

Review Introduction
Protein quantification data on drug metabolizing enzymes and transporters (collectively referred as DMET proteins) in human tissues are useful in predicting interindividual variability in drug disposition. While targeted proteomics is an emerging technique for quantification of DMET proteins, the methodology involves significant technical challenges especially when multiple samples are analyzed in a single study over a long period of time. Therefore, it is important to thoroughly address the critical variables that could affect DMET protein quantification.
Keywords: ADME, quantitative proteomics, drug metabolism, drug transport, interindividual variability
1. Introduction
Drug metabolism and drug transport are the primary mechanisms determining drug disposition in the body, for which the liver, intestine, kidney and brain play important roles. Interindividual variability in drug disposition poses a significant challenge during drug development and clinical practice. Demographic, biological and genetic factors such as age, sex, ethnicity, disease condition and genotype can affect activity or expression of drug metabolizing enzyme and transporter (DMET) proteins. Moreover, the effect of these population variables on DMET proteins is usually non-monotonic. For example, unlike the major cytochrome P450 (CYP) and uridine 5′-diphospho-glucuronosyltransferase (UGT) enzymes where age-dependent increase is observed in the protein abundance from neonates to adults (1, 2), CYP3A7 is a predominant fetal liver enzyme, protein abundance of which is steeply diminished after birth (3). Some enzymes such as CYP3A4 (4), CYP2B6 (5) and UGT2B17 (6) show gender-dependent activity. The enzyme activity or abundance of multiple hepatic DMEs are differentially regulated in disease status. For example, a significantly lower hepatic expression or activity of DMEs and transporters in patients with liver cirrhosis is reported as compared to the non-cirrhotic subjects (7, 8). Further, splice variants of DMET proteins such as CYP3A5 and CYP2C19 result in complete loss of activity (9, 10), while a non-synonymous single nucleotide polymorphism (SNP) of hepatic organic anion transporting polypeptide 1B1 (OATP1B1), c.463C>A, is associated with increased transporter function in human (11). These factors can affect safety and efficacy of narrow therapeutic window drugs justifying the need to characterize the inter-interindividual variability in DMET protein activity or abundance.
Because clinical studies to determine inter-individual variability in DMET protein activity are not routinely feasible, in vitro activity or quantification of DMET protein abundance in human tissues relevant to drug disposition could be used as an alternate to predict variability in drug pharmacokinetics when integrated into physiologically-based pharmacokinetic (PBPK) models (8, 11–14). However, specific probe substrates to quantify in vitro activity for most of the DMET proteins, other than CYPs, are not often available. Regarding protein quantification, the conventional methods such as Western blot are less selective, cumbersome and lack in high-throughput. Although Western blot could also be quantitative when combined with a calibration curve prepared from a purified protein, this practice is less common and relative quantification is generally used (15). mRNA expression analysis is simple, multiplexed and rapid, but protein activity or abundance often poorly correlate with the mRNA signal, particularly in banked tissue samples (16, 17). Considering these limitations, targeted proteomics has emerged as an important technique to determine interindividual variability in DMET protein abundance in tissues relevant to drug disposition (Table 1).
Table 1.
Representative published proteomics studies on quantification of DMEs/transporters to determine interindividual variability in human.
| Proteins | Tissue | Population/sample (number of samples) |
Reference |
|---|---|---|---|
| CYPs, UGTs, CYP reductase and transporters | Jejunum | Tissue from obese donors (24) | (18) |
| UGTs | Liver | Nontumorous liver samples (48) | (19) |
| CYPs, UGTs, CYP reductase and transporters | Liver | Livers (17) | (16) |
| UGTs | Liver, kidney, and intestine | HLM (9 individual donors) Pooled HLM, HKM and HIM HIM (3 individual donors) |
(20) |
| UGTs | Liver, intestinal, and kidney | Pooled HLM, | (21) |
| CYPs, UGTs | Liver | Hepatocellular carcinoma (HCC) tissue (15) and pooled HLM | (22) |
| CYPs and UGTs | Jejunum Liver |
Jejunum (3) Pooled HLM and HIM |
(23) |
| CYPs and UGTs | Liver | HLM pool (50)Individual HLM (23) | (24) |
| UGTs | Liver | Individual HLM (n = 60) individual S9 fractions (n = 59) |
(25) |
| CESs | Liver | Pediatric (136) and adults (35) | (26) |
| OATPs and P-gp | Liver | Tissue (5), hepatocytes (12) | (11) |
| CYP2C19 | Liver | HLM (347) | (10) |
| Transporters | Liver | Tissue (55) | (27) |
| MRP2 | Liver | Tissue (51) | (17) |
| BCRP | Liver | Tissue (65) | (28) |
| Transporters | Kidney | Cortex tissue (41) | (29) |
| Transporters | Intestine | Six organ donors (one female, five males) | (30) |
| Aldehyde oxidase | Liver | Pooled cytosol | (31) |
| Transporters | Liver and Kidney | Tissue | (25) |
| CYP2D6 | Liver | CYP2D6.1 and CYP2D6.10 supersomes and pooled HLM | (32) |
| CYPs | Liver | 12 individual HLM | (32, 33) |
| CYPs, UGTs, transporters | Liver, Kidney | Pooled HLM, HKM, and HIM (13) | (34) |
| CYP4F2 | Liver | Pooled HLM (n=50) | (35) |
| CYP3A7 | Liver | Fetal liver microsomes (16) | (36) |
| Transporters | Blood-brain barrier (BBB) | Brian microvessels (7) | (37) |
| Transporters and DMEs | BBB | Brian microvessels (12) | (38) |
| CYPs | Liver | Livers (100) | (39, 40) |
| FMOs | Liver | HLM pool, Individual adult HLM (9), Livers (7-fetal and 16 pediatric donors) | (41, 42) |
| Transporters | Intestine | Distal jejunum (n=3) and distal ileum (n=1) enterocyte membranes | (43) |
| CYP2D6 | Liver | HLM (30) | (44) |
| UGTs | Kidney | Kidney (12 normal and 14 neoplastic) | (45) |
| ADHs and ALDH1A1 | Liver | Cytosol, pediatric (137) and adults (57) | (46) |
The technique as shown in Figure 1, utilizes liquid chromatography coupled to tandem mass spectrometry (LC-MS/MS) that relies on selective quantification of the unique surrogate peptide(s) of the target protein in the protease-digested biological sample. The notable advantages of LC-MS/MS or selective or multiple reaction monitoring (SRM or MRM) proteomics are its ability to quantify multiple proteins in a sample (i.e., multiplexing), selectivity and reproducibility (47, 48). Besides its application in determining DMET protein interindividual variability, LC-MS/MS proteomics is proving to be an important technique in the field of drug metabolism and pharmacokinetics (DMPK), e.g., in vitro model validation (49, 50), in vitro to in vivo extrapolation (IVIVE) of drug clearance (51, 52), differential tissue or sub-cellular localization of proteins (48, 53), characterization of reactive metabolite-protein interaction (54) and protein biomarker discovery (e.g., exosome proteomics) (55). The approach has also been used to determine enzyme induction potential of drugs (56) and interspecies differences in protein abundance of DMET proteins, which affect animal model selection for drug toxicity studies (27, 57–59). Further, LC-MS/MS proteomics is a powerful tool to detect highly homologous proteins. For example, Mdr1a and Mdr1b are highly homologous proteins with >90% sequence similarity. However, by using specific peptides for each isoforms, NTTGALTTR (Mdr1a), NSTGSLTTR (Mdr1b), we selectively quantified these isoforms in mice placenta and brain (60). Similarly, highly homologous proteins such as human CYP3A4, CYP3A5 and CYP3A7, and UGT2B15 and UGT2B17 can be selectively quantified by using surrogate peptides (61).
Figure 1.
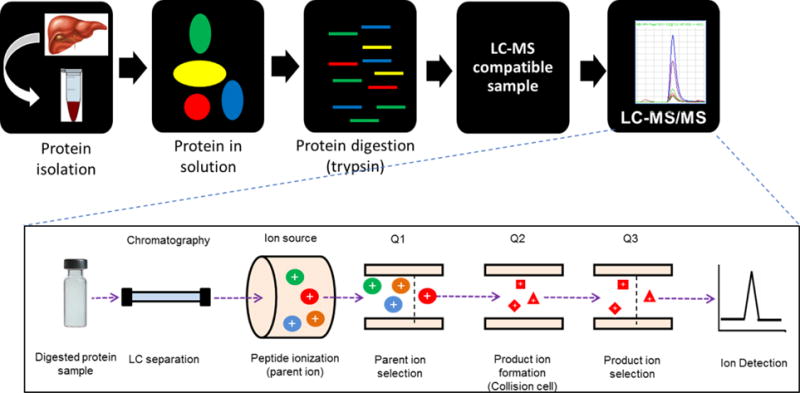
The LC-MS/MS tissue proteomics workflow. Tissues are homogenized, proteins are solubilized and digested. The digest is separated by LC, and the surrogate peptides are analyzed by triple quadrupole MS instrument in SRM mode (revised from Prasad et al. (47)).
