

Abstract
Heterogeneity amidst healthy individuals at genomic level is being widely acknowledged. This, in turn, is modulated by differential response to environmental cues and treatment regimens, necessitating the need for stratified/personalized therapy. We intend to understand the molecular determinants of Ayurvedic way (ancient Indian system of medicine) of endo-phenotyping individuals into distinct constitution types termed “Prakriti,” which forms the basis of personalized treatment. In this study, we explored and analyzed the healthy human gut microbiome structure within three predominant Prakriti groups from a genetically homogenous cohort to discover differentially abundant taxa, using 16S rRNA gene based microbial community profiling. We found Bacteroidetes and Firmicutes as major gut microbial components in varying composition, albeit with similar trend across Prakriti. Multiple species of the core microbiome showed differential abundance within Prakriti types, with gender specific signature taxons. Our study reveals that despite overall uniform composition of gut microbial community, healthy individuals belonging to different Prakriti groups have enrichment of specific bacteria. It highlights the importance of Prakriti based endo-phenotypes to explain the variability amongst healthy individuals in gut microbial flora that have important consequences for an individual's health, disease and treatment.
Keywords: Indian gut microbiome, Prakriti, precision medicine, ayurgenomics, ayurveda, 16S rRNA gene
Introduction
The human microbiome have been shown to have a functional role in the human physiology. Microbial flora and its dynamics are often associated with homeostasis within the body. Systemic characterization of the microbiota for comparison of microbial communities and their contribution to health and disease (Dominguez-Bello and Blaser, 2008; Rosenberg and Zilber-Rosenberg, 2011; Dave et al., 2012) have been carried out by the Human Microbiome Project (HMP) and Metagenomics of the Human Intestinal Tract (MetaHIT) consortium. These studies have provided insights into the composition of microbial community at various anatomical sites (Human Microbiome Project Consortium, 2012; Parfrey and Knight, 2012). The human microbiota has co-evolved closely with its host (Yatsunenko et al., 2012; Moeller et al., 2016) and is modulated by intrinsic and environmental factors. Recent studies have indicated that health and predisposition to various non-infectious diseases of humans are also determined by the genes coded by resident microbiome (Albenberg et al., 2012; Cho and Blaser, 2012; Gordon et al., 2012; Zhang et al., 2015). It is appreciated now that an understanding of human physiology is incomplete without the knowledge of the metagenome. Contemporary approaches have focused on investigating the microbial assemblage of the transient states observed over the course of specific diseases. However, the challenge is to elucidate whether the association between microbial community changes and pathology is causal in nature (Clemente et al., 2012; Haiser and Turnbaugh, 2012). Integrative analysis of human genome, physiology and microbiome will enable better understanding as to whether latter is involved in health and disease. However, there are variables contributed by human host as well as the microbiome, which could confound the observations. For example, the ethnicity and genetic background, age of the individual, dietary and lifestyle habits; all of which have been known to affect the human physiome and shape the microbiome (Fortenberry, 2013; Chong et al., 2015). Studies to identify association between microbial community structure based on ethnicity, diet, gender in healthy individuals, have met with limited success (Chong et al., 2015; Bhute et al., 2016). Absence of definitive patterns has been ascribed to genetic drift as well as population admixture. Therefore, it is a challenge to develop a population based catalog of human microbiome markers for predicting disease predisposition especially for a diverse Indian population. The subcontinent is home to more than one billion people with thousands of endogamous populations from different linguistic lineages and ethnic groups. Along with this diversity, individuals within these population/s have diverse food habits, digestive capabilities, and susceptibility to diseases (Bhute et al., 2016).
Ayurveda, the ancient Indian system of medicine documented and practiced for over 5000 years has an individualized approach to management of health and disease. According to this system, individuals can be classified on the basis of their constitution types termed “Prakriti” (Prasher et al., 2008, 2016, 2017; Sethi et al., 2011). Prakriti of an individual is determined at the time of birth and remains invariant throughout lifetime. It determines an individual's susceptibility, response to drug, diet and environment as well as prognosis for a disease. Prakriti is a consequence of relative proportions of three physiological entities (tridoshas) viz. Vata (V), Pitta (P), and Kapha (K), which govern different functions of transport, metabolism and storage, response to environment and homeostasis in the system. Perturbation of tridosha proportions from their homeostatic thresholds leads to disease state. Therefore, individuals based on their dominant proportions of doshas are called as Vata, Pitta, Kapha, Vata-Pitta, Vata-Kapha, Pitta- Kapha, and Vata-Pitta-Kapha Prakriti types. Amongst the seven constitution types, Vata, Pitta, and Kapha are the three phenotypic extremes with contrasting disease susceptibilities. Phenotypic assessment of Prakriti is carried out on the basis of examination of approximately 150 features comprising of anatomical, physiological, activity related attributes, and psychological parameters. For example, individuals of Pitta Prakriti would have better digestion and metabolic capacity whereas Kapha would have less and Vata with an irregular or unpredictable pattern. Recently, we have also been able to develop predictive models for Prakriti that recapitulate the Ayurveda constitution types through phenotypic traits of individuals (Prasher et al., 2008, 2016).
Many of the phenotypic attributes that are being associated with microbiome difference also differ between the constitution types. This includes desire and suitability for different diets, metabolic and digestive patterns, weight gain tendencies, gut motility and excretory patterns (Prasher et al., 2008, 2016). Besides, certain therapeutic modalities unique to Ayurveda that are aimed at maintenance of health and homeostasis lays emphasis on restoration of healthy flora (Prasher et al., 2016, 2017). Earlier, we and other groups have shown that healthy individuals of extreme Prakriti types, Vata, Pitta, and Kapha comprise 8–10% of a population and exhibit genome wide differences amongst constitution types, albeit from a genetically homogeneous background (Prasher et al., 2008; Aggarwal et al., 2010; Rotti et al., 2014; Govindaraj et al., 2015). These sub-types have underlying differences in genes that modulate pathways for apoptosis, metabolism, hypoxia response, haemostasis, and development. These differences can contribute to inter-individual variability in adaptation to high altitudes, susceptibility to high altitude pulmonary edema or thrombotic outcomes in hypoxia (Prasher et al., 2008; Aggarwal et al., 2010, 2015). Considering the significance of human microbiome in health and diseases, the current study is proposed to analyse gut microbiome in extreme constitution types to explore whether there could be prakriti specific microbial assemblage. The study was carried out in a genetically homogenous rural population comprising of healthy individuals of similar age group and dietary habits that were phenotypically stratified on the basis of Prakriti.
