

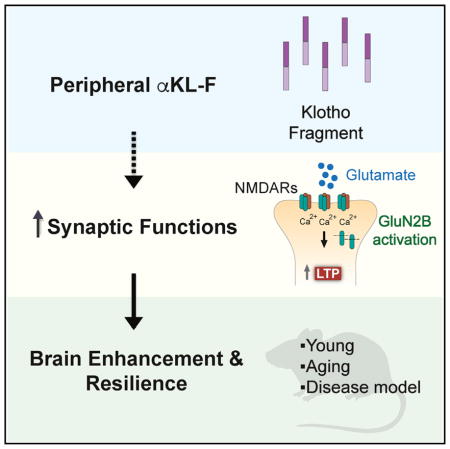
SUMMARY
Cognitive dysfunction and decreased mobility from aging and neurodegenerative conditions, such as Parkinson and Alzheimer diseases, are major biomedical challenges in need of more effective therapies. Increasing brain resilience may represent a new treatment strategy. Klotho, a longevity factor, enhances cognition when genetically and broadly overexpressed in its full, wild-type form over the mouse lifespan. Whether acute klotho treatment can rapidly enhance cognitive and motor functions or induce resilience is a gap in our knowledge of its therapeutic potential. Here, we show that an α-klotho protein fragment (αKL-F), administered peripherally, surprisingly induced cognitive enhancement and neural resilience despite impermeability to the blood-brain barrier in young, aging, and transgenic α-synuclein mice. αKL-F treatment induced cleavage of the NMDAR subunit GluN2B and also enhanced NMDAR-dependent synaptic plasticity. GluN2B blockade abolished αKL-F-mediated effects. Peripheral αKL-F treatment is sufficient to induce neural enhancement and resilience in mice and may prove therapeutic in humans.
In Brief
Klotho is a longevity factor associated with cognitive enhancement when genetically and widely overexpressed over the lifetime of mice. Leon et al. show that peripheral delivery of a klotho fragment, αKL-F, acutely enhances cognition and neural resilience in young, aging, and disease model mice, establishing its therapeutic relevance and dissecting its underlying mechanisms.
INTRODUCTION
Cognitive dysfunction and decreased mobility from aging and age-related neurodegenerative conditions such as Alzheimer disease (AD) and Parkinson disease (PD) are major biomedical challenges. Because more effective treatments are needed, and clinical trials targeting putative pathogenic proteins have failed, it is critical to develop alternate or complimentary therapeutic strategies. In light of this urgent medical need for our rapidly aging populations, delaying aging itself or increasing the function and resilience of the brain (Bennett, 2017; McEwen and Morrison, 2013) may represent new treatment strategies.
α-Klotho (klotho) is a pleiotropic protein that circulates as a hormone following cleavage from its transmembrane form. It regulates insulin (Kurosu et al., 2005), Wnt (Liu et al., 2007), and fibroblast growth factor (FGF) (Urakawa et al., 2006) signaling. Overexpression of klotho extends life in organisms (Château et al., 2010; Kurosu et al., 2005), whereas lowering klotho shortens it (Kuro-o et al., 1997). Elevated klotho levels in humans, resulting from genetic variation (Arking et al., 2002; Dubal et al., 2014; Yokoyama et al., 2017), also associate with lifespan (Arking et al., 2002; Invidia et al., 2010) in some populations. In model organisms and humans, levels of klotho decline with age (Duce et al., 2008; Semba et al., 2011), chronic stress (Prather et al., 2015), cognitive aging (Shardell et al., 2016), neurodegenerative disease (Semba et al., 2014), and models of neurodegenerative disease (Dubal et al., 2015; Massó et al., 2015).
We previously discovered that life-long, genetic overexpression of klotho causally enhances normal cognition and neural resilience independent of age and when broadly expressed in the mouse body and brain (Dubal et al., 2014, 2015). It does so, at least in part, by directly or indirectly optimizing synaptic functions through NMDA receptor (NMDAR)-dependent mechanisms (Dubal et al., 2014, 2015). Importantly, genetic, lifelong, and widespread klotho elevation also contributes to neural resilience in a human amyloid precursor protein (hAPP) model of neurodegenerative disease related to AD (Dubal et al., 2015); that is, it effectively counters cognitive and synaptic deficits despite high levels of pathogenic proteins, including Aβ, tau, and phospho-tau.
The relevance of klotho to brain health in humans is supported by the findings that elevated serum klotho, related to KLOTHO variation, are associated with better measures, including cognition (Dubal et al., 2014; Yokoyama et al., 2015), structural reserve of the prefrontal cortex in normal aging (Yokoyama et al., 2015), connectivity between cortical regions (Yokoyama et al., 2017), and physical performance in aging (Shardell et al., 2015), and that diminished klotho levels are associated with worse brain measures (Prather et al., 2015; Yokoyama et al., 2015, 2017). Furthermore, KLOTHO variation is associated with less cognitive decline and better cortical structure in another large cohort (de Vries et al., 2017), although positive genetic associations were not observed in other populations at particularly advanced ages (Almeida et al., 2017; Mengel-From et al., 2016).
Multiple proteins contribute to the pathogenesis of neurodegenerative diseases. α-Synuclein, a membrane protein whose overexpression inhibits mechanisms of exocytosis (Logan et al., 2017), is central to PD and other parkinsonian disorders and contributes to multi-etiology dementias such as AD. Transgenic mice that overexpress wild-type human α-synuclein (hSYN) simulate key aspects of neurodegenerative disease, including cognitive and motor dysfunction (Fleming et al., 2004; Hatami and Chesselet, 2015). Therapeutic strategies that induce resilience against common pathogenic proteins such as α-synuclein involved in multiple diseases with broad phenotypes (Montine et al., 2014) could positively affect the human condition.
Whether acute klotho elevation represents a strategy that can rapidly enhance cognition, motor functions, and/or induce brain resilience is a gap in our knowledge of its therapeutic potential. Here we show that αKL-F, a fragment of the α-klotho protein similar to its secreted form, can acutely improve cognitive and motor functions following peripheral administration. It does so despite apparent impermeability to the blood-brain barrier in young, aging, and hSYN transgenic mice. Further investigation of αKL-F-mediated molecular mechanisms revealed activation of glutamatergic signaling and enhancement of synaptic plasticity.
RESULTS
αKL-F Is a Recombinant Modified Fragment of Mouse Klotho that Does Not Penetrate into the Brain or Alter Endogenous Hippocampal Klotho Levels
We first characterized the recombinant, mouse αKL-F fragment (Kurosu et al., 2005) by mass spectrometry. αKL-F is a protein representing a truncated form of the endogenous α-klotho protein but lacking its transmembrane, N-terminal signal peptide, and intracellular domains (Figure 1A). This truncated peptide (954 amino acids [aa]) includes the KL-1 and KL-2 subdomains and a C-terminal 6-His tag sequence. It resembles the extracellular portion of α-klotho (or klotho) endogenously shed from the membrane by ADAM 10 and 17 (Bloch et al., 2009; Chen et al., 2007), leading to a protein of approximately 959 aa that includes some of the transmembrane domain (Chen et al., 2015) and circulates in mouse serum, human serum, and human cerebral spinal fluid (CSF) (Imura et al., 2004).
Figure 1. αKL-F Is a Recombinant Post-translationally Modified Fragment of Klotho, and Its Peripheral Administration Does Not Show BBB Crossing or Alteration of Endogenous Hippocampal Klotho Levels.

(A) Amino acid sequence of the endogenous α-klotho protein (top, aa 1–1014) and the recombinant α-klotho fragment (αKL-F, bottom, aa 35–982) followed by a His tag. N-T, N terminus; TR, transmembrane region; IC, intracellular domain.
(B) Locations of O-glycosylation and N-glycosylation sites of the recombinant protein made in Chinese hamster ovary (CHO) cells.
(C) αKL-F was not observed to cross into the brain 4 hr following i.p. injection of Veh or αKL-F (100 μg/kg). Western blot shows immunoprecipitation against His-tagged αKL-F (His-IP) in kidney (detected) and brain (not detected) lysates. αKL-F was added to the veh-treated lysates as a positive control for detection of His-tagged protein (His-Total).
(D) Representative western blot showing Klotho and GAPDH levels in homogenized whole hippocampus 4–5 hr following Veh or αKL-F (i.p., 10 μg/kg) treatment (n = 12/group; male; age, 4 months). Images were captured from the same gel.
(E) Quantitation of endogenous brain klotho levels showing no differences between Veh- and αKL-F-treated mice. NTG levels are arbitrarily defined as 1.0.
Data are mean ± SEM.
