 0.0007089 | 100 | M | AA | probably-damaging: 1.0 | 26.6 | |
| LRP4 | p.(R457C) | rs148856658 | FHS | 0.00006591 | 80 | M | EA | probably-damaging:0.999 | 35 |
| p.(A1130V) | rs138418874 | CHS | 0.00008247 | 91 | F | AA | possibly-damaging: 0.937 | 22.8 | |
| OSBPL1A | p.(R14G) | rs376849274 | FHS | 0.000008269 | 92 | F | EA | probably-damaging: 0.991 | 26.4 |
siblings from pedigree 1
siblings from pedigree 2
CHS: Cardiovascular Health Study
FHS: Framingham Heart Study
JHS: Jackson Heart Study
MESA: Multi-Ethnic Study of Atherosclerosis
EA: European American
AA: African American
Discussion
Integrating our familial studies with large population based studies, we identified rare nsSNPs in 3 novel gene candidates, UGT1A3, PLLP and PLEKHH1, associated with the HALP phenotype. The presence of identical, exceedingly rare nsSNPs that are likely deleterious to their gene product in siblings with HALP suggests a heritable gene product involved with cholesterol metabolism. These data were corroborated by identifying additional unrelated subjects from large population based studies who have likely deleterious nsSNPs within the same gene and the HALP phenotype. For example, p(R45W) within UGT1A3 was identified in siblings with HDL-C of 116 (male) and 158 (female) as well as a woman from MESA with an HDL-C of 84. Subsequently, this nsSNP was not shared with any individual within the low HDL-C control group. The allele frequencies of rs45625338 (UGT1A3), rs200119528 (PLEKHH1) and rs143221173 (PLLP) are likely even more rare than recorded given that sibling recurrence-risk ratio for a phenotype such as hyperalphalipoproteinemia will be greater than genotype relative risk within the general population. PolyPhen-2 and CADD have also identified this nsSNP as likely damaging to the gene product. While UGT1A3, PLLP and PLEKHH1 have not previously associated with HALP, PLEKHH1 has been suggested to play a role in cholesterol and fatty acid metabolism (29).
We also identified 4 novel genes, ANK2, DIS3L, ACACB and LRP4, associated with the HALP phenotype amongst unrelated subjects from the large population based studies. While no rare deleterious nsSNPs in these genes were identified within our related individuals, multiple rare deleterious nsSNPs within each of these genes that was only observed in subjects with HALP supports the notion that these were not random occurrences. Furthermore, none of these nsSNPs were identified in any individuals with low HDL-C. For instance, deleterious nsSNPs in ANK2 were identified within three unrelated individuals with HALP. Two of the three nsSNPs were identified in an American male of European ancestry (CHS) with an HDL-C of 115 mg/dL and African American female (JHS) with an HDL-C of 121 mg/dL. Interestingly, the LRP4 locus was previously found to be associated with HDL-C (30), supporting the notion that common as well as rare SNPs (identified in the present study) contribute to the high HDL-C phenotype. Finally, ACACB has been linked to obesity and diabetes mellitus (31), pathological processes that may adversely influence HDL-C metabolism and functionality (32).
One additional nsSNP was discovered within the OSBPL1A gene, but it remains noteworthy due to the gene’s known association with HDL-C metabolism (28). Direct measurement of cholesterol efflux in fibroblasts with this mutation and localization to apoA-1 containing hepatocytes suggests that OSBPL1A interacts specifically with the ABCA1 mediated pathway of cholesterol metabolism, possibly through direct interaction with apoA-1.
There are several limitations inherent to our study design that restrict the precision and power of our findings. Specifically, our data originate from several different protocols that utilized three distinct Illumina sequencing platforms. Consequently, the reproducibility of exome sequencing across platforms, though anticipated to be high, is not certain. Furthermore, like many genetic studies, our findings are also limited by an inherent non-randomized generation of data.