Materials and methods
Volunteer recruitment, Prakriti ascertainment, sample collection
The subjects were identified from a rural population in the Pune district of Western India. These participants belong to a cohort from Vadu Health and Demographic Surveillance System (Vadu HDSS) area who have been followed over years by the Vadu Rural Health Program, KEM Hospital Research Centre, Pune. The details of the sampling strategy and recruitment have been described earlier (Tiwari et al., 2017). Predominant Prakriti types, which comprises 8–10% of the population, were identified from randomly selected 10,100 individuals between the age group of 18–40 years. After preliminary screening using a questionnaire, 528 self reported healthy individuals were enrolled for detailed Prakriti evaluation using a questionnaire and methods developed in our earlier study (Prasher et al., 2008). Prakriti screening and clinical assessment was carried out by Ayurveda clinicians and a trained team of field research assistants. Assignment to Prakriti groups was carried out by two groups of physicians, one at field site and the other at CSIR-IGIB. Using unsupervised clustering approaches we have recently shown that these Prakriti groups form three natural clusters (Tiwari et al., 2017). The enrolled subjects were requested to provide fresh stool samples and it was ensured that they were not under any medication especially antibiotics. Field camps were organized in residential villages in the Vadu HDSS area. Two separate home visits were made by field teams—the first one, 8 days prior to camp to ensure availability of participants and the second, a day prior to camp to provide sterile containers along with the instructions to collect fresh stool samples. Standard operating procedures were strictly adhered to, while collecting samples, their storage at Vadu molecular lab, isolation of DNA, quality assurance and transportation of DNA aliquot to processing lab at CSIR-IGIB. A total of 135 extreme Prakriti individuals were identified, namely Kapha (n = 48), Pitta (n = 35), and Vata (n = 52).
The study population is relatively homogeneous in terms of ethnic and linguistic background as well as with respect to dietary and socio-cultural life style. In order to reaffirm the genetic homogeneity of the study population, we have earlier analyzed the genetic relatedness using unlinked and shared panel of 17675 SNP markers with the Indian Genome Variation Consortium (IGVC) diversity panel. Principal Component analysis (PCA) of the genotype data performed using EIGENSOFT 5.0 reaffirmed the homogeneity of the study population (Tiwari et al., 2017). This study has been carried out as per protocols approved by institutional ethics committee at CSIR-Institute of Genomics and Integrative Biology, Delhi and KEM Hospital Research Centre, Pune, India. Fresh stool samples were collected from subjects belonging to predominant Prakriti groups (Supplementary Table S1). Metagenomic DNA from stool samples was isolated using QIAamp DNA stool mini kit (Qiagen, Cat. No. 51504, USA).
16S rRNA gene amplicon sequencing
We amplified and sequenced V2-V6 region of 16S rRNA gene using metagenomic DNA of 135 individuals, inclusive of 70 females and 65 males using Roche GS FLX+ sequencing technology. At highest stringency of Q40, we got approximately 580 Mb per sequencing run with median read length of 800 bps.
Raw data processing and community compositional estimates
The Quantitative Insights into Microbial Ecology (QIIME) software package version 1.8.0 (Caporaso et al., 2010) was used to process and analyse raw sequencing data, separately for the male and female datasets. The split_library.py script was used in QIIME as a quality filtering step in which each sample was pre-processed with the maximum allowed one barcode error and two ambiguous bases (Ns). Sequences shorter than 300 or longer than 800 nucleotides with average quality <30 were removed from downstream analysis. In first pass of quality filtering, chimeric sequences were identified and removed by USEARCH 6.1 through QIIME's chimera processing scripts. Reads were clustered into operational taxonomic units (OTUs) with a sequence similarity threshold of 97% using UCLUST v1.2.22q (Edgar, 2010), within QIIME. Reads were assigned to OTUs using a closed reference OTU picking workflow (Caporaso et al., 2010) against GreenGenes 16S rRNA gene database (version 13_8), filtered at 97% sequence identity. In a closed-reference OTU picking, input sequences are aligned to pre-identified taxonomic clusters in a reference database. The input sequence is excluded if it does not match any reference sequence at user defined identity threshold. In further analysis, GreenGenes reference tree and taxonomic assignments were used. Microbial abundance were normalized to generate relative abundance of taxa present in each sample.
Gut microbiome diversity analysis
Alpha diversity (Shannon diversity and Observed species) for all samples were calculated using QIIME, to estimate species diversity, richness and evenness. Overall taxonomic differences and beta diversity were estimated through Principal Coordinates Analysis (PCoA) based on Bray-Curtis distances, using LabDSV (Roberts, 2013). Results of analyses were visualized using ggplot2 (Wickham, 2016) on R. The taxa of two highly abundant phyla, viz., Firmicutes and Bacteroidetes were removed in QIIME. The alpha diversity (Shannon diversity, richness and evenness) was calculated using “Vegan” package (Dixon, 2003) in R and beta diversity was calculated as mentioned above.
Estimation of core microbiome and biomarker discovery
Considering the variable nature of metagenomic compositional data, we also performed further analysis only for conserved taxons. Toward this, we estimated core microbial group within the samples with presence in at least 50% of the study samples. Taxonomic classification using alpha and beta diversity were analyzed for the core microbiome as explained above. LEfSe (Segata et al., 2011) was used to identify the microbiological markers associated with Prakriti by linear discriminate analysis (LDA) effect size of 2, and for multiclass analysis one-against-all option was used with default parameters (Goecks et al., 2010). Differentially abundant taxons were annotated to their genus and species level through manual Blast (NCBI web server) against the “refseq_rna” database, retaining the highest scoring hit. Since our objective was to identify signature taxa pertaining to Prakriti groups, we systematically removed redundancies keeping the OTU with highest LDA score and selected only those differential taxa that were exclusively present in a particular group.
qPCR validation of Prakriti specific microbial enterotypes
qPCR based validation of Prakriti specific dominant microbial enterotypes, Prevotella, Roseburia hominis, Eubacterium rectale, Blautia torques, and Blautia obeum was performed by absolute quantification of 16S rRNA gene copy number using genus specific primers (Supplementary Table S2). Total of 48 samples was used with proportional representation of Prakriti types. DNA template concentration for each sample was adjusted to 25 ng/μl. Amplification and detection were performed in a 10 μl reaction [5 μl 2x KAPA SYBR Green PCR Master Mix, 1 μl of each specific primer (10 μM), 1 μl template (25 ng/μl) and 2 μl molecular biology grade water] in triplicate using LC480 Real time PCR system (Roche, Switzerland). Amplification condition include one cycle of activation at 50°C for 2 min and denaturation at 95°C for 3 min, followed by 45 cycles at 95°C for 30 s, 58/64°C for 30 s, 72°C for 40 s; followed by extension at 72°C for 3 min. Melting curve was analyzed for non-specific products at 95°C for 5 s, 65°C for 1 min and 72°C continuous, followed by cooling at 37°C for 3 min. Group specific standard curves was generated from 10-fold serial dilutions of a known concentration of genomic DNA. Average Ct-values of the triplicate was used for estimating 16S rRNA gene copy numbers for each group using standard curves. Percentage abundance of each genus was obtained by calculating ratio of copy number of that genus to that of total bacteria (Eubacteria). Throughout the qPCR, efficiency was maintained above 90% with a correlation coefficient of >0.99. The statistical significance differences for Prakriti specific microbial enterotypes were determined by “Wilcoxon Rank Sum” test in R package.
