

Abstract
Introduction
We recently validated monoclonal antibody (mAb) 376.96 as an effective carrier for targeted α-particle radioimmunotherapy (RIT) with 212Pb in ovarian cancer mouse models. In this study, we tested the binding of radiolabeled mAb 376.96 to human pancreatic ductal adenocarcinoma (PDAC) cells and localization in xenografts in immune-deficient mice and evaluated 212Pb-labeled 376.96 (212Pb-376.96) for PDAC therapy.
Methods
In vitro Scatchard assays assessed the specific binding of 212Pb-376.96 to human PDAC3 adherent differentiated cells and non-adherent cancer initiating cells (CICs) dissociated from tumorspheres. In vitro clonogenic assays were used to measure the proliferation of adherent PDAC3 cells and CIC-enriched tumorspheres treated with 212Pb-376.96 or the irrelevant isotype-matched 212Pb-F3-C25. Mice bearing patient derived pancreatic cancer Panc039 xenografts were i.v. injected with 0.17–0.70 MBq 212Pb-376.96 or isotype control 212Pb-F3-C25, and used for biodistribution and tumor growth inhibition studies. Mice bearing orthotopic PDAC3 xenografts were i.v. co-injected with 99mTc-376.96 and 125I-F3-C25 and used for biodistribution studies.
Results
212Pb-376.96 specifically bound to PDAC3 adherent and dissociated tumorsphere CICs; Kd values averaged 9.0 and 21.7 nM, respectively, with 104-105 binding sites/cell. 212Pb-376.96 inhibited the clonogenic survival of PDAC3 cells or CICs dissociated from tumorspheres 3–6 times more effectively than isotype-matched control 212Pb-F3-C25. Panc039 s.c. tumors showed significantly higher uptake of 212Pb-376.96 (14.0 ± 2.1% ID/g) compared to 212Pb-F3-C25 (6.5 ± 0.9% ID/g, p<0.001) at 24 h after dosing. Orthotopic PDAC3 tumors showed significantly higher uptake of 99mTc-376.96 (6.4 ± 1.8% ID/g) compared to 125I-F3-C25 (3.9 ± 0.9% ID/g, p<0.05) at 24 h after dosing. Panc039 tumor growth was significantly inhibited by 212Pb-376.96 compared to 212Pb-F3-C25 or non-treated control tumors (p<0.05).
Conclusion
Our results provide evidence for the efficacy of B7-H3 targeted RIT against preclinical models of pancreatic ductal adenocarcinoma (PDAC) and support future studies with 212Pb-376.96 in combination with chemotherapy to potentiate efficacy against PDAC.
Keywords: Radioimmunotherapy, pancreatic ductal adenocarcinoma, 212Pb, B7-H3
INTRODUCTION
Pancreatic ductal adenocarcinoma (PDAC) is estimated to cause over 43,000 deaths in the United States during 2017 and is associated with a 5-year survival rate of 8% [1]. The majority of patients (>80%) are diagnosed with locally advanced or metastatic disease that is not effectively controlled through surgical resection. Currently approved first-line chemotherapies (gemcitabine and nab-paclitaxel or FOLFIRINOX) are associated with median survivals of less than 1 year [2]. The high rate of invasive dissemination, the resistance of PDAC cells to chemotherapy, and the dense tumor stroma are notable obstacles contributing to the failure of conventional therapeutic strategies against PDAC. Thus, there has been a clinical demand for better treatment of this devastating disease. Cancer initiating cells (CICs), a subpopulation of tumor cells that are resistant to standard chemotherapy and are highly tumorigenic in immunedeficient mice [3, 4], have been implicated in the initiation and metastasis of PDAC. PDAC CICs and differentiated cells that survive chemotherapy contribute to the poor prognosis associated with PDAC [5]. Therefore, novel therapies that effectively target both differentiated PDAC cells and CICs may have great potential to improve therapeutic efficacy for PDAC [6, 7].
Targeted radioimmunotherapy (RIT) approaches with various radioimmunoconjugates (RICs) have been assessed against PDAC in preclinical and clinical studies [8, 9], although none have yet advanced to large-scale trials. Targeted RIT with α-particle emitting radionuclides is a promising approach to eliminate microscopic clusters of malignant cells due to the short path length (50–80 μm), high linear energy transfer (LET; 100 keV/μm), and high relative biological effectiveness of α-particles [10–12]. α-particles are known to induce cell death or proliferation arrest regardless of the cell’s oxygen levels or sensitivity to chemotherapy or low LET (0.1–1 keV/μm) radiotherapy treatment (external beam or β-particle RIT) [11, 13]. These factors support the use of targeted α-particle RIT for locally advanced or metastatic PDACs that are resistant to first-line chemotherapies.
Targeted delivery of high LET radiation to both differentiated tumor cells and CICs represents a particularly attractive strategy to destroy malignant cells that survive alternative therapies while minimizing toxicities to normal tissues. The co-stimulatory protein B7-H3 (CD276) has been shown to be overexpressed in PDAC [14, 15], with higher expression correlating with aggressive and metastatic disease [16, 17]. PDAC cell lines and CICs express an extracellular B7-H3 epitope bound by monoclonal antibody (mAb) 376.96; this epitope is not significantly expressed in normal tissues [4, 18], suggesting mAb 376.96 as a promising carrier for RIT of PDAC. Recent studies have demonstrated the efficacy of mAb 376.96 labeled with 212Pb (t1/2 = 10.64 h), which decays to the α-particle emitter 212Bi (t1/2 = 60.5 min), for targeted RIT in ovarian cancer mouse models [19, 20]. The goals of this investigation were to test the specific binding of radiolabeled mAb 376.96 to PDAC cells and localization in xenografts, and to evaluate 212Pb-376.96 for pancreatic cancer therapy against PDAC mouse models.
MATERIALS AND METHODS
Reagents and instrumentation
All reagents were prepared from commercially available materials (Thermo Fisher, Sigma) unless specified otherwise. 212Pb in transient equilibrium with its daughter radionuclides was eluted as previously described [21] from a 224Ra/212Pb generator obtained from Oak Ridge National Laboratory (Oak Ridge, TN). Murine mAb 376.96 [22] and isotype-matched control murine mAb F3-C25, an anti-idiotypic mAb to the murine anti-HLA Class II mAb CR11-462 [23], were produced and characterized as previously described. mAbs were purified from mouse ascites fluid by protein affinity chromatography. The purity and activity of mAb preparations were monitored by SDS-PAGE and by specific reactivity with the corresponding antigens in binding assays. An energy and efficiency calibrated high-purity germanium (HPGe) detector (model GMX10P4-70; Ortec, Oak Ridge, TN) operated at −4000 V housed in a lead shield (Ortec) was used to determine the radionuclidic purity and radioactivity of 212Pb for all radiolabeling procedures. Spectra were processed using Gamma Vision-32 software (version 6.09; Ortec). Radioactivity measurements of samples from the in vitro and in vivo experiments were performed on calibrated Cobra II (Packard, Meriden, CT) or Wizard2 (Perkin Elmer, Shelton, CT) gamma counters using an energy window centered on the main gamma peaks from 212Pb (238.6 keV, 43.6%) or 212Bi (727.3 keV, 6.7%) after cross-calibrating the instruments with the HPGe detector. Radioactivity analyses were corrected for radioactive decay.
