

Rationale:
The balance between vascular prostacyclin, which is antithrombotic, and platelet thromboxane A2, which is prothrombotic, is fundamental to cardiovascular health. Prostacyclin and thromboxane A2 are formed after the concerted actions of cPLA2α (cytosolic phospholipase A2) and COX (cyclooxygenase). Urinary 2,3-dinor-6-keto-PGF1α (PGI-M) and 11-dehydro-TXB2 (TX-M) have been taken as biomarkers of prostacyclin and thromboxane A2 formation within the circulation and used to explain COX biology and patient phenotypes, despite concerns that urinary PGI-M and TX-M originate in the kidney.
Objective:
We report data from a remarkable patient carrying an extremely rare genetic mutation in cPLA2α, causing almost complete loss of prostacyclin and thromboxane A2, who was transplanted with a normal kidney resulting in an experimental scenario of whole-body cPLA2α knockout, kidney-specific knockin. By studying this patient, we can determine definitively the contribution of the kidney to the productions of PGI-M and TX-M and test their validity as markers of prostacyclin and thromboxane A2 in the circulation.
Methods and Results:
Metabolites were measured using liquid chromatography-tandem mass spectrometry. Endothelial cells were grown from blood progenitors. Before kidney transplantation, the patient’s endothelial cells and platelets released negligible levels of prostacyclin (measured as 6-keto-prostaglandin F1α) and thromboxane A2 (measured as TXB2), respectively. Likewise, the urinary levels of PGI-M and TX-M were very low. After transplantation and the establishment of normal renal function, the levels of PGI-M and TX-M in the patient’s urine rose to within normal ranges, whereas endothelial production of prostacyclin and platelet production of thromboxane A2 remained negligible.
Conclusions:
These data show that PGI-M and TX-M can be derived exclusively from the kidney without contribution from prostacyclin made by endothelial cells or thromboxane A2 by platelets in the general circulation. Previous work relying on urinary metabolites of prostacyclin and thromboxane A2 as markers of whole-body endothelial and platelet function now requires reevaluation.
Keywords: biomarkers, endothelial cells, kidney transplantation, phenotype, thromboxane A2
For over 40 years, the importance of balance in the production of prostanoids has been a central theme in the understanding of cardiovascular health. Attention has focused on prostacyclin derived from the vasculature, which is antithrombotic and a vasorelaxant, and thromboxane A2 derived from platelets, which is prothrombotic and a vasoconstrictor. Both prostacyclin and thromboxane A2 are formed after the concerted actions of cPLA2α (cytosolic phospholipase A2) and COX (cyclooxygenase). COX is present in 2 isoforms; COX-1 is constitutively expressed throughout the body,1–4 whereas COX-2 is present constitutively only in discreet regions of the body, which include the kidney.5–8 COX-2 is also expressed at the site of inflammation and in cancer and as such is the therapeutic target for the nonsteroidal anti-inflammatory group of drugs, which include aspirin, ibuprofen, and celecoxib.