Results
Uniform microbial taxonomic distribution and variability in Prakriti groups of healthy individuals
Using high throughput sequencing, we obtained 2,699,584 and 2,266,514 reads of 16S rRNA gene from 65 male and 70 female predominant Prakriti individuals respectively. Quality filtered raw sequences were taken forward for OTU identification and analyses. QIIME assisted chimera identification using USEARCH resulted in removal of 63,973 and 66,965 sequences from the male and female respectively, with remaining quality filtered set of 2,054,437 and 1,697,708 reads. GreenGenes based OTU identification through closed reference OTU picking protocol of QIIME resulted in identification of 3363 unique OTUs among males and 2882 within females. For stringency, reduced noise in the data and to ensure proper coverage of the entire gut microbial flora, we discarded samples with <4,000 reads. Our final dataset had 1,870,897 and 1,445,312 sequences from 50 male and 63 female subjects. We detected on average, 32,570 and 21,586 OTUs across male and female samples, indicating good coverage of microbial flora, although with variability (σmale = 25621.7 and σfemale = 14529.2). To overcome this variability, we normalized the absolute abundance counts to reflect relative abundances.
Taxonomic summary shows Bacteroidetes and Firmicutes to be the majority constituents at phylum level for both the male and female groups across Prakriti groups, together accounting for more than 98 percent of total abundance (σmale = 4.5%, σfemale = 1.9%) (Figure 1). To summarize diversities in individuals and Prakriti groups, we used the alpha and beta diversity metrics as introduced by R. H. Whittaker. Alpha diversity captures the richness, evenness and diversity of a sample. Our analyses showed that individuals have non-homogenous composition within their gut microbial assemblage, however with comparable alpha diversity variations across different Prakriti (Figure 2). Similarly, our beta diversity analysis to investigate community differences revealed that there was no clustering of individuals with respect to Prakriti (Supplementary Figure S1). We made similar observations across both genders. This is in consonance with previous findings of genomic and transcriptomic heterogeneity in healthy individuals (Cho and Blaser, 2012; Schwartz et al., 2012; Zhang et al., 2014). However, it needs to be highlighted that Bacteroidetes and Firmicutes are the two overwhelmingly dominant phyla in gut communities, which may have a masking effect on contributions of specific organisms of different phyla.
Figure 1.

Phyla level taxonomic summary of (A) male and (B) female gut microbial communities. Subjects have been grouped based on their corresponding Prakriti. Figure shows distribution of the two major gut microbial phyla Bacteroidetes and Firmicutes.
Figure 2.

Diversity estimates in gut microbial communities of healthy male and female subjects. (A) Diversity (Shannon), (B) Richness, and (C) Evenness in male samples. (D) Diversity (Shannon), (E) Richness, and (F) Evenness in female samples.
In order to access the roles of the lesser abundant taxa in Prakriti classifications, we excluded the top two abundant phyla, viz., Bacteroidetes and Firmicutes, and re-estimated the alpha and beta diversity. The alpha diversity within females showed that among the different Prakriti, Pitta individuals had lesser diversity and richness but higher evenness than the Vata and Kapha (Supplementary Figure S2). However, the diversity, richness and evenness was found to be similar in all the three Prakriti in the male samples (Supplementary Figure S2). The beta diversity analysis showed no Prakriti specific separation of male and female samples in PCoA plot even after the removal of the most highly abundant phyla (Supplementary Figure S2). Despite inter-individual heterogeneity, we observed overlapping variation across the Prakriti classes, which most likely is an outcome of similar genetic makeup and lifestyle habits.
Core microbial community and conserved diversity
Stable members of a microbial community often modulate physiology of the host-microbial symbiotic system (Tschop et al., 2009; Shade and Handelsman, 2012; D'Ainsworth et al., 2015). The dysbiosis or differential abundance is one of the primary determinants of health and disease spectrum. To investigate this, we estimated core microbiome by qualifying OTUs as core only if their presence was consistent across 50% of all samples. Using in-house custom scripts, we filtered taxons from the male and female groups, to identify 209 and 224 OTUs respectively (Supplementary Table S3). Taxonomic analysis of core groups showed that the phyla Bacteroidetes and Firmicutes follow similar trends of composition as that of the total microbiome. However, the core in female group was comprised of only Bacteroidetes, Firmicutes and Proteobacteria, whereas the core in males had additional members of the family Coriobacteriaceae of phylum Actinobacteria (Figure 3). We also observed abundant bacterial species to be present across all healthy individuals, thereby occupying majority space within the core microbiome group. Core microbiome also exhibited comparable alpha and beta diversity trends as that of the total microbiome.
Figure 3.

Family and genus that comprise the core microbiome. (A) Males and (B) Females.
Microbial taxons associated with Prakriti types
To control for sparseness associated with gut microbiome profiling, we looked for microbial organisms that were part of the core microbiome while being differentially abundant in a particular Prakriti using LEfSe. We performed statistical tests at multiple taxonomic levels and discovered 49 and four taxons across female and male respectively, to be significantly enriched in specific Prakriti categories. To control for potential false positives, we manually curated these differentially abundant species to arrive at a final set of 15 and two taxons that were associated with Prakriti and had no taxonomic overlap with other Prakriti specific bacteria (Table 1). To identify the correct taxonomic details of the signature OTUs, we used sequence alignment methods to identify the most probable candidate at the species level. It is important to note that these specific abundant species are present across most individuals in all Prakriti groups, however they are significantly enriched in a particular Prakriti, hence termed as signature species (Table 1, Figures 4A–F).
Table 1.
List of Prakriti specific signature taxa with details of their functional importance in the human gut.
| Signature Taxa | Gender | Prakriti | OTUID | p-value | LDA | Physiological relevance in human gut | References |
|---|---|---|---|---|---|---|---|
| Prevotella copri | Female | Kapha | 215670 | 0.006 | 5.620623 | Proinflammatory, onset of rheumatoid arthritis, insulin resistance | Wu et al., 2011; Scher et al., 2013 |
| Blautia luti | Female | Pitta | 178762 | 0.005 | 5.19693 | Butyrate producers, protect from graft versus host disease, restricts colonization of Vibrio cholera | Hsiao et al., 2014; Eren et al., 2015; Jenq et al., 2015 |
| Blautia obeum | Female | Pitta | 186748 | 0.018 | 4.759854 | Butyrate producers, protect from graft versus host disease, restricts colonization of Vibrio cholera | Hsiao et al., 2014; Eren et al., 2015; Jenq et al., 2015 |
| Blautia torques | Female | Pitta | 3272764 | 0.003 | 4.885697 | Butyrate producers, protect from graft versus host disease, restricts colonization of Vibrio cholera | Hsiao et al., 2014; Eren et al., 2015; Jenq et al., 2015 |
| Butyricicoccus pullicaecorum | Female | Pitta | 179826 | 0.001 | 5.158494 | Butyrate producers, protects from IBS, potential probiotic | Eeckhaut et al., 2013; Geirnaert et al., 2014 |
| Gemmiger formicilis | Female | Pitta | 341024 | 0.028 | 4.878518 | Induced during CTM treatment of T2D | Xu et al., 2015 |
| Incertae Sedis Mahella | Female | Pitta | 191783 | 0.026 | 4.828228 | – | – |
| Lachnospira eligens | Female | Pitta | 176269 | 0.005 | 4.822001 | – | – |
| Bacteroides vulgatus | Female | Vata | 184753 | 0.016 | 4.753459 | Induces insulin resistance, but found to protect from obseity in mice | Ridaura et al., 2013; Pedersen et al., 2016 |
| Blautia stercoris | Female | Vata | 185824 | 0.018 | 4.654206 | – | – |
| Butyrivibrio crossotus | Female | Vata | 4349261 | 0.001 | 5.137397 | Depleted in patients with Chronic Kidney Disease | Barros et al., 2015 |
| Clostridium indolis | Female | Vata | 338992 | 0.015 | 5.224559 | Carbohydrate metabolism | Biddle et al., 2014 |
| Eubacterium rectale | Female | Vata | 366794 | 0.001 | 5.658181 | Butyrate producer, depleted during ulcerative colitis | Vermeiren et al., 2012; Machiels et al., 2014; Cockburn et al., 2015; Riviere et al., 2015 |
| Oscillibacter valericigenes | Female | Vata | 175828 | 0.049 | 4.617719 | Oscillibacter related with bacterimia | Sydenham et al., 2014 |
| Roseburia hominis | Female | Vata | 198945 | 0.011 | 4.671769 | Butyrate producer, depleted during ulcerative colitis | Vermeiren et al., 2012; Machiels et al., 2014; Cockburn et al., 2015; Riviere et al., 2015 |
| Roseburia inulinivorans | Male | Pitta | 199091 | 0.004 | 4.526317 | Butyrate producer | Scott et al., 2006, 2011 |
| Fusicatenibacter saccharivorans | Male | Vata | 183401 | 0.045 | 4.705687 | – | – |
Figure 4.

