

Abstract
Mutations in the metabolic enzyme isocitrate dehydrogenase 1 (IDH1) are commonly found in gliomas. AGI-5198, a potent and selective inhibitor of the mutant IDH1 enzyme, was radiolabeled with radioiodine and fluorine-18. These radiotracers were evaluated as potential probes for imaging mutant IDH1 expression in tumors with positron emission tomography (PET). Radioiodination of AGI-5198 was achieved using a tin precursor in 79 ± 6% yield (n=9), and 18F-labeling was accomplished by the Ugi reaction in a decay-corrected radiochemical yield of 2.6 ± 1.6% (n=5). The inhibitory potency of the analogous nonradioactive compounds against mutant IDH1 (IDH1-R132H) was determined in enzymatic assays. Cell uptake studies using radiolabeled AGI-5198 analogues revealed somewhat higher uptake in IDH1-mutated cells than that in wild-type IDH1 cells. The radiolabeled compounds displayed favorable tissue distribution characteristics in vivo, and good initial uptake in IDH1-mutated tumor xenografts; however, tumor uptake decreased with time. Radioiodinated AGI-5198 exhibited higher tumor-to-background ratios compared with 18F-labeled AGI-5198; unfortunately, similar results were observed in wild-type IDH1 tumor xenografts as well, indicating lack of selectivity for mutant IDH1 for this tracer. These results suggest that AGI-5198 analogues are not a promising platform for radiotracer development. Nonetheless, insights gained from this study may help in design and optimization of novel chemical scaffolds for developing radiotracers for imaging the mutant IDH1 enzyme.
Keywords: IDH mutation, PET imaging, glioma, phenyl-glycine, IDH1 inhibitor
Graphical Abstract
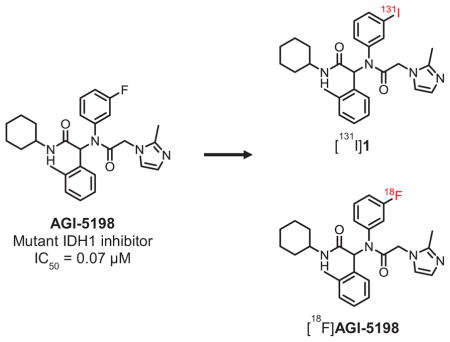
Malignant gliomas frequently harbor mutations in the gene encoding for the metabolic enzyme isocitrate dehydrogenase 1 (IDH1).1 Studies suggest that IDH1 mutations contribute to epigenetic dysregulation and widespread metabolic changes in tumor cells, including depletion of key cellular biochemicals such as glutamine and glutamine-derived metabolites.2, 3 Most of these effects are mediated by the metabolite D-2-hydroxyglutarate (D-2-HG), which is generated by mutated IDH1 and accumulates in tumor cells at levels ~100-fold higher than those found in wild-type cells.4, 5 IDH1 mutations impair the normal catalytic function of the enzyme - its ability to convert isocitrate to α-ketoglutarate (α-KG) - and confer a new function that enables the IDH1 mutant to recognize α-KG as a substrate and reduce it to D-2-HG.1, 4 IDH1 mutations are present in up to 80% of World Health Organization (WHO) grades II and III gliomas and in secondary glioblastoma, but are seldom found in primary (de novo) glioblastoma.1, 6 IDH mutations are also present in several other cancers including acute myeloid leukemia (AML),7 intrahepatic cholangiocarcinoma,8 Ollier disease and Maffucci syndrome,9 suggesting a causative role played by IDH mutations in cancers.