Editorial, see p 537
Meet the First Author, see p 534
It was found early on in prostanoid research that both prostacyclin and thromboxane A2 are very short lived within the circulation and that measurements of either of them or their immediate metabolites were relatively uninformative. The establishment of analytic techniques to measure 2,3-dinor-6-keto-PGF1α (PGI-M), a stable metabolite of prostacyclin, and 11-dehydro-TXB2 (TX-M), a stable metabolite of thromboxane A2 in urine, therefore, seemed to provide the possibility of useful biomarkers of cardiovascular health and of drug action. However, for PGI-M and TX-M to work as biomarkers, their levels in urine should reflect levels in the circulation, and although this idea has been suggested based on selective inhibition of urinary TX-M with aspirin,9 it has not been experimentally proven. Since this early work it has been generally assumed that PGI-M and TX-M measured in urine reflected levels in the cardiovascular system as a whole, dependent on prostacyclin production by endothelial cells and thromboxane A2 production by platelets.10–15 This assumption has now become dogma, and stable urinary metabolites of prostacyclin and thromboxane A2 have been used in many studies, for example, as of September 2017 an online search on PubMed with the terms urinary prostacyclin metabolite or urinary thromboxane metabolite returns over 300 and 400 papers, respectively, whereas search of clinicaltrials.gov with the term urinary prostanoid produces 48 entries. Results from these studies have apparently informed (1) drug action in clinical studies,11 (2) personal risk of cardiovascular disease in patient groups,14 and (3) a plethora of basic science relating to eicosanoids. A widely held concept derived from such studies is that prostacyclin released by endothelial cells is formed through the actions of COX-2, following from the observation that COX-2–selective drugs reduce PGI-M and relying on the assumption that PGI-M reflects the production of prostacyclin by endothelial cells.12,15,16 However, this idea is not universally accepted1,2,5,6,8,16 because conflicting observations indicate that COX-11–4,8,16 is the dominant isoform within the vasculature including endothelial cells leading some of us to suggest that urinary markers of prostacyclin can be derived from the kidney1 where COX-2 is highly expressed.5–7
To date there have been no definitive models in which the renal origin of PGI-M and TX-M can be tested. However, here, we present a report of a patient with inherited human group IV A cPLA2α deficiency,17 previously found by our group to almost completely lack the vital capacity to form several eicosanoids including endothelial prostacyclin and platelet thromboxane A2.17,18 In 2015, the patient underwent a kidney transplant receiving a normal cPLA2 sufficient organ. The transplant has resulted in the serendipitous generation of a remarkable experimental model akin to a human whole-body cPLA2α knockout, kidney-specific knockin. Now, for this patient, we can determine definitively the contribution of the kidney to the production of PGI-M and TX-M and so test the relevance of these measurements as markers of prostacyclin and thromboxane A2 in the circulation.
Methods
The authors declare that all supporting data are available within the article.
Patient Details
The patient (female, of Serbian heritage, born 1966) presented at the age of 2 years with peptic ulceration, bleeding, and pyloric stenosis, which required pyloroplasty and selective vagotomy. The patient went on to have a lifelong history of gastrointestinal disease17 cumulating in the diagnosis of cryptogenic multifocal ulcerous stenosing enteritis.17 In 2014, we reported that the patient carries a homozygous 4 bp deletion (g.155574_77delGTAA) in the PLA2G4A gene resulting in a frameshift of 10 amino acids before a premature stop codon (p.V707fsX10) and the loss of 43 amino acids (residues 707–749) at the C terminus of group IV A cPLA2α. This mutation results in a complete loss of cPLA2α protein expression. In line with loss of cPLA2α, generation of eicosanoids by whole blood,17 isolated platelets, peripheral blood monocytes, or blood outgrowth endothelial cells obtained from the patient18 was dramatically reduced. Plasma and urinary levels of most eicosanoids were also accordingly much lower than the normal range in samples from the patient.17,18 In 2014, renal function of the patient declined because of tubulointerstitial nephritis leading to end-stage renal failure requiring dialysis during which time the patient was producing ≈1 L/d of urine. In 2015, the patient underwent a renal transplant receiving a live unrelated spousal donor kidney. After the kidney transplant had stabilized, blood and urine samples were collected for analysis using liquid chromatography-tandem mass spectrometry at 1 to 3 months post-transplant. Blood outgrowth endothelial cells were also isolated after transplant and samples collected for eicosanoid measurements after stimulation in culture. The patient received tacrolimus as antirejection therapy.
Blood Collection and Ethics
Blood was collected by venepuncture, and urine by samples from midstream flow from healthy volunteers and the patient.
Whole Blood Stimulation
Heparin anticoagulated whole blood was incubated with vehicle (PBS) or Horm collagen (Nycomed, St Peter, Austria). Thromboxane B2 levels were measured by liquid chromatography-tandem mass spectrometry in the conditioned plasma.
Endothelial Cells
Blood outgrowth endothelial cells were grown out from progenitors in human blood as previously described.19–22 Once colonies emerged (between days 4 and 20), cells were expanded and maintained in Lonza EGM-2 media (Lonza, Slough, United Kingdom) +10% fetal bovine serum and experiments performed between passages 2 and 8.
