

Abstract
Objective
To determine molecular epidemiology of methicillin-resistant S. aureus in Tanzania using whole genome sequencing.
Methods
DNA from 33 Staphylococcus species was recovered from subcultured archived Staphylococcus isolates. Whole genome sequencing was performed on Illumina Miseq using paired-end 2 × 250 bp protocol. Raw sequence data were analyzed using online tools.
Results
Full susceptibility to vancomycin and chloramphenicol was observed. Thirteen isolates (43.3%) resisted cefoxitin and other antimicrobials tested. Multilocus sequence typing revealed 13 different sequence types among the 30 S. aureus isolates, with ST-8 (n = seven, 23%) being the most common. Gene detection in S. aureus stains were as follows: mecA, 10 (33.3%); pvl, 5 (16.7%); tst, 2 (6.7%). The SNP difference among the six Tanzanian ST-8 MRSA isolates ranged from 24 to 196 SNPs and from 16 to 446 SNPs when using the USA300_FPR3757 or the USA500_2395 as a reference, respectively. The mutation rate was 1.38 × 10−11 SNPs/site/year or 1.4 × 10−6 SNPs/site/year as estimated by USA300_FPR3757 or the USA500_2395, respectively.
Conclusion
S. aureus isolates causing infections in hospitalized patients in Moshi are highly diverse and epidemiologically unrelated. Temporal phylogenetic analysis provided better resolution on transmission and introduction of MRSA and it may be important to include this in future routines.
1. Introduction
Staphylococcus aureus is one of the most important causes of serious infections in humans worldwide [1]. In particular, methicillin-resistant S. aureus (MRSA) isolates have become common causes of infections in hospitals as well as community settings.
For quite some time, MRSA has been a worldwide leading cause of nosocomial infections [2, 3]. From the 1960s to early 1970s, MRSA infections were often confined to hospitals and healthcare facilities. During this time mostly the classical or hospital-associated MRSA (HA-MRSA) accounted for these infections. In the mid-1990s, a surge of MRSA infections was reported in community settings among healthy people who had not been in contact with hospital environments [4]. This change was regarded as a result of introduction of a unique MRSA strain, so-called community associated MRSA (CA-MRSA) [5]. The ability of CA-MRSA to cause frequent outbreaks, with high virulence and a high rate of dissemination to different geographical locations, has played a major role in increasing the emergence and distribution of staphylococcal infections globally [6].
Diagnosis of MRSA isolates has not been easy despite the advancement in medical technology. The cefoxitin disk diffusion method is a reliable test for MRSA; however it needs to be supplemented by several other tests [7]. The emergence of high throughput technologies such as whole genome sequencing (WGS) promises to revolutionize today's clinical microbiology practices [8], increasing insight into pathogens and hence coming up with correct diagnosis, treatment, and control measures for disease-causing pathogens [9]. Recently, bench-top WGS has become available and proven to be a very valuable tool in elucidating the epidemiology of bacterial species including MRSA [10–12]. However, the technology is currently still out of reach for clinical laboratories in developing countries [13] and deployed rarely in research setups in these countries [14]. Recently, this technology was introduced at the Kilimanjaro Christian Medical Centre (KCMC) in Moshi, Tanzania, and already during the first test run, nosocomial transmission of Enterococcus faecalis was identified [15].
The prevalence of MRSA infections in most African countries seems to be relatively low [2]. However, given the low research intensity on MRSA infections in Africa this data should not be neglected [16]. S. aureus and MRSA infections contribute to significant health challenges in Tanzania as well [17–19] despite the fact that the reports are few and hence the current prevalence may not be indicating the actual disease burden. It is therefore important to be able to explain the molecular epidemiology of the clones, which are locally circulating. This will contribute to the knowledge on infection transmission, and hence introduction of proper treatment and control programs. Here we report on molecular epidemiology of S. aureus in Tanzania using WGS tools located at KCMC. WGS was used to determine sequence types, phylogenetic relation, and comparison to genome sequences that were obtained from ENA (https://www.ebi.ac.uk/ena) and NCBI. Additionally, antimicrobial resistance genes as well as virulence factors were determined.
2. Material and Methods
2.1. Study Setting, Participants, and Sample Collection
The current study is based on the previously conducted project [20] at Kilimanjaro Christian Medical Centre (KCMC) tertiary care hospital in North-Eastern Tanzania, from August 2013 to August 2015. The study aimed to determine the pattern of bacterial pathogens associated with different health conditions among patients who were admitted to the medical and surgical departments of KCMC. For the study, informed consent was obtained from 575 patients and clinical samples were collected from these patients who were admitted to medical and surgical wards at KCMC during the period defined above. Demographic data of these patients were obtained from their patient hospital files. Relevant information was captured on designated case record forms (CRFs) and double entered in OpenClinica (OpenClinica LLC and collaborators, Waltham, MA, USA). Data was extracted and exported to STATA 13 (StataCorp LP, Texas 77845, USA) for data analyses.
2.2. Ethical Consideration
Ethical approval for the study was obtained from National Institute for Medical Research with Certificate number NIMR|HQ|R.8a|Vol.IX/2080 and from Kilimanjaro Christian Medical University College with Certificate RECC number 891.
2.3. Laboratory Methods and Data Analysis
In the previous study, the clinical samples collected from the 575 patients during routine clinical care were transported to the microbiology unit of the Kilimanjaro Clinical Research Institute (KCRI) Biotechnology Laboratory for routine classical microbiology processes. Both Gram negative and positive bacteria were observed. S. aureus was the most predominant among Gram positive isolates. Antimicrobial susceptibility testing was performed using disk diffusion techniques as described in the previous study [20]. All isolates from the previous study were stored at −80°C for whole genome sequencing (WGS). For the current study, we recovered 30 out of 35 S. aureus isolates upon subculturing. All 30 isolates were subjected to molecular biology lab for DNA isolation. We also retrieved coagulase negative staphylococci (CoNS) isolates from the archive. However, only CoNS of clinical importance were included in this study [21, 22]. Genomic DNA was extracted using MasterPure™ Gram Positive DNA Purification Kit Cat. No. MGP04100, Epicentre, Illumina. The quality and quantity of genomic DNA were confirmed using Qubit 2.0 fluorometer, (Thermal Fisher Scientific, Waltham, MA, USA). Library preparation (dual indexing) was done using NexteraXT DNA Preparation Kit (Illumina Inc., San Diego, CA, USA).
Whole genome sequencing of the library was completed on Illumina Miseq using a paired-end 2 × 250 bp protocol.
