

Abstract
Background
Metastasis of upper airway microbiota may have significant implications in the development of chronic lung disease. Here, we compare bacterial communities of matched sinus and lung mucus samples from cystic fibrosis (CF) subjects undergoing endoscopic surgery for treatment of chronic sinusitis.
Methods
Mucus from one maxillary sinus and expectorated sputum were collected from twelve patients. 16S rRNA gene sequencing was then performed on sample pairs to compare the structure and function of CF airway microbiota.
Results
Bacterial diversity was comparable between airway sites, though sinuses harbored a higher prevalence of dominant microorganisms. Ordination analyses revealed that samples clustered more consistently by airway niche rather than by individual. Finally, predicted metagenomes suggested that anaerobiosis was enriched in the lung.
Conclusions
Our findings indicate that while the lung may be seeded by individual sinus pathogens, airway microenvironments harbor distinct bacterial communities that should be considered in selecting antimicrobial therapies.
Keywords: Chronic sinusitis, microbiome, microbial ecology, mucus
INTRODUCTION
Defective CFTR ion transport at the sinus epithelium leads to decreased mucociliary clearance and obstruction of sinus ostia [1]. Secondary events such as impairment of host defenses and inflammatory processes render this niche susceptible to bacterial colonization and chronic rhinosinusitis (CRS)[1,2]. Notably, a striking incidence of CRS exists among CF subjects (71–100%[3]) relative to the general population (~16%[4]). This is particularly evident in patients with class I–III mutations, who, based on radiological evidence, have a CRS incidence rate of ~100% [1].
Culture-based studies have revealed that CF-associated CRS (CF-CRS) patients harbor distinct microbiota, including Staphylococcus aureus and Haemophilus influenzae in pediatric patients, followed by Pseudomonas aeruginosa and other pathogens as patients age [5]. This dynamic generally follows the same succession of CF lung microbiota [6] and several groups have demonstrated similarities in upper and lower airway bacteriology in CF subjects [7,8]. In fact, evidence has implicated the sinuses as infection foci for lung pathogens, where they first adapt to the host before descending into the lungs [9–15]. Genotypic analyses suggest a direct exchange between sites; P. aeruginosa isolates cultured simultaneously from sinuses and lungs were genetically identical in 38 of 40 subjects [10]. These data are supported by studies of CF lung transplants, where recipient allografts were re-colonized by the same P. aeruginosa clones found prior to transplantation [11,12]. Altogether, these observations support the notion of a sinus pathogen reservoir and a unified airway, in which treatment of CRS could have profound benefits for CF lung disease management.
While P. aeruginosa and S. aureus are commonly isolated from the CF sinuses [9], culture-independent studies also suggest colonization by anaerobes (e.g. Propionibacterium) and other non-canonical pathogens [13]. These data are consistent with microbiome profiles of CF lung disease, which is recognized to have a polymicrobial etiology [6,16]. Recent sequencing studies have evaluated relationships between oral, nasal and lung microbiota in adult CF patients [17,18], but to our knowledge, molecular approaches have not been used to assess relationships between sinuses and lungs at a microbial community level. From a clinical perspective, these data could be of considerable therapeutic benefit; sinus culture can be invasive and time consuming, and to date, sputum culture-guided therapies for CRS have been largely ineffective, with many subjects ultimately requiring surgical intervention [19,20]. Therefore, a deeper understanding of microbiological relationships between the sinuses and lungs may not only improve upon CF-CRS management, but may also motivate the use of less-invasive sinonasal sampling to help inform patient therapy [8,9,21].
Here, we used 16S rRNA gene sequencing to compare the bacterial composition of upper (sinus) and lower (lung) airways in CF patients undergoing endoscopic sinus surgery. The primary objective was to compare bacterial community diversity between sites, though we also performed predictive metagenomic profiling to assess differences in bacterial phenotypes throughout the airways. We discuss these findings in the context of using sputum versus sinus microbiota composition to steer targeted therapies for upper airway infection.