While LC-MS/MS proteomics is emerging, it is not routinely used by a majority of DMPK laboratories, especially due to the shortage of technical expertise in this area. There are many methodological challenges that need to be thoroughly addressed for optimum protein quantification, especially when the technique is applied to analyze a big cohort of samples over a long period. We have previously reported optimized quantitative proteomics method development protocol for DMET proteins (47, 61) and discussed the potential applications of quantitative proteomics data in the precision medicine (13). In this report, we discuss the critical issues and optimized practices for DMET protein quantification in human tissues or cells to detect subtle changes in the protein abundance due to the inter-individual or biological differences. Issues such as surrogate peptide quality, ion suppression in MS source, sample loss during processing, incomplete or inconsistent protein digestion, inter-day variability and tissue quality are discussed with case examples. The recommended best practice to address these issues is also summarized.
2. Importance of quantitative DMET proteomics in PBPK modeling
There are four major limitations of the current PBPK models, which could be resolved by using quantitative proteomics data, i.e., i), non-availability of protein abundance data for non-CYP enzymes such as alcohol dehydrogenases (ADHs), aldehyde dehydrogenases (ALDHs), and aldehyde oxidase (AO), ii) lack of comprehensive extra-hepatic DMET protein abundance data, iii) minimal information on interindividual variability in DMET protein abundance or activity, and iv) scarcity of abundance data on DMET proteins in preclinical species for allometric scaling purpose. Following section highlights the importance of quantitative DMET abundance data in predicting interindividual variability associated with age, sex, ethnicity, disease condition and genotype.
While a clinical PK study utilizing specific substrate is the gold-standard to characterize the variability, human studies are challenging to conduct because of the ethical and logistical hurdles such as lack of specific in vivo probes, potential side effects, medical costs, and low-throughput. Further, it is difficult to quantify in vitro activity of the transporters and a majority of the DMEs because of the non-availability of good in vitro models. Primary cells such as human hepatocytes isolated from the fresh tissues are also associated with altered expression or localization of these proteins (50, 62). Therefore, quantitative DMET proteomics data from banked human tissues could be used as an important alternate to predict interindividual variability in DMET protein activity. For example, since clinical trials are not routinely conducted during drug development on orphan populations such as children, DMET proteomics-based PBPK models could help in making appropriate decision when giving first-dose of a drug to such populations based on studies on adults. It has been previously demonstrated that drug transporters and DMEs can be precisely quantified in human tissues and the data obtained can be used to predict the effect of genetic variation (11), age (26) and disease condition (8).
Moreover, estimation of local tissue concentration is important to prediction of toxicity and efficacy of drugs. Since DMEs and transporters can dramatically affect tissue concentration without influencing systemic drug exposure (63–65), it is important that the tissue based PBPK models are developed. For that matter, when a drug is a substrate of uptake or efflux transporters, any drug-interaction affecting the transport function can cause differential effect on the plasma vs. tissue concentrations, e.g., inhibition of P-gp at the blood-brain barrier results differential effect on brain vs. plasma concentration of its substrate (66). Such hidden interactions and their variability across population could be predicted using population PBPK models by integrating proteomics data with the in vitro kinetics data. Such approaches would be particularly important in cancer precision therapeutics, where targeted delivery is warranted for optimum safety and efficacy (67).
3. Critical issues and optimized practices in DMET proteomics towards determination of interindividual variability
Tissue proteomics generally involves a number of processing steps such as homogenization, protein extraction, protein solubilization, denaturation, reduction of disulfide bonds, alkylation of cysteine residues, desalting, enzymatic digestion, LC separation, and analysis by MS (47). Each step encompasses various factors which if not controlled could add significant technical variability to the protein quantification results. Reducing technical variability is not only important in accurate estimation of biological variability, it is also essential for the accurate study design and estimating statistical power (68). The number of samples needed to obtain statistical significance increases considerably with the technical variability (Figure 2) and could become prohibitive to measure the inter-individual variability as study requires longer time for analysis, and increased study cost (68). Such technical variability can be controlled by considering critical methodological factors discussed in this section.
Figure 2.
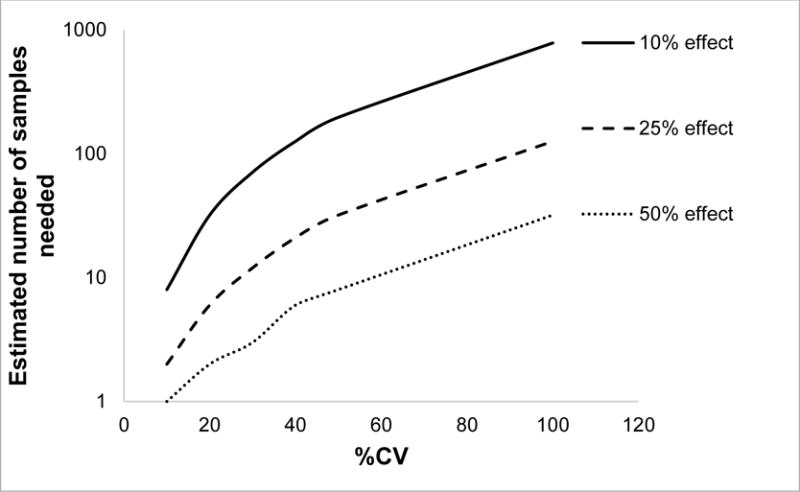
Effect of technical variability (%CV) on number of samples needed to observe 10, 25 and 50% effect with 80% power with alpha value of 0.05.
3.1. Surrogate peptide qualification
Selection of optimum surrogate peptide(s) is the first critical step in protein quantification. An ideal peptide should be unique, sensitive, stable, soluble and LC-MS compatible (Figure 3A) (47). The surrogate peptides should have optimum peptide length (6-22 amino acids). Reactive residues should be avoided; however, although cysteine (C) residue is highly reactive, it can be masked by iodoacetamide and therefore they should not present a significant technical issue. Peptides from transmembrane regions are difficult to extract and should be avoided. Peptides containing unstable residues (e.g., M and W), prone to autoxidation, should be given less priority. Peptide containing a residue susceptible to change due to non-synonymous single nucleotide polymorphism (SNPs) should be excluded unless they are the target of the study. For example, inclusion of a peptide with UGT2B15 SNP, rs1902023 could lead to erratic results (Figure 3B). UGT2B15 peptides, SVINDPVYK and NYLEDSLLK are unique, however, in the second peptide, tyrosine (Y) at 85th position can be replaced by aspartic acid (D) due to gene variation (69). Quantification of UGT2B15 using surrogate peptide, NYLEDSLLK (position 84 to 91) yielded half or zero protein abundance, as compared to the reference allele, in samples with heterozygous or homozygous variations, respectively (Figure 3B). Nevertheless, this information is useful for identifying non-synonymous variants. Highly homologous proteins, different only in leucine (L) or isoleucine (I) are not distinguished by LC-MS/MS due to similar mass-to-charge ratios of these peptides. For example, peptide, AGQLISELFTNR (a surrogate peptide of a non-hepatic carboxylesterase (CES) isoform, CES4), shows strong false positive signal in the liver digest as the latter has the same parent and the fragment ions to that of AGQLLSELFTNR representing hepatic CES isoform (CES1) (Figure 3C). Similarly, with respect to the liver and intestine, SAISIAEDEEWK is unique to CYP3A4 but CYP3A5 also contains isomeric proteotypic sequence SAISLAEDEEWK, which can cause overestimation of protein abundance. Thus, if the surrogate peptides contain L or I, a homology search should be done to ensure that there is no other peptide in the sample matrix which is only different in these isomeric amino acids. A similar trend can be seen in much rarer occasions with sequence variants containing lysine (K) and glutamine (Q), which also have very close molecular weights, especially if K is not cleaved by trypsin such as when followed by a proline (P). Post translational modification (PTMs), which can affect native protein quantification should also be avoided unless a specific study targeting stoichiometric dynamics of modification is desirable (70). If possible, exclude sites prone to missed cleavage, consecutive dibasic or tribasic residues (commonly referred as ragged ends, RR, KK, RK, and KR), C-terminal prolines to the peptide bond and proximity of the cleavage site to acidic residues (namely glutamic (E) and aspartic (D) acids) (47). Some issues with ragged ends (KR and KK) can be addressed if a prior digestive step using Lys-C is included. Lys-C is an enzyme which can efficiently target the C-terminal bonds of K and has been shown to be compatible with quantitative proteomic experiments (71). Unlike trypsin, Lys-C can cleave K followed by proline (P), making it ideal for sequential protein digestion followed by trypsin to decrease missed cleavages. Solubility and stability of peptide standards should be predicted while selecting peptide and these parameters should be validated by experimentally determining the same (47).