We assessed for post-translational modifications (PTMs) of αKL-F and detected six glycosylation sites (Figure 1B). The identified N- and O-glycosylation sites map to the KL1 and KL2 domains and could potentially affect many facets of the protein, including recognition, stability, and solubility. No regulatory PTMs, such as phosphorylation, were detected. Other physical properties of the protein, such as its aggregation, higher-form conformations, and their importance to cognition-related functions, remain to be determined.
To probe whether peripherally administered αKL-F penetrates the brain, we injected a high dose (10–40 times the doses used in our studies) of the His tag protein (100 μg/kg, intraperitoneally [i.p.]). We assessed for its presence in the kidney and brain 4 hr later, within its estimated 7.5-hr half-life (Hu et al., 2016), by immunoprecipitation of the His tag (Figure 1C). The protein was observed in the kidney but not in the brain. Consistent with previous findings using autoradiographic methods (Hu et al., 2016), our data suggest that peripheral treatment with αKL-F does not cross the blood-brain barrier (BBB).
Because the endogenous klotho protein is also produced and circulates within the BBB, we wondered whether peripheral αKL-F treatment causes shifts in brain klotho levels. Peripheral αKL-F did not alter the levels of endogenous klotho in the hippocampus (Figures 1D and 1E). These data suggest that the central activity of peripherally administered αKL-F probably engages signals transduced from the periphery to the CNS.
A Fragment of Klotho, αKL-F, Delivered Peripherally, Acutely Enhances Cognition in Young Mice
Whether klotho, or some form of it, can acutely enhance cognition or induce resilience is a gap in our knowledge of its biologic functions and therapeutic potential. To test the therapeutic potential of mouse αKL-F, we administered it peripherally (i.p.) in young mice.
We first tested spatial learning and memory in the Morris water maze. Young mice were treated with vehicle (Veh) or αKL-F (10 μg/kg, i.p.) daily prior to hidden platform training or a probe trial (Figure 2A). Remarkably, αKL-F treatment enhanced learning, as measured by decreased distance traveled to find the hidden platform (Figure 2B). αKL-F also enhanced spatial memory retention, as measured by increased time spent in the target quadrant (Figure 2C) and at the target site (Figure 2D) in the probe trial following hidden training. Mice treated with Veh or αKL-F showed no differences in their swim speeds (Figure 2E) or ability to find the target platform, identified by a visual cue (Figure 2F). Thus, similar to genetic, lifelong, and widespread overexpression of full-length mouse klotho (Dubal et al., 2014, 2015), peripheral delivery of mouse αKL-F was sufficient to enhance learning and memory, but acutely.
Figure 2. αKL-F, Delivered Peripherally, Acutely Enhances Cognition in Young Mice.

(A) Diagram of the experimental paradigm of Veh or αKL-F injection (i.p., 10 μg/kg) followed by spatial and working memory testing in the Morris water maze. Mice received daily treatment for 5 days, 16 or 4 hr prior to training and testing (n = 11–18/ group; sex-balanced; age, 4 months).
(B) Spatial learning curves (platform hidden) in the water maze. Data represent the daily average of distance traveled to the platform. αKL-F decreased the distance, indicating enhanced learning. Rank-sum test, αKL-F effect days 2–4, p < 0.05.
(C) Probe trial with the platform removed 24 hr after completion of hidden training. αKL-F increased the percentage of time spent in the target quadrant, indicating enhanced recall of spatial memory.
(D) Probe trial at 24 hr. αKL-F increased the duration of time spent at target, indicating enhanced recall of spatial memory.
(E) Velocity in the water maze did not differ between mice treated with Veh or αKL-F.
(F) The distance traveled to find a cued, visible platform in the water maze did not differ between mice treated with Veh or αKL-F.
(G–I) Diagram of the experimental paradigm of Veh or αKL-F injection (G, i.p., 10 μg/kg), followed by working memory testing in the small Y-maze (H and I). Mice (age, 4 months) received treatment 4 hr prior to testing.
(H) Percentages of alternations among arms during exploration of the Y-maze. αKL-F increased alternations, indicating enhanced working memory. In this cohort, mice (males, n = 12–13/group) were exposed to Veh or αKL-F 2 days before testing.
(I) Percentages of alternations among arms during exploration of the Y-maze. In a replicate independent cohort, αKL-F increased working memory. Mice (males, n = 9/group) were naive to previous treatments or testing.
*p < 0.05, **p < 0.01 (t test). Data are mean ± SEM.
See also Figure S1.
We then tested whether αKL-F improves another domain of cognition, working memory, in the small Y-maze (Figures 2H and 2I). In the same cohort and in several (over ten) independent, naive cohorts, young mice were treated with vehicle or αKL-F (10 μg/kg, i.p.) 4 hr prior to exploration in the small Y-maze. αKL-F acutely enhanced working memory, as measured by increased spontaneous alternations in the maze. αKL-F induced a threshold-dependent dose response on cognition, with the lowest effective dose detected at 2.5 μg/kg (Figure S1A). In addition, αKL-F-mediated cognitive enhancement combined with cognitive training persisted for at least 2 weeks after the last treatment (Figure S1B), suggesting organizational, longer-lasting, and beneficial effects on the synapse and brain. Collectively, these data show that peripheral administration of αKL-F is sufficient to induce enhancement of a normal brain.
Of note, in our experiments, we did not detect cognitive enhancement with human αKL-F (87% homology to mouse) administration in mice following the same acute treatment paradigms in the small Y-maze or water maze (data not shown). This suggests species specificity of the biologic action and additionally shows that a protein of the same size, but with a different sequence, did not arbitrarily enhance cognition.
αKL-F Enhances Cognition in the Aging Brain
Because aging is the primary risk factor for cognitive impairment, we wondered whether αKL-F could enhance cognition in the aging brain. To test this, aging mice (18 months) were treated once with Veh or αKL-F (10 μg/kg, i.p.) 1 day prior to training in the two-trial Y-maze (Figure 3A). This task probes both spatial and working memory by measuring exploration time in a novel compared with a familiar arm of the maze following context training (Dellu et al., 1992). During testing, Veh-treated, aged mice showed no preference for the novel arm. αKL-F induced preference for the novel arm, indicating that it enhanced spatial and working memory in aged mice (Figure 3B). Thus, a single peripheral injection of αKL-F boosted cognition in the aging brain.
Figure 3. αKL-F, Delivered Peripherally, Acutely Enhances Cognition in Aged Mice.

(A) Diagram of the experimental paradigm of Veh or αKL-F treatment (i.p., 10 μg/kg) and testing of aged mice in the two-trial Y-maze. (n = 8–10 mice/ group; sex-balanced groups; age, 18 months). Mice received an injection followed by training (24 hr after treatment) and then testing (40 hr after treatment).
(B) Duration of time spent in the novel compared with the familiar arm, expressed as a ratio to indicate novel arm preference. αKL-F increased spatial and working memory in aged mice over time. Two-way repeated measures ANOVA, αKL-F effect, **p < 0.01.
Data are mean ± SEM.
αKL-F Acutely Counters Motor and Cognitive Deficits in Transgenic hSYN Mice, an α-Synuclein Model of Neurodegenerative Disease
Next, we probed whether αKL-F can acutely counter neurodegenerative disease-related deficits and pathologies in mice that overexpress wild-type hSYN mice using motor and cognitive tasks (Figure 4A). We chose this model because the majority of dementias related to both PD and AD are multi-etiology, α-synuclein is commonly shared among them, and this model enabled testing of motor deficits. hSYN mice (Rockenstein et al., 2002) accumulate α-synuclein in neurons and synapses throughout the brain (Rockenstein et al., 2002) and exhibit motor and cognitive deficits (Fleming et al., 2004, 2006; Masliah et al., 2000; Morris et al., 2011).
Figure 4. αKL-F, Delivered Peripherally, Induces Neural Resilience by Acutely Countering Cognitive and Motor Deficits in Trans-genic hSYN Mice.

(A) Diagram of the experimental paradigm of Veh or αKL-F treatment (i.p., 2.5 μg/kg). Mice received single daily injections for 6 days. On day 4, NTG and hSYN mice were tested on the rotarod to assess motor performance. On day 7 (17 hr after the last injection), mice were tested in the two-trial Y-maze to assess spatial and working cognition.
(B) Motor learning and function measured by time on the rotarod during training. hSYN mice showed motor dysfunction compared with NTG mice. αKL-F treatment improved motor learning in hSYN mice (n = 10–15 mice/group; male; age, 3–6 months). Mixed model ANVOA: hSYN effect, p < 0.0001.
(C) Motor function measured by time on the rotarod during testing. hSYN mice showed motor dysfunction compared with NTG mice. Mixed model ANOVA: hSYN effect, p < 0.0001. αKL-F treatment increased time on the rotarod across sessions in hSYN mice. Two-way repeated measures ANOVA: αKL-F effect, *p < 0.05 as indicated by brackets.