MATERIALS AND METHODS
Patient cohort and specimen collection
Twelve participants with CF undergoing functional endoscopic sinus surgery (FESS) were recruited at the University of Minnesota Department of Otolaryngology. Informed consent was obtained from all subjects. Prior to FESS, each patient spit to discard saliva, followed by sputum expectoration into a collection tube. All samples were assessed macroscopically for salivary contamination, and none were rejected. Sinus secretions were obtained from a single maxillary sinus (middle meatal region) under endoscopic visualization by suction into mucus traps (Cardinal Health, Dublin, OH). Clinical data were also obtained (Table S1), including CFTR genotype, FESS procedures, clinical cultures, spirometry (FEV1%) and sinonasal outcome test (SNOT-22) scores. The UMN Institutional Review Board approved these studies (#1403M49021).
Quantitative PCR
Bacterial burden was estimated by quantifying 16S copy number from DNA extracted from clinical specimens using quantitative PCR. Reactions were prepared in triplicate using QuantiTect SYBR Green (Qiagen) and Universal 16S rRNA qPCR primers [22]. Details can be found in the supplemental data.
DNA sequencing and analysis
Genomic DNA was extracted from 300 μL of mucus using Powersoil Kits (MoBio, Carlsbad, CA) and was submitted to the UMN Genomics Center (UMGC) for 16S library preparation using a two-step PCR protocol [23]. The V4 region was amplified and sequenced using Illumina MiSeq TruSeq 2×300 paired-end technology. Water and reagent control samples were also submitted, but were below detection thresholds. Raw sequence files were deposited in the NCBI Sequence Read Archive (accession #PRJNA374847). Data were analyzed using a pipeline developed by the UMN Informatics Institute in collaboration with UMGC. Details are provided in the supplemental data.
Metagenomic prediction
Metagenomes were inferred from 16S rRNA data using Phylogenetic Investigation of Communities by Reconstruction of Unobserved States (PICRUSt)[24]. PICRUSt uses marker gene survey data to predict metagenome content through ancestral state reconstruction of quality-filtered 16S sequence data. We also used BugBase [25] to summarize predicted metagenomes by bacterial phenotype. BugBase scripts were run with default settings using filtered sequence data used in PICRUSt. Details are provided in the supplemental data.
Statistical analyses
Analyses were performed in GraphPad Prism 6.0 unless stated otherwise. Significance was assessed at the α=0.05 level. Hypothesis testing was conducted assuming a paired sample study design. Student’s t-tests were used where data passed the Kolmogorov-Smirnov test for normality. Statistics implemented in beta diversity and BugBase analyses are documented in the supplemental data.
RESULTS
Patient Cohort
Twelve CF adults with CRS undergoing functional endoscopic sinus surgery (FESS) each provided an expectorated sputum sample. Sinus mucus was collected at the beginning of FESS. These samples are herein referred to as “sinus” and “lung” samples, denoting their anatomical origin. Clinical data, including CF genotype, microbiology cultures, prior FESS procedures, spirometry and sino-nasal outcome test (SNOT-22) scores were also collected (Table S1). Six patients were homozygous ΔF508, five were heterozygous ΔF508, and two had heterozygous non-ΔF508 mutations. Sinus culture data were available for 11 of 12 sinus samples. Of these, 6 were positive for P. aeruginosa, and 5 were positive for S. aureus. Predominant lung pathogens detected by sputum culture were also recorded (Table S2).
Bacterial load
qPCR of 16S ribosomal RNA (rRNA) gene copy number was used to estimate bacterial load (Fig. 1). On average, sinus specimens contained 2.72×103 (IQR=2.69×102−1.72×103) 16S gene copies per ng genomic DNA, while lung sputum harbored 5.33×104 (IQR=6.18×102−5.02×104). These data suggest a modest difference in 16S gene abundance between sample types (Fig. 1A, P=0.0637). Interestingly, patient age was positively associated with 16S gene abundance in sinus samples, but this relationship was not observed for the lung (Fig. 1B). We also assessed the relationship between bacterial load and two clinical metrics, FEV1% and SNOT-22, neither of which significantly correlated with 16S copy number in either sample group (Fig. 1B). Taken together, these data suggest that the specific composition of each bacterial community rather than bacterial abundance contributes to disease states in both sinus and lung niches.