2.4. Multilocus Sequence Typing (MLST), Antimicrobial Resistance, and Virulence Genes
The raw reads were de novo assembled through the assembly pipeline (version 1.0) available from the Center for Genomic Epidemiology (CGE) (https://cge.cbs.dtu.dk/services/) which is based on the Velvet algorithms for de novo short reads assembly [23]. The assembled sequences were analyzed using bioinformatics tools to identify MLST sequence type (MLST version 1.7) [24], acquired antimicrobial resistance genes (ResFinder version 2.1) [25], and virulence genes (VirulenceFinder version 1.0) [26]. All tools were available online at https://cge.cbs.dtu.dk/services and https://www.ebi.ac.uk/ena.
2.5. Single Nucleotide Polymorphisms (SNPs) and Temporal Bayesian Phylogenetic Trees
Phylogenetic analyses were performed on 40 ST-8 S. aureus genomes. Six ST-8 S. aureus genomes were from the current study. These were found to harbor the mecA gene (hence MRSA) and it was therefore important to gain better understanding of their genetic relation. For global comparison, 34 ST-8 MRSA genomes were retrieved from ENA and NCBI. These 34 genomes were from five different studies [27–30]. A much larger number of genomes have been deposited, but we restricted the set to only include genomes for which epidemiological information was available. Raw sequence data of the 6 ST-8 S. aureus from our study has been submitted to the European Nucleotide Archive (https://www.ebi.ac.uk/ena) under study accession number PRJEB23314. Information pertaining to all 40 ST-8 MRSA used for construction of phylogenetic trees in this study is reported in the supplementary information (SI) appendix, Tables 1a and 1b.
2.5.1. Single Nucleotide Polymorphisms
SNPs were identified using the CSI phylogeny [31, 32] pipeline, available on CGE (http://www.genomicepidemiology.org). The raw reads were mapped to two reference genomes, S. aureus USA300_FPR3757 (accession number CP000255, chromosome length 2,917,469 bp) and USA500_2395 (accession number CP007499, chromosomal length 2,955,646) using BWA version 0.7.2 [33]. The mpileup tool from SAMTools version 0.1.18 [34] was applied to determine SNPs. SNPs were filtered out when not matching the following criteria: (1) a minimum distance of 10 bp between SNPs, (2) a minimum of 10% of the read depth at SNP positions, (3) mapping quality above 25, and (4) SNP quality above 30. All indels were excluded. The SNPs from each genome were concatenated to single alignment corresponding to position on the reference genome. The concatenated sequences were used for constructing a maximum likelihood tree using FastTree [35].
2.5.2. Temporal Bayesian Phylogenetic Tree
Prior to reconstructing a phylogenetic tree in BEAST, concatenated SNPs were examined for significant recombination sites. We used a novel hidden Markov model tool, RecHMM [36], to detect clusters of sequence diversity that mark recombination events within branches. SNPs in these regions were excluded in the temporal phylogenetic analysis. SNP sequences were then used for reconstructing a temporal phylogenetic tree using Bayesian Evolutionary Analysis Sampling Trees (BEAST) version 1.8.3 [37] to estimate mutation rate and divergence time. Test models with combinations of different population size change and molecular clock were evaluated to identify the best-fit model. This was achieved by performing different BEAST runs with different selected models. Comparison of the log files from all models in the output runs was done in Tracer, a program that compares all BEAST runs and gives a score (ACT score). The temporal tree with the highest posterior probability was then constructed using the best-fit model (random local clock and coalescent Bayesian skyline). The BEAST MCMC chains were simulated for 300 million steps and subsampled every 10,000 steps. The final maximum clade credibility (MCC) was determined using TreeAnnotator [37] with 10% of the MCMC steps discarded as burn-in. Statistical confidence was represented by the 95% highest posterior density (HPD) interval. Mutation rate and divergence time were estimated by BEAST and the population size over time was estimated using the Bayesian skyline plot implemented in Tracer [37]. The effective population size was inferred by the product of the interval size (γ i) and i(i − 1)/2, where i is the number of genealogical lineages in the interval [38, 39].
3. Results
3.1. Isolates Characteristics
Thirty S. aureus and three coagulase negative staphylococci (CoNS) isolates from patients who were admitted to wards of medical and surgical departments were whole genome sequenced. Of the thirty S. aureus isolates 8, 14, and 8 were isolated in 2013, 2014, and 2015, respectively. The three CoNS isolates were all isolated in year 2015. Twenty-nine bacterial isolates were recovered from wound swabs, 3 isolates from blood culture, and 1 from sputum. The causes of the wounds were as diverse as indicated in Table 1.
Table 1.
Isolate characteristics.
| Sample name | Species | Collection date | Diagnosis | Specimen | Ward | Room number | Bed number |
|---|---|---|---|---|---|---|---|
| 29 B | S. aureus | August 1, 2013 | Surgery (laparotomy) | Wound swab | Surgical ICU | 17 | 3 |
| 32 | S. aureus | August 1, 2013 | Bedsore | Wound swab | Surgical II | Corr | 4 |
| 37 B | S. aureus | August 2, 2013 | Injury-femur fracture | Wound Swab | Surgical II | 8 | 3 |
| 38 | S. aureus | August 2, 2013 | Bedsores | Wound swab | Surgical II | 8 | 2 |
| 56 | S. aureus | August 13, 2013 | Asthma + HIV | Blood | Medical I | 2 | 4 |
| 59 | S. aureus | August 15, 2013 | Tumor wound | Wound swab | Surgical I | Corr | 5 |
| 69 S | S. aureus | August 19, 2013 | Burn wound | Wound swab | Surgical I | 1 | 4 |
| 71 | S. aureus | August 20, 2013 | Abscess | Wound swab | Medical I | 2 | 2 |
| 108 | S. aureus | February 7, 2014 | Pleural effusion (fever) | Blood | Medical II | 11 | 3 |