Radiolabeling studies
mAbs 376.96 and F3-C25 were conjugated with the bifunctional chelator 2-(4-isothiocyanotobenzyl)-1,4,7,10-tetraaza-1,4,7,10-tetra-(2-carbamoylmethyl)-cyclododecane (TCMC; Macrocyclics, Plano, TX) following previously described procedures [24] to generate TCMC-mAb 376.96 and TCMC-mAb F3-C25. The average chelate/mAb ratios were determined by a spectrophotometric assay [25]. Radiolabeling and purification of the 212Pb-TCMC-mAb RICs (212Pb-376.96 or 212Pb-F3-C25) were performed as previously described [21] using 0.12–0.3 MBq of 212Pb in transient equilibrium with its daughter radionuclides per 1 μg of TCMC-mAb conjugate (18–44 GBq/μmol). Immediately after collecting the purified RICs, an aliquot was removed for additional characterization and 5 μL 0.1 mol/L EDTA and 50 μL 30% human serum albumin (Sigma-Aldrich, St. Louis, MO) were added to the RICs. The protein content of the removed aliquot was determined by Lowry analysis [26]. The radiochemical conversions and purities of the crude preparations and isolated RICs were determined as previously described [21]. mAb 376.96 containing 4.6 TCMC chelates per mAb was radiolabeled with 212Pb to produce 212Pb-376.96 in high yield and radiochemical purity (84% and 98%, respectively, n=6) as indicated by ITLC analysis. The control mAb F3-C25 containing 5.6 TCMC chelates per mAb molecule was radiolabeled to give 212Pb-F3-C25 with an average radiochemical purity of 93% (n=5). Procedures and results from in vitro stability analyses of 212Pb-376.96 are provided in the Supplementary data (Supplementary Table S1). Specific activities of 74–204 kBq/μg (11–30 GBq/μmol) for the 212Pb-RICs were used in the in vitro and in vivo studies described below. mAb 376.96 was conjugated with HYNIC-NHS and radiolabeled with 99mTc-tricine as previously described [27]; the isolated specific activity was 0.76 MBq/μg (81% radiochemical purity). mAb F3-C25 was radiolabeled with carrier-free Na125 I (MP Biomedicals, Solon, OH) in Pierce Pre-Coated Iodination Tubes (Thermo Scientific, Rockford, IL) according to manufacturer’s specifications; the isolated specific activity was 46.6 kBq/μg (86% radiochemical purity).
Human PDAC3 cell line and CICs
The PDAC3 cell line established from ascites fluid of a human patient with metastatic PDAC, has been previously described [28]. Cells grown as monolayers (“Adherent” conditions) were cultured with high glucose DMEM medium supplemented with 10% fetal bovine serum (FBS; HyClone, Logan, UT) in tissue culture treated plates (Corning Costar, Corning, NY). Cells grown as non-adherent tumorspheres were cultured with serum-free high glucose DMEM supplemented with 20 μg/mL bovine insulin, 20 ng/mL epidermal growth factor, 10 ng/mL basic fibroblast growth factor, 5 μg/mL transferrin, 5 ng/mL sodium selenite, 16 μg/mL putrescene, and 7.3 ng/mL progesterone in ultra-low attachment plates (Corning Costar) as previously described to enrich for cells with characteristics of CIC proliferation [29, 30]; at 3 days after seeding cells in these conditions, compact spheroids with diameters of 35–65 μm and high cell viability (>95%) had formed. Cell culture and in vitro experiments were performed at 37 °C in a 5% CO2 humidified atmosphere unless specified otherwise. Experiments were performed using cells under 15 passages from initial receipt and expansion; cells were discarded within 3 months of thawing the expanded frozen stocks.
In vitro binding assays
The binding of 212Pb-376.96 to adherent PDAC3 cells and to PDAC3 cells dissociated from non-adherent tumorspheres grown under CIC conditions was tested as previously described for ovarian cancer cell lines to calculate the binding affinity (Kd), binding sites per cell, and internalized fraction [20, 27].
In vitro clonogenic survival assays
Adherent clonogenic assays
PDAC3 cells were seeded at 100,000 cells/well in 24-well plates (Corning Costar) two days before treatment with RICs, defined as day 0 of the study. On day 0, cells were rinsed with PBS and incubated with serial dilutions of 212Pb-376.96 or 212Pb-F3-C25 in 0.4 mL assay medium (DMEM pH 7.4 with 30 mmol/L HEPES, 2 mmol/L L-glutamine, 1 mmol sodium pyruvate, 1% bovine serum albumin) for 2 h at 37 °C with gentle swirling. Cells were rinsed twice with PBS and incubated in fresh Adherent medium. Two days later (day 2), cells were trypsinized, counted, serially diluted in normal (Adherent) medium, and plated at 100 live cells/well in six replicate wells of a tissue culture treated 6-well plate (Corning Costar). The percent clonogenic survival relative to vehicle-treated controls was determined 10 days after plating (day 12) as previously described [20, 31].
CIC clonogenic assays
PDAC3 cells were seeded at 100,000 cells/well in 24-well ultra-low attachment plates in CIC conditions to form tumorspheres three days before treatment with RICs, defined as day 0 of the study. On day 0, intact tumorspheres (diameters of 35–65 μm) were collected, rinsed with PBS, and incubated with serial dilutions of 212Pb-376.96 or 212Pb-F3-C25 in 0.4 mL assay medium for 2 h at 37 °C with gentle swirling to keep tumorspheres in suspension. Tumorspheres were rinsed twice with PBS and incubated in new ultra-low attachment 24-well plates with fresh CIC medium. Two days later (day +2), tumorspheres were collected and dissociated into single cells with Accutase or trypsin, counted, and serially diluted in Adherent medium. Cells were plated and analyzed for clonogenic survival as in the Adherent clonogenic assays.
Animal subjects and husbandry
All in vivo studies were performed in 5–7 week old female athymic nude mice (Charles River, Wilmington, MA). Animal studies were approved by the University of Alabama at Birmingham Institutional Animal Care and Use Committee and performed in compliance with guidelines from the Public Health Service Policy and Animal Welfare Act of the United States. Sections (4×4×4 mm) of patient derived xenograft (PDX) Panc039 tumors propagated in the flank of nude mice as previously described [32] were implanted subcutaneously (s.c.) in mice. Radioactive biodistribution and therapy studies were initiated 4–5 weeks after implantation, when tumors had grown to 140–200 mm3. PDAC3 cells (2.5×106) were implanted orthotopically into the pancreas of mice using previously described techniques [33]; biodistribution studies were performed 3 months after pancreas implantation, when tumors were 1.4–2.0 g. Mice were given Nutra-Gel Diet (Bio-Serv, Flemington, NJ) in addition to standard chow for two weeks after administering the RICs. Mice were weighed twice per week and euthanized if >10% weight loss occurred or when tumors grew to 1.5 cm in diameter. Tumor volume was calculated by the formula (L×W2)/2 as previously described [34].