Cells were plated on 48- or 96-well plates. For eicosanoid measurements, endothelial cells were primed with interleukin-1β (IL-1β; 1 ng/mL; Invitrogen, Life Technologies, Paisley, United Kingdom) to upregulate COX pathways as described previously23 before being treated for 30 minutes with the calcium ionophore A23187 to activate PLA2.
Eicosanoid Analysis
Levels of prostanoids in urine, whole blood, and endothelial cell samples were determined by liquid chromatography-tandem mass spectrometry as previously described.1,18,24,25
Statistics and Data Analysis
Data are shown as individual data points.
Study Approval
All experiments were subject to written informed consent, local ethical approval (healthy volunteer samples for platelet/leukocyte studies; St Thomas’s Hospital Research Ethics Committee, reference 07/Q0702/24: endothelial cell studies; Royal Brompton and Harefield Hospital Research Ethics Committee, reference 08/H0708/69: patient samples; South East NHS Research Ethics Committee) and in accordance with Declaration of Helsinki principles.
Results
Before the kidney transplant, the patient had developed end-stage kidney failure with urine production of ≈1 L/d requiring hemodialysis 3× a week. Postoperative recovery after transplant was uneventful. Her renal function normalized with blood urea nitrogen of 6.6 and creatinine of 88 μmol/L by 4 weeks post-transplant.
In healthy volunteers, PGI-M and TX-M tend to be higher in females than males.18 However, due to her condition, before the transplant levels of PGI-M and TX-M in the patient’s urine were low and well below levels in control donors and the published26 normal ranges (Figure).17,18 Remarkably, the new kidney restored levels of urinary PGI-M and TX-M to the normal range. This phenomenon was found to be selective to the kidney because the ability of endothelial cells from the patient to produce prostacyclin and of platelets from the patient to produce thromboxane A2 remained low and unchanged by the kidney transplant (Figure). Similarly, there was no increase in the levels of PGI-M within the circulation, but rather a small reduction (25±3%), when plasma samples from after transplantation (n=5) were compared with those from before transplantation (n=8).
Figure.
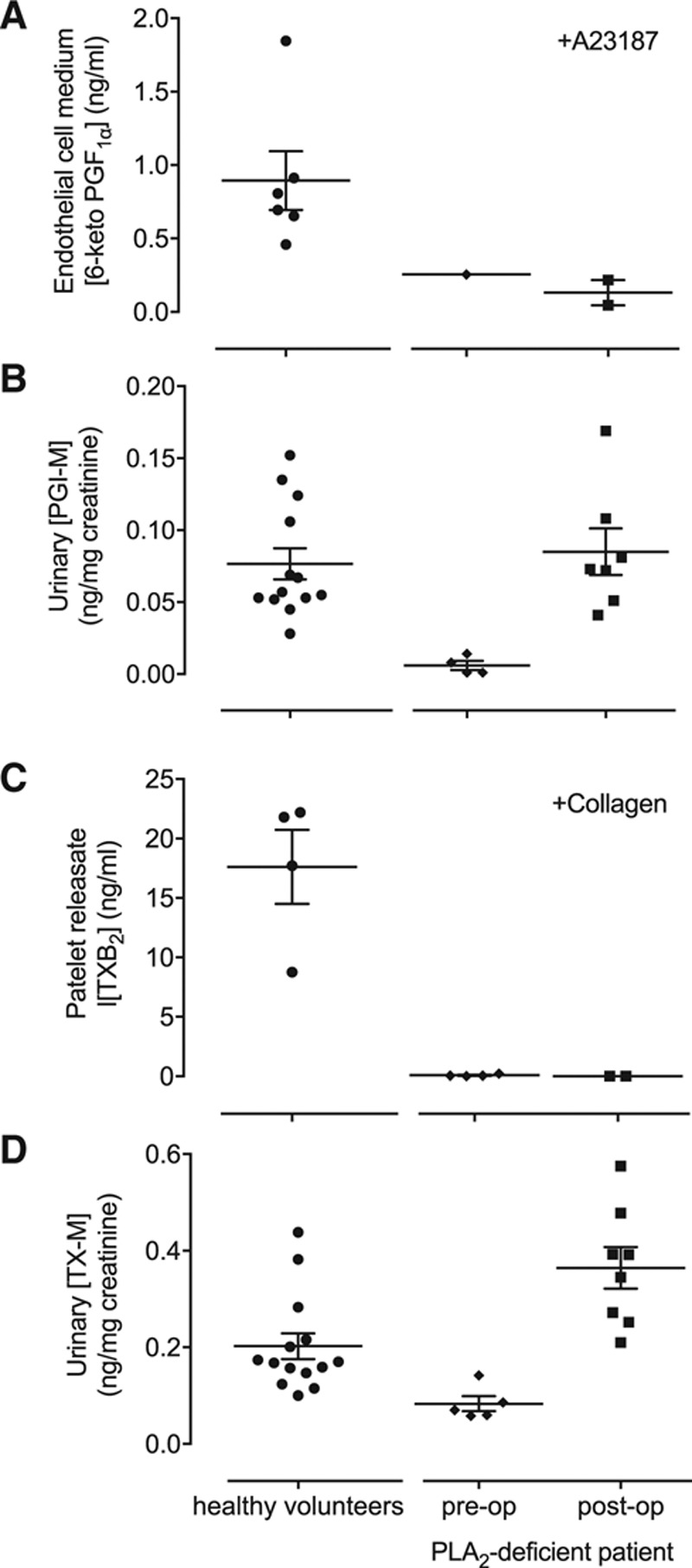