| 143 A | S. aureus | March 11, 2014 | Surgery (liver cirrhosis) | Wound swab | Medical I | 5 | 6 |
| 153 C | S. aureus | March 17, 2014 | Diabetes mellitus | Wound swab | Medical I | 5 | 9 |
| 159 A | S. aureus | March 20, 2014 | Abdominal tumor | Wound swab | Medical I | 5 | 10 |
| 166 | S. aureus | March 24, 2014 | MTA | Wound swab | Surgical I | Corr | - |
| 176 A | S. aureus | March 31, 2014 | Diabetes mellitus | Wound swab | Medical I | 5 | Extra bed |
| 196 | S. aureus | April 11, 2014 | Cellulitis | Wound swab | Surgical I | 5 | 3 |
| 201 A | S. aureus | April 11, 2014 | HIV (coughing) | Sputum | Medical I | 2 | Extra bed |
| 204 | S. aureus | April 15, 2014 | Septicemia | Blood | Medical ICU | - | 4 |
| 224 C | S. aureus | April 30, 2014 | Diabetes mellitus | Wound swab | Medical II | Corr | 6 |
| 260 | S. aureus | June 2, 2014 | Diabetes mellitus | Wound swab | Surgical I | 5 | 8 |
| 309 B | S. aureus | September 19, 2014 | Cellulitis | Wound swab | Surgical I | 5 | 6 |
| 319 D | S. aureus | September 30, 2014 | Burn | Wound swab | Surgical I | 1 | 3 |
| 323 B | S. aureus | October 1, 2014 | Diabetes mellitus | Wound swab | Surgical I | Corr | 5 |
| 348 B | S. aureus | November 18, 2014 | Tropical ulcer | Wound swab | Surgical I | 5 | 6 |
| 349 A | S. aureus | November 19, 2014 | Burn wound | Wound swab | Surgical I | 1 | 1 |
| 437 C | S. aureus | March 10, 2015 | Diabetes mellitus | Wound swab | Surgical I | 5 | 7 |
| 449 | S. aureus | March 31, 2015 | Diabetes mellitus | Wound swab | Medical I | 2 | 1 |
| 463 B | S. aureus | April 19, 2015 | Surgery (laparotomy) | Wound swab | Surgical I | 5 | 2 |
| 494 | S. aureus | May 19, 2015 | Burn wound | Wound swab | Surgical I | 1 | 1 |
| 547 | S. epidermidis | July 6, 2015 | Septic wound | Wound swab | Surgical I | 5 | 4 |
| 562 | S. haemolyticus | July 15, 2015 | HIV (cough) | Sputum | Medical I | 2 | 1 |
| 564 | S. haemolyticus | July 16, 2015 | Diabetes mellitus | Wound swab | Surgical I | 5 | 3 |
| 567 A | S. aureus | July 23, 2015 | Septic wound | Wound swab | Surgical 1 | Corr | 5 |
| 577 | S. aureus | August 3, 2015 | Diabetes mellitus | Wound swab | Surgical I | 2 | Extra bed |
3.2. Phenotypic Antimicrobial Resistance Pattern of S. aureus Isolates
All S. aureus tested were susceptible to vancomycin and chloramphenicol. Thirteen (43.3%) isolates were resistant to cefoxitin while resistance to trimethoprim-sulfa, erythromycin, and penicillin was observed for 16 (53.3%), 14 (46.7%), and 30 (100%) isolates, respectively, Table 2.
Table 2.
Phenotypic antimicrobial pattern of the 30 S. aureus isolates.
| PATIENT ID | ORG | CTX | C | E | P | SXT | VA |
|---|---|---|---|---|---|---|---|
| WGS 029 B | S. aureus | R | S | R | R | R | S |
| WGS 032 | S. aureus | R | S | R | R | R | S |
| WGS 037 B | S. aureus | S | S | S | R | S | S |
| WGS 038 | S. aureus | S | S | I | R | S | S |
| WGS 056 | S. aureus | R | S | R | R | R | S |
| WGS 059 | S. aureus | S | S | R | R | I | S |
| WGS 069 | S. aureus | R | S | R | R | R | S |
| WGS 071 | S. aureus | R | S | R | R | R | S |
| WGS 108 | S. aureus | R | S | R | R | R | S |
| WGS 143 A | S. aureus | R | S | S | R | S | S |
| WGS 153 C | S. aureus | R | S | S | R | R | S |
| WGS 159 A | S. aureus | S | S | S | R | S | S |
| WGS 166 | S. aureus | S | S | S | R | S | S |
| WGS 176 A | S. aureus | S | S | S | R | S | S |
| WGS 196 | S. aureus | S | S | R | R | R | S |
| WGS 201 A | S. aureus | S | S | I | R | S | S |
| WGS 204 | S. aureus | S | I | I | R | S | S |
| WGS 224 C | S. aureus | S | S | S | R | S | S |
| WGS 260 | S. aureus | S | S | S | R | S | S |
| WGS 309 B | S. aureus | R | S | R | R | R | S |
| WGS 319 D | S. aureus | R | S | R | R | R | S |
| WGS 323 A | S. aureus | S | S | R | R | S | S |
| WGS 348 B | S. aureus | R | S | R | R | R | S |
| WGS 349 A | S. aureus | R | S | S | R | R | S |
| WGS 437 | S. aureus | S | S | S | R | S | S |
| WGS 449 | S. aureus | S | S | S | R | S | S |
| WGS 463 B | S. aureus | S | S | S | R | R | S |
| WGS 494 | S. aureus | R | S | R | R | R | S |
| WGS 567A | S. aureus | S | S | S | R | R | S |
| WGS 577 | S. aureus | S | S | R | R | R | S |
| % resistance n (%) | 13 (43.3) | 0 (0) | 14 (46.7) | 30 (100) | 16 (53.3) | 0 (0) | |
C: Chloramphenicol, SXT: trimeth/sulfa, CXT: cefoxitin, E: erythromycin, P: penicillin G, VA: vancomycin, R: resistance, S: susceptible, and I: intermediate.
3.3. Multilocus Sequence Typing (MLST), Antimicrobial and Virulence Genes
In silico multilocus sequence typing revealed 13 different sequence types (ST) among the thirty S. aureus isolates. There were seven S. aureus isolates (23.3%) with ST-8, four (13.3%) with ST-1, and three (10%) with ST-152. The remaining isolates had diverse sequence types (see Figure 1).
Figure 1.
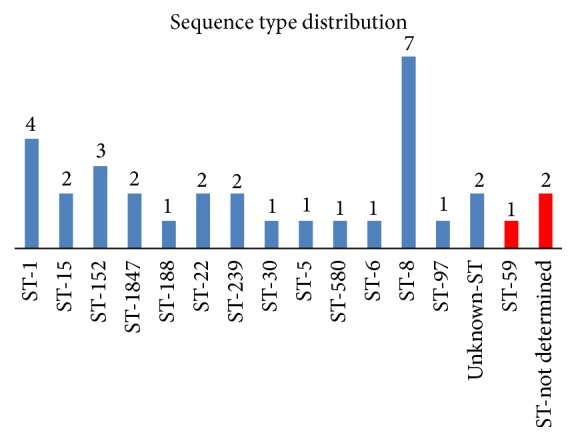
Distribution of sequence types (ST) among Staphylococcus aureus and coagulase negative Staphylococcus species (CoNS). Fourteen different sequence types were identified, with sequence type 8 being the most common. ST-59 and undetermined ST were observed among the CoNS (presented in red).
The three CoNS were one S. epidermidis with ST-59 and two S. haemolyticus of undetermined sequence type.
Presence of mecA was established in 13 (39.4%) of all Staphylococcus spp. isolates involved in this study. Ten (33.3%) of the thirty S. aureus isolates found to harbor mecA, thus classified as MRSA. Six out of the ten S. aureus isolates with mecA were ST-8, two were ST-239, and the remaining two had unknown sequence types. All three CoNS possessed the mecA gene as well. Additionally, five S. aureus isolates (16.7%) were found to harbor Panton-Valentine Leukocidin (pvl) virulence genes with absence of mecA. Two (6.7%) were found to have tst genes for toxic shock syndrome. Other antimicrobial and virulence genes possessed by all isolates are further described in Table 3.