Over the past few years, IDH1 mutations (IDH1-R132H, IDH1-R132C) have emerged as novel and highly promising drug targets in gliomas. Several pharmaceutical companies have active drug development programs targeting IDH1 mutations, and most of these programs are based on small-molecule IDH1 inhibitors representing a variety of chemical scaffolds.10 Preclinical studies using prototypical compounds from some of these chemical classes have established that small-molecule inhibitors can bind to the mutant IDH1 enzyme and inhibit its ability to produce D-2-HG, thereby reversing the effects of IDH mutations on cancer cells that harbor these mutations.11 Compounds based on a phenyl-glycine chemical scaffold were among the first chemical analogues that were identified as potent inhibitors of mutant IDH1 enzymes. High-throughput screening identified compounds with half-maximal inhibitory concentration (IC50) values of ≤ 0.1 μM for the most potent analogues against IDH1-R132H, the most commonly occurring IDH-mutation in glioma.12 The most extensively studied compound from the phenyl-glycine series, AGI-5198 (IC50: 0.07 μM), has been shown to inhibit D-2-HG production and have anti-tumor effects in IDH1-mutated glioma xenografts in vivo.11, 12 AGI-5198 also has very high selectivity for mutant IDH1 vs. wild-type IDH1 indicated by its lack of inhibitory potency against wild-type IDH1 (IC50: >100 μM)12. With the goal of developing radiolabeled compounds for noninvasive imaging of IDH1 mutations in gliomas with positron emission tomography (PET), we have synthesized radiolabeled analogues of AGI-5198 and evaluated their potential as radiotracers for this purpose. AGI-5198 was labeled with radioiodine or fluorine-18 and the labeled compounds were evaluated for their ability to bind to IDH1-mutated tumor cells in vitro and in vivo.
In addition to its high potency and selectivity for mutant IDH1, AGI-5198 offers additional advantage for radiolabeling - it has a fluorine atom which could be replaced with radioactive halogens, fluorine-18 (18F, t1/2: 110 min) or iodine-124 (124I, t1/2: 4.2 d), for PET imaging purposes. Structure-activity relationship (SAR) studies of phenyl-glycine analogues originally published by Popovici-Muller et al. showed that replacement of the 2-methylimidazole moiety in AGI-5198 with a phenyl or a heterocyclic function was well-tolerated in terms of inhibitory potency against IDH1-R132H.12 Thus, in addition to AGI-5198 and its iodo-analogue 1, compounds 2 and 3 were synthesized to explore the possibility of introducing 18F on a different substituent and also to better understand the SAR for radiotracer development (Scheme 1). For radiolabeling, the fluorine atom in compound 2 could be replaced with 18F by an aromatic nucleophilic substitution reaction on precursors containing a suitable leaving group (e.g., nitro, bromo).13 Similarly, 18F labeling of compound 4 could be achieved by using a sulfonate precursor or by a fluoroalkylation reaction using [18F]fluoroethyl bromide ([18F]FEtBr) on a phenol precursor.14 AGI-5198 as well as other nonradioactive reference compounds (1–3) were synthesized and evaluated in racemic form similar to previously published reports, and as shown in Scheme 1.12, 15 First, the chloroacetamide intermediate bearing a fluoro or iodo function on the meta-position of the phenyl ring was constructed by four-component Ugi reaction starting from 3-fluoroaniline or 3-iodoaniline, respectively. Next, the chloroacetamide intermediates were reacted with either 2-methylimidazole for AGI-5198 and 1, or the corresponding aromatic amine derivative for 2 and 3 to obtain the final compounds. The synthesis of the chloroacetamide derivatives in the Ugi-reaction was achieved in about 65% yield, and substitution of the chloro function with the corresponding amine analogue in compounds 1–3 was accomplished in 59–81% yield.
Scheme 1.

Synthesis scheme for the nonradioactive phenyl-glycine analogues.
The inhibitory activity of the unlabeled analogues against IDH1-R132H was evaluated in enzymatic assays as previously described using serial twofold dilutions (n=12) of the test compounds in the presence of the mutant IDH1 substrate α-KG, and by the diaphorase/resazurin detection system.14 The inhibitory potency (IC50) and maximal inhibitory capacity of the compounds were derived from the inhibitory response curves for each compound (Figure 1A, Table 1). These studies revealed a robust inhibition of mutant IDH1 by AGI-5198 and the iodo analogue 1, with IC50 values <0.05 μM for the two compounds. The inhibitory potency of AGI-5198 observed in the present study is in good agreement with that reported in the literature (IC50: 0.03 μM vs. 0.07 μM).12 Excellent retention of inhibitory activity for the iodo analogue indicates that replacement of fluorine with iodine on the phenyl ring is well-tolerated with respect to binding affinity for the mutant IDH1 enzyme. However, replacement of 2-methylimidazole in AGI-5198 with a 6-fluoropyridine-3-amine function in compound 2 decreased the inhibitory potency of the compound significantly (IC50: 1.4 μM). A further decrease in the inhibitory potency was observed for the fluoroethoxy analogue 3, which displayed relatively poor enzyme inhibition (IC50: 26.3 μM). At all tested concentrations, the inhibitory activity of the iodo analogue 1 was slightly more than that for the fluoro analogue AGI-5198, with enzyme inhibition reaching 100% at 6.12 μM compared with 12.25 μM for AGI-5198. For comparison, the fluoropyridine analogue 2 and the fluoroethoxy compound 3 displayed a maximal inhibition of 71.3 ± 5.3% (at 12.25 μM) and 34.3 ± 13.1% (at 98 μM), respectively, suggesting limited inhibitory potency for compounds 2 and 3 against the mutant IDH1 (Figure 1B). Based on their higher inhibitory potency, compounds 1 and 2 were selected for radiolabeling and further evaluation in mutant IDH tumor models.