Comparison of cellular production of eicosanoids and urinary levels of metabolites. A, Production of prostacyclin measured as 6-keto-prostaglandin F1α (6-keto-PGF1α) from endothelial cells stimulated with calcium ionophore (A23187); (B) urinary levels of the prostacyclin metabolite PGI-M (2,3-dinor-6-keto-PGF1α); (C) production of thromboxane A2 measured as thromboxane A2 (TXB2) from whole blood stimulated with collagen; and (D) urinary levels of the thromboxane metabolite TX-M (11-dehydro-TXB2). Measurements made in samples from healthy volunteers and from the patient before and after kidney transplantation. Results from healthy volunteers and cPLA2α (cytosolic phospholipase A2)-deficient patient pre-op includes data previously published.18
Discussion
Here, we describe a remarkable clinical and experimental situation, the serendipitous generation of a unique human model in which to explore the origins of the urinary eicosanoid metabolites of prostacyclin (PGI-M) and thromboxane (TX-M).
Because eicosanoids, including prostacyclin, protect the gastrointestinal tract and the kidney, the long-term clinical symptoms of the patient can be entirely explained by the genetic deficiency and the associated lack of cPLA2α activity, illustrating the powerful protective role that eicosanoids play in homeostasis. After receiving a genetically normal kidney, the patient continued to be almost entirely unable to produce prostacyclin and thromboxane A2 from her endothelial cells and platelets. However, despite the continuing absence of endothelial prostacyclin production and platelet thromboxane A2 production after transplant, the patient’s urine contains apparently normal26 levels of PGI-M and TX-M. Importantly, it has already been demonstrated that the use of tacrolimus to reduce organ rejection in renal transplant patients is not associated with changes in either PGI-M or TX-M.27 It is therefore impossible in this patient that PGI-M and TX-M were derived from, or reflective of, endothelial and platelet eicosanoid productions.
These results not only describe a unique clinical case of organ transplantation in a patient with an incredibly rare gene deletion but also show unequivocally that the kidney alone can support the production of normal levels of PGI-M and TX-M and that these cannot be assumed as markers for prostacyclin and thromboxane A2 production within the cardiovascular system as a whole. Although there may be concerns that the patient presented here has very particular pathologies, which may not speak for normal physiological function, the same can be said for any of the many patients across a wide range of diseases in which measurement of PGI-M and TX-M has been used to describe clinical conditions.