Relative abundances of Prakriti specific signature taxons in female subjects. Eubacteriumrectale (A) and Roseburia hominis (B) in Vata; Prevotella copri (C) in Kapha; Blautia luti (D), Butyricicoccus pullicaecoruma, (E) and Gemmigerformicilis (F) in Pitta.
The Pitta females have over representation of seven species, out of which three belong to the genus Blautia—Blautia luti, B. obeum, and B. torques (Figure 4D). Blautia is a newly classified genera in the order Clostridiales, and its species constitute a major fraction of the gut flora, often responsible for conversion of carbon and hydrogen to acetate (Liu et al., 2008). Blautia, a commensal group of bacteria associated with nutrition processing for the host (Eren et al., 2015) is also associated with protection from graft versus host disease (Jenq et al., 2015) and restricts colonization of Vibrio cholera, thereby aiding in recovery from disease (Hsiao et al., 2014). Few Blautia species have increased levels during diseases like irritable bowel syndrome (IBS), though their role in the pathology is yet to be ascertained (Rajilic-Stojanovic and de Vos, 2014; Taverniti and Guglielmetti, 2014). Pitta females also showed overabundance of Butyricicoccus pullicaecoruma, a butyrate producing beneficial bacteria (Figure 4E), which has a protective effect against IBS (Eeckhaut et al., 2013) and is being considered as a potential probiotic (Geirnaert et al., 2014). Another bacterium specifically enriched in Pitta females was Gemmiger formicilis (Figure 4F), a beneficial bacteria which has been shown to be induced by a Chinese traditional medicine treatment for type 2 diabetes (Xu et al., 2015). Additionally, Incertae Sedis Mahella and Lachnospira eligens were significantly overabundant in female subjects of Pitta Prakriti, however their role in the human gut remain unclear (Supplementary Figure S3).
Kapha females were characterized by overabundance of Prevotella copri (Figure 4C), a species of bacteria commonly associated with a plant rich diet (Wu et al., 2011). It is also shown to be strongly correlated with inflammation and rheumatoid arthritis (RA) (Scher et al., 2013). Very recently, P. copri was found to induce insulin resistance in humans, resulting in increased glucose insensitivity through biosynthesis of branched-chain amino acids (BCAA) (Pedersen et al., 2016).
In Vata females, we observed significant abundance of Bacteroides vulgatus, which along with Prevotella copri, is shown to induce insulin resistance (Pedersen et al., 2016). Interestingly, studies in mice have also highlighted its protective effect in obesity and related metabolic disorders (Ridaura et al., 2013). Signature species of Vata females included E. rectale and R. hominis (Figures 4A,B), butyrate producers. They are believed to be beneficial for the gut health as they have been observed to be depleted during ulcerative colitis in independent studies, similar to Faecalibacterium prausnitzii (Vermeiren et al., 2012; Machiels et al., 2014; Cockburn et al., 2015; Riviere et al., 2015). Vata females also show enrichment of Butyrivibrio crossotus which was found to be depleted in chronic kidney disease patients (Barros et al., 2015). Relative overabundance of these species suggest a healthy intestinal flora in the Vata subjects, however we also note excess of Oscillibacter valericigenes which belongs to the genera Oscillibacter that have been reported in a case of bacterimia (Sydenham et al., 2014). We identified two other signature species, Blautia stercoris and Clostridium indolis (Supplementary Figure S3), whose role in the human gut is not clear, though Clostridium indolis is believed to be involved in carbohydrate metabolism (Biddle et al., 2014). This indicates the importance of this species in gut and its abundance dynamics can have a protective as well as an adverse effect.
Though we detected more OTUs among male subjects (Supplementary Figure S4), we found very few Prakriti specific signature microbes that qualified our stringent analysis threshold. We observed two signature bacterial species, Roseburia inulinivorans and Fusicatenibacter saccharivorans belonging to Pitta and Vata subjects respectively. Though R. inulinivorans has been characterized to be a butyrate bacteria of potentially beneficial contribution to the intestinal health (Scott et al., 2006), there is limited information with respect to mechanism underlying as to how overabundance of these two species might affect the human gut. Our analysis suggests that the male gut communities are relatively more homogenous as compared to female counterpart, which may have resulted in detection of fewer differentially abundant species.
qPCR validation of Prakriti specific microbial enterotypes
To validate the 16S rRNA amplicon based identification of Prakriti specific microbial enterotypes, we carried out qPCR assays for absolute quantification of 16S rRNA gene copy number of differentially abundant microbial enterotypes in the study subjects (Supplementary Table S4). Relative abundance analysis of microbial enterotypes in different Prakriti has re-affirmed the insights from the 16S rRNA gene sequencing analysis (Figure 1). The differential abundance of signature species within each Prakriti type also show statistically significant difference based on qPCR. Prevotella group was significantly abundant in Kapha females compared to Pitta (p-0.0000635) and Vata (p-0.007), while Eubacterium rectale & Roseburia group was found significantly enriched (p-0.002 and p-0.035) within Vata females. Simultaneously, Blautia sp. was statistically enriched (p-0.006, p-0.02) within Pitta females (Figure 5). It would be important to understand and explore the functional role of these microbes vis-à-vis Prakriti types in subsequent studies.
Figure 5.