Recent results from Mendelian randomization studies have demonstrated that HDL-C-raising alleles at multiple loci encoding cholesterol metabolism genes do not protect against CHD (8). These findings have been disappointing in view of the wealth of epidemiologic data supporting HALP phenotype as a cardioprotective phenotype. Moreover, pharmacological interventions designed to raise HDL-C have also not translated into reduced CHD risk (33). However, failure to demonstrate the link between raising levels of HDL-C through one mechanism (i.e., CETP inhibition) does not necessarily rule out the possibility that other pathways exist enabling HDL to exert cardioprotective effects based on its inherent anti-inflammatory, anti-oxidant and cholesterol efflux properties (32). Using a hybrid model incorporating family and population-based studies, the present study identifies eight candidate genes associated with significantly elevated serum HDL-C in subjects without a history of CHD. While the extent to which possible confounders (e.g., LDL-C, triglycerides), might have favorably influenced these subjects’ apparent cardioprotection, these newly uncovered polymorphisms present an opportunity to further explore the complex molecular underpinnings of HDL-C and its putative role in reverse cholesterol transport and cardiovascular health.
Figure.
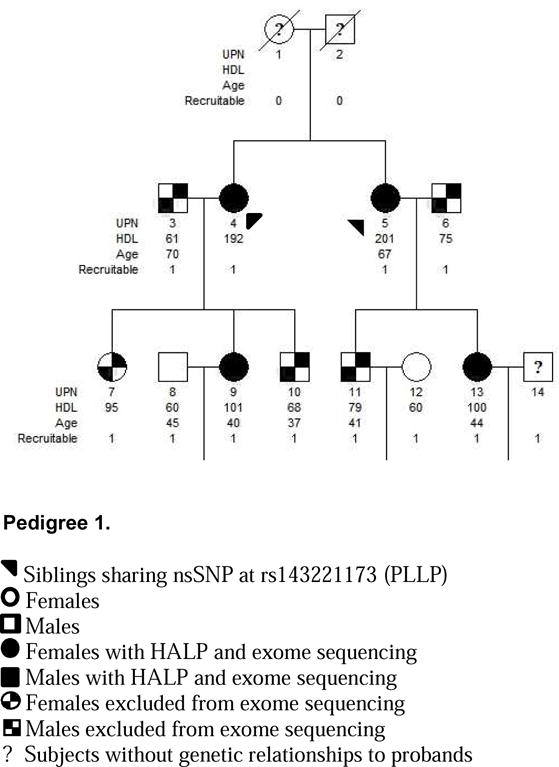
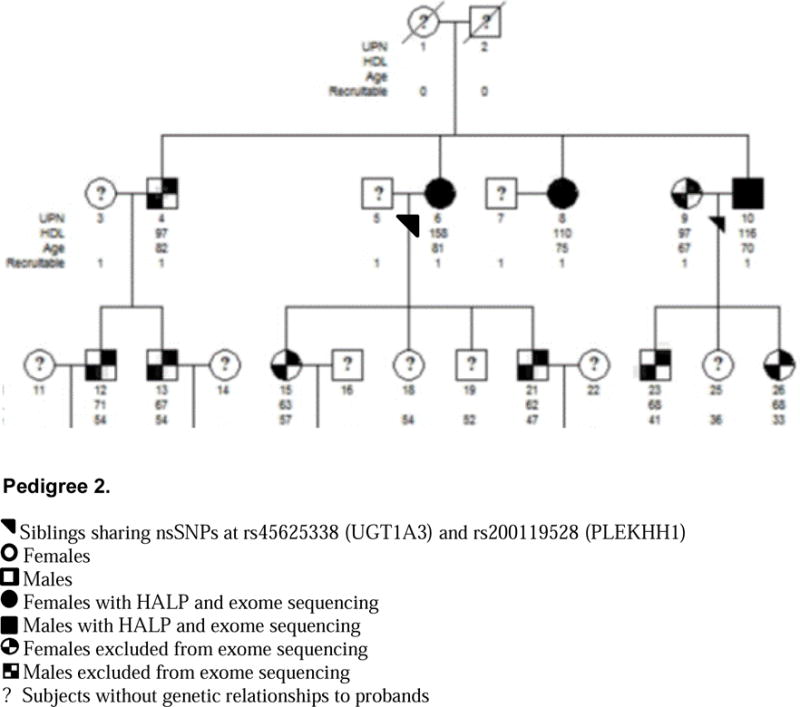
Two Families with the HALP Phenotype
Table 4.