Figure 3.
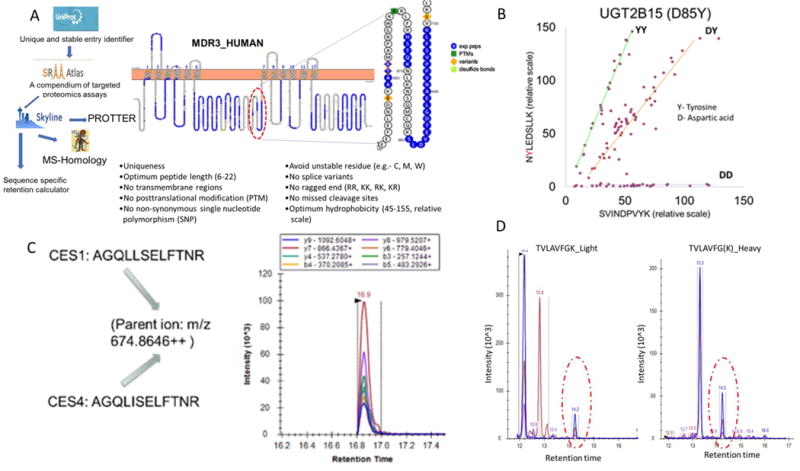
A. Generic peptide selection workflow and criterion used by our laboratory. First, Uniprot Entry number, i.e., unique and stable identifier, is obtained (http://www.uniprot.org). Using the Uniprot Entry number, MS-responsive peptides and corresponding SRM transitions are selected (http://www.srmatlas.org). These transitions are imported in Skyline software (https://skyline.ms/project/home/software/Skyline/begin.view), where topological location of peptides, reported SNPs, PTM sites and variants can be identified using Protter tool (http://wlab.ethz.ch/protter/start/). Surrogate peptides should be unique to the protein of interest, which can be confirmed by MS-homology search such as by using Protein Prospector (http://prospector.ucsf.edu/prospector/mshome.htm). By using sequence specific retention calculator (http://hs2.proteome.ca/SSRCalc/SSRCalcX.html) hydrophobicity and subsequent retention time can be estimated. For example, for a specific LC column, buffer composition and gradient flow, we derived a simple regression equation between hydrophobicity and actual retention time of known peptide standards that we use for our predictions of retention time. Critical peptide characteristics are summarized in the Figure 1A on the bottom inset (47, 61). B. Inclusion of peptide with non-synonymous SNP of UGT2B15 could lead erratic interpretation of interindividual variability in protein abundance. UGT2B15 peptides, SVINDPVYK and NYLEDSLLK are unique, however, in the second peptide, tyrosine (Y) at 85th position can be replaced by aspartic acid (D) due to genetic polymorphism. Relative quantification of UGT2B15 across samples using NYLEDSLLK resulted half or no abundance in donors harboring heterozygous or homozygous variants, respectively, as compared to the reference allele. C. Highly homologues proteins, different only in isomeric leucine or isoleucine are not distinguished by LC-MS/MS. For example, peptide, AGQLISELFTNR (a surrogate peptide of a non-hepatic carboxylesterase (CES) isoform, CES4), shows strong false positive signal in the liver digest as the latter has the same parent and the fragment ions to that of AGQLLSELFTNR representing hepatic CES isoform (CES1) (inset). D. Typical extracted ion chromatogram of light and peptide TVLAVFGK in trypsin digest of the kidney tissue sample. Peaks for multiple daughter ion coming from same parent ion should be superimposable and qualified by corresponding heavy peptide transition. For example, peak at retention time 14.2 minute qualifies the correct peak.
Once the surrogate peptide is selected, the corresponding heavy peptide should be procured. Then, the multiple daughter ions generated from same parent ion of a target peptide should be superimposable and qualified by corresponding heavy peptide transitions (Figure 3D). Contamination of light peptide in the heavy peptide standard should also be checked to avoid false positive results.
3.2. Matrix effect, ion suppression or enhancement, and sample loss during sample preparation
Because of the complexity of biological matrices, ion suppression or enhancement is very common in peptide quantification by conventional LC-MS/MS. Such effect is generally caused by lipids and other small molecules such as salts, buffers and peptides that co-elute and compete with peptides for ionization (72). Desalting by liquid-liquid extraction (26) or solid-phase extraction(73) before and/or after trypsin digestion reduces matrix complexity for the subsequent LC-MS/MS analysis. In addition, the mobile phase gradient can be set to elute all the polar buffer or salts and lipids to the waste before and after target peptide elution, respectively. For example, in a 30-min method, we usually divert the effluent to the waste for first five minutes and the last five minutes to avoid contamination of the MS source. Such flow diversion significantly helps in removing impurities affecting ionization.
While sample cleaning is an important step, the matrix effect can be assessed by using heavy labeled peptide as internal standard. Figure 4A shows correlation of a light peptide generated from bovine serum albumin (BSA) (the exogenous protein internal standard) and its corresponding heavy internal standard peptide. Since the known amounts of protein and peptide internal standards were added to all the samples, we expected a narrow range of distribution for these analytes. However, because of the differential ion suppression across samples, the correlation plot shows a wide linear distribution range indicating ion suppression which is addressed by the heavy peptide. Other than normalizing ion suppression and poor spray stability, addition of heavy peptides provides high degree of confidence because of the following additional reasons (74). First, heavy peptide is almost identical in the physicochemical properties and co-elutes with the light target peptide. Second, heavy peptide yields the corresponding fragment ions of the light peptide. Third, heavy peptide is generally identical in relative abundances of the fragment ions as the light peptide. And finally, the heavy peptide also serves as a control for sample concentration change due to evaporation or non-specific binding to the LC-MS/MS vial.
Figure 4.
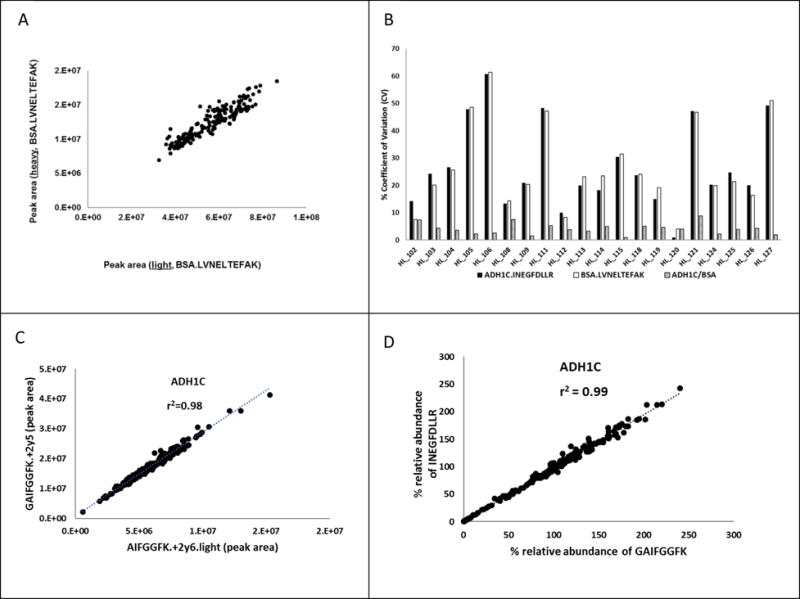
A. Effect of ion suppression or sample loss during processing on peak areas of light (x-axis) and heavy (y-axis) peptides of BSA added as an exogenous protein internal standard. Known amount of BSA and the corresponding heavy labeled peptide were added in each sample and the correlation plot was made between peak areas of light and heavy BSA peptide, LVNELTEFAK. Both spiked light (protein) and heavy (peptide) internal standards indicate sample loss, which can be corrected by normalizing response of the light peptide by the heavy peptide. B. % CV of a model protein, ADH1C analysis done on three consecutive dates on full processing replicates. BSA can address inter-day trypsin digestion variability and/or sample loss during the sample preparation. Y-axis is % coefficient of variation of peak area ratio of the ADH1C peptide, the BSA peptide and the ADH1C peptide normalize to the BSA peptide, respectively. X-axis is sample number. Normalization of the data with the BSA significantly reduces the % coefficient of variation. C. Correlation between peak areas of two daughter ions of the ADH1C surrogate peptide, GAIFGGFK. D. Correlation between % relative abundance (normalize to quality control sample) of two ADH1C surrogate peptides, GAIFGGFK and INEGFDLLR.