(D) Duration of time spent in the novel compared with the familiar arm during testing, expressed as a ratio to indicate novel arm preference at 5 min of exploration. αKL-F improved cognitive deficits in hSYN mice (n = 14–23 mice/group; male; age, 3–6 months). Two-way ANOVA: hSYN effect, p < 0.0001; αKL-F effect, p < 0.01.
(E–I) The hippocampus and cortex of Veh- and αKL-F-treated hSYN mice were analyzed after 3 days of daily injection (n = 5–6 mice/group; male; age, 3–7 months).
(E) Representative western blots showing hippocampal levels of total α-Syn, phosphorylated α-Syn (p-α-Syn, Ser-129), total mouse tau (T-Tau), phosphorylated mouse tau (p-Tau, Ser-396/404), Actin, and GAPDH in Veh- and αKL-F-treated hSYN mice.
(F) Quantitation of α-Syn levels, showing no differences between Veh- and αKL-F-treated hSYN mice.
(G) Quantitation of p-α-Syn (Ser-129) levels, showing no differences between Veh- and αKL-F-treated hSYN mice.
(H) Quantitation of T-Tau levels, showing no differences between Veh- and αKL-F-treated hSYN mice.
(I) Quantitation of p-Tau levels, showing no differences between Veh- and αKL-F-treated hSYN mice.
#p = 0.10, *p < 0.05 versus trial 1 (B, paired t tests) or as indicated by brackets (C, mean effect; D, unpaired t tests) via Bonferroni-Holm test. Data are mean ± SEM.
We first tested whether αKL-F counters motor deficits in hSYN mice. Motor dysfunction is a predominant deficit in hSYN mice and linked with clinical manifestations of α-synuclein toxicity in humans. Building on our findings that a lower dose of αKL-F enhanced function in the normal brain (Figure S1A), we injected Veh and a lower dose of αKL-F (2.5 μg/kg instead of 10 μg/kg, i.p.) daily for 2 days prior to rotarod testing in hSYN mice and nontransgenic (NTG) littermate controls (Figure 4A). As expected (Morris et al., 2011), all hSYN mice showed a motor deficit in training and testing, as measured by decreased time to fall off a fixed (Figure 4B) and accelerating (Figure 4C) rod. αKL-F increased motor learning in hSYN mice during training and mean motor performance during testing (Figures 4B and 4C). Thus, αKL-F improved motor learning and deficits, extending its therapeutic functions to another functional domain.
We then tested whether αKL-F counters cognitive deficits in hSYN mice. Because hSYN mice show motor deficits but move around normally, we used the two-trial Y-maze, which measures spatial and working cognition based on exploration without heavy reliance on motor strength. After rotarod testing, mice were injected with Veh and αKL-F (2.5 μg/kg, i.p.) daily for an additional 2 days (Figure 4A). Veh-treated hSYN mice showed cognitive deficits compared with NTG controls, as measured by decreased preference for the novel arm (Figure 4D). αKL-F improved novel arm preference (by main effect) in NTG and hSYN mice (Figure 4D). αKL-F countered cognitive deficits in hSYN mice (Figure 4D) despite equivalent levels of speed and movement among all experimental groups during training (data not shown). Thus, in addition to enhancing cognition in the normal and aging brain, peripheral treatment with αKL-F acutely improved cognitive deficits in an hSYN model.
αKL-F Induces Neural Resilience in hSYN Mice without Altering Pathogenic Protein Species Related to Neurodegenerative Disease
Because deficits in hSYN mice depend on α-synuclein expression, we assessed whether αKL-F reduces levels of the pathogenic protein. Acute treatment with αKL-F, with the same experimental paradigm that countered neural deficits, did not alter the steady-state levels of α-synuclein or its phosphorylated form (Figures 4E–4G) in the hippocampus or cortex. In addition, αKL-F did not alter the levels of potential co-pathogens of α-synuclein, such as tau or phospho-tau (Haggerty et al., 2011; Wills et al., 2010) in hSYN mice (Figures 4E, 4H, and 4I). Thus, acute and peripheral treatment with αKL-F induced neural resilience in hSYN mice, as defined by countering neural functions without altering the levels of pathogenic protein species.
αKL-F Acutely Increases GluN2B Cleavage without Altering Synaptic GluN2B Levels and Enriches Glutamate Receptor Signaling
To begin to explore the mechanisms underlying αKL-F-mediated cognitive enhancement and neural resilience, we focused subsequent studies on normal, young mice because effects of αKL-F were discovered in this age group, and these mice were more available. We assessed the GluN2B subunit of NMDARs because transgenic overexpression of klotho increases cognition through GluN2B-dependent functions (Dubal et al., 2014), transgenic elevation of GluN2B enhances cognition in mice and rats (Cao et al., 2007; Tang et al., 1999; Wang et al., 2009), and dysfunction of GluN2B contributes to cognitive decline in aging and neurodegenerative diseases (Ittner et al., 2010; Piggott et al., 1992; Sze et al., 2001; Zhao et al., 2009). We first assessed, in parallel with the effects of lifelong, transgenic klotho overexpression (Dubal et al., 2014, 2015), whether αKL-F treatment acutely increases the levels of GluN2B in synaptic fractions of the hippocampus. Synaptic levels of GluN2B in the hippocampus did not differ between Veh- and αKL-F-treated mice (Figures 5A–5C) despite αKL-F-mediated cognitive enhancement in mice. Thus, acute effects of αKL-F on cognition did not induce or require the synaptic increase of GluN2B levels observed in transgenic, klotho-overexpressing mice.
Figure 5. αKL-F Does Not Alter Synaptic Levels of HMW GluN2B.

(A–C) Synaptic membrane fraction 1 (PSD-enriched) and fraction 2 (non-PSD-enriched) isolated from the hippocampus of mice (age, 4 months; male) 4 hr after a single treatment followed by the small Y-maze or after 5 days of daily treatment with Veh or αKL-F (i.p., 10 μg/kg).
(A) Representative western blots from hippocampal membrane fractions from mice 4 hr (top) following a single treatment or 5 days following daily treatments (bottom). Only the HMW GluN2B band was observed.
(B) Quantitation of PSD-enriched (fraction 1) GluN2B protein levels 4 hr following treatment (n = 4/group).
(C) Quantitation of PSD-enriched (fraction 1) GluN2B protein levels 5 days following daily treatment (n = 10/group).
Data are mean ± SEM.
Synaptic proteins are dynamic structures with activity-dependent modification and turnover (Alvarez-Castelao and Schuman, 2015). Following stimulation, the GluN2B subunit of NMDARs can undergo cleavage by calpain, producing a low-molecular-weight (LMW) form of the subunit (~115 kDa) (Dong et al., 2004, 2006; Simpkins et al., 2003). We noticed that, in whole hippocampal homogenates, an LMW form of the GluN2B protein not detected in synaptic, post-synaptic density (PSD)-enriched fractions, differed between experimental groups upon western blotting (Figures 6A–6E). αKL-F treatment elevated LMW GluN2B by approximately 4 hr following treatment (Figures 6A and 6B), and this correlated with better cognitive ability in the small Y-maze (Figure 6C). αKL-F increased LMW GluN2B in multiple replicate cohorts (Figure 6D; data not shown) and tended to do so in the absence of a cognitive task following several days of treatment (albeit less intensely) (Figure 6E). Thus, acute treatment with αKL-F led to engagement of the GluN2B subunit of NMDARs, causing its increased cleavage.
Figure 6. αKL-F Acutely Increases an LMW Form of GluN2B.

(A–E) Whole hippocampal lysate isolated from mice treated with Veh or αKL-F (10 μg/kg i.p.) after 4 hr of a single treatment followed by the small Y-maze or 5 days of daily treatment and then assessed for GluN2B levels.
(A) Representative western blots from hippocampal total lysates (with 1% SDS) from mice with Veh or αKL-F 4 hr after treatment (top, images were captured from the same gel) or 5 days following daily treatment (bottom) (age, 4 months; male). HMW and LMW GluN2B forms were observed.
(B) Quantitation of LMW/total (HMW+LMW) GluN2B protein levels from the hippocampus 4 hr after treatment (n = 10/group).
(C) Correlation of relative LMW/total GluN2B fragment levels from mice shown in (D) with percentage alternations in the small Y-maze (R2 = 0.5, p < 0.03 with αKL-F treatment).
(D) Replicate independent cohort quantitation of LMW/total GluN2B protein levels from the hippocampus 4 hr after treatment (n = 21/group).
(E) Quantitation of LMW/total GluN2B protein levels from the hippocampus following 5 days of daily treatment in the absence of behavior studies (n = 11–12/group).