Table 3.
Multilocus sequence typing, antimicrobial resistance genes, and virulence genes.
| Sample name | Species | MLST | Resistance genes | Virulence factors |
|---|---|---|---|---|
| 260 | S. aureus | ST-1 | blaZ | splA, splB, splE, aur, hlb, lukE, lukD, seb, hlgB, hlgC, hlgA, sak, scn, se-h |
| 449 | S. aureus | ST-1 | blaZ | splA, splB, splE, aur, hlb, lukE, lukD, seb, hlgB, hlgC, hlgA, sak, scn, se-h |
| 494 | S. aureus | ST-1 | blaZ | splA, splB, splE, aur, hlb, lukE, lukD, seb, hlgB, hlgC, hlgA, sak, scn, se-h |
| 437 C | S. aureus | ST-1 | blaZ | |
| 577 | S. aureus | ST-15 | aac(6′)-aph(2′′), ant(6)-Ia, aph(3′)-III, blaZ, dfrG, erm(B), erm(C), lsa(A)-like, tet(K), tet(M) | splA, splB, splE, aur, hlb, lukE, lukD, hlgB, hlgA, hlgC, scn. |
| 463 B | S. aureus | ST-15 | blaZ, dfrG, erm(C), tet(K) | splA, splB, splE, aur, hlb, lukE, lukD, hlgB, hlgA, hlgC, scn. |
| 196 | S. aureus | ST-152 | blaZ-like, dfrG, erm(C), tet(K) | aur, hlb, hlgB, hlgA, sak, scan, lukF-PV, lukS-PV, edinB |
| 224 C | S. aureus | ST-152 | blaZ-like, dfrG, tet(K) | aur, hlb, hlgB, hlgA, sak, scan, lukF-PV, lukS-PV, edinB |
| 323 B | S. aureus | ST-152 | dfrG, erm(C), tet(K) | aur, hlb, hlgB, hlgA, sak, scan, lukF-PV, lukS-PV, edinB |
| 38 | S. aureus | ST-1847 | blaZ-like | nil |
| 37 B | S. aureus | ST-1847 | blaZ | splA, splB, splE, aur, hlb, sek, seq, lukE, lukD, hlgB, hlgC, hlgA, sak, scn, lukF-PV, lukS-PV, sea/sep, sen, tst. |
| 176 A | S. aureus | ST-188 | blaZ-like, tet(K)-like | splA, splB, splE, aur, hlb, lukE, lukD, hlgB, hlgA, sak, scn. |
| 204 | S. aureus | ST-22 | Not found | aur, hlb, hlgB, hlgA, hlgC, sak, scan, seo, sei, sen, seg, sem, tst, seu, |
| 201 A | S. aureus | ST-22 | Not found | aur, hlb, hlgB, hlgA, hlgC, sak, scan, seo, sei, sen, seg, sem, tst, enterotoxin, |
| 32 | S. aureus | ST-239 | aac(6′)-aph(2′′)-like, ant(6)-Ia-like, aph(3′)-III, erm(A), mecA-like, spc-like, tet(M) | Not found |
| 29 B | S. aureus | ST-239 | aac(6′)-aph(2′′)-like, ant(6)-Ia-like, aph(3′)-III, erm(A), mecA-like, spc-like, tet(M) | Not found |
| 143 A | S. aureus | ST-30 | blaZ | splE, aur, hlb, hlgB, hlgC, hlgA, sak, scn, lukF-PV, lukS-PV, seo, sei, seu, sen, seg, sem |
| 567 A | S. aureus | ST-5 | blaZ, dfrG, tet(K) | |
| 166 | S. aureus | ST-580 | blaZ | splA, splE, aur, hlb, lukE, lukD, hlgB, hlgA, hlgC, sak, scn. |
| 159 A | S. aureus | ST-6 | aadA2-like, aadB-like, blaCARB-2, blaZ, dfrA23, strA, strB, sul1, sul2, tet(31)-like, tet(G) | |
| 56 | S. aureus | ST-8 | aac(6′)-aph(2′′), blaZ-like, dfrG, erm(C), mecA | splA, splB, splE, aur, hlb, sek, seq, lukE, lukD, seb, hlgB, hlgC, hlgA, sak, scn. |
| 71 | S. aureus | ST-8 | aac(6′)-aph(2′′), blaZ, dfrG, erm(C), mecA | splA, splB, splE, aur, hlb, sek, seq, lukE, lukD, seb, hlgB, hlgC, hlgA, sak, scn. |
| 153 C | S. aureus | ST-8 | blaZ, dfrG, tet(K) | splA, splB, splE, aur, hlb, sek, seq, lukE, lukD, seb, hlgB, hlgC, hlgA, sak, scn, sea/sep. |
| 319 D | S. aureus | ST-8 | aac(6′)-aph(2′′), blaZ-like, dfrG, erm(C), mecA | splA, splB, splE, aur, hlb, sek, seq, lukE, lukD, seb, hlgB, hlgC, hlgA, sak, scn. |
| 348 B | S. aureus | ST-8 | aac(6′)-aph(2′′), blaZ-like, dfrG, erm(C), mecA | splA, splB, splE, aur, hlb, sek, seq, lukE, lukD, seb, hlgB, hlgC, hlgA, sak, scn. |
| 349 A | S. aureus | ST-8 | aac(6′)-aph(2′′), blaZ-like, dfrG, mecA | splA, splB, splE, aur, hlb, sek, seq, lukE, lukD, seb, hlgB, hlgC, hlgA, sak, scn. |
| 69 S | S. aureus | ST-8 | aac(6′)-aph(2′′), blaZ, dfrG, erm(C), mecA | splA, splB, splE, aur, hlb, sek, seq, lukE, lukD, seb, hlgB, hlgC, hlgA, sak, scn. |
| 59 | S. aureus | ST-97 | blaZ-like, tet(K) | splA, splB, splE, aur, hlb, lukE, lukD, seb, hlgB, hlgC, hlgA, sak, scn. |
| 108 | S. aureus | Unknown ST | aac(6′)-aph(2′′), blaZ-like, dfrG, erm(C), mecA-like | splA, splB, splE, aur, hlb, sek, seq, lukE, lukD, seb, hlgB, hlgC, hlgA, sak, scn, sea/sep. |
| 309 B | S. aureus | Unknown ST | aac(6′)-aph(2′′), blaZ-like, dfrG, erm(C), mecA-like | splA, splB, splE, aur, hlb, sek, seq, lukE, lukD, seb, hlgB, hlgC, hlgA, sak, scn, sea/sep. |
| 547 | S. epidermidis | ST-59 | aac(6′)-aph(2′′), blaZ, erm(C), fosA, lnu(A)-like, mecA-like, tet(K)-like | Not found |
| 562 | S. haemolyticus | ND | aac(6′)-aph(2′′), blaZ-like, dfrG, erm(C), mecA, tet(K)-like, tet(M) | Not found |
| 564 | S. haemolyticus | ND | aac(6′)-aph(2′′), blaZ, dfrG, mecA-like, mph(C), msr(A)-like | Not found |
3.4. Single Nucleotide Polymorphisms (SNPs)
The mapping of the raw reads of these 40 genomes to the reference genome, S. aureus USA300_FPR3757, detected 3,511 qualified SNPs. The maximum likelihood SNP tree was constructed using these 3,511 SNPs (Figure 2(a)). RecHMM detected 353 significant recombination SNPs. The remaining SNPs (n = 3,148, 90%) were used to construct a temporal phylogenetic tree (Figure 2(b)).