Sample loss during processing can be addressed by including an exogenous protein as an internal standard, which is spiked into the homogenized sample before protein denaturation (26, 46) Especially desalting by liquid-liquid extraction before trypsin digestion is a delicate step, where sample loss is generally unavoidable. As shown in Figure 4B, additional of BSA as an exogenous protein in the protocol resulted significant reduction of % coefficient of variance (%CV) in quantification of INEGFDLLR, a surrogate peptide for hepatic ADH1C (46).
The quality of peptide signal can be verified by plotting correlation between multiple fragments or SRM transitions generated from a peptide across different samples as long as the total protein concentration in the sample used for digestion was the same (Figure 4C). Since ion suppression can equally affect the fragment ions, ideally multiple peptides of a protein can be used to measure whether the variability in peak response reflects the true biological variability (Figure 4D).
3.3. Incomplete vs. inconsistent protein digestion and the concept of absolute scaling factor (ASF)
While most of the published data on DMET protein quantification are presented as absolute values e.g., pmol/mg total protein, it is important to acknowledge that these data are generally based on light or heavy peptide as a calibrator. This problem can only be addressed if a purified protein standard is available to generate calibration curve and undergoes the same processing steps as the samples (26). However, since analytical grade proteins are not routinely available, synthetic peptides are commonly used as a calibrator. If the latter is the case, it is important that the protein of interest is completely digested to its peptide fragments. While maximum digestion efficiency can be reached by changing digestion time or protease concentration, applying micro-wave assisted digestion, and adding different surfactant and solubilizing agents or protease type (Lyc-C plus trypsin) (21, 71, 75), it is usually difficult to prove whether digestion is complete (100% efficiency). Peptide stability is an important factor determining apparent digestion efficiency as some peptides degrade faster than others (76).
It is a common apprehension in the PBPK field that the protein quantification data, which is generated using a peptide calibrator may not be applicable to IVIVE or to predict interindividual variability as these are not absolute values due to incomplete digestion inefficiency (77). However, in our opinion irrespective of digestion efficiency, inter-system or interindividual variability (relative fold difference) in DMET protein abundance can be accurately measured if the extraction and digestion efficiency remain reproducible across samples. We propose the concept of absolute scaling factor (ASF) which relies on the fact that if the % extraction and digestion efficiency between samples remains consistent, the values obtained by using peptide calibrator will indicate true fold biological variability. Quantification of two peptides for OCT2, LNPSFLDLVR and SPGVAELSLR in HEK293 cells and kidney tissue homogenates yields the same absolute scaling factor (Expressionin vivo/Expressionin vitro) (Figure 5) for IVIVE, although absolute protein abundance values are different using peptides as calibrators. ASF can also be applied to compare transporter expression between different primary cells or cell-lines. This concept is essentially like relative expression factor (REF), which is generally used for the IVIVE of transporter mediated drug clearance (11, 78, 79).
Figure 5.
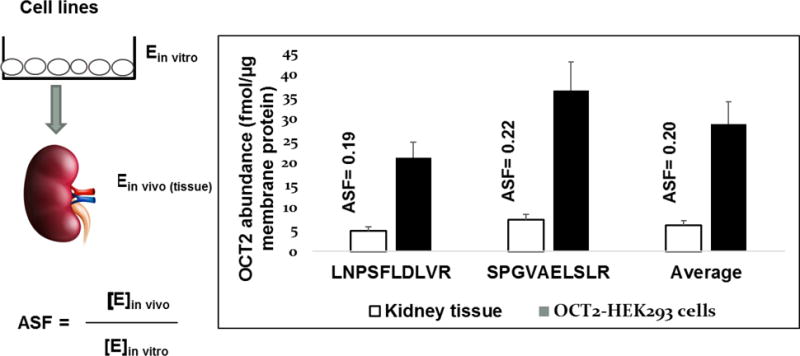
Concept of absolute scaling factor (ASF). While peptide yields due to differential trypsin digestion efficiency of individual peptides could be different, ASF remains constant. Therefore, irrespective of absolute quantification, ASF can reliably predict interindividual variability or extrapolated drug clearance from cell system to in vivo. For example, OCT2 abundance can differ between two peptides, however estimated ASF values (kidney tissue vs. HEK293 cells) will remain the same for IVIVE purpose and prediction of interindividual variability.
Pre-digestion variables such as sample matrix and reagent quality can also affect peptide yield. Arnold et al., demonstrated matrix-dependent effect on trypsin digestion and absolute quantification of ALDH1A1 (76). A comprehensive study by Walmsley et al., (80) revealed the origin of trypsin as a significant source of variability in proteomics. Notably, bovine trypsin produced a significantly higher number of missed cleavage peptides, whereas porcine trypsin resulted in more semi-tryptic peptides. On the other hand, recombinant trypsin is free of chymotrypsin and exhibits high specific activity (80). Moreover, variable digestion kinetics patterns are often observed when using different trypsin sources (80). Other variables such as protein loss during enrichment or desalting step (e.g., methanol-chloroform-water extraction) (46) and sample-to-sample trypsin digestion artifacts, should be considered. While the apparent trypsin digestion efficiency could be protein or peptide-specific, addition of exogenous protein such as BSA, beta-casein, beta-galactosidase (25, 29, 81) is an important control for pre-digestion variables (Supplementary Figure 1). Effect of surfactant, reducing or alkylating agents on digestion efficiency can be reduced by a desalting step such as solid-phase extraction (73) or the use chloroform-methanol-water extraction (26) before trypsin digestion.
3.4. Data analysis variables and recommended controls
SRM analysis of peptides is a three step process, which involves peptide parent ion selection in the first quadrupole (Q1), fragmentation of the selected ion in the second quadrupole (Q2) and selection of the specific fragments in the third quadrupole (Q3) (Figure 1). However, simultaneous monitoring of multiple SRM transitions increases the total cycle time which compromises the number of data points for each peptide signal. To avoid this problem, scheduled SRM approach involving multiple retention time windows based on peptide hydrophobicity, is recommended (47). The scheduled SRM approach enables analysis of significantly more number of peptides per LC-MS run. This is more useful during method development as retention time can be predicted by using theoretical sequence specific retention time calculation as previously discussed by us in detail (61). As a quality check, signals of multiple daughter ions or transitions of a parent should be superimposable, elute at predicted retention time and qualified by corresponding heavy transitions (Figure 3D). There could be other superimposable higher intensity signals (ghost peaks) in the specific retention time window, which should to be identified by using heavy labeled peptide to avoid false positive results (Figure 3D).
Ideally the lower limit of quantification (LLOQ) is established by spiking standards into blank matrices. However, blank matrix for qualification of endogenous proteins in a tissue sample is not available. The light peptide standards are usually spiked into a buffer to generate a calibration curve (11). Alternatively, calibration curve made by spiking of known heavy standards in to the matrix/buffer also provides estimates of dynamic range and LLOQ (Supplementary Table 1). For specific proteins, LLOQ can also be verified by using gene-deletion or splice variant samples. For example, in the case of UGT2B17, we estimated LLOQ based on light peptide response in the UGT2B17 gene deletion (del/del) sample (unpublished data).
Inter-day data variability can be caused by many small changes in the sample preparation and LC-MS/MS platform: different trypsin batches, different batches of buffer, decay in performance of columns, electrospray tips, inlet capillaries and instrument components, drifts in calibration and tuning, etc. Re-analysis of a pooled or specific sample across different batches addresses such inter-day variability. For example, for each day, the data can be normalized to the quality control. Instead of analysis of samples in triplicates in a single batch, we recommend repeating the whole experiment (digestion and LC-MS/MS analysis) independently on three different days. This establishes the robustness of the method. While the standard deviation in repeat analysis is high, there is more confidence on the data quality. Table 2 shows a systematic three-step data normalization strategy used in our laboratory to perform protein quantification data analysis.
Table 2.