(F) Western blot showing the specificity of the HMW and LMW GluN2B band by immunoprecipitation with and without preincubation with a GluN2B-blocking peptide harboring the GluN2B antibody epitope sequence.
#p < 0.10, *p < 0.05 versus Veh as indicated by brackets. Data are mean ± SEM. See also Figure S2.
To verify the specificity of the LMW band, we generated a GluN2B blocking peptide harboring the GluN2B antibody epitope sequence. We then immunoprecipitated GluN2B proteins from mouse hippocampal homogenates and preincubated them with and without the GluN2B-blocking peptide. The blocking peptide abrogated detection of the high-molecular-weight (HMW) and LMW bands, but not other non-specific bands, on western blotting (Figure 6F). Of note, the LMW form was not detected in synaptic fractions and surfaced clearly on western blotting of total hippocampal lysates only after increasing stringency with addition of 1% SDS to the homogenization buffer. Thus, GluN2B also exists in an LMW form, as reported previously (Dong et al., 2004, 2006; Simpkins et al., 2003), and αKL-F acutely increased its levels.
Because the klotho-derived fragment αKL-F probably engages multiple downstream pathways to enhance cognition, we probed broadly for possible targets with an independent and unbiased proteomic analysis of whole hippocampal homogenates from mice treated with Veh and αKL-F (10 μg/kg, i.p.) 4 hr prior to exploration in the small Y-maze. αKL-F induced glutamate receptor signaling as the top enriched canonical pathway by ingenuity pathway analysis, which identifies biologic patterns of molecular changes in a large dataset (Figure S2).
Taken together, two experimental approaches, hypothesis-and non-hypothesis-driven, independently converged on evidence for glutamatergic signaling as an important downstream target pathway of αKL-F-mediated functions. αKL-F modestly changed GluN2B in proteomic mass spectrometry studies (Figure S2); it tended to increase many, but not all, GluN2B peptides following trypsinization (Figure S2). This suggests that, instead of increasing overall protein levels, αKL-F may alter GluN2B cleavage, degradation, PTM, or other baseline cellular processes. This possibility is supported by our findings that αKL-F significantly increased the LMW form of GluN2B, but not the HMW form, on western blotting (Figure 6) and requires further study.
Downstream main effect analysis of all proteomic changes predicted that αKL-F activates synaptic transmission (Figure S2). We directly probed and validated this functional prediction.
αKL-F Acutely Enhances NMDAR-Dependent Synaptic Plasticity in the Hippocampus
Activation of glutamate receptor signaling is critical for synaptic functions and plasticity. Long-term potentiation (LTP) is a form of synaptic plasticity dependent on NMDAR activation and is a key cellular substrate of learning and memory (Morris et al., 1986; Nabavi et al., 2014; Nakazawa et al., 2004). To assess whether αKL-F activates synaptic transmission and enhances synaptic plasticity, we next measured LTP in acute hippocampal slices at the CA1 Schaffer collateral pathway synapse, largely mediated by NDMARs (Kauer et al., 1988). During the same time frame that it enhanced cognition and caused GluN2B cleavage (4 hr after i.p. treatment), αKL-F enhanced LTP, as determined by field excitatory postsynaptic potential (fEPSP) recordings (Figures 7A and 7B). Of note, the extent of synaptic enhancement with acute, peripheral αKL-F treatment was similar to that observed with lifetime, transgenic, full-length klotho over-expression in the body and brain, although a different hippocampal region was examined in the current study (Dubal et al., 2014, 2015)
Figure 7. αKL-F Acutely Enhances NMDAR-Dependent Synaptic Plasticity in the Hippocampus, and Selective Blockade of the NMDAR Subunit GluN2B Abolishes αKL-F Effects on Cognition.

(A and B) fEPSP recordings from acute hippocampal slices of 3-month-old male mice (n = 7–14 slices/group, 3 mice/group) treated with Veh or αKL-F 4h prior to brain slicing.
(A) LTP induction and potentiation in the CA1 region was monitored for 30 min following theta burst stimulation of the Schaffer collateral pathway.
(B) Average fEPSP slope averaged over 30 min in hippocampal slices of mice treated with Veh or αKL-F. Two-way repeated measures ANOVA: αKL-F effect, p < 0.01.
(C and D) Mice (n = 15/group; male; age, 4 months) were treated i.p. with Veh or αKL-F (10 μg/kg) 4 hr prior to testing, followed by i.p. saline (−) or Ro 25 (+) (5 mg/kg) 10 min prior to testing in the small Y-maze.
(C) Percentages of alternations among arms during exploration of the small Y-maze over 4 min. αKL-F increased alternations, and Ro 25 preferentially decreased αKL-F-mediated effects. Two-way ANOVA: Ro 25 by αKL-F interaction, p < 0.05.
(D) Percentage of decrease in alternations following Ro 25 treatment in Veh-and αKL-F-treated mice. Ro 25 preferentially decreased the percentage of alternations in mice with αKL-F treatment.
*p < 0.05, **p < 0.01 versus Veh or as indicated by brackets by t test (B and D) or Bonferroni-Holm test (C). Data are mean ± SEM. See also Figure S3.
Selective Blockade of NMDAR GluN2B Subunits Abolishes αKL-F Effects on Cognition
The dynamics of NMDAR channels are governed in part by their subunit compositions (Wang et al., 2008; Yashiro and Philpot, 2008). GluN2B-containing NMDARs deactivate more slowly than those with other subunit types, transducing longer-lasting calcium-dependent signals (Wang et al., 2008). Because αKL-F acutely increased measures of GluN2B activation, including increasing its LMW form and enhancing synaptic plasticity, we tested whether blocking GluN2B-containing NMDARs modulates the effects of αKL-F on cognition. We specifically tested whether blockade of GluN2B in an acute, transient, and broad manner preferentially abrogates αKL-F-mediated cognitive enhancement without disrupting normal cognition.
To test this, we used a low dose of Ro 25-6981 (Ro 25), a highly specific GluN2B antagonist (Lu et al., 2017; Paoletti and Neyton, 2007) that minimally affects normal cognition (Figure S3), as described previously (Dubal et al., 2014; Mathur et al., 2009). Mice treated with a single injection of Veh or αKL-F (10 μg/kg, i.p.) were tested in the small Y-maze 4 hr later. Ten minutes prior to Y-maze testing, they received saline or low-dose Ro 25 (5 mg/kg, i.p.) (Dubal et al., 2014; Mathur et al., 2009). As expected, in the absence of Ro 25, αKL-treated mice showed better working memory than Veh-treated controls (Figure 7C), consistent with their enhanced cognition and enhanced LTP. Ro 25 effectively and preferentially blocked αKL-F-mediated cognitive enhancement (Figures 7C and 7D). At higher doses, Ro 25 also inhibited normal cognition (Figure S3). Thus, in parallel with the effects observed in transgenic klotho-overexpressing mice (Dubal et al., 2014), a low dose of a specific GluN2B antagonist effectively and preferentially blocked the effects of peripheral, acute treatment with αKL-F.
DISCUSSION
Our cognitive, behavioral, biochemical, protein-based, electrophysiological, and pharmacological studies in mice reveal a role for the peripheral delivery of a post-translationally modified mouse klotho fragment, αKL-F, in acute enhancement of synaptic and brain functions. αKL-F enhanced normal cognition in young mice and boosted cognition in aging mice. Furthermore, αKL-F countered cognitive and motor dysfunction in hSYN mice that model aspects of neurodegenerative disease. It did so without altering the levels of α-synuclein or related co-pathogenic proteins, indicating that αKL-F increases neural resilience.
Further investigation into its mechanisms revealed that peripheral αKL-F delivery increased glutamatergic signaling. It increased the LMW form of the NMDAR subunit GluN2B on western blotting, a putative marker of its activation in the hippocampus (Dong et al., 2004, 2006; Simpkins et al., 2003) within 4 hr of delivery and cognitive testing. Furthermore, peripheral αKL-F acutely increased synaptic function, as measured by enhanced LTP, a cellular substrate of learning and memory. Antagonism of NMDAR function through acute blockade of GluN2B preferentially abrogated αKL-F-mediated enhancement of cognition.
Taken together, our findings suggest that peripheral treatment with αKL-F or a similarly derived and/or modified klotho fragment could enhance brain function and resilience and could represent a new therapeutic strategy for dysfunction related to aging, neurodegenerative diseases like PD and AD, and other brain diseases.