Figure 2.

(a) Maximum likelihood SNP phylogenetic tree of forty ST-8 S. aureus genomes. Raw reads from genomes were mapped to a reference genome of Staphylococcus aureus USA300_FPR3757 (accession number CP000255, chromosome length 2,917,469 bp) to generate 3511 qualified SNPs. SNPs generated from each genome were concatenated to single alignment corresponding to position of the reference genome. The concatenated sequences were used for constructing a maximum likelihood tree using FastTree. The tree was visualized by using FigTree version 1.4.0. Of the forty genomes, 6 were from Tanzania (TZ) presented in red; 23 from United States (USA) presented in blue; 6 from Switzerland (CH) presented in pink; one from Japan (JP), Denmark (DK), and Australia (AU) presented in magenta, green, and orange, respectively. USA500_2395 strain genome was included and presented in aqua blue. (b) Bayesian temporal phylogenetic tree of the 40 MRSA genomes from Tanzania (TZ), United States (USA), Switzerland (CH), Japan (JP), Australia (AU), and Denmark (DK). A total of 3148 (90%) SNP sequences were used for reconstructing Bayesian temporal phylogenetic tree using Bayesian Evolutionary Analysis Sampling Trees (BEAST) version 1.8.3. USA300_FPR3757 (accession number CP000255, chromosome length 2,917,469 bp) was used as reference during SNP generation. The mutation rate estimated by BEAST was 1.32 × 10−11 SNPs/site/year.
Mapping the 40 genomes to the second reference genome, S. aureus USA500_2395, detected 3,385 qualified SNPs. RecHMM detected 356 significant recombination SNPs. The maximum likelihood SNP tree was constructed using these 3385 SNPs (Figure 3(a)). The remaining SNPs (n = 3,029, 89%) were used to construct a temporal phylogenetic tree (Figure 3(b)). From the SNP phylogenetic trees, it seems that the six Tanzanian isolates form their own clade and are closely clustered together. The SNP difference among the six Tanzanian MRSA isolates ranged from 24 to 196 SNPs when using the USA300_FPR3757 as a reference, whereas with the USA500_2395 reference SNP differences ranged between 16 and 446 SNPs. However, the tree topology of the two SNP phylogenetic trees remains basically unchanged. Further information of the SNP differences among all genomes used in construction of the phylogenetic trees is indicated in supplementary material Tables 1a and 1b.
Figure 3.

(a) Maximum likelihood SNP phylogenetic tree of forty ST-8 S. aureus genomes. Raw reads from genomes were mapped to a reference genome of Staphylococcus aureus USA500_2395 (accession number CP007499, chromosomal length 2,955,646) to generate 3385 qualified SNPs. SNPs generated from each genome were concatenated to single alignment corresponding to position of the reference genome. The concatenated sequences were used for constructing a maximum likelihood tree using FastTree. The tree was visualized by using FigTree version 1.4.0. Of the forty genomes, 6 were from Tanzania (TZ) presented in red; 23 from United States (USA) presented in blue; 6 from Switzerland (CH) presented in pink; one from Japan (JP), Denmark (DK), and Australia (AU) presented in purple, green, and orange, respectively. USA300_FPR3757 strain genome was included and presented in aqua blue. (b) Bayesian temporal phylogenetic tree of the 40 MRSA genomes from Tanzania (Red), USA (Blue), Switzerland (CH), Japan (JP), Australia (AU), and Denmark (DK). A total of 3029 (89%) SNP sequences were used for reconstructing Bayesian temporal phylogenetic tree using Bayesian Evolutionary Analysis Sampling Trees (BEAST) version 1.8.3. Staphylococcus aureus USA500_2395 (accession number CP007499, chromosomal length 2,955,646) was used as reference during SNP generation. The mutation rate estimated by BEAST was 1.4 × 10−6 SNPs/site/year.
3.5. Temporal Bayesian Phylogenetic Trees
The temporal Bayesian phylogenetic tree using USA300_FPR3757 estimated the emergence of the most recent common ancestor to be in ~1942. Primarily the tree is divided into two main clusters in ~1954 (minor) and in ~1969 (major). The minor cluster shows less branching until ~1971 where two other clusters appear, one cluster composing the Tanzanian isolates and the other the USA500 isolate. Further evolution took place to form another lineage in ~2009 when the most recent common ancestor of Tanzanian MRSA isolates seemed to emerge. Subsequent lineages further emerged in ~2010, 2011, and 2012 among Tanzanian MRSA isolates. The 1969 major cluster is more complex compared to the minor cluster. From 1969 further evolution took place in ~1989 and in ~1994. However, from the tree it is predicted that most of the lineages that evolved in the USA and Switzerland emerged around 1990s (Figure 1a in supplementary material).
Using USA500_2395 as the reference, the tree estimates that the most recent common ancestor emerged around ~1960. The tree is also divided into two major clusters in ~1965 (major cluster) and ~2009 (minor). The major cluster is further divided to form subsequent lineages. In ~1992 this major cluster was divided into two other clusters forming in ~2001 and ~2002. The 2001 cluster is mainly composed of USA strains and few from Switzerland, while the 2002 cluster contains only Switzerland strains. The minor (2009) lineage is composed of Tanzanian strains only, which shows introduction of new strains in early and late ~2011 and thereafter further evolution in ~2012 (Figure 1b in supplementary material). However, the infection observed in the hospital in 2013 was due to strains evolved in early and late ~2011 while 2014 infections was due to strains evolved in both 2011 and 2012. Bayesian analysis also shows that the common ancestor between Tanzanian isolates and USA500_2395 emerged around 1971, while with USA300_FPR3757 this emerged around 1961. The mutation rates estimated from the BEAST were 1.38 × 10−11 SNPs/site/year and 1.4 × 10−6 SNPs/site/year using USA300 and USA500 as references, respectively.