In vivo biodistribution studies
Mice bearing s.c. PDX Panc039 tumors were injected i.v. with ~0.74 MBq (~4.0 μg) 212Pb-376.96 or 212Pb-F3-C25 (n = 4 mice/group) in 0.2 mL PBS. Mice bearing orthotopic PDAC3 tumors were intended for use in parallel biodistribution studies with 212Pb-376.96 or 212Pb-F3-C25, although variable rates of tumor development precluded using this PDAC xenograft model during the useable lifetime of the 224Ra/212Pb generator (see Discussion below). To test the localization of radiolabeled mAb 376.96 in the PDAC3 xenografts, mice bearing established tumors (n = 5 mice) were injected i.v. with a solution containing 99mTc-376.96 (6.02 MBq, 10.5 μg) and 125I-F3-C25 (22.39 kBq, 10.6 μg) in 0.2 mL PBS. Mice were euthanized at 24 h post injection and selected tissues were removed and counted in a gamma counter. Percent uptake of the injected dose per gram (% ID/g) in all experiments was calculated by comparing the tissue activity to solutions with known activity of the isotope of interest. 125I was counted after 99mTc had fully decayed.
In vivo therapy studies
Groups of mice (n = 10/group) bearing s.c. Panc039 tumors were given a single i.v. dose of 212Pb-376.96 or 212Pb-F3-C25 (0.36–0.73 MBq) or received no treatment (control). Tumor growth was calculated as mean percent change in volume with standard error relative to the initial volume at the time of dosing.
Statistical analyses
Data were analyzed using Microsoft Excel or GraphPad Prism (Version 5.02). When comparing multiple groups, one-way or two-way ANOVA tests were performed, followed by Bonferroni tests to compare differences between individual groups. All p values correspond to two-tailed tests. p<0.05 was considered significant.
RESULTS
212Pb-376.96 binds to differentiated human PDAC cells and human PDAC CICs
In vitro Scatchard binding assays showed 212Pb-376.96 bound to adherent PDAC3 cells and to cells dissociated from non-adherent CIC tumorspheres with affinities of 9.0 ± 1.1 nM (n=6) and 21.7 ± 0.7 nM (n=3), respectively; specific binding was 73–76%. Representative binding results and Scatchard plots are shown in Figure 1. Significantly more binding sites/cell were present on the dissociated CICs compared to adherent cells (Table 1; p<0.05).
Figure 1.
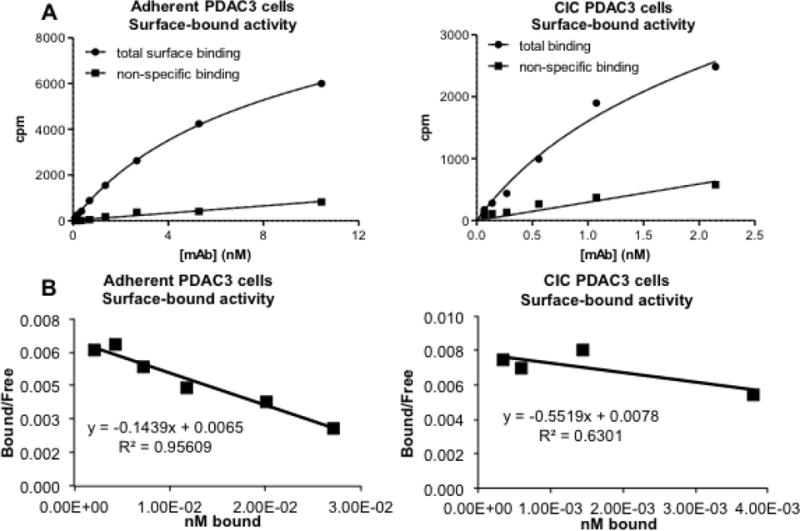
Representative graphs showing in vitro binding of 212Pb-376.96 to PDAC3 cells and CICs (A) and Scatchard plots of the data (B).
Table 1.
In vitro binding analysis of 212Pb-376.96 to PDAC3 cells.
| Kd ± S.E.M. (nmol/L) | Binding sites/cell ± S.E.M. (×104) | Percent internalized | |
|---|---|---|---|
| Adherent PDAC3 | 9.0 ± 1.1 | 3.3 ± 0.6 | 44 |
| CIC PDAC3 | 21.7 ± 0.7 | 18.9 ± 2.6 | 40 |
212Pb-376.96 inhibits the clonogenic survival of differentiated human PDAC cells and human PDAC CICs in vitro
Adherent PDAC3 cells treated in vitro with 212Pb-376.96 showed significantly lower clonogenic survival compared to cells treated with 212Pb-F3-C25 (Table 2, p<0.05). Similarly, PDAC3 cells obtained from CIC tumorspheres were more sensitive to 212Pb-376.96 than to 212Pb-F3-C25 (p=0.18). Clonogenic survival following treatment with 212Pb-F3-C25 was more variable than after treatment with 212Pb-376.96. As shown in Table 2, the clonogenic survival of PDAC3 differentiated cells and CICs was inhibited 3–6 times more effectively by 212Pb-376.96 than by 212Pb-F3-C25.
Table 2.
In vitro inhibition of PDAC3 adherent cells or CIC clonogenic survival by 212Pb-376.96 or 212Pb-F3-C25.
| Cells | aIC50 ± S.E.M. (kBq/mL) | |
|---|---|---|
| 212Pb-376.96 | 212Pb-F3-C25 | |
| Adherent PDAC3 | 41 ± 14 | 120 ± 12 |
| CIC PDAC3 | 26 ± 17 | 180 ± 150 |
Data are presented as the mean ± S.E.M. of 2–4 individual experiments, with each experiment performed with 4–6 replicate wells.
212Pb-376.96 targets human PDAC xenograft tumors in vivo
Mice with established s.c. PDX Panc039 tumors showed significantly higher uptake of the injected 212Pb dose at 24 h after administration of 212Pb-376.96 compared to the isotype control 212Pb-F3-C25 (14.0 ± 2.1% and 6.5 ± 0.9% ID/g, respectively; p<0.001) (Figure 2A); the level of 212Bi in the tumors was consistent with the activity of 212Pb observed in both groups (Supplementary Tables S2 and S3). Mice in the 212Pb-376.96 group showed uptake of 212Pb in normal organs including spleen, liver, kidneys, and blood (18.2%, 9.8%, 10–14%, and 9.6% ID/g, respectively); mice in the 212Pb-F3-C25 group showed comparable retention of 212Pb in liver, kidneys, and blood (Figure 2A, Supplementary Tables S2 and S3). Mice in both groups showed higher retention of 212Pb than 212Bi in the liver, while more 212Bi than 212Pb was retained in the spleen, kidneys, blood, and femurs of mice from both groups (Supplementary Tables S2 and S3).
Figure 2.