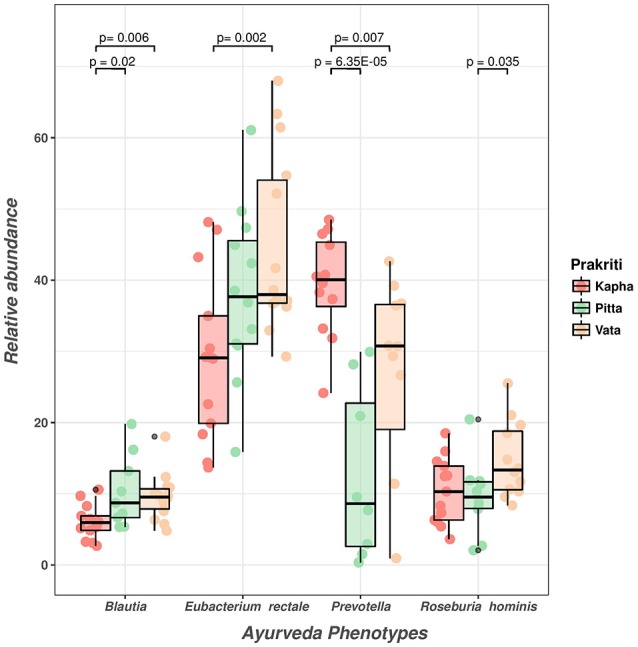
qPCR analysis to validate relative abundance of differentially abundant microbial enterotypes in Vata, Pitta, and Kapha. Statistical significance of Prakriti specific microbial enterotypes were determined by “Wilcoxon Rank Sum” test.
Discussion
Human health is the manifestation of a perpetual interaction between the human body and its surroundings. Increasing evidence suggests an underlying molecular heterogeneity in healthy individuals, which result in differential response to disease and their treatments. Over the past decade, we have increasingly realized the importance of microbiome and its role in human health (Cho and Blaser, 2012; Zhang et al., 2014). Advances in genomic technologies have facilitated significant discoveries in the area of metagenomics and some have already made their way into clinical practices. Thus it is important to understand the variability in the microbial flora among healthy individuals and its role in disease predisposition, protection and prognostic markers (Parfrey and Knight, 2012). Ayurveda begins with identification of an individual's intrinsic constitution “Prakriti,” whereas diseased state “Vikriti” is considered to be a deviation from the baseline. The restorative regimen addresses the cause and baseline in an individualized manner and hence closely resembles precision medicine (Prasher et al., 2008, 2017; Dey and Pahwa, 2014).
In this study, we examined the gut microbial community of 113 male and female volunteers with predominant Prakriti phenotypes from the Vadu HDSS population (western part of India), in an effort to catalog the gut microbial diversity among healthy individuals. Our results show that genetically homogenous population with similar cultural and dietary habits, can have subtle yet important variations within the gut microbial community. Alpha and beta diversity analyses revealed marked differences in community composition among subjects, albeit, at Prakriti level, we observe a homogenous gut microbiome across male and female. Less than one-tenth of the total flora show conservation in each group, indicating a dynamic microbiome in a homogenous population. We queried for Prakriti associated differences in the core microbiome to avoid artificial abundance differences that may arise due to sampling bias. The core microbiome resembled that of the total set, with Bacteroidetes and Firmicutes accounting for majority of the phyla detected. We investigated the core microbiome for both genders to search for Prakriti associated taxons representative of true differential abundance. Our analysis showed two OTUs to be differentially abundant across Prakriti categories in males, whereas females had 15 taxonomic groups, out of 209 and 224 OTUs respectively, which formed the core microbiome.
Our investigation across both genders found microbial taxa indicative of a healthy gut flora reiterating the health status of our study volunteers. The Pitta individuals showed enrichment of several butyrate producing microbes, which have been shown to be protective against inflammation, IBS (Geirnaert et al., 2014) and graft-versus-host-disease (Jenq et al., 2015). These suggest that Pitta have a robust flora and a healthier gut. Amongst the three extremes, Pitta Prakriti has been described to have good digestion and metabolism capacity, regular bowel habit with tendencies for loose motions (Prasher et al., 2008, 2016; Dey and Pahwa, 2014). Additionally, Pitta individual are more prone to inflammation (Juyal et al., 2012). Since the study has been carried out on healthy individuals, the presence of bacteria that are protective against inflammation and disease like IBS might suggest their role in maintaining homeostasis. These observations also corroborate with our earlier reported observations of higher expression of immune response genes in Pitta compared to Vata (Scott et al., 2006).
On the contrary, we observed significant overabundance of P. copri among female Kapha individuals. Prevotella and P. copri specifically have been found to compromise host health, and have been associated with rheumatoid arthritis (Juyal et al., 2012; Scher et al., 2013) and insulin resistance (Pedersen et al., 2016). Phenotypes associated with insulin resistance like obesity, susceptibility for type 2 diabetes and atherosclerosis have been described for Kapha Prakriti (Prasher et al., 2008; Govindaraj et al., 2015; Doddoli et al., 2016). Besides, our earlier study has also revealed higher levels of lipids in Kapha individuals compared to other Prakriti types (Prasher et al., 2008; Doddoli et al., 2016). The enrichment of Prevotella in Kapha might to some extent explain the descriptions of Prakriti. This provides an opportunity toward in-depth investigation of gut flora of Kapha individuals to assess potential predisposition to these disease states. Vata individuals showed a mix of beneficial bacteria and otherwise in their gut flora. In addition to presence of hostile organisms like B. vulgatus and Oscillibacter valericigenes, we also detected several anti-inflammatory butyrate producing species like Eubacterium rectale and R. hominis. These observations indicate toward Vata individuals being predisposed to health risks pertaining to presence of detrimental microbes, however it also has enrichment of several beneficial bacteria. The combination of non-beneficial and protective species in Vata might be able to fine balance health, as is visible in healthy subjects. However, change in their proportions might lead to different outcomes to which Vata individuals may be susceptible. Vata individuals have been described to have irregular and unpredictable digestion, metabolism and bowel functions (Prasher et al., 2008, 2017; Dey and Pahwa, 2014). They have also been shown to have lower immune responses in our earlier study (Prasher et al., 2008; Dey and Pahwa, 2014). Preventive regimes may be targeted toward maintenance and enhancement of healthy flora specifically in these groups of individuals, highlighting the importance of personalized approach in preventive medicine based on this analysis.
Conclusions
Using genomic techniques and the principles of Ayurveda, our group has previously elucidated the link between adaptation to low oxygen environment, high altitude pulmonary edema and Prakriti. In this study, we embarked to measure the variability in the gut metagenome of healthy individuals and its potential effect on disease vulnerability and natural protection. We discovered several important bacterial species with beneficial as well as detrimental association with human health, being selectively enriched in the gut flora of healthy individuals. Insights obtained from this study provide fundamental understanding of underlying metagenomic heterogeneity and its potential application toward personalized therapy.
Data availability
NGS sequence reads for samples included in this study can be accessed using the following link https://figshare.com/s/e981faa54cc3347999d9.
Ethics statement
This study was carried out in accordance with the recommendations of Indian Council of Medical Research, India guidelines for biomedical research, with written informed consent from all subjects. All subjects gave written informed consent in accordance with the Declaration of Helsinki. The protocol was approved by the Institutional Human ethics committees of K.E.M. Hospital and Research Center, Pune as well as Institute of Genomics and Integrative Biology, Delhi, India.