4. Discussion
S. aureus is an important causative agent of hospital-associated infections worldwide. Studying the pathogen at its molecular level has the potential to bring about breakthroughs in diagnosis, treatment, and infection control. In the current study we determined the molecular epidemiology of S. aureus isolates using the next generation whole genome sequencing facility available at KCMC-KCRI Biotechnology Laboratory in Moshi, Tanzania.
Multilocus sequence typing revealed 13 different sequence types among the 30 S. aureus isolates. This diversity suggests that S. aureus infections in the hospitalized patients that were sampled were not epidemiologically related. This is in line with similar findings in other studies in Africa [40] and other parts of the world [41]. The high genetic diversity of S. aureus lineages confers their high adaptability to different environments resulting in widespread distribution [42]. Sequence type 8 MRSA which has been observed to be the predominant strain in our hospital is also considered to be the most studied CA-MRSA [43]. The strain has been observed in other parts of the world [44–46] as a cause of infections in both hospital and community settings [43].
Resistance of S. aureus to the antimicrobial agent cefoxitin can be used to predict existence of MRSA [47]. This study reports resistance to cefoxitin to be 43.3% by using classical microbiology techniques. The prevalence was further narrowed down to 33.3% when detecting mecA using whole genome sequencing. These findings are not far from what has been reported in other studies in Tanzania, as it seems that the prevalence of MRSA in Tanzania is above 10% but less than 50% [17–19, 48–51] in hospitalized patients and ranges between 1.5% and 2.1% in healthy individuals [19, 52]. The prevalence of MRSA infections observed in most African countries ranges between 25% and 50% [2], which is lower than what is seen on other continents [53].
Several virulence genes including pvl and tst were observed in the isolates studied. Some of these genes may have influenced the severity of the S. aureus infections. It then becomes necessary to properly identify the genes for the purpose of controlling and monitoring their spread. In our study we reported five isolates identified to carry pvl genes with one of them also possessing the tst gene. All five isolates were methicillin susceptible S. aureus. Such findings correlate with other studies, which indicate that approximately 50% of MSSA isolates from Africa carry pvl genes [54]. However the findings are in contrast with a study conducted in Tanzania whereby ST-88 and ST-1797 were found to be dominant MRSA clones, and pvl genes were detected in ST-88 MRSA isolates [50].
Regarding determination of antimicrobial resistance and virulence genes in S. aureus, tracing genetic relation of clones circulating in a certain locality is vital key for determining the evolution history [55]. Phylogenetic analysis additionally provides insight into the current and possible future composition of the organism's population. The phylogenetic analysis of ST-8 strains from Tanzania using two different references clearly demonstrated that they belong to their own clade in global epidemiology. The two maximum likelihood SNP trees did not differ significantly in topology. However, the temporal phylogenetic analysis suggests that the USA500_2395 is closer related to Tanzanian ST-8 MRSA than USA300_FPR3757 despite the fact that these two strains are closely related [28]. The temporal phylogenetic analysis based on the USA500-2395 reference suggests that the MRSA infection observed at KCMC hospital was due to multiple introduction events. Three strains emerged at the beginning and end of ~2011; two caused infection in 2013, and one stayed without causing infection in that year. The third together with the one emerged in 2012 caused infections in 2014.
These results illustrate the value of temporal phylogeny in phylogenetic analysis. It provides a framework for examining evolution and molecular diversity in a more comprehensive way, by suggesting approximate times at which divergence occurred. However, the analysis is computationally expensive, taking several days.
5. Conclusion
This study showed that S. aureus isolates causing infections in hospitalized patients in Moshi, Tanzania, are highly diverse and likely not epidemiologically related. Using temporal phylogenetic analysis provided a higher resolution insight into the transmission and introduction of MRSA, and it may be valuable to include this analysis in future routine analyses despite its computational cost. This study demonstrates the feasibility and value of high throughput technologies in a resource-constrained setting. We propose that technologies such as NGS should be deployed in particular in infectious disease-endemic settings. This will improve diagnosis, treatment, and control of infections in these areas. Just as most of Africa skipped land-line telephony to jump straight to ubiquitous mobile phone use, next generation sequencing technology may provide a similar leapfrog in infectious disease diagnosis, treatment, and control.
Acknowledgments
This work was supported by DANIDA (DFC no. 12-007 DTU). The authors would like to thank all the participants who took part in this study. They also express their special thanks to Eliangikaya Mangowi for sample collection. Thanks and appreciations go to KCMC management for allowing sample collection from the hospital. Finally, thanks go to all KCRI Biotechnology Laboratory and DTU-Food staff members for their contributions.
Conflicts of Interest
The authors declare that they have no conflicts of interest.
Supplementary Materials
Figure 1a. BEAST tree using USA300_FPR37357 as a reference. Node labels indicate years.
Figure 1b. BEAST using USA500_2395 as a reference. Node labels indicate years.
Table 1a. Information of the six ST-8 MRSA genomes from Tanzania.
Table 1b. Information of the 34 ST-8 MRSA genomes from other parts of the world.