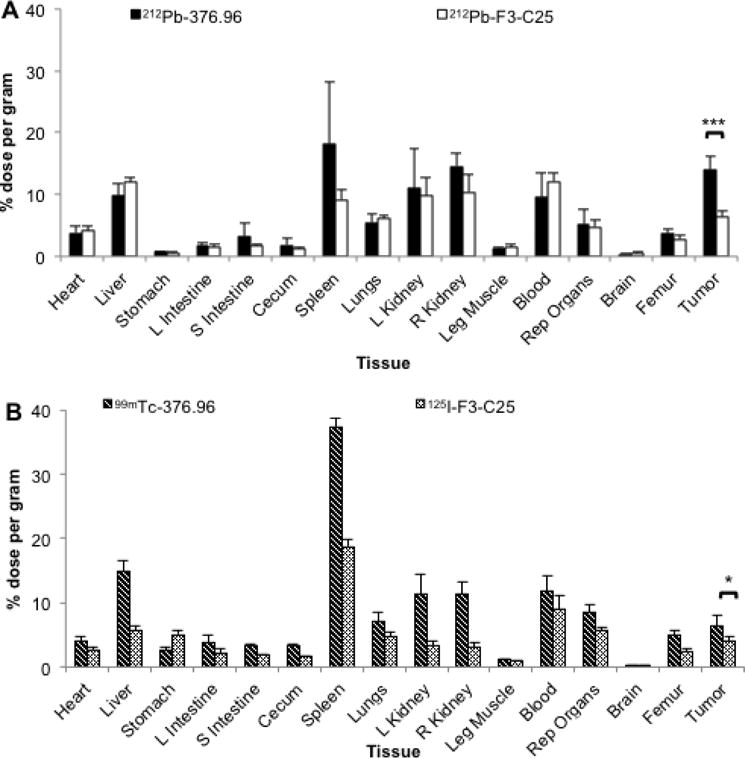
Biodistribution analyses in mice with PDAC xenograft tumors. (A) Biodistribution at 24 h after i.v. injection of 212Pb-376.96 or 212Pb-F3-C25 (0.74 MBq) in groups of athymic nude mice (n = 4/group) bearing s.c. PDX Panc039 tumors. (B) Biodistribution at 24 h after i.v. co-injection of 99mTc-376.96 (6.02 MBq) and 125I-F3-C25 (22.39 kBq) in athymic nude mice (n = 5 mice) bearing orthotopic PDAC3 tumors; 125I was counted after 99mTc had fully decayed. After euthanization of mice, tissues were collected, weighed, and counted to determine the percent of the injected dose per gram (% ID/g) of tissue. Data are presented as mean ± standard deviation. *p<0.05; ***p<0.001
Orthotopic PDAC3 tumors showed significantly higher uptake of 99mTc-376.96 compared to 125I-F3-C25 (6.4 ± 1.8% and 3.9 ± 0.9% ID/g, respectively, p<0.05) at 24 h after i.v. co-injection of the two radiolabeled antibodies (Figure 2B). The spleen showed high uptake of 99mTc-376.96, which correlated with the locally invasive growth of the orthotopic PDAC3 tumors into and around the spleen at the time of necropsy (data not shown). Biodistribution data for normal tissues and tumors are provided in Supplementary Table S4. These biodistribution results in different tumor models confirmed the effective and specific targeting of human PDAC with radiolabeled mAb 376.96.
212Pb-376.96 significantly inhibits the growth of human PDX PDAC tumors in vivo
The uptake and targeting specificity of 212Pb-376.96 in established Panc039 tumors suggested the relevant application of this RIT agent for treating solid PDAC tumors. Preliminary studies indicated that a single i.v. dose of 0.2–0.5 MBq 212Pb-376.96 was well tolerated in mice and inhibited the growth of s.c. Panc039 tumors relative to non-treated controls (Supplementary Figure S1). For the experiments discussed here, tumor growth inhibition studies were performed in groups of s.c. Panc039 tumor-bearing mice given a single i.v. injection of 0.36, 0.54, or 0.73 MBq 212Pb-376.96. Another group of tumor-bearing mice received a single i.v. injection of an intermediate dose of 212Pb-F3-C25 (0.46 MBq) to determine the effects of non-targeted RIT against Panc039 tumors. All RIT treatments resulted in significant tumor growth inhibition relative to non-treated controls as early as day 9 (p<0.05) after injecting the RICs into mice, indicating the potency of 212Pb RIT against this tumor model (Figure 3). The two higher doses of 212Pb-376.96 (0.54 and 0.73 MBq) were significantly more effective at slowing tumor growth than the low dose (0.36 MBq) of 212Pb-376.96 and 0.46 MBq dose of 212Pb-F3-C25 as early as day 12 (p<0.05). This level of tumor growth inhibition continued throughout the period of the experiment, although no complete tumor regressions were observed. 212Pb-376.96 at 0.36 MBq and the higher dose (0.46 MBq) of the control 212Pb-F3-C25 were equally effective against Panc039 tumor growth (Figure 3). These results confirmed the growth inhibitory effects of B7-H3 targeted α-particle RIT against PDAC xenografts and also indicated the challenge of eradicating these established solid tumors under the conditions explored.
Figure 3.
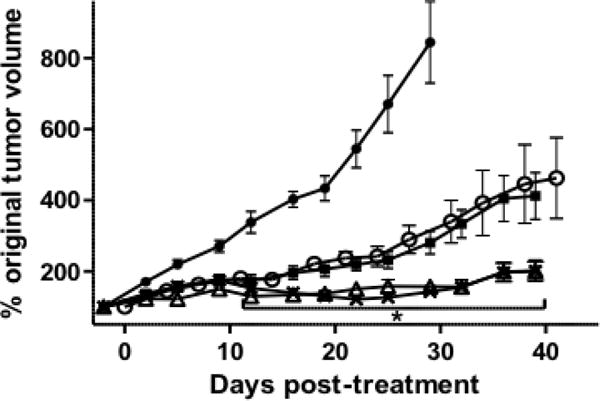
Tumor growth curves showing the effects of 212Pb-376.96 or 212Pb-F3-C25 on PDX Panc039 tumor growth inhibition in vivo. Groups of mice bearing established s.c. Panc039 tumors (140–200 mm3) either were left untreated (filled circles) or injected i.v. with 0.46 MBq 212Pb-F3-C25 (open circles) or with 0.36 MBq (filled squares), 0.54 MBq (open triangles), or 0.73 MBq (× marks) 212Pb-376.96 (n = 10/group). Data were plotted as mean percent change (± standard error of the mean) in normalized tumor volume over time relative to the initial volume at the time of injection with the RIC. *p<0.05 vs. 212Pb-F3-C25 for the time points indicated.
All treatments caused a transient drop in mice body weight; all groups of mice had recovered to their initial weight before treatment within 2 weeks after injecting the RICs (Supplementary Figure S2). At the end of the study period, the average percent increase in weights of mice treated with 0.54 or 0.73 MBq was slightly less than in mice from the non-treated control group (increase of 6.8 ± 4.0%, 10.0 ± 5.3%, and 16.3 ± 4.5%, respectively). These results indicate RIT with 212Pb-376.96 was generally well tolerated at the dose levels and time frames explored, although toxicology effects on blood parameters or normal organs have yet to be characterized.