Figure 1.

(A) Inhibitory activity curves for AGI-5198 and the synthesized analogues against IDH1-R132H in enzymatic assays. (B) Inhibitory efficiency shown as the maximal enzyme inhibition observed for the nonradioactive analogues in experiments depicted in A. Data are presented as mean ± SEM.
Table 1.
Structural characteristics and inhibitory potency against R132H mutant IDH1 for the synthesized compounds.
| Compound | M. Wt. | clogP | Polar Surface Area (PSA) | IC50 vs. IDH1-R132H (μM) |
|---|---|---|---|---|
| AGI-5198 | 462.6 | 4.7 | 65.0 | 0.03 |
| 1 | 570.5 | 5.7 | 65.0 | <0.01 |
| 2 | 492.6 | 5.8 | 73.8 | 1.39 |
| 3 | 535.6 | 6.4 | 70. 7 | 26.31 |
Radioiodination of 1 was achieved from a tin precursor, which was synthesized from the nonradioactive iodo compound 1 following a previously reported method.16 The tin precursor was purified by preparative TLC twice, and aliquots of 100 μg were used for radiolabeling by an electrophilic substitution reaction using hydrogen peroxide as the oxidizing agent in the presence of acetic acid (Scheme 2A).16 Compound 1 was labeled with 125I (t1/2: 59.4 d) for in vitro studies and 131I (t1/2: 8 d) for in vivo studies, with a radiolabeling efficiency of 79 ± 6% (n=9; results for two radionuclides combined). For 18F labeling of AGI-5198 by aromatic nucleophilic substitution, our efforts to synthesize a diaryliodonium salt precursor using a trimethyltin compound and Koser’s reagent17 did not yield the desired product in reasonable yields or sufficient purity for radiolabeling. Hence, [18F]AGI-5198 was constructed by the Ugi reaction similar to that described for the chloroacetamide intermediates, and a modification of a previously reported method for the radio-Ugi reaction.12, 18 For this reaction, 3-[18F]fluoroaniline19 was used as the prosthetic group and was reacted with the other Ugi components at 100°C in ethanol (Scheme 2B). After radiolabeling, the Ugi reaction mixture was purified by semi-preparative HPLC to obtain [18F]AGI-5198 in a decay-corrected radiochemical yield of 2.6 ± 1.6% (n=5) and with >98% purity (Supplementary Figures 1 and 2).
Scheme 2.

Synthesis scheme for labeling AGI-5198 with radioiodine (A) and fluorine-18 (B).