Figure 1. 16S gene copies are greater in CF lung sputum compared to sinus samples, and correlate with patient age.
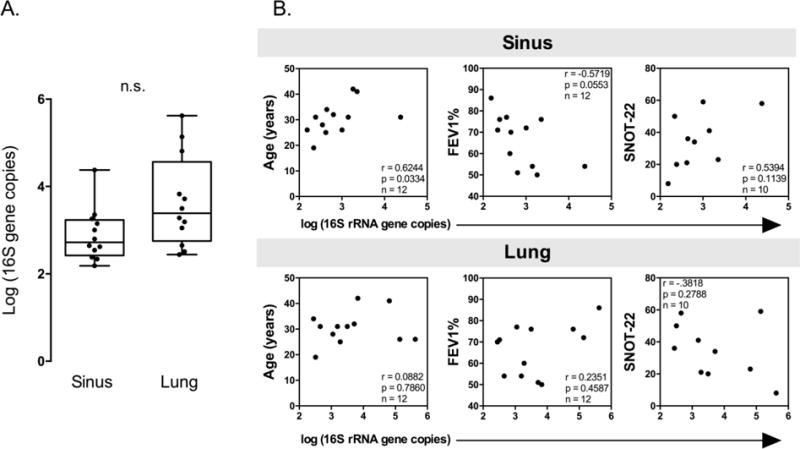
Quantitative PCR was used to enumerate 16S rRNA gene copies ng of genomic DNA isolated from sinus and lung samples. A. Comparison of 16S rRNA gene copies in sinus and lung sample pairs by qPCR. (Paired Student’s t-test P=0.0637). B. Spearman correlations with 16S rRNA gene copies and patient clinical data. In sinus samples, 16S copies are positively and significantly correlated with patient age.
Bacterial community membership varies with location
Bacterial composition was profiled using 16S rRNA gene sequencing. After filtering for quality and subsampling to an even depth, 51 genera were identified across samples (Fig. 2). To investigate genera that accounted for the majority of sequences, we adopted the definition of a dominant genus (the most abundant genus with over twice the abundance of the second most abundant genus)[16]. A dominant genus was present in 100% of sinus samples but only 33% of lung samples. Pseudomonas and Staphylococcus were dominant genera in five sinus samples each (42%), while Streptococcus and Burkholderia were each dominant in a single sample (Fig. 2). As expected, only P. aeruginosa and S. aureus were dominant lung pathogens. Species-level identification for abundant pathogens was determined by culture data, when available (Table S2). The median relative abundance of the predominant genus was 0.88 (IQR=0.75–0.99) in each sinus sample, and 0.42 (IQR=0.34–0.78) for lung samples. The most abundant sinus OTUs were assigned to Pseudomonas, Staphylococcus, and Streptococcus. By contrast, lung samples harbored an abundance of Pseudomonas, Veillonella, and Prevotella, consistent with previous studies [6,16].
Figure 2. Bacterial community composition of the upper and lower airways.
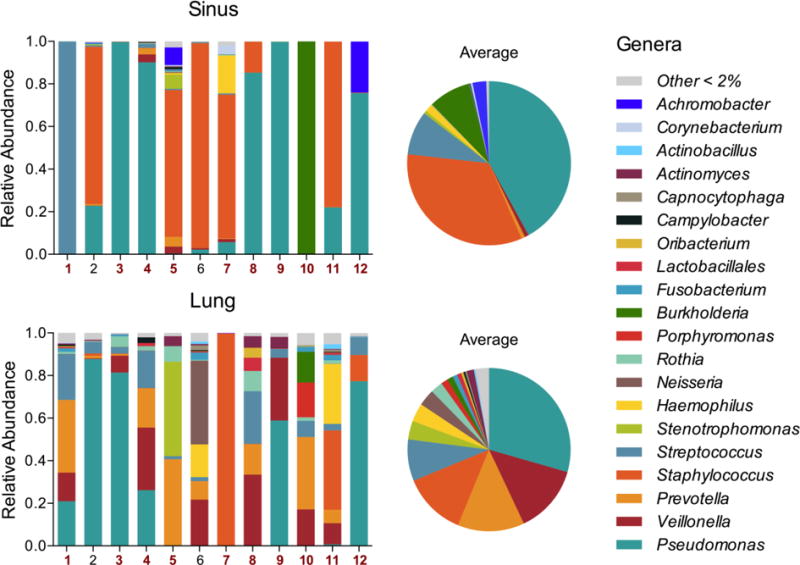
Stacked bar plots of relative abundances of genera in paired sinus and lung samples demonstrate dominance of Pseudomonas, Staphylococcus, and Streptococcus genera. Red patient numbers indicate samples where bacterial membership was significantly correlated within pairs. Data for these relationships is presented in Table S2. Pie charts show the average abundance of genera for each sample type across the patient cohort.