DISCUSSION
The studies presented here are unique in that the RIC targets a tumor-specific B7-H3 antigen expressed on CICs and differentiated tumor cells from multiple models of PDAC [4, 18]. 212Pb-376.96 bound to PDAC3 cells with similar affinities as previously shown in ovarian cancer cell lines, although fewer binding sites per cell were present on the PDAC3 cells compared to ovarian cancer cells [20]. Of interest, the dissociated cells from CIC tumorspheres exposed to 212Pb-376.96 showed lower clonogenic survival than adherent PDAC3 cells exposed to 212Pb-376.96. This could be due to the higher number of binding sites on the CICs relative to adherent cells and to the bystander effect of α-particles emitted from an antigen-bound RIC into the highly compact CIC tumorsphere, as a single α-particle would be expected to traverse the entire diameter of the PDAC3 tumorsphere (<70 μm). The 3–6 fold greater sensitivity of PDAC3 cells to 212Pb-376.96 relative to the control 212Pb-F3-C25 is comparable to that observed with 212Pb-trastuzumab relative to a control RIC in alternative PDAC cell lines [10]. PDAC3 adherent cells and CICs were slightly less sensitive than ovarian cancer cells to 212Pb-376.96 [20]. These differences are likely due to disparities in antigen expression, cell size, and activation of repair mechanisms between the cell lines.
The biodistribution studies in two PDAC models showed significantly higher tumor accumulation of 212Pb-376.96 or 99mTc-376.96 compared to the isotype control RICs. These results verified the utility of mAb 376.96 for targeting the B7-H3 epitope in these PDAC models. Both the PDAC3 and Panc039 tumor models were intended for use in parallel biodistribution studies with 212Pb-376.96, although the variable rate of PDAC3 tumor growth prohibited experiments with these tumors during the usable lifetime of the 224Ra/212Pb generator. 203Pb (t1/2 = 51.9 h; 279.2 keV, 80.9%) has favorable properties for SPECT imaging and could be used as a matched-pair diagnostic radionuclide for 212Pb RIT applications; however, this radionuclide was not available at the time this study was carried out. Biodistribution studies in mice with PDAC3 tumors used 99mTc and 125I due to their low cost and well-characterized radiolabeling properties. It is recognized that using different radionuclides could impact the relative biodistribution of the RICs. The lower uptake of 99mTc-376.96 in the PDAC3 tumors compared to 212Pb-376.96 in the Panc039 tumors was likely due to the large size of the PDAC3 tumors (>1.5 g) at the time of the study.
The previous TCMC-mAb 376.96 conjugate used in the studies with intraperitoneal (i.p.) models of ovarian cancer [20] had a high chelate/mAb ratio (12.7) that was similar to the chelate/mAb ratio (14.0) of the 212Pb-TCMC-trastuzumab conjugate used for i.p. injection in the non-human primate and human clinical trials [35–38]. As the 212Pb-RICs in the current studies were intended for i.v. rather than i.p. injection, lower chelate/mAb ratios (4.6–5.6) were used to reduce potential retention of the RICs in the reticuloendothelial system (RES) after systemic injection [39, 40]. The uptake of 212Pb and 212Bi in the spleens of mice in the current biodistribution studies with 212Pb-376.96 was comparable to that observed in our previous study for this IgG2a isotype RIC [20]. The observed differences in normal tissue retention between 212Pb and its daughter 212Bi are consistent with previous studies that systemically administered 212Pb-labeled compounds [41, 42]; ~30% of 212Bi has been shown to be released from the original 212Pb-chelate complex due to internal conversion during decay of 212Pb [43], which would allow distribution of free 212Bi and its daughter radionuclides during circulation of the 212Pb-RICs. Several strategies could be employed to minimize the amount of systemically released 212Bi emitted from 212Pb-conjugates, such as employing small targeting carriers that have rapid pharmacokinetics when i.v. injections are required [41, 42], targeting intracavitary (e.g., i.p.) lesions with local administrations when using mAbs as targeting vectors [10, 35, 36, 44], or other approaches to control recoiling daughter radionuclides that emit α-particles [45].
Prior studies have not explored α-particle RIC biodistribution and therapeutic efficacy using a macroscopic PDX PDAC tumor model in vivo. The high Panc039 tumor uptake and relatively low normal tissue retention of 212Pb-376.96 relative to the control RIC in the present studies supported the potential efficacy of targeted RIT against this PDAC model with tolerable levels of 212Pb-376.96. The partial growth inhibition of the Panc039 tumors by the control RIC suggests efficacy may be due to disruption of tumor vasculature by α-particles emitted from circulating 212Pb-RICs, or to killing of tumor and stromal cells from 212Pb-RICs that extravasated through leaky vessels within the tumors. Several studies have shown inhibition of tumor growth from control mAbs labeled with 212Pb [46, 47] or other radionuclides [48]. Dosimetry analyses would be helpful in estimating the relative efficacy of the targeted or control 212Pb-RICs in future studies. In addition to target antigen expression on PDAC cells, it is possible that the B7-H3 antigen targeted by mAb 376.96 is also expressed in the vasculature of the Panc039 xenografts; B7-H3 expression has been characterized in the tumor vasculature of several malignancies [49–51], although antigen expression in PDAC vasculature has not yet been confirmed. Future work that explores the effects of 212Pb-376.96 on PDAC tumor vasculature would help elucidate the mechanism of this RIT strategy beyond killing small clusters of tumor cells.
Several studies have investigated the efficacy of α-particle RIT against preclinical models of PDAC [8, 10, 52–56]. Studies that initiated treatment soon (<3 days) after s.c. PDAC xenograft implantation before macroscopic tumor burden had developed showed that tolerable doses of targeted 213Bi-RICs were able to prevent tumor formation, indicating therapeutic efficacy in these scenarios of sub-clinical PDAC foci [53–55]. Studies that initiated treatment with targeted 212Pb- or 213Bi-RICs at later time points (>6 days) after i.p. xenograft implantation when disseminated PDAC burden was established showed moderate efficacy and specificity relative to non-targeted RICs; PDAC tumor burden progressed in all animals under the conditions explored [10, 52]. Studies using targeted 213Bi-RIT or brachytherapy with 224Raimplanted rods in macroscopic s.c. PDAC tumors showed delayed tumor growth due to therapy, although tumor progression was not eradicated in these studies [56, 57]. The RIT results presented above with 212Pb-376.96 against macroscopic PDAC tumors are consistent with these previous studies, indicating the significant challenge to eradicate established PDAC with a single administration of α-particle RICs. While the limited tissue penetration of α-particles is not ideal for treating large solid tumors, preclinical studies have proven the concept that fractionated RIT with short-lived α-particle radionuclides can eradicate macroscopic human xenograft tumors in mice [58]. These results support future investigations of fractionated dosing schedules and combinatorial chemotherapy regimens with 212Pb-376.96 against PDAC.