Cardiovascular and Metabolic Health Data in HALP subjects with Novel nsSNPs
| Gene | AA | HDL | Sex | Race | Age | Lipid Med | LDL | TG | BMI | Waist | DM med | DM | Sys/Dia BP | HTN med | HTN | MI | Fm Hx Stroke | Smoke |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| UGT1A3 | p.(R45W) | 84 | F | EA | 48 | 0 | 56 | 49 | N/A | 97 | 0 | 0 | 106/73 | 0 | 0 | 0 | 0 | 1 |
| p.(R45W) | 116 | M* | EA | 70 | 0 | 72 | 48 | N/A | N/A | 0 | 0 | N/A | 0 | 0 | 0 | 0 | 0 | |
| p.(R45W) | 152 | F* | EA | 81 | 0 | 116 | 80 | N/A | N/A | 0 | 0 | N/A | 0 | 0 | 0 | 0 | 0 | |
| PLEKHH1 | p.(K1311Q) | 93 | F | AA | 65 | 0 | 125 | 71 | 35.3 | 113 | 0 | 0 | 143/73 | 1 | 1 | 0 | 0 | 1 |
| p.(R1179C) | 116 | M* | EA | 70 | 0 | 72 | 48 | N/A | N/A | 0 | 0 | N/A | 0 | 0 | 0 | 0 | 0 | |
| p.(R1179C) | 152 | F* | EA | 81 | 0 | 116 | 80 | N/A | N/A | 0 | 0 | N/A | 0 | 0 | 0 | 0 | 0 | |
| PLLP | p.(A53V) | 101 | F | EA | 63 | 0 | 117 | 86 | 23.3 | 84 | 0 | 0 | 103/67 | 1 | 1 | 0 | 1 | 0 |
| p.(L114R) | 92 | M | EA | 58 | 0 | 136 | 47 | 24.7 | 88 | 0 | 0 | 124/78 | 0 | 0 | 0 | 1 | 0 | |
| p.(A141T) | 192 | Fˆ | EA | 69 | 0 | 125 | 71 | N/A | N/A | 0 | 0 | N/A | 0 | 0 | 0 | 0 | 0 | |
| p.(A141T) | 201 | Fˆ | EA | 67 | 0 | 130 | 48 | N/A | N/A | 0 | 0 | N/A | 0 | 0 | 0 | 0 | 0 | |
| ANK2 | p.(T527A) | 97 | F | EA | 52 | 0 | 78 | 137 | 23.3 | 86 | 0 | 0 | 104/69 | 0 | 0 | 0 | 0 | 0 |
| p.(E1458G) | 115 | M | EA | 73 | 0 | 79 | 69 | 21.6 | 76 | 0 | 0 | 167/78 | 0 | 1 | 0 | 0 | 1 | |
| p.(T2242M) | 121 | F | AA | 44 | 0 | 154 | 47 | 26.3 | 88 | 0 | 0 | 97/63 | 1 | 1 | 0 | 0 | 1 | |
| DIS3L | p.(E130A) | 90 | F | AA | 48 | 1 | 90 | 68 | 50.9 | 123 | 0 | 1 | 136/84 | 1 | 1 | 0 | 1 | 0 |
| p.(R928Q) | 102 | F | AA | 68 | 0 | 60 | 45 | 21.4 | 78 | 0 | 0 | 97/61 | 0 | 0 | 0 | 0 | 1 | |
| ACACB | p.(G646D) | 92 | F | EA | 45 | 0 | 123 | 34 | 20.3 | 60 | 0 | 0 | 172/110 | 0 | 1 | 0 | 0 | 0 |
| p.(R1119C) | 83 | F | EA | 69 | 0 | 42 | 81 | 25.3 | 77 | 0 | 0 | 132/70 | 0 | 0 | 0 | 0 | 0 | |
| p.(R1586H) | 100 | M | AA | 79 | 0 | 130 | 112 | 25.3 | 97 | 0 | 0 | 126/67 | 0 | 1 | 0 | 0 | 1 | |
| LRP4 | p.(R457C) | 80 | M | EA | 52 | 1 | N/A | 513 | 23.9 | 96 | 0 | 0 | 154/99 | 1 | 1 | 0 | 0 | 1 |
| p.(A1130V) | 91 | F | AA | 75 | 0 | 44 | 169 | 25.4 | 80 | 0 | 0 | 173/59 | 1 | 1 | 0 | 1 | 1 | |
| OSBPL1A | p.(R14G) | 92 | F | EA | 45 | 0 | 123 | 34 | 20.3 | 60 | 0 | 0 | 172/110 | 0 | 1 | 0 | 0 | 0 |
siblings from pedigree 1
siblings from pedigree 2
N/A: not available
Highlights.