Peripheral αKL-F Elevation Causes Central Cognitive Enhancement and Phenocopies Lifelong, Widespread, Genetic Klotho Overexpression
Surprisingly, a fragment of klotho, αKL-F, acutely enhanced cognition in young mice when delivered peripherally, thus phenocopying, in part, the functional effects we found with genetic, lifelong, and widespread overexpression of full-length, wild-type klotho (Dubal et al., 2014). This was unexpected because the fragment is impermeable to the BBB by prediction (Pardridge, 2005), direct radiography (Hu et al., 2016), and our immunoprecipitation studies. Because the αKL-F effects were acute, we conclude that neurodevelopmental effects of klotho, or sustained and high levels of wild-type klotho in the brain over a long period, are not required for neural enhancement. How peripheral αKL-F transmits a signal to the CNS is a fascinating question that remains to be answered. Possibilities include induction or modification of other factors that cross or interact with the BBB and direct or indirect alteration of central klotho levels, although we did not find evidence for the latter. Importantly, the ability of peripheral αKL-F to induce central effects is of therapeutic value because it bypasses the need to deliver protein directly into the CNS and, thus, evades a primary limitation of developing brain therapies. Our study does not rule out that klotho might also act with the CNS but, rather, establishes the relevance of its acute, peripheral elevation, particularly in a truncated form.
αKL-F, Lacking Several Structural Domains, Causes Better Cognitive Functions
The cognition-enhancing effects of αKL-F define an important segment of the klotho protein required for brain benefits. Because αKL-F lacks the transmembrane, intracellular, and N-terminal regions, our findings identify the cognition-enhancing effects within amino acids 35–982 comprising the KL1 and KL2 domains. How this post-translationally modified protein, which exists endogenously in a similar form (sometimes called soluble klotho), and its interactions with other proteins lead to signals than converge upon glutamatergically mediated cognitive enhancement remain to be determined.
Because wild-type, full-length klotho is a pleiotropic protein that engages multiple signaling peripheral pathways, including insulin (Kurosu et al., 2005), Wnt (Liu et al., 2007), and FGF (Urakawa et al., 2006), narrowing the fragment(s) necessary for cognitive enhancement may help to further target neural functions that may be independent, or not, of klotho’s putative actions on aging-related mechanisms.
The effects of peripheral αKL-F treatment on cognition largely phenocopied the effects of widespread, transgenic klotho over-expression; however, direct assessment of whether one is more robust is limited by the variability inherent to behavioral assays and the divergent nature of pharmacologic and genetic strategies. Acute pharmacologic elevation, potentially relevant for therapeutic development, may also be influenced by various factors affecting the potency and detection of neural enhancement, such as klotho aggregation and oligomerization (Imura et al., 2004) or precipitation; protein production, handling, or stability; differing influences of subtle stressors on the complex measurement of cognition (Sorge et al., 2014); or some combination of factors. In our studies, αKL-F effects were replicated in well over ten independent mouse cohorts of diverse characteristics (young, aging, and disease model mice) with dosing of a fresh preparation. How the fragment can be optimized by dose, composition, and biologic action may increase our understanding of how to enhance the brain-targeted effects of klotho.
αKL-F Improves Cognition in the Aging Brain
Peripheral delivery of αKL-F acutely enhanced cognition in aging mice. This is important because it demonstrates that αKL-F can increase spatial and working memory in the naturally aged brain and, therefore, can effectively target vulnerable substrates of cognition in aging. The long-lasting effect in aged mice induced better cognitive functions nearly 2 days following treatment, after predicted elimination of αKL-F (Hu et al., 2016). In further studies of young mice, a long-lasting αKL-F effect combined with cognitive training was observed over 2 weeks later. Together, our data suggest that αKL-F may harbor therapeutic benefits for the young and aging brain in a long-lasting and organizational manner. Because the synapse is a target of aging and enhanced by αKL-F, we speculate that the beneficial effects occur through synaptic remodeling.
αKL-F Causes Neural Resilience against Motor and Cognitive Dysfunction without Altering Pathogenic Protein Levels
Peripheral αKL-F acutely improved motor dysfunction in an α-synuclein model of neurodegenerative diseases, extending the beneficial effects of the klotho-derived fragment from cognition to motor functions. Because transgenic klotho elevation does not modify measures of muscle function (Phelps et al., 2013), our findings raise the possibility that αKL-F acutely counters motor problems by facilitating cognitive motor learning or boosting motor regions of the brain, probably through substrates of synaptic plasticity. Of note, we did not find effects of αKL-F on total movements in the open field or time spent in the open arms of an elevated plus maze (data not shown), suggesting that the protein improves motor function without causing hyperactivity or anxiety.
Peripheral αKL-F acutely counteracted cognitive deficits in a disease model with overexpression of the wild-type α-synuclein human protein, which plays a major role in α-synucleinopathies, including Parkinson disease, multiple system atrophy, and Lewy body dementia. Because dementias are multi-etiology and share common pathogenic proteins, including α-synuclein (Montine et al., 2014), the protein is also relevant to AD. Of note, the immediate beneficial cognitive effect of αKL-F described here is similar to effects of genetic, lifelong, and widespread overexpression of full-length wild-type klotho in the body and brain of hAPP mice, another disease model related to AD (Dubal et al., 2015).
αKL-F induced motor and cognitive benefit in hSYN mice without altering the levels of pathogenic or putative co-pathogenic proteins, α-synuclein, phospho-α-synuclein, tau, and phospho-tau. These data support growing lines of evidence that klotho induces neural resilience or the capacity to acutely counter or recover from cognitive and motor dysfunction. We speculate that klotho, and more specifically αKL-F, modulates common downstream targets, such as glutamatergic signaling at the synapse, in the pathogenesis of aging, AD, PD, and other diseases.
αKL-F Acutely Activates Glutamatergic Signaling to Enhance Cognition and Synaptic Plasticity
Hypothesis- and non-hypothesis-driven approaches independently converged on evidence for glutamate receptor signaling as an important downstream pathway of αKL-F-mediated functions. αKL-F acutely caused enriched levels of the NMDAR subunit GluN2B in the normal, young brain. However, instead of synaptic enrichment of HMW levels observed in transgenic mice chronically overexpressing klotho (Dubal et al., 2014), we found that acute αKL-F treatment increased LMW GluN2B in whole hippocampal homogenate within 4 hr. This probably represents calpain-mediated cleavage linked with GluN2B activation (Simpkins et al., 2003; Wu and Lynch, 2006) rather than increased transcription, translation, or trafficking of the subunit to the synapse. From a temporal perspective, it is possible that the immediate effects of klotho activate GluN2B-containing NMDARs by cleavage and that the lifelong, chronic overexpression effects of klotho cause compensatory GluN2B increases to ultimately offset klotho-mediated, ongoing cleavage and turnover.
NMDAR channel dynamics are governed by their subunit compositions (Yashiro and Philpot, 2008); GluN2B-containing NMDARs deactivate more slowly and transduce longer-lasting intracellular signals (Wang et al., 2008). Increased GluN2B-dependent NMDAR functions cause enhanced cognitive and synaptic functions (Cao et al., 2007; Tang et al., 1999; Wang et al., 2009). Thus, our findings support the hypothesis that αKL-F-mediated signals preferentially activate GluN2B-containing NMDARs. Consistent with this hypothesis, peripheral delivery of αKL-F enhanced cognition and synaptic plasticity, and selective blockade of GluN2B preferentially abrogated αKL-F-mediated effects. Of note, a study of direct application of a klotho fragment to hippocampal slices did not observe increased hippocampal LTP (Li et al., 2017), potentially reflecting hypoactive protein, induction protocol differences, and/or a requirement for peripheral rather than central klotho delivery to elicit synaptic effects.
The direct target of αKL-F, the relevant and causal peripheral mechanisms linked to that target, and the processes by which these core peripheral events reach the brain to affect the NMDAR and other synaptic functions remain to be determined. It is interesting to speculate that immune signals, other known klotho-related mechanisms, or some combination of pathways could converge onto glutamatergic signaling to induce better synaptic functions underlying cognitive and motor enhancement and neural resilience.
Our findings highlight a role for αKL-F in promoting optimal synaptic functions in the normal brain and to boost “synaptic health” (Morrison and Baxter, 2014) in aging and disease-states. Because synaptic health may confer resilience against the effects of aging and a myriad of aging-and non-aging related neurologic and psychiatric diseases, the potential to enhance it may be relevant to the human condition. Synaptic enhancement induced by αKL-F might be non-specific or, in contrast, could directly relate to abnormal mechanisms involved in aging and disease. Future studies will need to determine whether αKL-F specifically reduces detrimental synaptic mechanisms caused by aging, α-synuclein, and other pathogenic proteins.
αKL-F-mediated enhancement of synaptic functions and brain resilience with peripheral treatment in mice is potentially promising for paths toward human therapeutics in aging and disease.