Interestingly, although many taxa were shared between sample pairs, the absence of a pathogen (e.g. Pseudomonas, Achromobacter) in one sample was not predictive of its absence in its paired sample sequence data. For example, subjects 5 and 12 harbored Achromobacter in their upper airways, despite being undetectable in sputum. This was also true of Staphylococcus in subject 6. Conversely, subjects 1 and 11 harbored an abundance of known CF pathogens (Pseudomonas and Haemophilus, respectively) in the lung that were not associated with upper airway infection. These examples demonstrate that infections at either site can be perpetuated by different pathogen within an individual, and that sputum cultures and 16S rRNA sequence data are not always representative of upper airway infection.
Based on these examples, we then determined the extent to which genera were shared between sites. Spearman correlations between genera in matched pairs revealed that within-patient similarities allowed for significant positive correlation between sites in ten of twelve sample pairs (average Spearman ρ=0.45)(Table S3). However, a group-wise comparison of paired samples showed a weaker correlation (Spearman ρ=0.32, P=0.001). These results highlight the potential for bacterial communities of the upper and lower airways to be similar within a given patient, yet the group correlation underlines the general dissimilarity in microbiota between sinus and lung microenvironment.
Bacterial diversity varies between upper and lower airways
As described above, few taxa dominated most sequences in each sample. To investigate this further, two alpha diversity metrics, Observed operational taxonomic units (OTUs) and Shannon diversity, were used as measures of richness (biodiversity) and evenness (equitability)[26]. Observed OTUs in lung samples were greater than in sinuses, indicating greater richness, though the difference was not significant (Fig. 3A). Using the Shannon diversity index, sinuses were characterized by greater unevenness relative to lung samples, consistent with the high prevalence of dominant sinus genera (Fig. 3B). When ordered by rank, an average of 10 and 20 genera accounted for 99% of sequences in sinus and lung samples, respectively (Fig. 3C). These data indicate that the lung bacterial community is greater in both richness and evenness relative to the sinuses.
Figure 3. Alpha diversity of CF maxillary sinuses differs from CF lung sputum.