Author contributions
DD, MM, and BP designed the project. RuP, DA, BG, AS, and SaJ performed volunteer recruitment and sample collection. NC, VA, and RaP performed experiments and NGS sequencing. AM, SG, NC, RaP, TS, FM, and VS performed data analyses. SwJ, NC, MV, performed qPCR experiments and analysis. AM, SG, NC, MV, and RaP wrote the manuscript; MM, DD, and BP reviewed the data and manuscript. All authors have read and approved the manuscript.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Authors acknowledge contribution from all field staff and medical/para medical/administrative staff who worked painstakingly on the study. Authors acknowledge study population from KEMHRC-VADU, Pune, areas for their participation in the study. TRISUTRA also acknowledges CSIR-IGIB for administrative, infrastructure and IT support. AM is supported by Department of Biotechnology-BINC Senior Research Fellowship. TPS acknowledges DBT Ramalingaswamy Fellowship and Indian Institute of Technology, Mandi for support.
Footnotes
Funding. The work was supported by grant (MLP3601 and MLP901) from Council of Scientific and Industrial Research (CSIR) Govt. of India for the project entitled “Setting up of a CSIR Unit- TRISUTRA (Translational Research and Innovative Science ThRough Ayurgenomics)”.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2018.00118/full#supplementary-material
PCoA plot of beta diversity calculated using Bray-Curtis distance for (A) males and (B) female samples.
{kind=link}
Diversity panel of Prakriti after removing two highly abundant phyla, viz., Firmicutes and Bacteroidetes. (A) Diversity (Shannon), (B) Richness, and (C) Evenness in male samples. (E) Diversity (Shannon), (F) Richness, and (G) Evenness in female samples. PCoA plot of beta diversity calculated using Bray-Curtis distance (D) in males and (H) in female samples.
{kind=link}
Relative abundances of Prakriti specific signature taxons Bacteroides vulgatus (A); Blautia obeum (B); Blautia stercoris (C); Blautia torques (D); Butyrivibrio crossotus (E); Clostridium indolis (F); Incertae Sedis Mahella (G); Lachnospira eligens (H); Oscillibacter valericigenes (I) in female subjects.
{kind=link}
Relative abundances of Prakriti specific signature taxons i.e., Roseburia inulinivorans (A) and Fusicatenibacter saccharivorans (B) in male subjects.
{kind=link}
Details of volunteers enrolled in this study.
List of Primers used for qPCR analysis.
Core Microbiome in male and female groups.
qPCR analysis data to validate relative abundance of differentially abundant microbial enterotypes in Vata, Pitta, and Kapha.
References
- Aggarwal S., Gheware A., Agrawal A., Ghosh S., Prasher B., Mukerji M., et al. (2015). Combined genetic effects of EGLN1 and VWF modulate thrombotic outcome in hypoxia revealed by Ayurgenomics approach. J. Transl. Med. 13, 184. 10.1186/s12967-015-0542-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aggarwal S., Negi S., Jha P., Singh P. K., Stobdan T., Pasha M. A. Q., et al. (2010). EGLN1 involvement in high-altitude adaptation revealed through genetic analysis of extreme constitution types defined in Ayurveda. Proc. Natl. Acad. Sci. U.S.A. 107, 18961–18966. 10.1073/pnas.1006108107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albenberg L. G., Lewis J. D., Wu G. D. (2012). Food and the gut microbiota in inflammatory bowel diseases: a critical connection. Curr. Opin. Gastroenterol. 28, 314–320. 10.1097/MOG.0b013e328354586f [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barros A. F., Borges N. A., Ferreira D. C., Carmo F. L., Rosado A. S., Fouque D., et al. (2015). Is there interaction between gut microbial profile and cardiovascular risk in chronic kidney disease patients? Future Microbiol. 10, 517–526. 10.2217/fmb.14.140 [DOI] [PubMed] [Google Scholar]
- Bhute S., Pande P., Shetty S. A., Shelar R., Mane S., Kumbhare S. V., et al. (2016). Molecular characterization and meta-analysis of gut microbial communities illustrate enrichment of prevotella and megasphaera in Indian subjects. Front. Microbiol. 7:660. 10.3389/fmicb.2016.00660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biddle A. S., Leschine S., Huntemann M., Han J., Chen A., Kyrpides N., et al. (2014). The complete genome sequence of Clostridium indolis DSM 755(T.). Stand. Genomic Sci. 9, 1089–1104. 10.4056/sigs.5281010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caporaso J. G., Kuczynski J., Stombaugh J., Bittinger K., Bushman F. D., Costello E. K., et al. (2010). QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7, 335–336. 10.1038/nmeth.f.303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho I., Blaser M. J. (2012). The human microbiome: at the interface of health and disease. Nat. Rev. Genet. 13, 260–270. 10.1038/nrg3182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong C. W., Ahmad A. F., Lim Y. A. L., Teh C. S. J., Yap I. K. S., Lee S. C., et al. (2015). Effect of ethnicity and socioeconomic variation to the gut microbiota composition among pre-adolescent in Malaysia. Sci. Rep. 5:13338. 10.1038/srep13338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clemente J. C., Ursell L. K., Parfrey L. W., Knight R. (2012). The impact of the gut microbiota on human health: an integrative view. Cell 148, 1258–1270. 10.1016/j.cell.2012.01.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cockburn D. W., Orlovsky N. I., Foley M. H., Kwiatkowski K. J., Bahr C. M., Maynard M., et al. (2015). Molecular details of a starch utilization pathway in the human gut symbiont Eubacterium rectale. Mol. Microbiol. 95, 209–230. 10.1111/mmi.12859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Ainsworth T., Krause L., Bridge T., Torda G., Raina J.-B., Zakrzewski M., et al. (2015). The coral core microbiome identifies rare bacterial taxa as ubiquitous endosymbionts. ISME J. 9, 2261–2274. 10.1038/ismej.2015.39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dave M., Higgins P. D., Middha S., Rioux K. P. (2012). The human gut microbiome: current knowledge, challenges, and future directions. Transl. Res. J. Lab. Clin. Med. 160, 246–257. 10.1016/j.trsl.2012.05.003 [DOI] [PubMed] [Google Scholar]
- Dey S., Pahwa P. (2014). Prakriti and its associations with metabolism, chronic diseases, and genotypes: possibilities of new born screening and a lifetime of personalized prevention. J. Ayurveda Integr. Med. 5, 15–24. 10.4103/0975-9476.128848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon P. (2003). VEGAN, a package of R functions for community ecology. J. Veg. Sci. 14, 927–930. 10.1111/j.1654-1103.2003.tb02228.x [DOI] [Google Scholar]