EXPERIMENTAL PROCEDURES
Mice
All studies were conducted in a blinded manner in age-matched and sex-balanced, congenic C57BL/6 mice unless indicated otherwise. Specifically, mice were randomly allocated to each group, and the experimenter was blinded to their treatment. Aged mice were obtained from the National Institute on Aging (NIA) mouse colony. Transgenic hSYN mice (Rockenstein et al., 2002) overexpress human wild-type α-synuclein under the Thy-1 promoter. Because the α-synuclein transgene is inserted on the X chromosome in this line and randomly inactivates in females, we utilized only males from this line. Mice were kept on a 12-hr light/dark cycle with ad libitum access to food (Picolab Rodent Diet 20) and water. All studies were approved by the Institutional Animal Care and Use Committee of the University of California, San Francisco and conducted in compliance with NIH guidelines. See the Supplemental Experimental Procedures for further details.
Mass Spectrometry for PTMs of the Klotho Fragment
Mouse klotho fragment peptide (R&D Systems, 1819-KL) was trypsin-digested and subjected to liquid chromatography-tandem mass spectrometry using a NanoAcquity (Waters) ultra-performance liquid chromatography (UPLC) system interfaced to a linear trap quadrupole (LTQ) Orbitrap Velos (Thermo Scientific) mass spectrometer. Data were analyzed using Protein Prospector version 5.19 (Chalkley et al., 2008). See the Supplemental Experimental Procedures for further details.
Cognition and Behavior
Mice were tested as described previously in the Morris water maze (Dubal et al., 2014), small Y-maze (Dubal et al., 2014), two-trial Y-maze (Dellu et al., 1992), and rotarod (Morris et al., 2011). Drug treatments were administered as indicated. See the Supplemental Experimental Procedures for further details.
Synaptic Membrane Fractionation
Synaptic membrane fractions were separated for protein analysis as described (Dubal et al., 2014; Dubal et al., 2015).
Western Blot Analyses
Protein studies on the hippocampus and cortex were performed and quantified as described previously (Dubal et al., 2015). Additionally, α-synuclein (1:1,000, Abcam) and phosphorylated α-synuclein-Ser129 (1:1,000, Wako Pure Chemicals Industries) were measured. SDS (1%) was included in homogenates for optimal detection of the LMW GluN2B protein.
Immunoprecipitation
Immunoprecipitation (IP) of klotho was performed using modification of methods described previously (Bonifacino et al., 2001). In brief, hippocampal lysates were incubated with magnetic Dynabeads protein G (Thermo Fisher Scientific) conjugated with GluN2B antibody (2 μg, 06600, Millipore) For specificity of bands identified by IP, GluN2B antibody (Ab) was incubated with a blocking peptide (KFNGSSNGHVYEKLSSIESDV, 21 aa, generated by Watsonbio Sciences) harboring the GluN2B antibody epitope sequence prior to conjugation with Dynabeads. Beads were magnetically pulled, washed, and eluted. The precipitates were resolved on SDS-PAGE gel. See the Supplemental Experimental Procedures for further details.
Electrophysiology
Coronal brain slices of 300-μm thickness from 3-month-old mice were obtained as described previously (Dubal et al., 2014; Dubal et al., 2015) with modifications. Recordings were performed using the Med64-Quad II multi-electrode array (Alpha MED Scientific) in the CA1 region following stimulation of the Schaffer collateral path. See the Supplemental Experimental Procedures for further details.
Statistical Analyses
Experimenters were blinded to the treatment. Statistical analyses were performed as described previously (Dubal et al., 2014, 2015) using GraphPad Prism 7 for t tests. R Studio (v 2.0) was used for rank-sum tests as described previously (Possin et al., 2016) and post hoc tests. All t tests were two-tailed unless indicated otherwise. Post hoc tests were conducted with the Bonferroni-Holm correction (R) to control for a family-wise error rate at α = 0.05. Error bars represent SEM, and null hypotheses were rejected at or below a p value of 0.05.
See the Supplemental Experimental Procedures for further details regarding all experimental methods.
Supplementary Material
Highlights.
A klotho fragment (αKL-F) enhances cognition in young and aging mice
αKL-F counters deficits in α-synuclein mice without altering pathogenic protein levels
αKL-F induces GluN2B cleavage and increases NMDAR-dependent synaptic plasticity
Selective NMDAR blockade of GluN2B subunits abolishes acute αKL-F effects
Acknowledgments
We thank Stephen Hauser for discussions, Ruth Huttenhain for expertise with proteomic analyses, Frederic Hopf for input on electrophysiology, and Emily Davis and Elena Minones-Moyano for assistance with tissue collection and discussions. Behavioral data were obtained in part using the Gladstone Institutes Neurobehavioral Core. This study was funded by grants from NINDS (R01 NS092918 to D.B.D.), the National Center for Advancing Translational Sciences, National Institutes of Health, through UCSF-CTSI (UL1 TR001872 to D.B.D.), the American Federation for Aging Research (to D.B.D.), the Glenn Medical Foundation (to D.B.D.), and the Dr. Miriam and Sheldon G. Adelson Medical Research Foundation (to A.L.B.). The study was also funded by gifts from Unity Biotechnology (to D.B.D.), the Bakar Foundation (to D.B.D.), the Bradley Foundation (to D.B.D.), and the Coulter-Weeks Foundation (to D.B.D.). Portions of this work are the subject of a provisional patent application held by the Regents of the University of California.
Footnotes
Supplemental Information includes Supplemental Experimental Procedures and three figures and can be found with this article online at http://dx.doi.org/10.1016/j.celrep.2017.07.024.
AUTHOR CONTRIBUTIONS
J.L., D.W., B.I.G., R.J.C., A.L.B., A.J.M., and D.B.D. designed and conducted experiments and analyzed data. J.L. and D.B.D. wrote the manuscript. All authors edited and reviewed the manuscript.
References
- Almeida OP, Morar B, Hankey GJ, Yeap BB, Golledge J, Jablensky A, Flicker L. Longevity Klotho gene polymorphism and the risk of dementia in older men. Maturitas. 2017;101:1–5. doi: 10.1016/j.maturitas.2017.04.005. [DOI] [PubMed] [Google Scholar]
- Alvarez-Castelao B, Schuman EM. The Regulation of Synaptic Protein Turnover. J Biol Chem. 2015;290:28623–28630. doi: 10.1074/jbc.R115.657130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arking DE, Krebsova A, Macek M, Sr, Macek M, Jr, Arking A, Mian IS, Fried L, Hamosh A, Dey S, McIntosh I, Dietz HC. Association of human aging with a functional variant of klotho. Proc Natl Acad Sci USA. 2002;99:856–861. doi: 10.1073/pnas.022484299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett DA. Mixed pathologies and neural reserve: Implications of complexity for Alzheimer disease drug discovery. PLoS Med. 2017;14:e1002256. doi: 10.1371/journal.pmed.1002256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloch L, Sineshchekova O, Reichenbach D, Reiss K, Saftig P, Kuro-o M, Kaether C. Klotho is a substrate for alpha-, beta- and gamma-secretase. FEBS Lett. 2009;583:3221–3224. doi: 10.1016/j.febslet.2009.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonifacino JS, Dell’Angelica EC, Springer TA. Immunoprecipitation. Curr Protoc Mol Biol. 2001;Chapter 10(Unit 10):16. doi: 10.1002/0471142727.mb1016s48. [DOI] [PubMed] [Google Scholar]
- Cao X, Cui Z, Feng R, Tang YP, Qin Z, Mei B, Tsien JZ. Maintenance of superior learning and memory function in NR2B transgenic mice during ageing. Eur J Neurosci. 2007;25:1815–1822. doi: 10.1111/j.1460-9568.2007.05431.x. [DOI] [PubMed] [Google Scholar]