Representative table showing three-step data normalization approach. First, average light peak areas for specific peptide daughter fragments were divided by corresponding average heavy peak areas. This ratio was further divided by BSA light/heavy area ratio. For each day, these data were normalized by the average quality control values. Average ± SD (%CV) without and with normalization in the sub-table at the bottom, which shows importance of IS, BSA and QC normalization steps.
| Day | Sample number | Light peptide | Heavy peptide | Area ratio | BSA AR | Ratio of ratio | Average QC | Normalized to QCs |
|---|---|---|---|---|---|---|---|---|
| Day 1 | Sample 1 | 11500000 | 2080000 | 5.5 | 4.3 | 1.3 | 140.1 | |
| Sample 2 | 8126667 | 2090000 | 3.9 | 4.3 | 0.9 | 99.4 | ||
| Sample 3 | 12726667 | 2033333 | 6.3 | 4.4 | 1.4 | 155.4 | ||
| QC1 | 6106667 | 1560000 | 3.9 | 4.4 | 0.9 | 0.9 | 96.8 | |
| QC2 | 6516667 | 1680000 | 3.9 | 4.2 | 0.9 | 99.9 | ||
| QC3 | 5890000 | 1553333 | 3.8 | 4.0 | 0.9 | 103.3 | ||
| Day 2 | Sample 1 | 12776667 | 2026667 | 6.3 | 4.4 | 1.4 | 163.6 | |
| Sample 2 | 9313333 | 1510000 | 6.2 | 6.2 | 1.0 | 113.6 | ||
| Sample 3 | 13330000 | 1327667 | 10.0 | 6.7 | 1.5 | 170.9 | ||
| QC1 | 4930000 | 1397333 | 3.5 | 4.1 | 0.9 | 0.9 | 98.5 | |
| QC2 | 5193333 | 1763333 | 2.9 | 3.5 | 0.8 | 96.7 | ||
| QC3 | 5486667 | 1448333 | 3.8 | 4.1 | 0.9 | 104.8 | ||
| Day 3 | Sample 1 | 7210000 | 983000 | 7.3 | 4.9 | 1.5 | 157.7 | |
| Sample 2 | 4700000 | 1044333 | 4.5 | 4.7 | 1.0 | 102.0 | ||
| Sample 3 | 7996667 | 1180667 | 6.8 | 4.4 | 1.5 | 162.3 | ||
| QC1 | 3730000 | 888000 | 4.2 | 4.4 | 1.0 | 0.9 | 101.1 | |
| QC2 | 3826667 | 976667 | 3.9 | 4.3 | 0.9 | 97.4 | ||
| QC3 | 3766667 | 1013333 | 3.7 | 3.9 | 1.0 | 101.6 |
| Average ± SD (%CV) without and with normalization
| |||
|---|---|---|---|
| Sample 1 | Sample 2 | Sample 3 | |
| 1. Average (Light transitions) | 14e6±4e6 (21) | 15e6±4e6 (25) | 11e6±1e6 (10) |
| 2. Average (IS normalization) | 2.61±0.33 (12.6) | 2.04±0.49 (24.2) | 3.10±0.84 (27.1) |
| 3. Average (IS and BSA normalization) | 0.57±0.03 (5.5) | 0.40±0.02 (4.8) | 0.60±0.03 (5.0) |
| 4. Average (IS, BSA and QC normalization) | 1.6±0.05 (3) | 1.12±0.06 (5.6) | 1.67±0.575 (3.4) |
IS= heavy peptide internal standards
3.5 Additional challenges in quantitative DMET proteomics
Quantification of proteins in a non-homogenous tissue (kidney, brain, and intestine) requires information on location of the dissection region. For example, different brain cortex regions are perfused differentially and vary in neuroanatomy, which can result in differential protein abundance. If such information is available, intersegment variability in the protein abundance can be quantified as a confounding factor in the interindividual variability determination (29).
It is important for transporter quantification in primary cells or overexpression cell-lines to address that the transporters are correctly expressed in the plasma membrane. Intracellular accumulation of non-functional transporters due to dysfunctional trafficking quantified by the crude membrane fractions, could be misleading when these data are used for IVIVE. Isolation of pure plasma membrane fractions, though difficult, could be achieved (82). Variability in subcellular fraction purity, yield and enrichment could be confounding in determining interindividual variability. Use of marker proteins specific to each subcellular fraction can be promising. For example, although Na+/K+−ATPase is expressed only in the basolateral membrane, it can serve as a plasma membrane marker to evaluate if drug transporters are co-expressed with the marker (49).
The ultimate success of protein measurement can be established by evaluating correlation of protein abundance with activity of specific substrates. Ohtsuki et al. showed that in 17 liver biopsy samples, the protein expression levels of CYP3A4, CYP2B6, CYP2C8 and CYP2C19, CYP2E1, CYP1A2, CYP2C9 and CYP2D6 reasonably correlate with the corresponding enzyme activity (r2=0.68 to 0.95) (16). In our hands, CYP3A4 expression data correlate well with 4-OH-midazolam formation (Supplementary Figure 2, unpublished data). Recently, Kumar et al. showed that proteomics data correlate well with activity of OATP1B1 and BCRP in cell lines expressing the proteins (49). Principal component analysis (PCA) of multiple proteins in a data set can identify poor quality samples, considering that the quality of sample affects all the proteins in the same manner (Supplementary Figure 3).
Inter-laboratory variability in absolute quantification of DMET proteins (83) highlights the major limitation of the technique. All the variable affecting technical variability which we have discussed may differ laboratory-to-laboratory. Purity of protein or peptide standards provided by suppliers is crucial for absolute quantification. Another major challenge is to verify quality of archived tissue samples. Information on procurement, storage and handling of tissue samples as well as detailed demographics should be available to tease out some confounding factors to interindividual variability determination.
Inherently low sensitivity of LC-MS/MS detection (as compared to fluorescence detection), cost of instrument and non-availability of skilled manpower in the field are other challenges. Moreover, invasive nature of tissue sample procurement restricts application of tissue proteomics to the available samples from tissue banks.
6. Recommended best practices
Hypothesis-driven LC-MS/MS experiment offers high selectivity, dynamic range, large capacity for multiplexing (up to hundreds of proteins per analysis in scheduled SRM), cost-effective transition from assay development to tissue sample analysis. During method development, basic reliability of assay can be achieved by i) ensuring complete co-elution or overlap of the entire set of monitored fragment ions for both heavy and light for each analyte, ii) establishing that the m/z and relative ratios of precursor and fragment ions for the analyte measured compared with those of an authentic standard spectrum, iii) correlating the observed retention of the peptide to the predicted (or previously observed) retention of an authentic peptide, and iii) ensuring reproducibility of the results across multiple biological or full process replicates (72). We propose a systemic strategy as outlined in Figure 6. Briefly, full-process triplicates, addition of external protein standard in the sample, addition of pooled heavy internal standards in each sample and use of pooled quality control samples to obtain robust proteomics data are critical steps for optimum results. While the targeted SRM or MRM proteomics was the focus of this review, the best practices discussed above can be applied to other emerging quantitative proteomics approaches such as data-independent analysis sequential window acquisition of all theoretical fragment ion spectra (SWATH) (84), and parallel reaction monitoring (PRM) (85).
Figure 6.
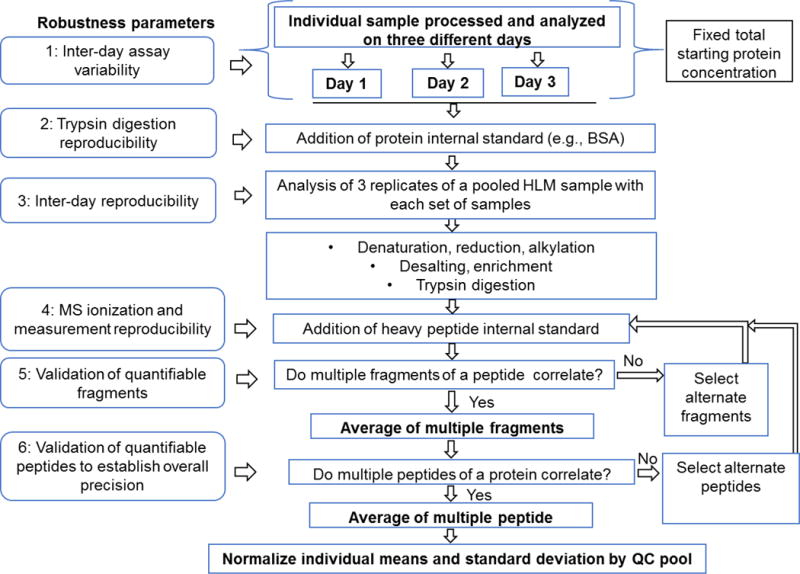
Optimized workflow of quantitative proteomics analysis applicable to large cohort of biological samples for accurate determination of interindividual (biological) variability.