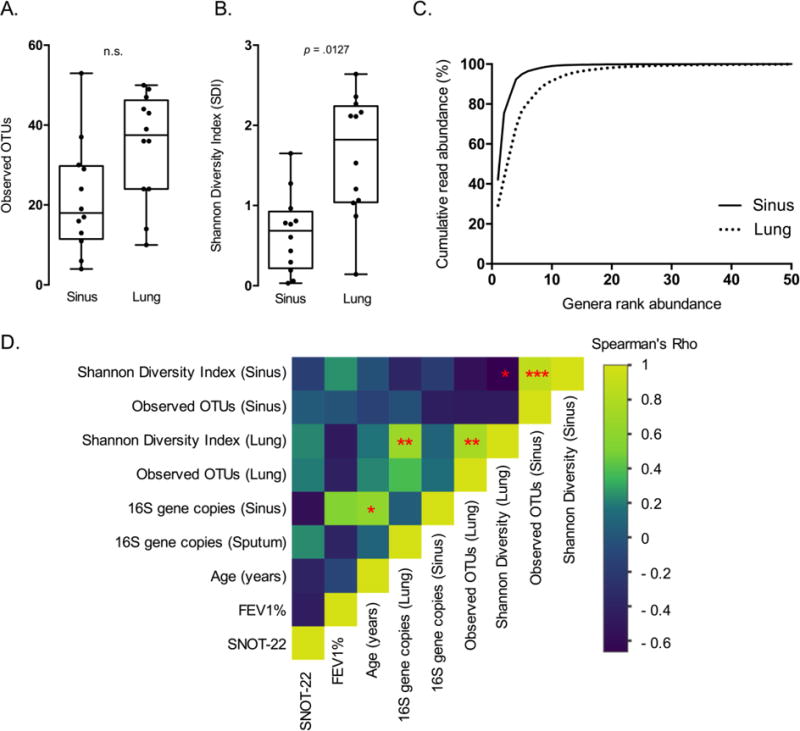
A. Observed OTUs are modestly greater in lung samples compared to the sinuses. Lung samples display significantly greater evenness in OTU distribution relative to sinus samples (Paired Student’s t-test, P=0.0127). B. Rank abundance curves reveal that both sinus and lung bacterial communities are dominated by a few organisms. 10 and 20 genera represent 99% of the sequences for sinus and lung samples, respectively. C. Spearman correlation heat map shows association of bacterial diversity (Observed OTUs, Shannon), bacterial 16S gene abundance, and clinical factors. (* P ≤ 0.05, ** P ≤ 0.01, *** P ≤ 0.001).
Spearman correlations were then calculated to assess relationships between bacterial load, alpha diversity and patient clinical data (Fig. 3D). These data revealed a significant inverse correlation between Shannon diversity in the sinus and lung (ρ=−0.664, P=0.022). Because the data show a similar richness between sites, and both sinus and lung Shannon diversity revealed positive relationships with observed OTUs (sinus ρ=0.881, P=3.35×10−4; lung, ρ=0.774, P=0.007), we infer that diversity differences are driven by evenness in these niches. Lung Shannon diversity was also positively correlated with lung 16S rRNA copy number (ρ=0.678, P=0.019), and these data reiterate the positive correlation between sinus 16S rRNA gene copy number and patient age (ρ=0.624, P=0.033; Fig. 1B). There was no correlation found between alpha diversity metrics, or relative abundance of Staphylococcus/Pseudomonas and antibiotics prescribed three days prior to surgery for both sample types (Table S4). Altogether, these results demonstrate that bacterial diversity differs between sinus and lung niches, and that a decrease in diversity at either site is associated with the decrease in even distribution of bacterial taxa.
Airway location as a descriptor of phylogenetic variance
Beta diversity of airway microbiota was then visualized using ordination. Of interest was whether bacterial communities would cluster more closely by patient or sampling site. Previous studies characterizing upper and lower airway microbiota revealed shared taxa between sites, but also inter-individual variation [8,17]. Evidence also suggests that bacterial metastasis between the sinuses and lung is commonplace [9]. Based on these previous studies, we hypothesized that samples would cluster more closely by patient, rather than by sampling site.
To address this hypothesis, we compared samples utilizing weighted Unifrac, an abundance-sensitive, phylogenetically-relevant diversity metric [28]. This metric was calculated to determine phylogenetic pairwise distances between each sample, then plotted using principal coordinates analysis (PCoA). Contrary to our hypothesis, within-patient sample pairs did not cluster together nearly as strongly as they did by sampling site (Fig. 4A), which rejected the null hypothesis the groupings (sinuses and lungs) had the same centroid (PERMANOVA, P=0.019). Lung samples also demonstrated considerable phylogenetic variation when compared to sinus samples (Fig. 4A). When relative abundances from dominant taxa (Pseudomonas and Staphylococcus) were overlaid, there was a strong association with the PCoA sample distribution (Fig. 4B). These analyses demonstrate that overall community structure is largely driven by dominant genera, and that upper and lower airways select for unique bacterial community structures, despite sharing individual taxa.
Figure 4. Ordination of weighted Unifrac distances shows clustering by sample type and dominant organism.