-
-
Exome sequencing data were analyzed in 1645 subjects from diverse populations.
-
-
Functional polymorphisms were identified in genes not previously associated with HDL.
-
-
Novel candidate genes may contribute to the hyperalpha (HALP) phenotype.
Acknowledgments
This study was funded by NIH grants HL094980 and HL61369. Authors’ contributions: J.R., J.O., B.M. and M.M. contributed to the design of the study. C.O., D.M., N.B., J.R., J.O. performed data collection and genetic analysis. C.O., J.R., M.M. and B.M. contributed to data interpretation. All authors contributed to the drafting, editing and final approval of the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Financial Disclosure
The authors have no financial relationships to disclose as relevant to the contents of the manuscript.
References
- 1.Miller GJ, Miller NE. Plasma-high-density-lipoprotein concentration and development of ischaemic heart-disease. Lancet. 1975;1:16–19. doi: 10.1016/s0140-6736(75)92376-4. [DOI] [PubMed] [Google Scholar]
- 2.Castelli WP, Doyle JT, Gordon T, Hames CG, Hjortland MC, Hulley SB, Kagan A, Zukel WJ. HDL cholesterol and other lipids in coronary heart disease. The cooperative lipoprotein phenotyping study. Circulation. 1977;55:767–772. doi: 10.1161/01.cir.55.5.767. [DOI] [PubMed] [Google Scholar]
- 3.Miller M, Seidler A, Kwiterovich PO, Pearson TA. Long-term predictors of subsequent cardiovascular events with coronary artery disease and ‘desirable’ levels of plasma total cholesterol. Circulation. 1992;86:1165–70. doi: 10.1161/01.cir.86.4.1165. [DOI] [PubMed] [Google Scholar]
- 4.Rye K, Barter P. Cardioprotective functions of HDLs. J Lipid Res. 2014;55:168–179. doi: 10.1194/jlr.R039297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Glueck CJ, Gartside PS, Steiner PM, Miller M, Todhunter T, Haaf J, Pucke M, Terrana M, Fallat RW, Kashyap ML. Hyperalpha- and hypobeta-lipoproteinemia in octogenarian kindreds. Atherosclerosis. 1977;27:387–406. doi: 10.1016/0021-9150(77)90159-9. [DOI] [PubMed] [Google Scholar]
- 6.Milman S, Atzmon G, Crandall J, Barzilai N. Phenotypes and genotypes of high density lipoprotein cholesterol in exceptional longevity. Curr Vasc Pharmacol. 2014;12:690–7. doi: 10.2174/1570161111666131219101551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Larach DB, Cuchel M, Rader DJ. Monogenic causes of elevated HDL cholesterol and implications for development of new therapeutics. Clin Lipidol. 2013;8:635–648. doi: 10.2217/clp.13.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Voight BF, Peloso GM, Orho-Melander M, et al. Plasma HDL cholesterol and risk of myocardial infarction: a mendelian randomisation study. Lancet. 2012;380:572–80. doi: 10.1016/S0140-6736(12)60312-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lin GM, Li YH, Han CL. Elevated concentrations of high-density lipoprotein cholesterol and cardiovascular risk paradox in patients with coronary heart disease and the equivalents. Int J Cardiol. 2014;176:1316–7. doi: 10.1016/j.ijcard.2014.07.153. [DOI] [PubMed] [Google Scholar]