Importantly, as mentioned above, PGI-M has been used to define the idea that endothelial cells produce prostacyclin through the action of COX-215 because selective inhibitor drugs, such as celecoxib, reduce urinary PGI-M.11 However, this idea is not universally accepted1,2,8,28 and has not been supported by direct evidence, which instead identifies the ubiquitously expressed constitutive form, COX-1 as the principle driver of prostacyclin in the circulation.1,2,28 Similarly, there have been anomalies in the rationale that TX-M accurately reflects thromboxane A2 in the circulation. For example, early studies demonstrated that platelet thromboxane A2 production could be strongly inhibited without a concomitant reduction in urinary TX-M.29 At the time, this was taken as indicating the need for substantial platelet COX inhibition to reduce in vivo platelet activation. Our data now provide definitive proof for the alternative, and simpler, conclusion that both urinary PGI-M and TX-M originate from the kidney and are not necessary reflective of prostacyclin and thromboxane A2 in the circulation.
In conclusion, we now need to reconsider the many studies and clinical trials that have used measures of PGI-M and TX-M to construct some of the fundamental concepts of eicosanoid biology and to characterize various patient groups. This is particularly important in the areas of aspirin therapy and COX-2 biology where urinary markers have been used to inform discussions on the mechanisms associated with nonsteroidal anti-inflammatory group of drugs and cardiovascular risk.12,15,16 In the light of our findings, which prove that urinary PGI-M can originate from the kidney, we may conclude that earlier studies showing COX-2 inhibitor drugs to reduce PGI-M simply confirm the kidney as a prime site for constitutive COX-2 expression and add to the idea that blockade of the production of protective COX-2–derived prostanoids in the kidney contributes to nonsteroidal anti-inflammatory group of drug–induced cardiovascular side effects.
Sources of Funding
This work was supported by the British Heart Foundation (FS/12/53/29643, FS/16/1/31699 and PG/15/47/31591), the Wellcome Trust (0852551Z108/Z), and the Intramural Research Program of the US National Institutes of Health (NIH) National Institute of Environmental Health Sciences (grant no. Z01-025034). R.B. Knowles, N.S. Kirkby, D.M. Reed, M.L. Edin, W.E. White, M.V. Chan, and G.L. Milne performed the research; J.A. Mitchell, R.B. Knowles, N.S. Kirkby, D.M. Reed, M.L. Edin, G.L. Milne, D.C. Zeldin, and T.D. Warner analyzed the data; R.B. Knowles, N.S. Kirkby, D.M. Reed, M.L. Edin, W.E. White, M.V. Chan, H. Longhurst, M.M. Yaqoob, G.L. Milne, and D.C. Zeldin edited the article; J.A. Mitchell, M.M. Yaqoob, and T.D. Warner designed the research; and J.A. Mitchell and T.D. Warner wrote the article.
Disclosures
None.
Nonstandard Abbreviations and Acronyms
- COX
- cyclooxygenase
- cPLA2α
- cytosolic phospholipase A2
- IL-1β
- interleukin-1β
- PGI-M
- 2,3-dinor-6-keto-PGF1α
- TXB2
- thromboxane A2
- TX-M
- 11-dehydro-TXB2
In November 2017, the average time from submission to first decision for all original research papers submitted to Circulation Research was 11.99 days.
Brief UltraRapid Communications are designed to be a format for manuscripts that are of outstanding interest to the readership, report definitive observations, but have a relatively narrow scope. Less comprehensive than Regular Articles but still scientifically rigorous, BURCs present seminal findings that have the potential to open up new avenues of research. A decision on BURCs is rendered within 7 days of submission.
Novelty and Significance
What Is Known?
The balance of formation of the COX (cyclooxygenase)-derived eicosanoids, vascular prostacyclin, which is antithrombotic, and platelet thromboxane, A2, which is prothrombotic, lies at the center of cardiovascular health.
It has been widely assumed that the production of these 2 short-lived mediators can be followed by the measurement of stable urinary metabolites, 2,3-dinor-6-keto-PGF1α (PGI-M) for prostacyclin and 11-dehydro-TXB2 (TX-M) for thromboxane A2, and these measurements have been used to explain COX biology and patient phenotypes.
What New Information Does This Article Contribute?
Study of the urinary metabolites of a unique patient lacking the ability to form vascular prostacyclin and platelet thromboxane A2 who received a kidney transplant demonstrated unequivocally that PGI-M and TX-M can be derived exclusively from the kidney.