Supplementary Material
Acknowledgments
This research is supported by Eunice Kennedy Shriver National Institute of Child Health and Human Development (NICHD) grant number, R01HD081299-03.
Abbreviations
- DME
Drug-metabolizing enzyme
- DMET
drug metabolizing enzyme and transporter
- LC-MS/MS
liquid chromatography coupled with tandem mass spectrometry
- MRM
multiple reaction monitoring
- PK
pharmacokinetics
- PBPK
physiologically based pharmacokinetic
- SRM
selective reaction monitoring
References
- 1.Hines RN, McCarver DG. The ontogeny of human drug-metabolizing enzymes: phase I oxidative enzymes. J Pharmacol Exp Ther. 2002;300:355–60. doi: 10.1124/jpet.300.2.355. [DOI] [PubMed] [Google Scholar]
- 2.McCarver DG, Hines RN. The ontogeny of human drug-metabolizing enzymes: phase II conjugation enzymes and regulatory mechanisms. J Pharmacol Exp Ther. 2002;300:361–6. doi: 10.1124/jpet.300.2.361. [DOI] [PubMed] [Google Scholar]
- 3.Vyhlidal CA, Bi C, Ye SQ, Leeder JS. Dynamics of Cytosine Methylation in the Proximal Promoters of CYP3A4 and CYP3A7 in Pediatric and Prenatal Livers. Drug Metab Dispos. 2016;44:1020–6. doi: 10.1124/dmd.115.068726. [DOI] [PubMed] [Google Scholar]
- 4.Wolbold R, et al. Sex is a major determinant of CYP3A4 expression in human liver. Hepatology. 2017;38:978–88. doi: 10.1053/jhep.2003.50393. [DOI] [PubMed] [Google Scholar]
- 5.Lamba V, et al. Hepatic CYP2B6 expression: gender and ethnic differences and relationship to CYP2B6 genotype and CAR (constitutive androstane receptor) expression. J Pharmacol Exp Ther. 2003;307:906–22. doi: 10.1124/jpet.103.054866. [DOI] [PubMed] [Google Scholar]
- 6.Gallagher CJ, Balliet RM, Sun D, Chen G, Lazarus P. Sex Differences in UDP-Glucuronosyltransferase 2B17 Expression and Activity. Drug Metab Dispos. 2010;38:2204–9. doi: 10.1124/dmd.110.035345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Johnson TN, Boussery K, Rowland-Yeo K, Tucker GT, Rostami-Hodjegan A. A semi-mechanistic model to predict the effects of liver cirrhosis on drug clearance. Clin Pharmacokinet. 2010;49:189–206. doi: 10.2165/11318160-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 8.Wang L, et al. Transporter Expression in Liver Tissue from Subjects with Alcoholic or Hepatitis C Cirrhosis Quantified by Targeted Quantitative Proteomics. Drug Metab Dispos. 2016;44:1752–8. doi: 10.1124/dmd.116.071050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hustert E, et al. The genetic determinants of the CYP3A5 polymorphism. Pharmacogenetics. 2001;11:773–9. doi: 10.1097/00008571-200112000-00005. [DOI] [PubMed] [Google Scholar]
- 10.Shirasaka Y, et al. Interindividual variability of CYP2C19-catalyzed drug metabolism due to differences in gene diplotypes and cytochrome P450 oxidoreductase content. Pharmacogenomics J. 2016;16:375–87. doi: 10.1038/tpj.2015.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Prasad B, et al. Interindividual variability in hepatic organic anion-transporting polypeptides and P-glycoprotein (ABCB1) protein expression: quantification by liquid chromatography tandem mass spectroscopy and influence of genotype, age, and sex. Drug Metab Dispos. 2014;42:78–88. doi: 10.1124/dmd.113.053819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jiang XL, Zhao P, Barrett JS, Lesko LJ, Schmidt S. Application of physiologically based pharmacokinetic modeling to predict acetaminophen metabolism and pharmacokinetics in children. CPT Pharmacometrics Syst Pharmacol. 2013;2:e80. doi: 10.1038/psp.2013.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Prasad B, Vrana M, Mehrotra A, Johnson K, Bhatt DK. The Promises of Quantitative Proteomics in Precision Medicine. J Pharm Sci. 2017;106:738–44. doi: 10.1016/j.xphs.2016.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Johnson TN, Rostami-Hodjegan A, Tucker GT. Prediction of the clearance of eleven drugs and associated variability in neonates, infants and children. Clin Pharmacokinet. 2006;45:931–56. doi: 10.2165/00003088-200645090-00005. [DOI] [PubMed] [Google Scholar]
- 15.Heidebrecht F, Heidebrecht A, Schulz I, Behrens SE, Bader A. Improved semiquantitative Western blot technique with increased quantification range. Journal of immunological methods. 2009;345:40–8. doi: 10.1016/j.jim.2009.03.018. [DOI] [PubMed] [Google Scholar]
- 16.Ohtsuki S, et al. Simultaneous absolute protein quantification of transporters, cytochromes P450, and UDP-glucuronosyltransferases as a novel approach for the characterization of individual human liver: comparison with mRNA levels and activities. Drug Metab Dispos. 2012;40:83–92. doi: 10.1124/dmd.111.042259. [DOI] [PubMed] [Google Scholar]
- 17.Deo AK, Prasad B, Balogh L, Lai Y, Unadkat JD. Interindividual variability in hepatic expression of the multidrug resistance-associated protein 2 (MRP2/ABCC2): quantification by liquid chromatography/tandem mass spectrometry. Drug Metab Dispos. 2012;40:852–5. doi: 10.1124/dmd.111.043810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Miyauchi E, et al. Quantitative Atlas of Cytochrome P450, UDP-Glucuronosyltransferase, and Transporter Proteins in Jejunum of Morbidly Obese Subjects. Mol Pharm. 2016;13:2631–40. doi: 10.1021/acs.molpharmaceut.6b00085. [DOI] [PubMed] [Google Scholar]
- 19.Margaillan G, et al. Multiplexed Targeted Quantitative Proteomics Predicts Hepatic Glucuronidation Potential. Drug Metab Dispos. 2015;43:1331–5. doi: 10.1124/dmd.115.065391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Harbourt DE, et al. Quantification of Human Uridine-Diphosphate Glucuronosyl Transferase (UGT) 1A Isoforms in Liver, Intestine and Kidney using nanoLC-MS/MS. Anal Chem. 2012;84:98–105. doi: 10.1021/ac201704a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sato Y, et al. Optimized methods for targeted peptide-based quantification of human uridine 5′-diphosphate-glucuronosyltransferases in biological specimens using liquid chromatography-tandem mass spectrometry. Drug Metab Dispos. 2014;42:885–9. doi: 10.1124/dmd.113.056291. [DOI] [PubMed] [Google Scholar]
- 22.Yan T, et al. Significantly decreased and more variable expression of major CYPs and UGTs in liver microsomes prepared from HBV-positive human hepatocellular carcinoma and matched pericarcinomatous tissues determined using an isotope label-free UPLC-MS/MS method. Pharmaceutical research. 2015;32:1141–57. doi: 10.1007/s11095-014-1525-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Groer C, et al. Absolute protein quantification of clinically relevant cytochrome P450 enzymes and UDP-glucuronosyltransferases by mass spectrometry-based targeted proteomics. Journal of pharmaceutical and biomedical analysis. 2014;100:393–401. doi: 10.1016/j.jpba.2014.08.016. [DOI] [PubMed] [Google Scholar]
- 24.Achour B, Russell MR, Barber J, Rostami-Hodjegan A. Simultaneous quantification of the abundance of several cytochrome P450 and uridine 5′-diphospho-glucuronosyltransferase enzymes in human liver microsomes using multiplexed targeted proteomics. Drug Metab Dispos. 2014;42:500–10. doi: 10.1124/dmd.113.055632. [DOI] [PubMed] [Google Scholar]
- 25.Fallon JK, Neubert H, Hyland R, Goosen TC, Smith PC. Targeted quantitative proteomics for the analysis of 14 UGT1As and -2Bs in human liver using NanoUPLC-MS/MS with selected reaction monitoring. Journal of proteome research. 2013;12:4402–13. doi: 10.1021/pr4004213. [DOI] [PubMed] [Google Scholar]
- 26.Boberg M, et al. Age-Dependent Absolute Abundance of Hepatic Carboxylesterases (CES1 and CES2) by LC-MS/MS Proteomics: Application to PBPK Modeling of Oseltamivir In Vivo Pharmacokinetics in Infants. Drug Metab Dispos. 2017;45:216–23. doi: 10.1124/dmd.116.072652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang L, et al. Interspecies variability in expression of hepatobiliary transporters across human, dog, monkey, and rat as determined by quantitative proteomics. Drug Metab Dispos. 2015;43:367–74. doi: 10.1124/dmd.114.061580. [DOI] [PubMed] [Google Scholar]