Cell uptake studies with radioiodinated 1 and [18F]AGI-5198 were conducted in tumor cell lines expressing the IDH1-R132H mutation. Two cell lines were studied; the HCT116 colorectal carcinoma cell line genetically engineered to express the heterozygous IDH1-R132H mutant enzyme (IDH1R132H/WT), and an IDH1-mutated astrocytoma cell line (IMA) containing a native IDH1-R132H mutation (IDH1R132H/WT).20, 21 Isogeneic cell lines expressing only wild-type IDH1 (WT-IDH1) were used as controls.14 As reported previously, these mutant IDH1 cell lines have 80–100-fold elevated levels of D-2-HG compared to those in the corresponding WT-IDH1 cell line, confirming the functional activity of mutant IDH1 in these cell lines.20, 21 Cell uptake studies revealed higher uptake of radioiodinated 1 in IDH1-mutated tumor cell lines to some extent (left panels, Figure 2). Uptake of radioactivity was 1.7–2.1-fold higher in the IMA cell line and 1.3–1.5-fold higher in the IDH1-mutated HCT-116 cell line, compared to the corresponding WT-IDH1 cell line controls. For [18F]AGI-5198, the uptake in the IDH1-mutated HCT116 cell line was significantly higher at all time points with uptake ratios increasing from about 1.6 at 5 min to 2.0 at 30 min, remaining at that level until 120 min. However, in the IMA cell line, the uptake of [18F]AGI-5198 was higher only at 5 min but not thereafter (right panels, Figure 2). Blocking studies conducted with radioiodinated 1 conducted in parallel revealed an increase in uptake of the labeled compound in the presence of an excess of nonradioactive 1 (50 μM) in both IDH1-mutated and WT-IDH1 cell lines. In general, the uptake increased by a factor of 1.4–1.7 for the IMA cell line and 1.5–1.8 for its isogenic WT-IDH1 cell line, but the uptake ratios between the mutant IDH1 and WT-IDH1 cell lines remained very similar (1.8–2.0) to those for the no-carrier-added experiments (Supplementary Figure 3).
Figure 2.

Uptake of radioiodinated 1 ([125I]1) and 18F-labeled AGI-5198 in isogenic tumor cell lines expressing IDH1-R132H or wild-type IDH1. Upper panels: Uptake of labeled compounds in the IDH1-mutated HCT116 cell line; Lower panels: uptake in the IDH1-mutated astrocytoma (IMA) cell line. Data are mean ± SEM for three wells. *P < 0.05 (Student’s t-test).
Although these results were unexpected, the modest uptake ratios and the increased uptake of radioiodinated 1 in the presence of nonradioactive 1 is consistent with possible competitive inhibition of the radiolabeled compounds for binding to mutant IDH1 by either endogenous substrates or ligands. It has been shown previously that the mode of binding of phenyl-glycine-based inhibitors to IDH1-R132H is reversible and competitive with respect to the endogenous substrate α-KG, and uncompetitive with respect to the co-factor NADPH.12, 15 Additionally, a recent report suggests that phenyl-glycine analogues also may bind to an allosteric metal binding pocket on the mutant IDH1 enzyme (R132H). Moreover, the binding is competitive with respect to the catalytically-essential magnesium ion (Mg2+).22 At radiotracer levels, the molar concentration of radioiodinated 1 or [18F]AGI-5198 in cells is expected to be much lower than the concentration of magnesium ion (0.1–1 mM)22 or α-KG (11–161 μmol per gram in glioma)4. Thus, it is likely that radiolabeled phenyl-glycine analogues synthesized by no-carrier-added radiosynthetic methods cannot compete at the mutant IDH1 binding pocket(s) with the substrate α-KG or magnesium ion. Although this hypothesis needs to be tested further, this could explain, at least in part, the lower uptake levels and uptake ratios of radioiodinated 1 and [18F]AGI-5198 in cell uptake studies, despite their high inhibitory potency against mutant IDH1 (IC50 < 0.05 μM). In view of their limited uptake and uptake ratios in the cell uptake studies, we have not attempted to achieve enantiomeric separation of the radiolabeled compounds by chiral-HPLC. However, it has been reported previously for other phenyl-glycine analogues that the (S)-enantiomer has higher potency, essentially responsible for all of the enzymatic activity, compared to the (R)-enantiomer,12, 15 suggesting that enantioselective synthesis and evaluation might improve the uptake and/or uptake ratios for these tracers by a factor of 2, assuming similar non-specific binding for the two stereoisomers.