- Doddoli S., Shete S., Kulkarni D., Bhogal R. (2016). Effect of yoga training on lipid metabolism in industrial workers with reference to body constitution (Prakriti). J. Tradit. Complement. Med. 7, 322–326. 10.1016/j.jtcme.2016.08.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominguez-Bello M. G., Blaser M. J. (2008). Do you have a probiotic in your future? Microbes Infect. Inst. Pasteur. 10, 1072–1076. 10.1016/j.micinf.2008.07.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar R. C. (2010). Search and clustering orders of magnitude faster than BLAST. Bioinforma. Oxf. Engl. 26, 2460–2461. 10.1093/bioinformatics/btq461 [DOI] [PubMed] [Google Scholar]
- Eeckhaut V., Machiels K., Perrier C., Romero C., Maes S., Flahou B., et al. (2013). Butyricicoccus pullicaecorum in inflammatory bowel disease. Gut 62, 1745–1752. 10.1136/gutjnl-2012-303611 [DOI] [PubMed] [Google Scholar]
- Eren A. M., Sogin M. L., Morrison H. G., Vineis J. H., Fisher J. C., Newton R. J., et al. (2015). A single genus in the gut microbiome reflects host preference and specificity. ISME J. 9, 90–100. 10.1038/ismej.2014.97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortenberry J. D. (2013). The uses of race and ethnicity in human microbiome research. Trends Microbiol. 21, 165–166. 10.1016/j.tim.2013.01.001 [DOI] [PubMed] [Google Scholar]
- Geirnaert A., Steyaert A., Eeckhaut V., Debruyne B., Arends J. B. A., Van Immerseel F., et al. (2014). Butyricicoccus pullicaecorum, a butyrate producer with probiotic potential, is intrinsically tolerant to stomach and small intestine conditions. Anaerobe 30, 70–74. 10.1016/j.anaerobe.2014.08.010 [DOI] [PubMed] [Google Scholar]
- Goecks J., Nekrutenko A., Taylor J., Galaxy Team (2010). Galaxy: a comprehensive approach for supporting accessible, reproducible, and transparent computational research in the life sciences. Genome Biol. 11:R86. 10.1186/gb-2010-11-8-r86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon J. I., Dewey K. G., Mills D. A., Medzhitov R. M. (2012). The human gut microbiota and undernutrition. Sci. Transl. Med. 4:137ps12. 10.1126/scitranslmed.3004347 [DOI] [PubMed] [Google Scholar]
- Govindaraj P., Nizamuddin S., Sharath A., Jyothi V., Rotti H., Raval R., et al. (2015). Genome-wide analysis correlates Ayurveda Prakriti. Sci. Rep. 5:15786. 10.1038/srep15786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haiser H. J., Turnbaugh P. J. (2012). Is it time for a metagenomic basis of therapeutics? Science 336, 1253–1255. 10.1126/science.1224396 [DOI] [PubMed] [Google Scholar]
- Hsiao A., Ahmed A. M. S., Subramanian S., Griffin N. W., Drewry L. L., Petri W. A. J., et al. (2014). Members of the human gut microbiota involved in recovery from Vibrio cholerae infection. Nature 515, 423–426. 10.1038/nature13738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Human Microbiome Project Consortium (2012). Structure, function and diversity of the healthy human microbiome. Nature 486, 207–214. 10.1038/nature11234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenq R. R., Taur Y., Devlin S. M., Ponce D. M., Goldberg J. D., Ahr K. F., et al. (2015). Intestinal blautia is associated with reduced death from graft-versus-host disease. Biol. Blood Marrow Transplant. 21, 1373–1383. 10.1016/j.bbmt.2015.04.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juyal R. C., Negi S., Wakhode P., Bhat S., Bhat B., Thelma B. K. (2012). Potential of ayurgenomics approach in complex trait research: leads from a pilot study on rheumatoid arthritis. PLoS ONE 7:e45752. 10.1371/journal.pone.0045752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C., Finegold S. M., Song Y., Lawson P. A. (2008). Reclassification of Clostridium coccoides, Ruminococcus hansenii, Ruminococcus hydrogenotrophicus, Ruminococcus luti, Ruminococcus productus and Ruminococcus schinkii as Blautia coccoides gen. nov., comb. nov., Blautia hansenii comb. nov., Blautia hydrogenotrophica comb. nov., Blautia luti comb. nov., Blautia producta comb. nov., Blautia schinkii comb. nov. and description of Blautia wexlerae sp. nov., isolated from human faeces. Int. J. Syst. Evol. Microbiol. 58, 1896–1902. 10.1099/ijs.0.65208-0 [DOI] [PubMed] [Google Scholar]
- Machiels K., Joossens M., Sabino J., De Preter V., Arijs I., Eeckhaut V., et al. (2014). A decrease of the butyrate-producing species Roseburia hominis and Faecalibacterium prausnitzii defines dysbiosis in patients with ulcerative colitis. Gut 63, 1275–1283. 10.1136/gutjnl-2013-304833 [DOI] [PubMed] [Google Scholar]
- Moeller A. H., Caro-Quintero A., Mjungu D., Georgiev A. V., Lonsdorf E. V., Muller M. N., et al. (2016). Cospeciation of gut microbiota with hominids. Science 353, 380–382. 10.1126/science.aaf3951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parfrey L. W., Knight R. (2012). Spatial and temporal variability of the human microbiota. Clin. Microbiol. Infect. Off. Publ. Eur. Soc. Clin. Microbiol. Infect. Dis. 18(Suppl. 4), 8–11. 10.1111/j.1469-0691.2012.03861.x [DOI] [PubMed] [Google Scholar]
- Pedersen H. K., Gudmundsdottir V., Nielsen H. B., Hyotylainen T., Nielsen T., Jensen B. A. H., et al. (2016). Human gut microbes impact host serum metabolome and insulin sensitivity. Nature 535, 376–381. 10.1038/nature18646 [DOI] [PubMed] [Google Scholar]
- Prasher B., Gibson G., Mukerji M. (2016). Genomic insights into ayurvedic and western approaches to personalized medicine. J. Genet. 95, 209–228. 10.1007/s12041-015-0607-9 [DOI] [PubMed] [Google Scholar]
- Prasher B., Negi S., Aggarwal S., Mandal A. K., Sethi T. P., Deshmukh S. R., et al. (2008). Whole genome expression and biochemical correlates of extreme constitutional types defined in Ayurveda. J. Transl. Med. 6:48. 10.1186/1479-5876-6-48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasher B., Varma B., Kumar A., Khuntia B. K., Pandey R., Narang A., et al. (2017). Ayurgenomics for stratified medicine: TRISUTRA consortium initiative across ethnically and geographically diverse Indian populations. J. Ethnopharmacol. 197, 274–293. 10.1016/j.jep.2016.07.063 [DOI] [PubMed] [Google Scholar]
- Rajilic-Stojanovic M., de Vos W. M. (2014). The first 1000 cultured species of the human gastrointestinal microbiota. FEMS Microbiol. Rev. 38, 996–1047. 10.1111/1574-6976.12075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridaura V. K., Faith J. J., Rey F. E., Cheng J., Duncan A. E., Kau A. L., et al. (2013). Gut microbiota from twins discordant for obesity modulate metabolism in mice. Science 341:1241214. 10.1126/science.1241214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivière A., Gagnon M., Weckx S., Roy D., De Vuyst L. (2015). Mutual Cross-Feeding Interactions between Bifidobacterium longum subsp. longum NCC2705 and Eubacterium rectale ATCC 33656 explain the bifidogenic and butyrogenic effects of arabinoxylan oligosaccharides. Appl. Environ. Microbiol. 81, 7767–7781. 10.1128/AEM.02089-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts D. (2013). labdsv: Ordination and Multivariate Analysis for Ecology. R Package version 1.6-1. Available online at: https://cran.r-project.org/web/packages/labdsv.