- 28.Prasad B, Lai Y, Lin Y, Unadkat JD. Interindividual variability in the hepatic expression of the human breast cancer resistance protein (BCRP/ABCG2): effect of age, sex, and genotype. J Pharm Sci. 2013;102:787–93. doi: 10.1002/jps.23436. [DOI] [PubMed] [Google Scholar]
- 29.Prasad B, et al. Abundance of Drug Transporters in the Human Kidney Cortex as Quantified by Quantitative Targeted Proteomics. Drug Metab Dispos. 2016;44:1920–4. doi: 10.1124/dmd.116.072066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Drozdzik M, et al. Protein abundance of clinically relevant multidrug transporters along the entire length of the human intestine. Mol Pharm. 2014;11:3547–55. doi: 10.1021/mp500330y. [DOI] [PubMed] [Google Scholar]
- 31.Barr JT, Jones JP, Joswig-Jones CA, Rock DA. Absolute Quantification of Aldehyde Oxidase Protein in Human Liver Using Liquid Chromatography-Tandem Mass Spectrometry. Mol Pharm. 2013;10 doi: 10.1021/mp4003046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yu AM, Qu J, Felmlee MA, Cao J, Jiang XL. Quantitation of human cytochrome P450 2D6 protein with immunoblot and mass spectrometry analysis. Drug Metab Dispos. 2009;37:170–7. doi: 10.1124/dmd.108.024166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu X, et al. Quantitative analysis of cytochrome P450 isoforms in human liver microsomes by the combination of proteomics and chemical probe-based assay. Proteomics. 2014;14:1943–51. doi: 10.1002/pmic.201400025. [DOI] [PubMed] [Google Scholar]
- 34.Nakamura K, et al. Large-scale multiplex absolute protein quantification of drug-metabolizing enzymes and transporters in human intestine, liver, and kidney microsomes by SWATH-MS: Comparison with MRM/SRM and HR-MRM/PRM. Proteomics. 2016;16:2106–17. doi: 10.1002/pmic.201500433. [DOI] [PubMed] [Google Scholar]
- 35.Edson KZ, et al. Cytochrome P450-dependent catabolism of vitamin K: omega-hydroxylation catalyzed by human CYP4F2 and CYP4F11. Biochemistry. 2013;52:8276–85. doi: 10.1021/bi401208m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shuster DL, et al. Identification of CYP3A7 for glyburide metabolism in human fetal livers. Biochem Pharmacol. 2014;92:690–700. doi: 10.1016/j.bcp.2014.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Uchida Y, et al. Quantitative targeted absolute proteomics of human blood-brain barrier transporters and receptors. Journal of neurochemistry. 2011;117:333–45. doi: 10.1111/j.1471-4159.2011.07208.x. [DOI] [PubMed] [Google Scholar]
- 38.Shawahna R, et al. Transcriptomic and quantitative proteomic analysis of transporters and drug metabolizing enzymes in freshly isolated human brain microvessels. Mol Pharm. 2011;8:1332–41. doi: 10.1021/mp200129p. [DOI] [PubMed] [Google Scholar]
- 39.Zhang HF, et al. Correlation of Cytochrome P450 Oxidoreductase Expression with the Expression of 10 Isoforms of Cytochrome P450 in Human Liver. Drug Metab Dispos. 2016;44:1193–200. doi: 10.1124/dmd.116.069849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang HF, et al. Correlation of Cytochrome P450 Oxidoreductase Expression with the Expression of 10 Isoforms of Cytochrome P450 in Human Liver. 2016 doi: 10.1124/dmd.116.069849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen Y, Zane NR, Thakker DR, Wang MZ. Quantification of Flavin-containing Monooxygenases 1, 3, and 5 in Human Liver Microsomes by UPLC-MRM-Based Targeted Quantitative Proteomics and Its Application to the Study of Ontogeny. Drug Metab Dispos. 2016;44:975–83. doi: 10.1124/dmd.115.067538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chen Y, Zane NR, Thakker DR, Wang MZ. Quantification of Flavin-containing Monooxygenases 1.3 and 5 in Human Liver Microsomes by UPLC-MRM-based Targeted Quantitative Proteomics and Its Application to the Study of Ontogeny. 2016 doi: 10.1124/dmd.115.067538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Harwood MD, Achour B, Russell MR, Carlson GL, Warhurst G, Rostami-Hodjegan A. Application of an LC-MS/MS method for the simultaneous quantification of human intestinal transporter proteins absolute abundance using a QconCAT technique. Journal of pharmaceutical and biomedical analysis. 2015;110:27–33. doi: 10.1016/j.jpba.2015.02.043. [DOI] [PubMed] [Google Scholar]
- 44.Langenfeld E, Zanger UM, Jung K, Meyer HE, Marcus K. Mass spectrometry-based absolute quantification of microsomal cytochrome P450 2D6 in human liver. Proteomics. 2009;9:2313–23. doi: 10.1002/pmic.200800680. [DOI] [PubMed] [Google Scholar]
- 45.Margaillan G, et al. Quantitative profiling of human renal UDP-glucuronosyltransferases and glucuronidation activity: a comparison of normal and tumoral kidney tissues. Drug Metab Dispos. 2015;43:611–9. doi: 10.1124/dmd.114.062877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bhatt DK, Gaedigk A, Pearce RE, Leeder JS, Prasad B. Age-dependent protein abundance of cytosolic alcohol and aldehyde dehydrogenases in human liver. Drug Metab Dispos. 2017 doi: 10.1124/dmd.117.076463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Prasad B, Unadkat JD. Optimized approaches for quantification of drug transporters in tissues and cells by MRM proteomics. Aaps j. 2014;16:634–48. doi: 10.1208/s12248-014-9602-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Vrana M, et al. Database of Optimized Proteomic Quantitative Methods for Human Drug Disposition‐Related Proteins for Applications in Physiologically Based Pharmacokinetic Modeling. CPT: Pharmacometrics & Systems Pharmacology. 2017;6:267–76. doi: 10.1002/psp4.12170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kumar V, et al. Quantitative transporter proteomics by liquid chromatography with tandem mass spectrometry: addressing methodologic issues of plasma membrane isolation and expression-activity relationship. Drug Metab Dispos. 2015;43:284–8. doi: 10.1124/dmd.114.061614. [DOI] [PubMed] [Google Scholar]
- 50.Qiu X, Bi YA, Balogh LM, Lai Y. Absolute measurement of species differences in sodium taurocholate cotransporting polypeptide (NTCP/Ntcp) and its modulation in cultured hepatocytes. J Pharm Sci. 2013;102:3252–63. doi: 10.1002/jps.23582. [DOI] [PubMed] [Google Scholar]
- 51.Bi YA, et al. Quantitative assessment of the contribution of sodium-dependent taurocholate co-transporting polypeptide (NTCP) to the hepatic uptake of rosuvastatin, pitavastatin and fluvastatin. Biopharm Drug Dispos. 2013;34:452–61. doi: 10.1002/bdd.1861. [DOI] [PubMed] [Google Scholar]
- 52.Vildhede A, et al. Mechanistic Modeling of Pitavastatin Disposition in Sandwich-Cultured Human Hepatocytes: A Proteomics-Informed Bottom-Up Approach. Drug Metab Dispos. 2016;44:505–16. doi: 10.1124/dmd.115.066746. [DOI] [PubMed] [Google Scholar]
- 53.Drissi R, Dubois ML, Boisvert FM. Proteomics methods for subcellular proteome analysis. The FEBS journal. 2013;280:5626–34. doi: 10.1111/febs.12502. [DOI] [PubMed] [Google Scholar]
- 54.Yang GX, Li X, Snyder M. Investigating metabolite-protein interactions: an overview of available techniques. Methods. 2012;57:459–66. doi: 10.1016/j.ymeth.2012.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Duijvesz D, et al. Proteomic profiling of exosomes leads to the identification of novel biomarkers for prostate cancer. PloS one. 2013;8:e82589. doi: 10.1371/journal.pone.0082589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Williamson BL, et al. Quantitative protein determination for CYP induction via LC-MS/MS. Proteomics. 2011;11:33–41. doi: 10.1002/pmic.201000456. [DOI] [PubMed] [Google Scholar]