- Chalkley RJ, Baker PR, Medzihradszky KF, Lynn AJ, Burlingame AL. In-depth analysis of tandem mass spectrometry data from disparate instrument types. Mol Cell Proteomics. 2008;7:2386–2398. doi: 10.1074/mcp.M800021-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Château MT, Araiz C, Descamps S, Galas S. Klotho interferes with a novel FGF-signalling pathway and insulin/Igf-like signalling to improve longevity and stress resistance in Caenorhabditis elegans. Aging. 2010;2:567–581. doi: 10.18632/aging.100195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CD, Podvin S, Gillespie E, Leeman SE, Abraham CR. Insulin stimulates the cleavage and release of the extracellular domain of Klotho by ADAM10 and ADAM17. Proc Natl Acad Sci USA. 2007;104:19796–19801. doi: 10.1073/pnas.0709805104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CD, Li H, Liang J, Hixson K, Zeldich E, Abraham CR. The Anti-Aging and Tumor Suppressor Protein Klotho Enhances Differentiation of a Human Oligodendrocytic Hybrid Cell Line. J Mol Neurosci. 2015;55:76–90. doi: 10.1007/s12031-014-0336-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Vries CF, Staff RT, Harris SE, Chapko D, Williams DS, Reichert P, Ahearn T, McNeil CJ, Whalley LJ, Murray AD. Klotho, APOEε4, cognitive ability, brain size, atrophy, and survival: a study in the Aberdeen Birth Cohort of 1936. Neurobiol Aging. 2017;55:91–98. doi: 10.1016/j.neurobiolaging.2017.02.019. [DOI] [PubMed] [Google Scholar]
- Dellu F, Mayo W, Cherkaoui J, Le Moal M, Simon H. A two-trial memory task with automated recording: study in young and aged rats. Brain Res. 1992;588:132–139. doi: 10.1016/0006-8993(92)91352-f. [DOI] [PubMed] [Google Scholar]
- Dong YN, Waxman EA, Lynch DR. Interactions of postsynaptic density-95 and the NMDA receptor 2 subunit control calpain-mediated cleavage of the NMDA receptor. J Neurosci. 2004;24:11035–11045. doi: 10.1523/JNEUROSCI.3722-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong YN, Wu HY, Hsu FC, Coulter DA, Lynch DR. Developmental and cell-selective variations in N-methyl-D-aspartate receptor degradation by calpain. J Neurochem. 2006;99:206–217. doi: 10.1111/j.1471-4159.2006.04096.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubal DB, Yokoyama JS, Zhu L, Broestl L, Worden K, Wang D, Sturm VE, Kim D, Klein E, Yu GQ, et al. Life extension factor klotho enhances cognition. Cell Rep. 2014;7:1065–1076. doi: 10.1016/j.celrep.2014.03.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubal DB, Zhu L, Sanchez PE, Worden K, Broestl L, Johnson E, Ho K, Yu GQ, Kim D, Betourne A, et al. Life extension factor klotho prevents mortality and enhances cognition in hAPP transgenic mice. J Neurosci. 2015;35:2358–2371. doi: 10.1523/JNEUROSCI.5791-12.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duce JA, Podvin S, Hollander W, Kipling D, Rosene DL, Abraham CR. Gene profile analysis implicates Klotho as an important contributor to aging changes in brain white matter of the rhesus monkey. Glia. 2008;56:106–117. doi: 10.1002/glia.20593. [DOI] [PubMed] [Google Scholar]
- Fleming SM, Salcedo J, Fernagut PO, Rockenstein E, Masliah E, Levine MS, Chesselet MF. Early and progressive sensorimotor anomalies in mice overexpressing wild-type human α-synuclein. J Neurosci. 2004;24:9434–9440. doi: 10.1523/JNEUROSCI.3080-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleming SM, Salcedo J, Hutson CB, Rockenstein E, Masliah E, Levine MS, Chesselet MF. Behavioral effects of dopaminergic agonists in transgenic mice overexpressing human wildtype α-synuclein. Neuroscience. 2006;142:1245–1253. doi: 10.1016/j.neuroscience.2006.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haggerty T, Credle J, Rodriguez O, Wills J, Oaks AW, Masliah E, Sidhu A. Hyperphosphorylated Tau in an α-synuclein-overexpressing transgenic model of Parkinson’s disease. Eur J Neurosci. 2011;33:1598–1610. doi: 10.1111/j.1460-9568.2011.07660.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatami A, Chesselet MF. Transgenic rodent models to study alpha-synuclein pathogenesis, with a focus on cognitive deficits. Curr Top Behav Neurosci. 2015;22:303–330. doi: 10.1007/7854_2014_355. [DOI] [PubMed] [Google Scholar]
- Hu MC, Shi M, Zhang J, Addo T, Cho HJ, Barker SL, Ravikumar P, Gillings N, Bian A, Sidhu SS, et al. Renal Production, Uptake, and Handling of Circulating αKlotho. J Am Soc Nephrol. 2016;27:79–90. doi: 10.1681/ASN.2014101030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imura A, Iwano A, Tohyama O, Tsuji Y, Nozaki K, Hashimoto N, Fujimori T, Nabeshima Y. Secreted Klotho protein in sera and CSF: implication for post-translational cleavage in release of Klotho protein from cell membrane. FEBS Lett. 2004;565:143–147. doi: 10.1016/j.febslet.2004.03.090. [DOI] [PubMed] [Google Scholar]
- Invidia L, Salvioli S, Altilia S, Pierini M, Panourgia MP, Monti D, De Rango F, Passarino G, Franceschi C. The frequency of Klotho KL-VS polymorphism in a large Italian population, from young subjects to centenarians, suggests the presence of specific time windows for its effect. Biogerontology. 2010;11:67–73. doi: 10.1007/s10522-009-9229-z. [DOI] [PubMed] [Google Scholar]
- Ittner LM, Ke YD, Delerue F, Bi M, Gladbach A, van Eersel J, Wölfing H, Chieng BC, Christie MJ, Napier IA, et al. Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer’s disease mouse models. Cell. 2010;142:387–397. doi: 10.1016/j.cell.2010.06.036. [DOI] [PubMed] [Google Scholar]
- Kauer JA, Malenka RC, Nicoll RA. A persistent postsynaptic modification mediates long-term potentiation in the hippocampus. Neuron. 1988;1:911–917. doi: 10.1016/0896-6273(88)90148-1. [DOI] [PubMed] [Google Scholar]
- Kuro-o M, Matsumura Y, Aizawa H, Kawaguchi H, Suga T, Utsugi T, Ohyama Y, Kurabayashi M, Kaname T, Kume E, et al. Mutation of the mouse klotho gene leads to a syndrome resembling ageing. Nature. 1997;390:45–51. doi: 10.1038/36285. [DOI] [PubMed] [Google Scholar]
- Kurosu H, Yamamoto M, Clark JD, Pastor JV, Nandi A, Gurnani P, McGuinness OP, Chikuda H, Yamaguchi M, Kawaguchi H, et al. Suppression of aging in mice by the hormone Klotho. Science. 2005;309:1829–1833. doi: 10.1126/science.1112766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Vo HT, Wang J, Fox-Quick S, Dobrunz LE, King GD. Klotho regulates CA1 hippocampal synaptic plasticity. Neuroscience. 2017;347:123–133. doi: 10.1016/j.neuroscience.2017.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Fergusson MM, Castilho RM, Liu J, Cao L, Chen J, Malide D, Rovira II, Schimel D, Kuo CJ, et al. Augmented Wnt signaling in a mammalian model of accelerated aging. Science. 2007;317:803–806. doi: 10.1126/science.1143578. [DOI] [PubMed] [Google Scholar]
- Logan T, Bendor J, Toupin C, Thorn K, Edwards RH. α-Synuclein promotes dilation of the exocytotic fusion pore. Nat Neurosci. 2017;20:681–689. doi: 10.1038/nn.4529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu W, Du J, Goehring A, Gouaux E. Cryo-EM structures of the triheteromeric NMDA receptor and its allosteric modulation. Science. 2017;355 doi: 10.1126/science.aal3729. Published online February 23, 2017. http://dx.doi.org/10.1126/science.aal3729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masliah E, Rockenstein E, Veinbergs I, Mallory M, Hashimoto M, Takeda A, Sagara Y, Sisk A, Mucke L. Dopaminergic loss and inclusion body formation in α-synuclein mice: implications for neurodegenerative disorders. Science. 2000;287:1265–1269. doi: 10.1126/science.287.5456.1265. [DOI] [PubMed] [Google Scholar]
- Massó A, Sánchez A, Gimenez-Llort L, Lizcano JM, Cañete M, García B, Torres-Lista V, Puig M, Bosch A, Chillon M. Secreted and Transmembrane αKlotho Isoforms Have Different Spatio-Temporal Profiles in the Brain during Aging and Alzheimer’s Disease Progression. PLoS ONE. 2015;10:e0143623. doi: 10.1371/journal.pone.0143623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathur P, Graybeal C, Feyder M, Davis MI, Holmes A. Fear memory impairing effects of systemic treatment with the NMDA NR2B subunit antagonist, Ro 25-6981, in mice: attenuation with ageing. Pharmacol Biochem Behav. 2009;91:453–460. doi: 10.1016/j.pbb.2008.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McEwen BS, Morrison JH. The brain on stress: vulnerability and plasticity of the prefrontal cortex over the life course. Neuron. 2013;79:16–29. doi: 10.1016/j.neuron.2013.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mengel-From J, Soerensen M, Nygaard M, McGue M, Christensen K, Christiansen L. Genetic Variants in KLOTHO Associate With Cognitive Function in the Oldest Old Group. J Gerontol A Biol Sci Med Sci. 2016;71:1151–1159. doi: 10.1093/gerona/glv163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montine TJ, Koroshetz WJ, Babcock D, Dickson DW, Galpern WR, Glymour MM, Greenberg SM, Hutton ML, Knopman DS, Kuzmichev AN, et al. ADRD. Conference Organizing Committee (2014). Recommendations of the Alzheimer’s disease-related dementias conference. Neurology. 2013;83:851–860. doi: 10.1212/WNL.0000000000000733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris RG, Anderson E, Lynch GS, Baudry M. Selective impairment of learning and blockade of long-term potentiation by an N-methyl-D-aspartate receptor antagonist, AP5. Nature. 1986;319:774–776. doi: 10.1038/319774a0. [DOI] [PubMed] [Google Scholar]