Next, radioiodinated 1 ([131I]1) and [18F]AGI-5198 were evaluated in separate groups of athymic mice bearing IDH1-mutated HCT116 tumor xenografts to investigate their biodistribution characteristics in an IDH1-R132H-expressing tumor model. Figure 3 shows the tissue distribution results obtained between 0.5 and 4 h post injection (n=5 per time point), expressed as percentage injected dose per gram (% ID/g). Comparison of the tissue distribution data for [131I]1 and [18F]AGI-5198 revealed similar distribution profiles for the two compounds, with both exhibiting rapid clearance of radioactivity from blood and major organs with time. Although radioiodinated 1 showed somewhat lower blood activity at 30 min (0.35 ± 0.03% vs. 0.88 ± 0.16% ID/g for [18F]AGI-5198), the radioactivity levels in the blood were very similar for the two compounds at 4 h (~0.1% ID/g). As would be expected for lipophilic small molecules, the labeled phenyl-glycine analogues were eliminated primarily by the hepatobiliary system, with liver and intestines accounting for 61 ± 18% of the injected dose (% ID) for [131I]1 and 87 ± 7% ID for [18F]AGI-5198 at 4 h. The initial tumor uptake was also very similar for the two labeled compounds - 0.93 ± 0.24 % ID/g for [131I]1 and 0.84 ± 0.18 % ID/g for [18F]AGI-5198 at 0.5 h. The tumor uptake decreased at later time points, to 0.22 ± 0.11% ID/g for [131I]1 and 0.33 ± 0.08% ID/g for [18F]AGI-5198 at 4 h. With [131I]1, the tumor uptake was significantly higher than that for the blood and muscle, resulting in peak tumor-to-blood ratios (TBR) of 4.6 ± 2.0 at 2 h, and peak tumor-to-muscle ratios (TMR) of 5.0 ± 1.3 at 4 h. In comparison, [18F]AGI-5198 exhibited peak TBRs of 3.0 ± 1.3 (4 h) and TMRs of 2.5 ± 1.0 (2 h), somewhat lower than those for [131I]1. Consistent with the lack of blood-brain-barrier (BBB) permeability reported for other phenyl-glycine analogues (e.g., ML-309) in normal mice,15 the uptake and retention of the labeled compounds in brain was very low. The low bone uptake for [18F]AGI-5198 and low thyroid uptake for [131I]1 indicated that these compounds are stable against dehalogenation in vivo (Figure 3).
Figure 3.

Tissue distribution of radioiodinated 1 ([131I]1) (A) and [18F]AGI-5198 (B) in athymic mice bearing HCT116 IDH1-R132H tumor xenografts in the flank region. Data are presented as mean ± SEM for five animals at each time point.
In order to examine the selectivity of tracer uptake in mutant-IDH1 HCT116 tumor xenografts, [131I]1 also was evaluated in WT-IDH1 tumor- bearing mice at 1–4 h (Figure 4A). Comparison of the [131I]1 % ID/g values observed in the mutant-IDH1- and WT-IDH1 HCT116 tumor xenografts revealed no significant differences between the two tumor models (Figure 4B). The tumor-to-muscle ratios were also very similar for the mutant- and WT-IDH1 tumors at 1–4 h postinjection (Figure 4C), suggesting a lack of selectivity of [131I]1 for IDH1-R132H in this xenograft model. These results, combined with the in vitro data, indicate that [131I]1 (or its labeled catabolite) likely binds to the WT-IDH1 enzyme as well in tumors. Although the reported IC50 of AGI-5198 against WT-IDH1 is much higher than that for mutant IDH1 (>100 μM vs. 0.07 μM against IDH1-R132H), it is worth noting that the IC50 values are derived from different types of assays – the IC50 reflect the ability of the compound to inhibit enzymatic activity (i.e. isocitrate→α-KG conversion in case of WT-IDH1 assays and α-KG→2-HG conversion in mutant IDH1 assays)12, 14 while the cell-based assays used in the present study reveal the direct binding of the radiolabeled AGI-5198 analogues to mutant- and WT-IDH1 enzyme expressing tumor cell lines.
Figure 4.

(A) Tissue distribution of radioiodinated 1 ([131I]1) in WT-IDH1 tumor xenografts. (B) Comparison of tumor uptake (% ID/g) for radioiodinated 1 in mutant IDH1 vs. WT-IDH1 tumors (HCT116). (C) Comparison of tumor-to-muscle ratios for radioiodinated 1 in in mutant IDH1 vs. WT-IDH1 tumors. Data are presented as mean ± SEM for five animals at each time point.