- 10.Vigna GB, Satta E, Bernini F, et al. Flow-mediated dilation, carotid wall thickness and HDL function in subjects with hyperalphalipoproteinemia. Nutr Metab Cardiovasc Dis. 2014;24:777–83. doi: 10.1016/j.numecd.2014.02.010. [DOI] [PubMed] [Google Scholar]
- 11.Zanoni P, Khetarpal SA, Larach DB, et al. CHD Exome+ Consortium; CARDIoGRAM Exome Consortium; Global Lipids Genetics Consortium Rare variant in scavenger receptor BI raises HDL cholesterol and increases risk of coronary heart disease. Science. 2016;351:1166–71. doi: 10.1126/science.aad3517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Barter PJ, Caulfield M, Eriksson M, et al. ILLUMINATE Investigators Effects of torcetrapib in patients at high risk for coronary events. N Engl J Med. 2007;357:2109–22. doi: 10.1056/NEJMoa0706628. [DOI] [PubMed] [Google Scholar]
- 13.Schwartz GG, Olsson AG, Abt M, et al. dal-OUTCOMES Investigators Effects of dalcetrapib in patients with a recent acute coronary syndrome. N Engl J Med. 2012;367:2089–99. doi: 10.1056/NEJMoa1206797. [DOI] [PubMed] [Google Scholar]
- 14.Lincoff AM, Nicholls SJ, Riesmeyer JS, et al. Evacetrapib and Cardiovascular Outcomes in High-Risk Vascular Disease. N Engl J Med. 2017;376:1933–1942. doi: 10.1056/NEJMoa1609581. [DOI] [PubMed] [Google Scholar]
- 15.Nicholls SJ, Ruotolo G, Brewer HB, Kane JP, Wang MD, Krueger KA, Adelman SJ, Nissen SE, Rader DJ. Cholesterol Efflux Capacity and Pre-Beta-1 HDL Concentrations Are Increased in Dyslipidemic Patients Treated With Evacetrapib. J Am Coll Cardiol. 2015;66:2201–10. doi: 10.1016/j.jacc.2015.09.013. [DOI] [PubMed] [Google Scholar]
- 16.Cefalù AB, Noto D, Magnolo L, Pinotti E, Gomaraschi M, Martini S, Vigna GB, Calabresi L, Tarugi P, Averna MR. Novel mutations of CETP gene in Italian subjects with hyperalphalipoproteinemia. Atherosclerosis. 2009;204:202–7. doi: 10.1016/j.atherosclerosis.2008.08.031. [DOI] [PubMed] [Google Scholar]
- 17.Miller M. HDL cholesterol in CHD Risk Assessment. In: Ballantyne CM, editor. Clinical Lipidology: Companion to Braunwald’s Heart Disease. 2008. pp. 119–129. [Google Scholar]
- 18.Psaty BM, Kuller LH, Bild D, Burke GL, Kittner SJ, Mittelmark M, Price TR, Rautaharju PM, Robbins J. Methods of assessing prevalent cardiovascular disease in the Cardiovascular Health Study. Ann Epidemiol. 1995;5:270–277. doi: 10.1016/1047-2797(94)00092-8. [DOI] [PubMed] [Google Scholar]
- 19.Riestra P, Gebreab SY, Xu R, Khan RJ, Bidulescu A, Correa A, Tekola-Ayele F, Davis SK. Gender-specific associations between ADIPOQ gene polymorphisms and adiponectin levels and obesity in the Jackson Heart Study cohort. BMC Med Genet. 2015;16:65. doi: 10.1186/s12881-015-0214-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vargas JD, Manichaikul A, Wang XQ, Rich SS, Rotter JI, Post WS, Polak JF, Budoff MJ, Bluemke DA. Common genetic variants and subclinical atherosclerosis: The Multi-Ethnic Study of Atherosclerosis (MESA) Atherosclerosis. 2016;245:230–6. doi: 10.1016/j.atherosclerosis.2015.11.