Performing the in vitro assays with a single PDAC cell line is a limitation noted for the studies above; previous work has shown that several PDAC cell lines express the B7-H3 antigen bound by mAb 376.96 [4, 18], suggesting additional cell lines would be suitable for inclusion in future studies with 212Pb-376.96. Limitations for the in vivo studies include the modest specificity of tumor growth inhibition with 212Pb-376.96 relative to 212Pb-F3-C25. The relatively large starting size of the tumors, limited tissue extravasation of mAbs, and the short tissue penetration of α-particles, including those emitted from RICs bound to the tumor antigen, are factors that could have detracted from therapeutic efficacy of the targeted RIC relative to the control RIC. It should be noted that a lower dose of 212Pb-376.96 produced an equivalent inhibition in Panc039 tumor growth as a higher dose of 212Pb-F3-C25. Such a benefit of targeted 212Pb RIT relative to non-specific RIT was greater than that observed in a previous study that used a large, established s.c. ovarian tumor model [34]. Dosimetry and toxicology analyses were beyond the scope of the present studies, which were intended as the initial step to investigate the potential for mAb 376.96 as a carrier for 212Pb RIT of PDAC. Future studies will define dosimetry and toxicology parameters and will determine if fractionated doses of 212Pb-376.96 alone or in combination with chemotherapy and inhibitors of CIC proliferation can cause tumor regression in preclinical models of human PDAC. These additional preclinical studies would be essential prior to clinical assessment of mAb 376.96 as a targeting agent for RIT. As murine mAbs typically induce immunogenic responses in humans, humanization of mAb 376.96 would be needed prior to assessment of fractionated dosing regimens in humans.
CONCLUSION
The results from the above studies support the concept for B7-H3 targeted RIT with 212Pb-376.96 against preclinical models of PDAC. 212Pb-376.96 specifically bound to and inhibited the growth of PDAC cells and CICs in vitro as well as established patient derived PDAC tumor xenografts in vivo. The aberrant overexpression of B7-H3 in multiple malignancies [59, 60] suggests using mAb 376.96 as a carrier for targeted RIT would be worthy of exploration beyond the ovarian and PDAC models examined to date.
Supplementary Material
Acknowledgments
Jeffrey Sellers, Sharon Samuel, Sheila Bright, Cherlene Hardy, Debbie Della Manna, Quentin Whittsit, Sivaram Sridharan, Jillian Richter, Carey Hickerson, Marie Warren, Patsy Oliver, and Karim Budhwani are gratefully acknowledged for their contributions. The 212Pb used in this research was obtained from a 224Ra/212Pb generator supplied by the United States Department of Energy Office of Science by the Isotope Program in the Office of Nuclear Physics.
Financial support: Funding was provided by NIH grant R21CA173120 and the Comprehensive Cancer Center at the University of Alabama at Birmingham.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflicts of interest: The authors declare no potential conflicts of interest.
References
- 1.American Cancer Society. Cancer Facts and Figures 2017. 2017. [Google Scholar]
- 2.Ghosn M, Ibrahim T, Assi T, El Rassy E, Kourie HR, Kattan J. Dilemma of first line regimens in metastatic pancreatic adenocarcinoma. World J Gastroenterol. 2016;22:10124–30. doi: 10.3748/wjg.v22.i46.10124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature. 2001;414:105–11. doi: 10.1038/35102167. [DOI] [PubMed] [Google Scholar]
- 4.Wang Y, Sabbatino F, Yu L, Favoino E, Wang X, Ligorio M, et al. Tumor antigen-specific monoclonal antibody-based immunotherapy, cancer initiating cells and disease recurrence. In: Bonavida B, editor. Resistance to Immunotherapeutic Antibodies in Cancer. Springer; New York: 2013. pp. 25–47. [Google Scholar]
- 5.Ercan G, Karlitepe A, Ozpolat B. Pancreatic cancer stem cells and therapeutic approaches. Anticancer Res. 2017;37:2761–75. doi: 10.21873/anticanres.11628. [DOI] [PubMed] [Google Scholar]
- 6.Huang EH, Heidt DG, Li CW, Simeone DM. Cancer stem cells: a new paradigm for understanding tumor progression and therapeutic resistance. Surgery. 2007;141:415–9. doi: 10.1016/j.surg.2006.12.015. [DOI] [PubMed] [Google Scholar]
- 7.Krantz SB, Shields MA, Dangi-Garimella S, Munshi HG, Bentrem DJ. Contribution of epithelial-to-mesenchymal transition and cancer stem cells to pancreatic cancer progression. J Surg Res. 2012;173:105–12. doi: 10.1016/j.jss.2011.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sahlin M, Bauden MP, Andersson R, Ansari D. Radioimmunotherapy—a potential novel tool for pancreatic cancer therapy? Tumor Biol. 2015;36:4053–62. doi: 10.1007/s13277-015-3479-y. [DOI] [PubMed] [Google Scholar]
- 9.Shah M, Da Silva R, Gravekamp C, Libutti SK, Abraham T, Dadachova E. Targeted radionuclide therapies for pancreatic cancer. Cancer Gene Ther. 2015;22:375–9. doi: 10.1038/cgt.2015.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Milenic DE, Garmestani K, Brady ED, Albert PS, Ma D, Abdulla A, et al. α-particle radioimmunotherapy of disseminated peritoneal disease using a 212Pb-labeled radioimmunoconjugate targeting HER2. Cancer Biother Radiopharm. 2005;20:557–68. doi: 10.1089/cbr.2005.20.557. [DOI] [PubMed] [Google Scholar]
- 11.Elgqvist J, Frost S, Pouget J-P, Albertsson P. The potential and hurdles of targeted alpha therapy - clinical trials and beyond. Front Oncol. 2014;3:324. doi: 10.3389/fonc.2013.00324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Palm S, Bäck T, Haraldsson B, Jacobsson L, Lindegren S, Albertsson P. Biokinetic modeling and dosimetry for optimizing intraperitoneal radioimmunotherapy of ovarian cancer microtumors. J Nucl Med. 2016;57:594–600. doi: 10.2967/jnumed.115.167825. [DOI] [PubMed] [Google Scholar]