The aim of this study was to investigate the potential usefulness of the phenyl-glycine scaffold for developing radiotracers to image IDH1-R132H. We selected AGI-5198 as the lead compound for radiotracer design and development because of its high inhibitory potency against IDH1-R132H.12 Moreover, previous studies have demonstrated the ability of AGI-5198 to decrease D-2-HG production and inhibit tumor growth in IDH1-R132H mutant glioma xenografts,11, 12 indicating cellular target engagement of mutant IDH1 by AGI-5198 in vivo. In view of its higher inhibitory potency, higher uptake ratios in the clinically relevant IMA cell line, and the better tumor-to-background ratios obtained with [131I]1, further evaluation of this compound in astrocytoma tumor models in vivo might be warranted. However, our data suggests that compounds with binding to the mutant IDH1 enzyme that is competitive with respect to the substrate α-KG or other cellular species (e.g., Mg2+) are not ideal for development as radiotracers for mutant IDH1 imaging. In addition, the candidate radiotracers for imaging mutant IDH1 expression in gliomas should fulfill the general criteria for CNS PET radiotracers, including optimal lipophilicity (e.g., logD = 2.0–3.5), ability to enter brain in sufficient concentration, high binding affinity for the mutant IDH1 enzyme (KD ideally <10 nM), adequate metabolic stability in vivo, low nonspecific binding, and minimal or no binding to off-target proteins in the brain.23–25 Based on insights gained from this study, we are currently evaluating different chemical scaffolds whose analogues have been shown to bind to an allosteric binding pocket on mutant IDH1 and exert noncompetitive inhibition with respect to the substrate α-KG. This should circumvent potential interference from the substrate for radiotracer binding to the mutant enzyme in IDH-mutated gliomas.26, which we have shown to be problematic with the phenyl-glycine scaffold in the current study. Results with these new scaffolds will be reported separately.
Supplementary Material
Acknowledgments
We thank Genglin Jin and Christopher G. Duncan for derivation of the cell lines used in this work, and Dr. Xiao-Guang Zhao for help with the tissue distribution studies. This work was supported by the Southeastern Brain Tumor Foundation and the National Institutes of Health (CA182025).
Footnotes
Conflicts of Interest
None
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Yan H, Parsons DW, Jin GL, et al. IDH1 and IDH2 Mutations in Gliomas. N Engl J Med. 2009;360:765–773. doi: 10.1056/NEJMoa0808710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Reitman ZJ, Jin G, Karoly ED, et al. Profiling the effects of isocitrate dehydrogenase 1 and 2 mutations on the cellular metabolome. Proc Natl Acad Sci USA. 2011;108:3270–3275. doi: 10.1073/pnas.1019393108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Waitkus MS, Diplas BH, Yan H. Isocitrate dehydrogenase mutations in gliomas. Neuro Oncol. 2016;18:16–26. doi: 10.1093/neuonc/nov136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dang L, White DW, Gross S, et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature. 2009;462:739–744. doi: 10.1038/nature08617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gross S, Cairns RA, Minden MD, et al. Cancer-associated metabolite 2-hydroxyglutarate accumulates in acute myelogenous leukemia with isocitrate dehydrogenase 1 and 2 mutations. J Exp Med. 2010;207:339–344. doi: 10.1084/jem.20092506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Watanabe T, Nobusawa S, Kleihues P, Ohgaki H. IDH1 mutations are early events in the development of astrocytomas and oligodendrogliomas. Am J Pathol. 2009;174:1149–1153. doi: 10.2353/ajpath.2009.080958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mardis ER, Ding L, Dooling DJ, et al. Recurring Mutations Found by Sequencing an Acute Myeloid Leukemia Genome. N Engl J Med. 2009;361:1058–1066. doi: 10.1056/NEJMoa0903840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang P, Dong Q, Zhang C, et al. Mutations in isocitrate dehydrogenase 1 and 2 occur frequently in intrahepatic cholangiocarcinomas and share hypermethylation targets with glioblastomas. Oncogene. 2013;32:3091–3100. doi: 10.1038/onc.2012.315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pansuriya TC, van Eijk R, d’Adamo P, et al. Somatic mosaic IDH1 and IDH2 mutations are associated with enchondroma and spindle cell hemangioma in Ollier disease and Maffucci syndrome. Nat Genet. 2011;43:1256–1261. doi: 10.1038/ng.1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dang L, Su SSM. Isocitrate Dehydrogenase Mutation and (R)-2-Hydroxyglutarate: From Basic Discovery to Therapeutics Development. Annu Rev Biochem. 2017;86:305–331. doi: 10.1146/annurev-biochem-061516-044732. [DOI] [PubMed] [Google Scholar]