These findings suggest reevaluation of the urinary metabolites of prostacyclin and thromboxane A2 as markers of whole-body endothelial and platelet function.
The balance between vascular prostacyclin, which is antithrombotic, and platelet thromboxane A2, which is prothrombotic, is fundamental to cardiovascular health. However, both these mediators are very short lived and cannot be directly measured in circulation. Hence, researchers have relied on PGI-M and TX-M as urinary biomarkers. These measures have been used to explain COX biology despite the lack of definitive proof of their sources. We studied a unique patient carrying an extremely rare genetic mutation in group IV A cPLA2α (cytosolic phospholipase A2), causing almost complete loss of prostacyclin and thromboxane A2, who was transplanted with a normal kidney. Before kidney transplantation, the patient’s endothelial cells and platelets released negligible levels of prostacyclin and thromboxane A2, respectively, accompanied with very low levels of PGI-M and TX-M. After transplantation, the levels of PGI-M and TX-M in the patient’s urine rose to within normal ranges without any increases in the production of prostacyclin and thromboxane A2. These findings demonstrate that PGI-M and TX-M can be derived exclusively from the kidney and suggest that literature relying on the measurement of these metabolites needs to be reexamined.
References
- 1.Kirkby NS, Lundberg MH, Harrington LS, Leadbeater PD, Milne GL, Potter CM, Al-Yamani M, Adeyemi O, Warner TD, Mitchell JA. Cyclooxygenase-1, not cyclooxygenase-2, is responsible for physiological production of prostacyclin in the cardiovascular system. Proc Natl Acad Sci USA. 2012;109:17597–17602. doi: 10.1073/pnas.1209192109. doi: 10.1073/pnas.1209192109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Flavahan NA. Balancing prostanoid activity in the human vascular system. Trends Pharmacol Sci. 2007;28:106–110. doi: 10.1016/j.tips.2007.01.003. doi: 10.1016/j.tips.2007.01.003. [DOI] [PubMed] [Google Scholar]
- 3.Liu B, Luo W, Zhang Y, Li H, Zhu N, Huang D, Zhou Y. Involvement of cyclo-oxygenase-1-mediated prostacyclin synthesis in the vasoconstrictor activity evoked by ACh in mouse arteries. Exp Physiol. 2012;97:277–289. doi: 10.1113/expphysiol.2011.062034. doi: 10.1113/expphysiol.2011.062034. [DOI] [PubMed] [Google Scholar]
- 4.Luo W, Liu B, Zhou Y. The endothelial cyclooxygenase pathway: insights from mouse arteries. Eur J Pharmacol. 2016;780:148–158. doi: 10.1016/j.ejphar.2016.03.043. doi: 10.1016/j.ejphar.2016.03.043. [DOI] [PubMed] [Google Scholar]
- 5.Kirkby NS, Zaiss AK, Urquhart P, Jiao J, Austin PJ, Al-Yamani M, Lundberg MH, MacKenzie LS, Warner TD, Nicolaou A, Herschman HR, Mitchell JA. LC-MS/MS confirms that COX-1 drives vascular prostacyclin whilst gene expression pattern reveals non-vascular sites of COX-2 expression. PLoS One. 2013;8:e69524. doi: 10.1371/journal.pone.0069524. doi: 10.1371/journal.pone.0069524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kirkby NS, Chan MV, Zaiss AK, Garcia-Vaz E, Jiao J, Berglund LM, Verdu EF, Ahmetaj-Shala B, Wallace JL, Herschman HR, Gomez MF, Mitchell JA. Systematic study of constitutive cyclooxygenase-2 expression: role of NF-κB and NFAT transcriptional pathways. Proc Natl Acad Sci USA. 2016;113:434–439. doi: 10.1073/pnas.1517642113. doi: 10.1073/pnas.1517642113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Harris RC. COX-2 and the kidney. J Cardiovasc Pharmacol. 2006;47(suppl 1):S37–S42. doi: 10.1097/00005344-200605001-00007. [DOI] [PubMed] [Google Scholar]