- 13.Yong K, Brechbiel M. Application of 212Pb for targeted alpha-particle therapy (TAT): Pre-clinical and mechanistic understanding through to clinical translation. AIMS Med Sci. 2015;2:228–45. doi: 10.3934/medsci.2015.3.228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Loos M, Hedderich DM, Ottenhausen M, Giese NA, Laschinger M, Esposito I, et al. Expression of the costimulatory molecule B7-H3 is associated with prolonged survival in human pancreatic cancer. BMC Cancer. 2009;9:463. doi: 10.1186/1471-2407-9-463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen Y, Sun J, Zhao H, Zhu D, Zhi Q, Song S, et al. The coexpression and clinical significance of costimulatory molecules B7-H1, B7-H3, and B7-H4 in human pancreatic cancer. Onco Targets Ther. 2014;7:1465–72. doi: 10.2147/OTT.S66809. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 16.Yamato I, Sho M, Nomi T, Akahori T, Shimada K, Hotta K, et al. Clinical importance of B7-H3 expression in human pancreatic cancer. Br J Cancer. 2009;101:1709–16. doi: 10.1038/sj.bjc.6605375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhao XIN, Li D-C, Zhu X-G, Gan W-J, Li ZHI, Xiong F, et al. B7-H3 overexpression in pancreatic cancer promotes tumor progression. Int J Mol Med. 2013;31:283–91. doi: 10.3892/ijmm.2012.1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sabbatino F, Wang Y, Wang X, Schwab JH, Ferrone S, Ferrone CR. Novel tumor antigen-specific monoclonal antibody-based immunotherapy to eradicate both differentiated cancer cells and cancer-initiating cells in solid tumors. Semin Oncol. 2014;41:685–99. doi: 10.1053/j.seminoncol.2014.08.007. [DOI] [PubMed] [Google Scholar]
- 19.Kasten B, Katre A, Kim H, Arend R, Fan J, Ferrone S, et al. Targeted radioimmunotherapy with B7-H3-specific 212Pb-mAb 376.96 in models of human ovarian cancer. J Nucl Med. 2016;57:55. [Google Scholar]
- 20.Kasten BB, Arend RC, Katre AA, Kim H, Fan J, Ferrone S, et al. B7-H3-targeted 212Pb radioimmunotherapy of ovarian cancer in preclinical models. Nucl Med Biol. 2017;47:23–30. doi: 10.1016/j.nucmedbio.2017.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Baidoo KE, Milenic DE, Brechbiel MW. Methodology for labeling proteins and peptides with lead-212 (212Pb) Nucl Med Biol. 2013;40:592–9. doi: 10.1016/j.nucmedbio.2013.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Imai K, Wilson BS, Bigotti A, Natali PG, Ferrone S. A 94,000-dalton glycoprotein expressed by human melanoma and carcinoma cells. J Natl Cancer Inst. 1982;68:761–9. [PubMed] [Google Scholar]
- 23.Perosa F, Ferrone S. Syngeneic anti-idiotypic antisera to murine anti-HLA Class II monoclonal antibodies. J Immunol. 1987;139:1232–9. [PubMed] [Google Scholar]
- 24.Chappell LL, Dadachova E, Milenic DE, Garmestani K, Wu C, Brechbiel MW. Synthesis, characterization, and evaluation of a novel bifunctional chelating agent for the lead isotopes 203Pb and 212Pb. Nucl Med Biol. 2000;27:93–100. doi: 10.1016/s0969-8051(99)00086-4. [DOI] [PubMed] [Google Scholar]
- 25.Dadachova E, Chappell LL, Brechbiel MW. Spectrophotometric method for determination of bifunctional macrocyclic ligands in macrocyclic ligand-protein conjugates. Nucl Med Biol. 1999;26:977–82. doi: 10.1016/s0969-8051(99)00054-2. [DOI] [PubMed] [Google Scholar]
- 26.Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the folin phenol reagent. J Biol Chem. 1951;193:265–75. [PubMed] [Google Scholar]
- 27.Kim H, Zhai G, Liu Z, Samuel S, Shah N, Helman EE, et al. EMMPRIN as a novel target for pancreatic cancer therapy. Anticancer Drugs. 2011;22:864–74. doi: 10.1097/CAD.0b013e328349311e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Perera RM, Stoykova S, Nicolay BN, Ross KN, Fitamant J, Boukhali M, et al. Transcriptional control of autophagy-lysosome function drives pancreatic cancer metabolism. Nature. 2015;524:361–5. doi: 10.1038/nature14587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dalerba P, Cho RW, Clarke MF. Cancer stem cells: Models and concepts. Annu Rev Med. 2007;58:267–84. doi: 10.1146/annurev.med.58.062105.204854. [DOI] [PubMed] [Google Scholar]
- 30.Schultz MJ, Holdbrooks AT, Chakraborty A, Grizzle WE, Landen CN, Buchsbaum DJ, et al. The tumor-associated glycosyltransferase ST6Gal-I regulates stem cell transcription factors and confers a cancer stem cell phenotype. Cancer Res. 2016;76:3978–88. doi: 10.1158/0008-5472.CAN-15-2834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bose RN, Maurmann L, Mishur RJ, Yasui L, Gupta S, Grayburn WS, et al. Non-DNA-binding platinum anticancer agents: Cytotoxic activities of platinum–phosphato complexes towards human ovarian cancer cells. Proc Natl Acad Sci U S A. 2008;105:18314–9. doi: 10.1073/pnas.0803094105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kim H, Samuel S, Lopez-Casas P, Grizzle W, Hidalgo M, Kovar J, et al. SPARC-independent delivery of nab-paclitaxel without depleting tumor stroma in patient-derived pancreatic cancer xenografts. Mol Cancer Ther. 2016;15:680–8. doi: 10.1158/1535-7163.MCT-15-0764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kim H, Folks KD, Guo L, Sellers JC, Fineberg NS, Stockard CR, et al. Early therapy evaluation of combined cetuximab and irinotecan in orthotopic pancreatic tumor xenografts by dynamic contrast-enhanced magnetic resonance imaging. Mol Imaging. 2011;10:153–67. [PMC free article] [PubMed] [Google Scholar]
- 34.Horak E, Hartmann F, Garmestani K, Wu C, Brechbiel M, Gansow OA, et al. Radioimmunotherapy targeting of HER2/neu oncoprotein on ovarian tumor using lead-212-DOTA-AE1. J Nucl Med. 1997;38:1944–50. [PubMed] [Google Scholar]
- 35.Meredith R, Torgue J, Shen S, Fisher DR, Banaga E, Bunch P, et al. Dose escalation and dosimetry of first-in-human α radioimmunotherapy with 212Pb-TCMC-trastuzumab. J Nucl Med. 2014;55:1636–42. doi: 10.2967/jnumed.114.143842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Meredith RF, Torgue J, Azure MT, Shen S, Saddekni S, Banaga E, et al. Pharmacokinetics and imaging of 212Pb-TCMC-trastuzumab after intraperitoneal administration in ovarian cancer patients. Cancer Biother Radiopharm. 2014;29:12–7. doi: 10.1089/cbr.2013.1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kasten BB, Azure MT, Schoeb TR, Fisher DR, Zinn KR. Imaging, biodistribution, and toxicology evaluation of 212Pb-TCMC-trastuzumab in nonhuman primates. Nucl Med Biol. 2016;43:391–6. doi: 10.1016/j.nucmedbio.2016.04.001. [DOI] [PubMed] [Google Scholar]