- Morris M, Koyama A, Masliah E, Mucke L. Tau reduction does not prevent motor deficits in two mouse models of Parkinson’s disease. PLoS ONE. 2011;6:e29257. doi: 10.1371/journal.pone.0029257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison JH, Baxter MG. Synaptic health. JAMA Psychiatry. 2014;71:835–837. doi: 10.1001/jamapsychiatry.2014.380. [DOI] [PubMed] [Google Scholar]
- Nabavi S, Fox R, Proulx CD, Lin JY, Tsien RY, Malinow R. Engineering a memory with LTD and LTP. Nature. 2014;511:348–352. doi: 10.1038/nature13294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakazawa K, McHugh TJ, Wilson MA, Tonegawa S. NMDA receptors, place cells and hippocampal spatial memory. Nat Rev Neurosci. 2004;5:361–372. doi: 10.1038/nrn1385. [DOI] [PubMed] [Google Scholar]
- Paoletti P, Neyton J. NMDA receptor subunits: function and pharmacology. Curr Opin Pharmacol. 2007;7:39–47. doi: 10.1016/j.coph.2006.08.011. [DOI] [PubMed] [Google Scholar]
- Pardridge WM. The blood-brain barrier: bottleneck in brain drug development. NeuroRx. 2005;2:3–14. doi: 10.1602/neurorx.2.1.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phelps M, Pettan-Brewer C, Ladiges W, Yablonka-Reuveni Z. Decline in muscle strength and running endurance in klotho deficient C57BL/6 mice. Biogerontology. 2013;14:729–739. doi: 10.1007/s10522-013-9447-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piggott MA, Perry EK, Perry RH, Court JA. [3H]MK-801 binding to the NMDA receptor complex, and its modulation in human frontal cortex during development and aging. Brain Res. 1992;588:277–286. doi: 10.1016/0006-8993(92)91586-4. [DOI] [PubMed] [Google Scholar]
- Possin KL, Sanchez PE, Anderson-Bergman C, Fernandez R, Kerchner GA, Johnson ET, Davis A, Lo I, Bott NT, Kiely T, et al. Cross-species translation of the Morris maze for Alzheimer’s disease. J Clin Invest. 2016;126:779–783. doi: 10.1172/JCI78464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prather AA, Epel ES, Arenander J, Broestl L, Garay BI, Wang D, Dubal DB. Longevity factor klotho and chronic psychological stress. Transl Psychiatry. 2015;5:e585. doi: 10.1038/tp.2015.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rockenstein E, Mallory M, Hashimoto M, Song D, Shults CW, Lang I, Masliah E. Differential neuropathological alterations in transgenic mice expressing alpha-synuclein from the platelet-derived growth factor and Thy-1 promoters. J Neurosci Res. 2002;68:568–578. doi: 10.1002/jnr.10231. [DOI] [PubMed] [Google Scholar]
- Semba RD, Cappola AR, Sun K, Bandinelli S, Dalal M, Crasto C, Guralnik JM, Ferrucci L. Plasma klotho and mortality risk in older community-dwelling adults. J Gerontol A Biol Sci Med Sci. 2011;66:794–800. doi: 10.1093/gerona/glr058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semba RD, Moghekar AR, Hu J, Sun K, Turner R, Ferrucci L, O’Brien R. Klotho in the cerebrospinal fluid of adults with and without Alzheimer’s disease. Neurosci Lett. 2014;558:37–40. doi: 10.1016/j.neulet.2013.10.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shardell M, Semba RD, Kalyani RR, Hicks GE, Bandinelli S, Ferrucci L. Serum 25-Hydroxyvitamin D, Plasma Klotho, and Lower-Extremity Physical Performance Among Older Adults: Findings From the InCHIANTI Study. J Gerontol A Biol Sci Med Sci. 2015;70:1156–1162. doi: 10.1093/gerona/glv017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shardell M, Semba RD, Rosano C, Kalyani RR, Bandinelli S, Chia CW, Ferrucci L. Plasma Klotho and Cognitive Decline in Older Adults: Findings From the InCHIANTI Study. J Gerontol A Biol Sci Med Sci. 2016;71:677–682. doi: 10.1093/gerona/glv140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpkins KL, Guttmann RP, Dong Y, Chen Z, Sokol S, Neumar RW, Lynch DR. Selective activation induced cleavage of the NR2B subunit by calpain. J Neurosci. 2003;23:11322–11331. doi: 10.1523/JNEUROSCI.23-36-11322.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorge RE, Martin LJ, Isbester KA, Sotocinal SG, Rosen S, Tuttle AH, Wieskopf JS, Acland EL, Dokova A, Kadoura B, et al. Olfactory exposure to males, including men, causes stress and related analgesia in rodents. Nat Methods. 2014;11:629–632. doi: 10.1038/nmeth.2935. [DOI] [PubMed] [Google Scholar]
- Sze C, Bi H, Kleinschmidt-DeMasters BK, Filley CM, Martin LJ. N-Methyl-D-aspartate receptor subunit proteins and their phosphorylation status are altered selectively in Alzheimer’s disease. J Neurol Sci. 2001;182:151–159. doi: 10.1016/s0022-510x(00)00467-6. [DOI] [PubMed] [Google Scholar]
- Tang YP, Shimizu E, Dube GR, Rampon C, Kerchner GA, Zhuo M, Liu G, Tsien JZ. Genetic enhancement of learning and memory in mice. Nature. 1999;401:63–69. doi: 10.1038/43432. [DOI] [PubMed] [Google Scholar]
- Urakawa I, Yamazaki Y, Shimada T, Iijima K, Hasegawa H, Okawa K, Fujita T, Fukumoto S, Yamashita T. Klotho converts canonical FGF receptor into a specific receptor for FGF23. Nature. 2006;444:770–774. [Google Scholar]
- Wang H, Stradtman GG, 3rd, Wang XJ, Gao WJ. A specialized NMDA receptor function in layer 5 recurrent microcircuitry of the adult rat prefrontal cortex. Proc Natl Acad Sci USA. 2008;105:16791–16796. doi: 10.1073/pnas.0804318105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D, Cui Z, Zeng Q, Kuang H, Wang LP, Tsien JZ, Cao X. Genetic enhancement of memory and long-term potentiation but not CA1 long-term depression in NR2B transgenic rats. PLoS ONE. 2009;4:e7486. doi: 10.1371/journal.pone.0007486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wills J, Jones J, Haggerty T, Duka V, Joyce JN, Sidhu A. Elevated tauopathy and alpha-synuclein pathology in postmortem Parkinson’s disease brains with and without dementia. Exp Neurol. 2010;225:210–218. doi: 10.1016/j.expneurol.2010.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu HY, Lynch DR. Calpain and synaptic function. Mol Neurobiol. 2006;33:215–236. doi: 10.1385/MN:33:3:215. [DOI] [PubMed] [Google Scholar]
- Yashiro K, Philpot BD. Regulation of NMDA receptor subunit expression and its implications for LTD, LTP, and metaplasticity. Neuropharmacology. 2008;55:1081–1094. doi: 10.1016/j.neuropharm.2008.07.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoyama JS, Sturm VE, Bonham LW, Klein E, Arfanakis K, Yu L, Coppola G, Kramer JH, Bennett DA, Miller BL, Dubal DB. Variation in longevity gene KLOTHO is associated with greater cortical volumes. Ann Clin Transl Neurol. 2015;2:215–230. doi: 10.1002/acn3.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoyama JS, Marx G, Brown JA, Bonham LW, Wang D, Coppola G, Seeley WW, Rosen HJ, Miller BL, Kramer JH, Dubal DB. Systemic klotho is associated with KLOTHO variation and predicts intrinsic cortical connectivity in healthy human aging. Brain Imaging Behav. 2017;11:391–400. doi: 10.1007/s11682-016-9598-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X, Rosenke R, Kronemann D, Brim B, Das SR, Dunah AW, Magnusson KR. The effects of aging on N-methyl-D-aspartate receptor subunits in the synaptic membrane and relationships to long-term spatial memory. Neuroscience. 2009;162:933–945. doi: 10.1016/j.neuroscience.2009.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.