- 8.Ahmetaj-Shala B, Kirkby NS, Knowles R, Al’Yamani M, Mazi S, Wang Z, Tucker AT, Mackenzie L, Armstrong PC, Nüsing RM, Tomlinson JA, Warner TD, Leiper J, Mitchell JA. Evidence that links loss of cyclooxygenase-2 with increased asymmetric dimethylarginine: novel explanation of cardiovascular side effects associated with anti-inflammatory drugs. Circulation. 2015;131:633–642. doi: 10.1161/CIRCULATIONAHA.114.011591. doi: 10.1161/CIRCULATIONAHA.114.011591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.FitzGerald GA, Oates JA, Hawiger J, Maas RL, Roberts LJ, II, Lawson JA, Brash AR. Endogenous biosynthesis of prostacyclin and thromboxane and platelet function during chronic administration of aspirin in man. J Clin Invest. 1983;71:676–688. doi: 10.1172/JCI110814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.FitzGerald GA, Brash AR, Falardeau P, Oates JA. Estimated rate of prostacyclin secretion into the circulation of normal man. J Clin Invest. 1981;68:1272–1276. doi: 10.1172/JCI110373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McAdam BF, Catella-Lawson F, Mardini IA, Kapoor S, Lawson JA, FitzGerald GA. Systemic biosynthesis of prostacyclin by cyclooxygenase (COX)-2: the human pharmacology of a selective inhibitor of COX-2. Proc Natl Acad Sci USA. 1999;96:272–277. doi: 10.1073/pnas.96.1.272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Capone ML, Tacconelli S, Rodriguez LG, Patrignani P. NSAIDs and cardiovascular disease: transducing human pharmacology results into clinical read-outs in the general population. Pharmacol Rep. 2010;62:530–535. doi: 10.1016/s1734-1140(10)70310-8. [DOI] [PubMed] [Google Scholar]
- 13.Catella F, FitzGerald GA. Paired analysis of urinary thromboxane B2 metabolites in humans. Thromb Res. 1987;47:647–656. doi: 10.1016/0049-3848(87)90103-4. [DOI] [PubMed] [Google Scholar]
- 14.Eikelboom JW, Hirsh J, Weitz JI, Johnston M, Yi Q, Yusuf S. Aspirin-resistant thromboxane biosynthesis and the risk of myocardial infarction, stroke, or cardiovascular death in patients at high risk for cardiovascular events. Circulation. 2002;105:1650–1655. doi: 10.1161/01.cir.0000013777.21160.07. [DOI] [PubMed] [Google Scholar]
- 15.Funk CD, FitzGerald GA. COX-2 inhibitors and cardiovascular risk. J Cardiovasc Pharmacol. 2007;50:470–479. doi: 10.1097/FJC.0b013e318157f72d. doi: 10.1097/FJC.0b013e318157f72d. [DOI] [PubMed] [Google Scholar]
- 16.Mitchell JA, Warner TD. COX isoforms in the cardiovascular system: understanding the activities of non-steroidal anti-inflammatory drugs. Nat Rev Drug Discov. 2006;5:75–86. doi: 10.1038/nrd1929. doi: 10.1038/nrd1929. [DOI] [PubMed] [Google Scholar]
- 17.Brooke MA, Longhurst HJ, Plagnol V, Kirkby NS, Mitchell JA, Rüschendorf F, Warner TD, Kelsell DP, MacDonald TT. Cryptogenic multifocal ulcerating stenosing enteritis associated with homozygous deletion mutations in cytosolic phospholipase A2-α. Gut. 2014;63:96–104. doi: 10.1136/gutjnl-2012-303581. doi: 10.1136/gutjnl-2012-303581. [DOI] [PubMed] [Google Scholar]
- 18.Kirkby NS, Reed DM, Edin ML, Rauzi F, Mataragka S, Vojnovic I, Bishop-Bailey D, Milne GL, Longhurst H, Zeldin DC, Mitchell JA, Warner TD. Inherited human group IV A cytosolic phospholipase A2 deficiency abolishes platelet, endothelial, and leucocyte eicosanoid generation. FASEB J. 2015;29:4568–4578. doi: 10.1096/fj.15-275065. doi: 10.1096/fj.15-275065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Martin-Ramirez J, Hofman M, van den Biggelaar M, Hebbel RP, Voorberg J. Establishment of outgrowth endothelial cells from peripheral blood. Nat Protoc. 2012;7:1709–1715. doi: 10.1038/nprot.2012.093. doi: 10.1038/nprot.2012.093. [DOI] [PubMed] [Google Scholar]