- 38.Meredith RF, Torgue JJ, Rozgaja TA, Banaga EP, Bunch PW, Alvarez RD, et al. Safety and outcome measures of first-in-human intraperitoneal α radioimmunotherapy with 212Pb-TCMC-trastuzumab. Am J Clin Oncol. 2016 doi: 10.1097/COC.0000000000000353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kukis DL, DeNardo GL, DeNardo SJ, Mirick GR, Miers LA, Greiner DP, et al. Effect of the extent of chelate substitution on the immunoreactivity and biodistribution of 2IT-BAT-Lym-1 immunoconjugates. Cancer Res. 1995;55:878–84. [PubMed] [Google Scholar]
- 40.Al-Ejeh F, Darby JM, Thierry B, Brown MP. A simplified suite of methods to evaluate chelator conjugation of antibodies: Effects on hydrodynamic radius and biodistribution. Nucl Med Biol. 2009;36:395–402. doi: 10.1016/j.nucmedbio.2009.01.001. [DOI] [PubMed] [Google Scholar]
- 41.Miao Y, Hylarides M, Fisher DR, Shelton T, Moore H, Wester DW, et al. Melanoma therapy via peptide-targeted α-radiation. Clin Cancer Res. 2005;11:5616–21. doi: 10.1158/1078-0432.CCR-05-0619. [DOI] [PubMed] [Google Scholar]
- 42.Su FM, Beaumier P, Axworthy D, Atcher R, Fritzberg A. Pretargeted radioimmunotherapy in tumored mice using an in vivo 212Pb/212Bi generator. Nucl Med Biol. 2005;32:741–7. doi: 10.1016/j.nucmedbio.2005.06.009. [DOI] [PubMed] [Google Scholar]
- 43.Mirzadeh S, Kumar K, Gansow Otto A. The Chemical Fate of 212Bi-DOTA Formed by β− Decay of 212Pb(DOTA)2−. Radiochim Acta. 1993:1–10. [Google Scholar]
- 44.Milenic D, Molinolo A, Solivella M, Banaga E, Torgue J, Besnainou S, et al. Toxicological studies of 212Pb intravenously or intraperitoneally injected into mice for a phase 1 trial. Pharmaceuticals. 2015;8:416–34. doi: 10.3390/ph8030416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.de Kruijff RM, Wolterbeek HT, Denkova AG. A critical review of alpha radionuclide therapy—how to deal with recoiling daughters? Pharmaceuticals. 2015;8:321–36. doi: 10.3390/ph8020321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Milenic DE, Baidoo KE, Shih JH, Wong KJ, Brechbiel MW. Evaluation of platinum chemotherapy in combination with HER2-targeted α-particle radiation. Cancer Biother Radiopharm. 2013;28:441–9. doi: 10.1089/cbr.2012.1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yong KJ, Milenic DE, Baidoo KE, Brechbiel MW. Sensitization of tumor to 212Pb radioimmunotherapy by gemcitabine involves initial abrogation of G2 arrest and blocked DNA damage repair by interference with Rad51. Int J Radiat Oncol, Biol, Phys. 2013;85:1119–26. doi: 10.1016/j.ijrobp.2012.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sharkey RM, Karacay H, Govindan SV, Goldenberg DM. Combination radioimmunotherapy and chemoimmunotherapy involving different or the same targets improves therapy of human pancreatic carcinoma xenograft models. Mol Cancer Ther. 2011;10:1072–81. doi: 10.1158/1535-7163.MCT-11-0115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Seaman S, Stevens J, Yang MY, Logsdon D, Graff-Cherry C, St Croix B. Genes that distinguish physiological and pathological angiogenesis. Cancer Cell. 2007;11:539–54. doi: 10.1016/j.ccr.2007.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Crispen PL, Sheinin Y, Roth TJ, Lohse CM, Kuntz SM, Frigola X, et al. Tumor cell and tumor vasculature expression of B7-H3 predict survival in clear cell renal cell carcinoma. Clin Cancer Res. 2008;14:5150–7. doi: 10.1158/1078-0432.CCR-08-0536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zang X, Sullivan PS, Soslow RA, Waitz R, Reuter VE, Wilton A, et al. Tumor associated endothelial expression of B7-H3 predicts survival in ovarian carcinomas. Mod Pathol. 2010;23:1104–12. doi: 10.1038/modpathol.2010.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Milenic DE, Garmestani K, Brady ED, Albert PS, Ma D, Abdulla A, et al. Targeting of HER2 antigen for the treatment of disseminated peritoneal disease. Clin Cancer Res. 2004;10:7834–41. doi: 10.1158/1078-0432.CCR-04-1226. [DOI] [PubMed] [Google Scholar]
- 53.Qu CF, Song EY, Li Y, Rizvi SMA, Raja C, Smith R, et al. Pre-clinical study of 213Bi labeled PAI2 for the control of micrometastatic pancreatic cancer. Clin Exp Metastasis. 2005;22:575–86. doi: 10.1007/s10585-005-5788-9. [DOI] [PubMed] [Google Scholar]
- 54.Qu CF, Song YJ, Rizvi SMA, Li Y, Smith R, Perkins A, et al. In vivo and in vitro inhibition of pancreatic cancer growth by targeted alpha therapy using 213Bi-CHX.A”-C595. Cancer Biol Ther. 2005;4:848–53. doi: 10.4161/cbt.4.8.1892. [DOI] [PubMed] [Google Scholar]
- 55.Allen BJ, Abbas Rizvi SM, Qu CF, Smith RC. Targeted alpha therapy approach to the management of pancreatic cancer. Cancers. 2011;3:1821–43. doi: 10.3390/cancers3021821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bryan RA, Jiang Z, Jandl T, Strauss J, Koba W, Onyedika C, et al. Treatment of experimental pancreatic cancer with 213-bismuth-labeled chimeric antibody to single-strand DNA. Expert Rev Anticancer Ther. 2014;14:1243–9. doi: 10.1586/14737140.2014.952285. [DOI] [PubMed] [Google Scholar]
- 57.Horev-Drori G, Cooks T, Bittan H, Lazarov E, Schmidt M, Arazi L, et al. Local control of experimental malignant pancreatic tumors by treatment with a combination of chemotherapy and intratumoral 224Radium-loaded wires releasing alpha-emitting atoms. Transl Res. 2012;159:32–41. doi: 10.1016/j.trsl.2011.08.009. [DOI] [PubMed] [Google Scholar]
- 58.Bäck T, Chouin N, Lindegren S, Kahu H, Jensen H, Albertsson P, et al. Cure of human ovarian carcinoma solid xenografts by fractionated α-radioimmunotherapy with 211At-MX35-F(ab′)2: Influence of absorbed tumor dose and effect on long-term survival. J Nucl Med. 2017;58:598–604. doi: 10.2967/jnumed.116.178327. [DOI] [PubMed] [Google Scholar]
- 59.Picarda E, Ohaegbulam KC, Zang X. Molecular pathways: Targeting B7-H3 (CD276) for human cancer immunotherapy. Clin Cancer Res. 2016;22:3425–31. doi: 10.1158/1078-0432.CCR-15-2428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Castellanos JR, Purvis IJ, Labak CM, Guda MR, Tsung AJ, Velpula KK, et al. B7-H3 role in the immune landscape of cancer. Am J Clin Exp Immunol. 2017;6:66–75. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.