- 57.Salphati L, et al. Evaluation of organic anion transporting polypeptide 1B1 and 1B3 humanized mice as a translational model to study the pharmacokinetics of statins. Drug Metab Dispos. 2014;42:1301–13. doi: 10.1124/dmd.114.057976. [DOI] [PubMed] [Google Scholar]
- 58.Hoshi Y, Uchida Y, Tachikawa M, Inoue T, Ohtsuki S, Terasaki T. Quantitative atlas of blood-brain barrier transporters, receptors, and tight junction proteins in rats and common marmoset. J Pharm Sci. 2013;102:3343–55. doi: 10.1002/jps.23575. [DOI] [PubMed] [Google Scholar]
- 59.Ito K, et al. Quantitative membrane protein expression at the blood-brain barrier of adult and younger cynomolgus monkeys. J Pharm Sci. 2011;100:3939–50. doi: 10.1002/jps.22487. [DOI] [PubMed] [Google Scholar]
- 60.Liao MZ, et al. P-gp/ABCB1 exerts differential impacts on brain and fetal exposure to norbuprenorphine. Pharmacological research. 2017;119:61–71. doi: 10.1016/j.phrs.2017.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Vrana M, Whittington D, Nautiyal V, Prasad B. A database of optimized proteomic quantitative methods for 284 human drug disposition related proteins for applications in PBPK modeling. CPT Pharmacometrics Syst Pharmacol. 2017 doi: 10.1002/psp4.12170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Leth-Larsen R, Lund RR, Ditzel HJ. Plasma Membrane Proteomics and Its Application in Clinical Cancer Biomarker Discovery*. Mol Cell Proteomics. 2010;9:1369–82. doi: 10.1074/mcp.R900006-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chung FS, et al. Positron emission tomography imaging of tissue P-glycoprotein activity during pregnancy in the non-human primate. British journal of pharmacology. 2010;159:394–404. doi: 10.1111/j.1476-5381.2009.00538.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Patilea-Vrana G, Unadkat JD. Transport vs. Metabolism: What Determines the Pharmacokinetics and Pharmacodynamics of Drugs? Insights From the Extended Clearance Model. Clin Pharmacol Ther. 2016;100:413–8. doi: 10.1002/cpt.437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hsiao P, et al. Verapamil P-glycoprotein transport across the rat blood-brain barrier: cyclosporine, a concentration inhibition analysis, and comparison with human data. J Pharmacol Exp Ther. 2006;317:704–10. doi: 10.1124/jpet.105.097931. [DOI] [PubMed] [Google Scholar]
- 66.Hsiao P, Bui T, Ho RJ, Unadkat JD. In vitro-to-in vivo prediction of P-glycoprotein-based drug interactions at the human and rodent blood-brain barrier. Drug Metab Dispos. 2008;36:481–4. doi: 10.1124/dmd.107.018176. [DOI] [PubMed] [Google Scholar]
- 67.Nyquist MD, Prasad B, Mostaghel EA. Harnessing Solute Carrier Transporters for Precision Oncology. Molecules (Basel, Switzerland) 2017;22(4) doi: 10.3390/molecules22040539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Piehowski PD, et al. Sources of Technical Variability in Quantitative LC-MS Proteomics: Human Brain Tissue Sample Analysis. Journal of proteome research. 2013;12:2128–37. doi: 10.1021/pr301146m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Court MH, et al. UDP-glucuronosyltransferase (UGT) 2B15 pharmacogenetics: UGT2B15 D85Y genotype and gender are major determinants of oxazepam glucuronidation by human liver. J Pharmacol Exp Ther. 2004;310:656–65. doi: 10.1124/jpet.104.067660. [DOI] [PubMed] [Google Scholar]
- 70.Johnson H, Eyers CE. Analysis of post-translational modifications by LC-MS/MS. Methods in molecular biology (Clifton, NJ) 2010;658:93–108. doi: 10.1007/978-1-60761-780-8_5. [DOI] [PubMed] [Google Scholar]
- 71.Saveliev S, Bratz M, Zubarev R, Szapacs M, Budamgunta H, Urh M. Trypsin/Lys-C protease mix for enhanced protein mass spectrometry analysis. Nature Methods. 2013;10 [Google Scholar]
- 72.Carr SA, et al. Targeted Peptide Measurements in Biology and Medicine: Best Practices for Mass Spectrometry-based Assay Development Using a Fit-for-Purpose Approach*. Mol Cell Proteomics. 2014;13:907–17. doi: 10.1074/mcp.M113.036095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Guo X, Kristal BS. The use of under-loaded C18 solid-phase extraction plates increases reproducibility of analysis of tryptic peptides from unfractionated human plasma. Analytical biochemistry. 2012;426:86–90. doi: 10.1016/j.ab.2012.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Abbatiello SE, et al. Large-Scale Interlaboratory Study to Develop, Analytically Validate and Apply Highly Multiplexed, Quantitative Peptide Assays to Measure Cancer-Relevant Proteins in Plasma*. Mol Cell Proteomics. 2015;14:2357–74. doi: 10.1074/mcp.M114.047050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Reddy PM, Hsu WY, Hu JF, Ho YP. Digestion completeness of microwave-assisted and conventional trypsin-catalyzed reactions. Journal of the American Society for Mass Spectrometry. 2010;21:421–4. doi: 10.1016/j.jasms.2009.11.006. [DOI] [PubMed] [Google Scholar]
- 76.Arnold SL, Stevison F, Isoherranen N. Impact of Sample Matrix on Accuracy of Peptide Quantification: Assessment of Calibrator and Internal Standard Selection and Method Validation. Anal Chem. 2016;88:746–53. doi: 10.1021/acs.analchem.5b03004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Chen B, et al. Strategies of Drug Transporter Quantitation by LC-MS: Importance of Peptide Selection and Digestion Efficiency. Aaps j. 2017 doi: 10.1208/s12248-017-0106-4. [DOI] [PubMed] [Google Scholar]
- 78.Harwood MD, Achour B, Neuhoff S, Russell MR, Carlson G, Warhurst G. In Vitro-In Vivo Extrapolation Scaling Factors for Intestinal P-Glycoprotein and Breast Cancer Resistance Protein: Part I: A Cross-Laboratory Comparison of Transporter-Protein Abundances and Relative Expression Factors in Human Intestine and Caco-2 Cells. Drug Metab Dispos. 2016;44:297–307. doi: 10.1124/dmd.115.067371. [DOI] [PubMed] [Google Scholar]
- 79.Emoto C, Fukuda T, Johnson TN, Neuhoff S, Sadhasivam S, Vinks AA. Characterization of Contributing Factors to Variability in Morphine Clearance Through PBPK Modeling Implemented With OCT1 Transporter. CPT Pharmacometrics Syst Pharmacol. 2017;6:110–9. doi: 10.1002/psp4.12144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Walmsley SJ, Rudnick PA, Liang Y, Dong Q, Stein SE, Nesvizhskii AI. Comprehensive analysis of protein digestion using six trypsins reveals the origin of trypsin as a significant source of variability in proteomics. Journal of proteome research. 2013;12:5666–80. doi: 10.1021/pr400611h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Rardin MJ, et al. MS1 Peptide Ion Intensity Chromatograms in MS2 (SWATH) Data Independent Acquisitions. Improving Post Acquisition Analysis of Proteomic Experiments*. Mol Cell Proteomics. 2015;14:2405–19. doi: 10.1074/mcp.O115.048181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Weekes MP, Antrobus R, Lill JR, Duncan LM, Hör S, Lehner PJ. Comparative Analysis of Techniques to Purify Plasma Membrane Proteins. J Biomol Tech. 2010;21:108–15. [PMC free article] [PubMed] [Google Scholar]
- 83.Harwood MD, et al. In Vitro-In Vivo Extrapolation Scaling Factors for Intestinal P-glycoprotein and Breast Cancer Resistance Protein: Part II. The Impact of Cross-Laboratory Variations of Intestinal Transporter Relative Expression Factors on Predicted Drug Disposition. Drug Metab Dispos. 2016;44:476–80. doi: 10.1124/dmd.115.067777. [DOI] [PubMed] [Google Scholar]
- 84.Schubert OT, et al. Building high-quality assay libraries for targeted analysis of SWATH MS data. Nature protocols. 2015;10:426–41. doi: 10.1038/nprot.2015.015. [DOI] [PubMed] [Google Scholar]
- 85.Peterson AC, Russell JD, Bailey DJ, Westphall MS, Coon JJ. Parallel reaction monitoring for high resolution and high mass accuracy quantitative, targeted proteomics. Molecular & cellular proteomics : MCP. 2012;11:1475–88. doi: 10.1074/mcp.O112.020131. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.