- Rosenberg E., Zilber-Rosenberg I. (2011). Symbiosis and development: the hologenome concept. Birth Defects Res. Part C Embryo Today Rev. 93, 56–66. 10.1002/bdrc.20196 [DOI] [PubMed] [Google Scholar]
- Rotti H., Raval R., Anchan S., Bellampalli R., Bhale S., Bharadwaj R., et al. (2014). Determinants of prakriti, the human constitution types of Indian traditional medicine and its correlation with contemporary science. J. Ayurveda Integr. Med. 5, 167–175. 10.4103/0975-9476.140478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scher J. U., Sczesnak A., Longman R. S., Segata N., Ubeda C., Bielski C., et al. (2013). Expansion of intestinal Prevotella copri correlates with enhanced susceptibility to arthritis. Elife 2:e01202. 10.7554/eLife.01202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz S., Friedberg I., Ivanov I. V., Davidson L. A., Goldsby J. S., Dahl D. B., et al. (2012). A metagenomic study of diet-dependent interaction between gut microbiota and host in infants reveals differences in immune response. Genome Biol. 13:r32. 10.1186/gb-2012-13-4-r32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott K. P., Martin J. C., Campbell G., Mayer C.-D., Flint H. J. (2006). Whole-genome transcription profiling reveals genes up-regulated by growth on fucose in the human gut bacterium “Roseburia inulinivorans”. J. Bacteriol. 188, 4340–4349. 10.1128/JB.00137-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott K. P., Martin J. C., Chassard C., Clerget M., Potrykus J., Campbell G., et al. (2011). Substrate-driven gene expression in Roseburia inulinivorans: importance of inducible enzymes in the utilization of inulin and starch. Proc. Natl. Acad. Sci. U.S.A. 108(Suppl. 1), 4672–4679. 10.1073/pnas.1000091107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segata N., Izard J., Waldron L., Gevers D., Miropolsky L., Garrett W. S., et al. (2011). Metagenomic biomarker discovery and explanation. Genome Biol. 12:R60. 10.1186/gb-2011-12-6-r60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sethi T. P., Prasher B., Mukerji M. (2011). Ayurgenomics: a new way of threading molecular variability for stratified medicine. ACS Chem. Biol. 6, 875–880. 10.1021/cb2003016 [DOI] [PubMed] [Google Scholar]
- Shade A., Handelsman J. (2012). Beyond the Venn diagram: the hunt for a core microbiome. Environ. Microbiol. 14, 4–12. 10.1111/j.1462-2920.2011.02585.x [DOI] [PubMed] [Google Scholar]
- Sydenham T. V., Arpi M., Klein K., Justesen U. S. (2014). Four cases of bacteremia caused by Oscillibacter ruminantium, a newly described species. J. Clin. Microbiol. 52, 1304–1307. 10.1128/JCM.03128-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taverniti V., Guglielmetti S. (2014). Methodological issues in the study of intestinal microbiota in irritable bowel syndrome. World J. Gastroenterol. 20, 8821–8836. 10.3748/wjg.v20.i27.8821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiwari P., Kutum R., Sethi T., Shrivastava A., Girase B., Aggarwal S., et al. (2017). Recapitulation of Ayurveda constitution types by machine learning of phenotypic traits. PLoS ONE 12:e0185380. 10.1371/journal.pone.0185380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tschöp M. H., Hugenholtz P., Karp C. L. (2009). Getting to the core of the gut microbiome. Nat. Biotechnol. 27, 344–346. 10.1038/nbt0409-344 [DOI] [PubMed] [Google Scholar]
- Vermeiren J., Van den Abbeele P., Laukens D., Vigsnaes L. K., De Vos M., Boon N., et al. (2012). Decreased colonization of fecal Clostridium coccoides/Eubacterium rectale species from ulcerative colitis patients in an in vitro dynamic gut model with mucin environment. FEMS Microbiol. Ecol. 79, 685–696. 10.1111/j.1574-6941.2011.01252.x [DOI] [PubMed] [Google Scholar]
- Wickham H. (2016). ggplot2: Elegant Graphics for Data Analysis. New York, NY: Springer. [Google Scholar]
- Wu G. D., Chen J., Hoffmann C., Bittinger K., Chen Y.-Y., Keilbaugh S. A., et al. (2011). Linking long-term dietary patterns with gut microbial enterotypes. Science 334, 105–108. 10.1126/science.1208344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J., Lian F., Zhao L., Zhao Y., Chen X., Zhang X., et al. (2015). Structural modulation of gut microbiota during alleviation of type 2 diabetes with a Chinese herbal formula. ISME J. 9, 552–562. 10.1038/ismej.2014.177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yatsunenko T., Rey F. E., Manary M. J., Trehan I., Dominguez-Bello M. G., Contreras M., et al. (2012). Human gut microbiome viewed across age and geography. Nature 486, 222–227. 10.1038/nature11053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X., Zhang D., Jia H., Feng Q., Wang D., Liang D., et al. (2015). The oral and gut microbiomes are perturbed in rheumatoid arthritis and partly normalized after treatment. Nat. Med. 21, 895–905. 10.1038/nm.3914 [DOI] [PubMed] [Google Scholar]
- Zhang Z., Geng J., Tang X., Fan H., Xu J., Wen X., et al. (2014). Spatial heterogeneity and co-occurrence patterns of human mucosal-associated intestinal microbiota. ISME J. 8, 881–893. 10.1038/ismej.2013.185 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PCoA plot of beta diversity calculated using Bray-Curtis distance for (A) males and (B) female samples.
Diversity panel of Prakriti after removing two highly abundant phyla, viz., Firmicutes and Bacteroidetes. (A) Diversity (Shannon), (B) Richness, and (C) Evenness in male samples. (E) Diversity (Shannon), (F) Richness, and (G) Evenness in female samples. PCoA plot of beta diversity calculated using Bray-Curtis distance (D) in males and (H) in female samples.
Relative abundances of Prakriti specific signature taxons Bacteroides vulgatus (A); Blautia obeum (B); Blautia stercoris (C); Blautia torques (D); Butyrivibrio crossotus (E); Clostridium indolis (F); Incertae Sedis Mahella (G); Lachnospira eligens (H); Oscillibacter valericigenes (I) in female subjects.
Relative abundances of Prakriti specific signature taxons i.e., Roseburia inulinivorans (A) and Fusicatenibacter saccharivorans (B) in male subjects.
Details of volunteers enrolled in this study.
List of Primers used for qPCR analysis.
Core Microbiome in male and female groups.
qPCR analysis data to validate relative abundance of differentially abundant microbial enterotypes in Vata, Pitta, and Kapha.
Data Availability Statement
NGS sequence reads for samples included in this study can be accessed using the following link https://figshare.com/s/e981faa54cc3347999d9.