- 20.Reed DM, Foldes G, Gatheral T, et al. Pathogen sensing pathways in human embryonic stem cell derived-endothelial cells: role of NOD1 receptors. PLoS One. 2014;9:e91119. doi: 10.1371/journal.pone.0091119. doi: 10.1371/journal.pone.0091119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Starke RD, Ferraro F, Paschalaki KE, Dryden NH, McKinnon TA, Sutton RE, Payne EM, Haskard DO, Hughes AD, Cutler DF, Laffan MA, Randi AM. Endothelial von Willebrand factor regulates angiogenesis. Blood. 2011;117:1071–1080. doi: 10.1182/blood-2010-01-264507. doi: 10.1182/blood-2010-01-264507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Starke RD, Paschalaki KE, Dyer CE, Harrison-Lavoie KJ, Cutler JA, McKinnon TA, Millar CM, Cutler DF, Laffan MA, Randi AM. Cellular and molecular basis of von Willebrand disease: studies on blood outgrowth endothelial cells. Blood. 2013;121:2773–2784. doi: 10.1182/blood-2012-06-435727. doi: 10.1182/blood-2012-06-435727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Boutaud O, Aronoff DM, Richardson JH, Marnett LJ, Oates JA. Determinants of the cellular specificity of acetaminophen as an inhibitor of prostaglandin H(2) synthases. Proc Natl Acad Sci USA. 2002;99:7130–7135. doi: 10.1073/pnas.102588199. doi: 10.1073/pnas.102588199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Daniel VC, Minton TA, Brown NJ, Nadeau JH, Morrow JD. Simplified assay for the quantification of 2,3-dinor-6-keto-prostaglandin F1 alpha by gas chromatography-mass spectrometry. J Chromatogr B Biomed Appl. 1994;653:117–122. doi: 10.1016/0378-4347(93)e0432-p. [DOI] [PubMed] [Google Scholar]
- 25.Rauzi F, Kirkby NS, Edin ML, Whiteford J, Zeldin DC, Mitchell JA, Warner TD. Aspirin inhibits the production of proangiogenic 15(S)-HETE by platelet cyclooxygenase-1. FASEB J. 2016;30:4256–4266. doi: 10.1096/fj.201600530R. doi: 10.1096/fj.201600530R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Adler DH, Cogan JD, Phillips JA, III, Schnetz-Boutaud N, Milne GL, Iverson T, Stein JA, Brenner DA, Morrow JD, Boutaud O, Oates JA. Inherited human cPLA(2alpha) deficiency is associated with impaired eicosanoid biosynthesis, small intestinal ulceration, and platelet dysfunction. J Clin Invest. 2008;118:2121–2131. doi: 10.1172/JCI30473. doi: 10.1172/JCI30473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jespersen B, Thiesson HC, Henriksen C, Therland K, Falk C, Poulsen T, Fogh B, Madsen K, Walther S, Jensen BL. Differential effects of immunosuppressive drugs on COX-2 activity in vitro and in kidney transplant patients in vivo. Nephrol Dial Transplant. 2009;24:1644–1655. doi: 10.1093/ndt/gfp004. doi: 10.1093/ndt/gfp004. [DOI] [PubMed] [Google Scholar]
- 28.Zhou Y, Luo W, Zhang Y, Li H, Huang D, Liu B. Cyclo-oxygenase-1 or -2-mediated metabolism of arachidonic acid in endothelium-dependent contraction of mouse arteries. Exp Physiol. 2013;98:1225–1234. doi: 10.1113/expphysiol.2013.072017. doi: 10.1113/expphysiol.2013.072017. [DOI] [PubMed] [Google Scholar]
- 29.Reilly IA, FitzGerald GA. Inhibition of thromboxane formation in vivo and ex vivo: implications for therapy with platelet inhibitory drugs. Blood. 1987;69:180–186. [PubMed] [Google Scholar]