References
- 1.Kuroda M., Ohta T., Uchiyama I., et al. Whole genome sequencing of meticillin-resistant Staphylococcus aureus. The Lancet. 2001;357:1225–1240. doi: 10.1016/s0140-6736(00)04403-2. [DOI] [PubMed] [Google Scholar]
- 2.Falagas M. E., Karageorgopoulos D. E., Leptidis J., Korbila I. P. MRSA in Africa: filling the global map of antimicrobial resistance. PLoS ONE. 2013;8(7) doi: 10.1371/journal.pone.0068024.e68024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.de Kraker M. E. A., Davey P. G., Grundmann H. Mortality and hospital stay associated with resistant Staphylococcus aureus and Escherichia coli bacteremia: estimating the burden of antibiotic resistance in Europe. PLoS Medicine. 2011;8(10) doi: 10.1371/journal.pmed.1001104.e1001104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.David M. Z., Daum R. S. Community-associated methicillin-resistant Staphylococcus aureus: epidemiology and clinical consequences of an emerging epidemic. Clinical Microbiology Reviews. 2010;23(3):616–687. doi: 10.1128/CMR.00081-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mediavilla J. R., Chen L., Mathema B., Kreiswirth B. N. Global epidemiology of community-associated methicillin resistant Staphylococcus aureus (CA-MRSA) Current Opinion in Microbiology. 2012;15(5):588–595. doi: 10.1016/j.mib.2012.08.003. [DOI] [PubMed] [Google Scholar]
- 6.Ray P., Gautam V., Singh R. Methicillin-resistant Staphylococcus aureus (MRSA) in developing and developed countries: implications and solutions. Regional Health Forum. 2011;15:74–82. [Google Scholar]
- 7.Datta P., Gulati N., Singla N., et al. Evaluation of various methods for the detection of meticillin-resistant Staphylococcus aureus strains and susceptibility patterns. Journal of Medical Microbiology. 2011;60(11):1613–1616. doi: 10.1099/jmm.0.032219-0. [DOI] [PubMed] [Google Scholar]
- 8.Köser C. U., Ellington M. J., Cartwright E. J. P., et al. Routine use of microbial whole genome sequencing in diagnostic and public health microbiology. PLoS Pathogens. 2012;8(8) doi: 10.1371/journal.ppat.1002824.e1002824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Didelot X., Bowden R., Wilson D. J., Peto T. E. A., Crook D. W. Transforming clinical microbiology with bacterial genome sequencing. Nature Reviews Genetics. 2012;13(9):601–612. doi: 10.1038/nrg3226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.SenGupta D. J., Cummings L. A., Hoogestraat D. R., et al. Whole-genome sequencing for high-resolution investigation of methicillin-resistant Staphylococcus aureus epidemiology and genome plasticity. Journal of Clinical Microbiology. 2014;52(8):2787–2796. doi: 10.1128/JCM.00759-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Salipante S. J., SenGupta D. J., Cummings L. A., Land T. A., Hoogestraat D. R., Cookson B. T. Application of whole-genome sequencing for bacterial strain typing in molecular epidemiology. Journal of Clinical Microbiology. 2015;53(4):1072–1079. doi: 10.1128/JCM.03385-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Köser C. U., Holden M. T., Ellington M. J., et al. Rapid whole-genome sequencing for investigation of a neonatal MRSA outbreak. The New England Journal of Medicine. 2012;366(24):2267–2275. doi: 10.1056/NEJMoa1109910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kingsmore S. F., Lantos J. D., Dinwiddie D. L., et al. Next-generation community genetics for low- and middle-income countries. Genome Medicine. 2012;4(3, article no. 25) doi: 10.1186/gm324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Helmy M., Awad M., Mosa K. A. Limited resources of genome sequencing in developing countries: Challenges and solutions. Applied and Translational Genomics. 2016;9:15–19. doi: 10.1016/j.atg.2016.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sonda T., Kumburu H., Zwetselaar M. V., et al. Benchtop whole-genome sequencing for identification of nosocomial outbreaks in Tanzania. Infection Control and Hospital Epidemiology. 2016;37(5):622–623. doi: 10.1017/ice.2016.28. [DOI] [PubMed] [Google Scholar]
- 16.Schaumburg F., Alabi A. S., Peters G., Becker K. New epidemiology of Staphylococcus aureus infection in Africa. Clinical Microbiology and Infection. 2014;20(7):589–596. doi: 10.1111/1469-0691.12690. [DOI] [PubMed] [Google Scholar]
- 17.Mushi M. F., Mwalutende A. E., Gilyoma J. M., et al. Predictors of disease complications and treatment outcome among patients with chronic suppurative otitis media attending a tertiary hospital, Mwanza Tanzania Ear disorders. BMC Ear, Nose, and Throat Disorders. 2016;16(1, article no. 1) doi: 10.1186/s12901-015-0021-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mawalla B., Mshana S. E., Chalya P. L., Imirzalioglu C., Mahalu W. Predictors of surgical site infections among patients undergoing major surgery at Bugando Medical Centre in Northwestern Tanzania. BMC Surgery. 2011;11, article 21 doi: 10.1186/1471-2482-11-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Geofrey A., Abade A., Aboud S. Methicillin-resistant staphylococcus aureus (MRSA) colonization among Intensive Care Unit (ICU) patients and health care workers at Muhimbili national hospital, Dar Es Salaam, Tanzania, 2012. Pan African Medical Journal. 2015;21, article 211 doi: 10.11604/pamj.2015.21.211.4207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kumburu H. H., Sonda T., Mmbaga B. T., et al. Patterns of infections, aetiological agents and antimicrobial resistance at a tertiary care hospital in northern Tanzania. Tropical Medicine & International Health. 2017;22(4):454–464. doi: 10.1111/tmi.12836. [DOI] [PubMed] [Google Scholar]
- 21.Otto M. Staphylococcus epidermidis—the "accidental" pathogen. Nature Reviews: Microbiology. 2009;7(8):555–567. doi: 10.1038/nrmicro2182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Barros E. M., Ceotto H., Bastos M. C. F., Dos Santos K. R. N., Giambiagi-deMarval M. Staphylococcus haemolyticus as an important hospital pathogen and carrier of methicillin resistance genes. Journal of Clinical Microbiology. 2012;50(1):166–168. doi: 10.1128/jcm.05563-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zerbino D. R., Birney E. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Research. 2008;18(5):821–829. doi: 10.1101/gr.074492.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Larsen M. V., Cosentino S., Rasmussen S., et al. Multilocus sequence typing of total-genome-sequenced bacteria. Journal of Clinical Microbiology. 2012;50(4):1355–1361. doi: 10.1128/JCM.06094-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zankari E., Hasman H., Cosentino S., et al. Identification of acquired antimicrobial resistance genes. Journal of Antimicrobial Chemotherapy. 2012;67(11):2640–2644. doi: 10.1093/jac/dks261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hasman H., Saputra D., Sicheritz-Ponten T., et al. Rapid whole-genome sequencing for detection and characterization of microorganisms directly from clinical samples. Journal of Clinical Microbiology. 2014;52(1):139–146. doi: 10.1128/JCM.02452-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Baba T., Takeuchi F., Kuroda M., et al. Genome and virulence determinants of high virulence community-acquired MRSA. The Lancet. 2002;359(9320):1819–1827. doi: 10.1016/s0140-6736(02)08713-5. [DOI] [PubMed] [Google Scholar]
- 28.Benson M. A., Ohneck E. A., Ryan C., et al. Evolution of hypervirulence by a MRSA clone through acquisition of a transposable element. Molecular Microbiology. 2014;93(4):664–681. doi: 10.1111/mmi.12682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sowash M. G., Uhlemann A. NIH Public Access. 2014.