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Eicher JD, Chen MH, Pitsillides AN, Lin H, Veeraraghavan N, Brody JA, Metcalf GA, Muzny DM, Gibbs RA, Becker DM, Becker LC, Faraday N, Mathias RA, Yanek LR, Boerwinkle E, Cupples LA, Johnson AD. Whole exome sequencing in the Framingham Heart Study identifies rare variation in HYAL2 that influences platelet aggregation. Thromb Haemost. 2017;117:1083–1092. doi: 10.1160/TH16-09-0677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pollin TI, Damcott CM, Shen H, Ott SH, Shelton J, Horenstein RB, Post W, McLenithan JC, Bielak LF, Peyser PA, Mitchell BD, Miller M, O’Connell JR, Shuldiner AR. A null mutation in human APOC3 confers a favorable plasma lipid profile and apparent cardioprotection. Science. 2008;322:1702–1705. doi: 10.1126/science.1161524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cole JW, Stine OC, Liu X, et al. Rare variants in ischemic stroke: an exome pilot study. PLoS One. 2012;7:e35591. doi: 10.1371/journal.pone.0035591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dong C, Wei P, Jian X, Gibbs R, et al. Comparison and integration of deleteriousness prediction methods for nonsynonymous SNVs in whole exome sequencing studies. Hum Mol Genet. 2015;24:2125–2137. doi: 10.1093/hmg/ddu733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Butler MG, Rafi SK, Hossain W, Stephan DA, Manzardo AM. Whole exome sequencing in females with autism implicates novel and candidate genes. Int J Mol Sci. 2015;16:1312–35. doi: 10.3390/ijms16011312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Adzhubei IA, Schmidt S, Peshkin L, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kircher M, Witten DM, Jain P, et al. A general framework for estimating the relative pathogenicity of human genetics. Nat Genet. 2014;46(3):310–315. doi: 10.1038/ng.2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Motazacker MM, Pirhonen J, van Capelleveen JC, et al. A loss-of-function variant in OSBPL1A predisposes to low plasma HDL cholesterol levels and impaired cholesterol efflux capacity. Atherosclerosis. 2016;249:140–147. doi: 10.1016/j.atherosclerosis.2016.04.005. (2016) [DOI] [PubMed] [Google Scholar]
- 29.Mirkov S, Myers JL, Ramírez J, Liu W. Transcription and Lipid Accumulation in the Liver. Metabolism. 2012;61:2–6. doi: 10.1016/j.metabol.2012.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Teslovich T, Musunuru K, Smith AE, et al. Biological, clinical and population relevance of 95 loci for blood lipids. Nature. 2010;466:707–713. doi: 10.1038/nature09270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ma L, Murea M, Snipes JA, et al. An ACACB variant implicated in diabetic nephropathy associates with body mass index and gene expression in obese subjects. PLoS One. 2013;8:e5619. doi: 10.1371/journal.pone.0056193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Toth PP, Barter PJ, Rosenson RS, et al. High-density lipoproteins: a consensus statement from the National Lipid Association. J Clin Lipidol. 2013;7:484–525. doi: 10.1016/j.jacl.2013.08.001. [DOI] [PubMed] [Google Scholar]
- 33.Doggrell SA. No cardiovascular benefit with evacetrapib - is this the end of the road for the ‘cetrapibs’? Expert Opin Pharmacother. 2017;18:1439–1442. doi: 10.1080/14656566.2017.1365838. [DOI] [PubMed] [Google Scholar]