- 11.Rohle D, Popovici-Muller J, Palaskas N, et al. An inhibitor of mutant IDH1 delays growth and promotes differentiation of glioma cells. Science. 2013;340:626–630. doi: 10.1126/science.1236062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Popovici-Muller J, Saunders JO, Salituro FG, et al. Discovery of the First Potent Inhibitors of Mutant IDH1 That Lower Tumor 2-HG in Vivo. ACS Med Chem Lett. 2012;3:850–855. doi: 10.1021/ml300225h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dolci L, Dolle F, Jubeau S, Vaufrey F, Crouzel C. 2-[F-18]fluoropyridines by no-carrier-added nucleophilic aromatic substitution with [F-18]FK-K-222 - A comparative study. J Label Compd Radiopharm. 1999;42:975–985. [Google Scholar]
- 14.Chitneni SK, Reitman ZJ, Gooden DM, Yan H, Zalutsky MR. Radiolabeled inhibitors as probes for imaging mutant IDH1 expression in gliomas: Synthesis and preliminary evaluation of labeled butyl-phenyl sulfonamide analogs. Eur J Med Chem. 2016;119:218–230. doi: 10.1016/j.ejmech.2016.04.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Davis MI, Gross S, Shen M, et al. Biochemical, cellular, and biophysical characterization of a potent inhibitor of mutant isocitrate dehydrogenase IDH1. J Biol Chem. 2014;289:13717–13725. doi: 10.1074/jbc.M113.511030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vaidyanathan G, White B, Affleck DJ, McDougald D, Zalutsky MR. Radioiodinated O-6-Benzylguanine derivatives containing an azido function. Nucl Med Biol. 2011;38:77–92. doi: 10.1016/j.nucmedbio.2010.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pike VW, Butt F, Shah A, Widdowson DA. Facile synthesis of substituted diaryliodonium tosylates by treatment of aryltributylstannanes with Koser’s reagent. J Chem Soc, Perkin Trans 1. 1999:245–248. [Google Scholar]
- 18.Li L, Hopkinson MN, Yona RL, Bejot R, Gee AD, Gouverneur V. Convergent F-18 radiosynthesis: A new dimension for radiolabelling. Chem Sci. 2011;2:123–131. [Google Scholar]
- 19.Vasdev N, Dorff PN, O’Neil JP, Chin FT, Hanrahan S, VanBrocklin HF. Metabolic stability of 6,7-dialkoxy-4-(2-, 3-and 4-[F-18]fluoroanilino)quinazolines, potential EGFR imaging probes. Bioorganic Med Chem. 2011;19:2959–2965. doi: 10.1016/j.bmc.2011.03.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Duncan CG, Barwick BG, Jin G, et al. A heterozygous IDH1R132H/WT mutation induces genome-wide alterations in DNA methylation. Genes Dev. 2012;22:2339–2355. doi: 10.1101/gr.132738.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jin G, Reitman ZJ, Duncan CG, et al. Disruption of wild-type IDH1 suppresses D-2-hydroxyglutarate production in IDH1-mutated gliomas. Cancer Res. 2013;73:496–501. doi: 10.1158/0008-5472.CAN-12-2852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Deng G, Shen J, Yin M, et al. Selective inhibition of mutant isocitrate dehydrogenase 1 (IDH1) via disruption of a metal binding network by an allosteric small molecule. J Biol Chem. 2015;290:762–774. doi: 10.1074/jbc.M114.608497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pike VW. PET radiotracers: crossing the blood-brain barrier and surviving metabolism. Trends Pharmacol Sci. 2009;30:431–440. doi: 10.1016/j.tips.2009.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chitneni SK. SM Radiol J. 2016. IDH1 Mutations in Glioma: Considerations for Radiotracer Development; p. 2. [PMC free article] [PubMed] [Google Scholar]
- 25.Eckelman WC, Kilbourn MR, Mathis CA. Discussion of targeting proteins in vivo: in vitro guidelines. Nuc Med Biol. 2006;33:449–451. doi: 10.1016/j.nucmedbio.2006.03.008. [DOI] [PubMed] [Google Scholar]
- 26.Yen K, Travins J, Wang F, et al. AG-221, a First-in-Class Therapy Targeting Acute Myeloid Leukemia Harboring Oncogenic IDH2 Mutations. Cancer Discov. 2017;7:478–493. doi: 10.1158/2159-8290.CD-16-1034. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.