- 30.Von Dach E., Diene S. M., Fankhauser C., Schrenzel J., Harbarth S., François P. Comparative genomics of community-associated methicillin-resistant Staphylococcus aureus shows the emergence of clone ST8-USA300 in Geneva, Switzerland. The Journal of Infectious Diseases. 2016;213(9):1370–1379. doi: 10.1093/infdis/jiv489. [DOI] [PubMed] [Google Scholar]
- 31.Leekitcharoenphon P., Kaas R. S., Thomsen M. C. F., Friis C., Rasmussen S., Aarestrup F. M. snpTree—a web-server to identify and construct SNP trees from whole genome sequence data. BMC Genomics. 2012;13(supplement, article S6) doi: 10.1186/1471-2164-13-S7-S6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kaas R. S., Leekitcharoenphon P., Aarestrup F. M., Lund O. Solving the problem of comparing whole bacterial genomes across different sequencing platforms. PLoS ONE. 2014;9(8) doi: 10.1371/journal.pone.0104984.e104984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li H., Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25(14):1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li H., Handsaker B., Wysoker A., et al. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25(16):2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Price M. N., Dehal P. S., Arkin A. P. Fasttree: computing large minimum evolution trees with profiles instead of a distance matrix. Molecular Biology and Evolution. 2009;26(7):1641–1650. doi: 10.1093/molbev/msp077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhou Z., McCann A., Weill F.-X., et al. Transient darwinian selection in salmonella enterica serovar paratyphi a during 450 years of global spread of enteric fever. Proceedings of the National Acadamy of Sciences of the United States of America. 2014;111(33):12199–12204. doi: 10.1073/pnas.1411012111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Drummond A. J., Suchard M. A., Xie D., Rambaut A. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Molecular Biology and Evolution. 2012;29(8):1969–1973. doi: 10.1093/molbev/mss075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tajima F. Evolutionary relationship of DNA sequences in finite populations. Genetics. 1983;105(2):437–460. doi: 10.1093/genetics/105.2.437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ho S. Y. W., Shapiro B. Skyline-plot methods for estimating demographic history from nucleotide sequences. Molecular Ecology Resources. 2011;11(3):423–434. doi: 10.1111/j.1755-0998.2011.02988.x. [DOI] [PubMed] [Google Scholar]
- 40.Abdulgader S. M., Shittu A. O., Nicol M. P., Kaba M. Molecular epidemiology of Methicillin-resistant Staphylococcus aureus in Africa: A systematic review. Frontiers in Microbiology. 2015;6, article no. 348 doi: 10.3389/fmicb.2015.00348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rolo J., Miragaia M., Turlej-Rogacka A., et al. High genetic diversity among community-associated Staphylococcus aureus in Europe: results from a multicenter study. PLoS ONE. 2012;7(4) doi: 10.1371/journal.pone.0034768.e34768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ebruke C., Dione M. M., Walter B., et al. High genetic diversity of Staphylococcus aureus strains colonising the nasopharynx of Gambian villagers before widespread use of pneumococcal conjugate vaccines. BMC Microbiology. 2016;16(1, article no. 661) doi: 10.1186/s12866-016-0661-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Khokhlova O. E., Hung W.-C., Wan T.-W., et al. Healthcare- and community-associated methicillin-resistant Staphylococcus aureus (MRSA) and fatal pneumonia with pediatric deaths in Krasnoyarsk, Siberian Russia: Unique MRSA's multiple virulence factors, genome, and stepwise evolution. PLoS ONE. 2015;10(6):p. 1. doi: 10.1371/journal.pone.0128017.e0128017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yamamoto T., Takano T., Higuchi W., et al. Comparative genomics and drug resistance of a geographic variant of ST239 methicillin-resistant Staphylococcus aureus emerged in Russia. PLoS ONE. 2012;7(1) doi: 10.1371/journal.pone.0029187.e29187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hsu L.-Y., Harris S. R., Chlebowicz M. A., et al. Evolutionary dynamics of methicillin-resistant Staphylococcus aureus within a healthcare system. Genome Biology. 2015;16(1, article no. 81) doi: 10.1186/s13059-015-0643-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jones M. B., Montgomery C. P., Boyle-Vavra S., et al. Genomic and transcriptomic differences in community acquired methicillin resistant Staphylococcus aureus USA300 and USA400 strains. BMC Genomics. 2014;15(1, article no. 1145) doi: 10.1186/1471-2164-15-1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jain A., Agarwal A., Verma R. K. Cefoxitin disc diffusion test for detection of meticillin-resistant staphylococci. Journal of Medical Microbiology. 2008;57(8):957–961. doi: 10.1099/jmm.0.47152-0. [DOI] [PubMed] [Google Scholar]
- 48.Manyahi J., Matee M. I., Majigo M., Moyo S., Mshana S. E., Lyamuya E. F. Predominance of multi-drug resistant bacterial pathogens causing surgical site infections in Muhimbili National Hospital, Tanzania. BMC Research Notes. 2014;7(1, article 500) doi: 10.1186/1756-0500-7-500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kayange N., Kamugisha E., Mwizamholya D. L., Jeremiah S., Mshana S. E. Predictors of positive blood culture and deaths among neonates with suspected neonatal sepsis in a tertiary hospital, Mwanza-Tanzania. BMC Pediatrics. 2010;10(1, article 39) doi: 10.1186/1471-2431-10-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Moremi N., Mshana S. E., Kamugisha E., et al. Predominance of methicillin resistant Staphylococcus aureus -ST88 and new ST1797 causing wound infection and abscesses. The Journal of Infection in Developing Countries. 2012;6(8):620–625. doi: 10.3855/jidc.2093. [DOI] [PubMed] [Google Scholar]
- 51.Morgan E., David M. Z. Do citation trends reflect epidemiologic patterns? Assessing MRSA, emerging and re-emerging pathogens, 1963-2014. BMC Infectious Diseases. 2015;15(1, article no. 460) doi: 10.1186/s12879-015-1182-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Okamo B., Moremi N., Seni J., Mirambo M. M., Kidenya B. R., Mshana S. E. Prevalence and antimicrobial susceptibility profiles of Staphylococcus aureus nasal carriage among pre-clinical and clinical medical students in a Tanzanian University Microbiology. BMC Research Notes. 2016;9(1, article no. 47) doi: 10.1186/s13104-016-1858-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chen C.-J., Huang Y.-C. New epidemiology of Staphylococcus aureus infection in Asia. Clinical Microbiology and Infection. 2014;20(7):605–623. doi: 10.1111/1469-0691.12705. [DOI] [PubMed] [Google Scholar]
- 54.Breurec S., Fall C., Pouillot R., et al. Epidemiology of methicillin-susceptible Staphylococcus aureus lineages in five major African towns: High prevalence of Panton-Valentine leukocidin genes. Clinical Microbiology and Infection. 2011;17(4):633–639. doi: 10.1111/j.1469-0691.2010.03320.x. [DOI] [PubMed] [Google Scholar]
- 55.Soltis D. E., Soltis P. S. The role of phylogenetics in comparative genetics. Plant Physiology. 2003;132(4):1790–1800. doi: 10.1104/pp.103.022509. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure 1a. BEAST tree using USA300_FPR37357 as a reference. Node labels indicate years.
Figure 1b. BEAST using USA500_2395 as a reference. Node labels indicate years.
Table 1a. Information of the six ST-8 MRSA genomes from Tanzania.
Table 1b. Information of the 34 ST-8 MRSA genomes from other parts of the world.