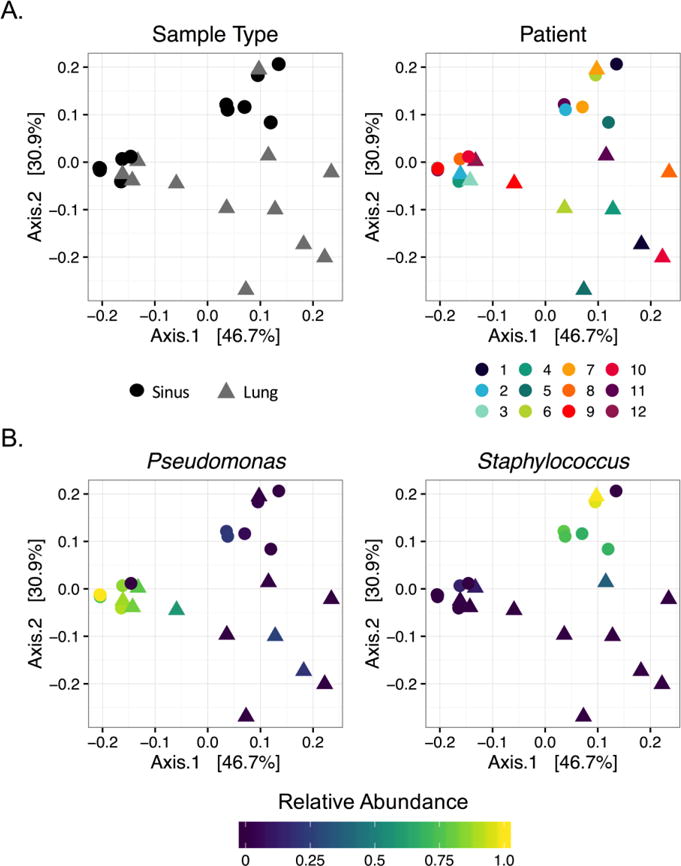
A. Samples show more similarity by sample type rather than sampled individual (PERMANOVA, P=0.019). Color and shape denote patient and sample type, respectively. Sinus and lung samples do not cluster by patient, but do show clustering by sample type. B. PCoA colored by relative abundance of dominant organisms (defined in text), shows sinus sample grouping is highly dependent on relative abundance of Pseudomonas and Staphylococcus.
Predicted metagenomes show phenotypic conservation between sites
To gain insight into bacterial community function, PICRUSt[23] was implemented to infer bacterial metagenomes based on 16S rRNA gene content. Sequences derived from each sample had a low Nearest Sequenced Taxon Index average of 0.017, indicating a high relatedness between bacteria found in the samples to sequenced genomes, and a high prediction accuracy for the dataset. Twenty unique KEGG pathways were represented among inferred metagenomes, and showed striking similarity between sinus and lung samples (Fig. S1). This suggests that the functional capacity of lung and sinus bacterial communities are relatively similar, despite taxonomic differences.
To further summarize PICRUSt output, we utilized BugBase, a bioinformatics tool that infers community-wide phenotypes from predicted metagenomes[24]. BugBase identified that gene functions associated with anaerobiosis were enriched in lung samples (P=0.01), and could be attributed to three genera: Veillonella, Prevotella, and Porphyromonas (Fig. 5A). Gram-negative ultrastructure and biofilm formation are two bacterial phenotypes often associated with airway pathogenicity, however, our analysis showed that these phenotypes did not differ significantly between sites. Gram-negative genera contributing to this phenotype were more varied in lung samples and included Veillonella, Prevotella, Neisseria, Stenotrophomonas, supporting the increased richness observed in these samples. Biofilm-forming bacteria were observed in both sinus and lung predicted metagenomes. This phenotype was influenced by the presence of Pseudomonas in both airway sites, but Burkholderia and Achromobacter both differentiated sinus from lung samples, while Neisseria and Stenotrophomonas were more highly represented in lung samples. Altogether, these data point towards the lung environment being a more anaerobic niche, but that other phenotypes classically linked to bacterial pathogenicity, such as biofilm formation, are similar between sites.
Figure 5. BugBase analysis of PICRUSt-predicted metagenomes.
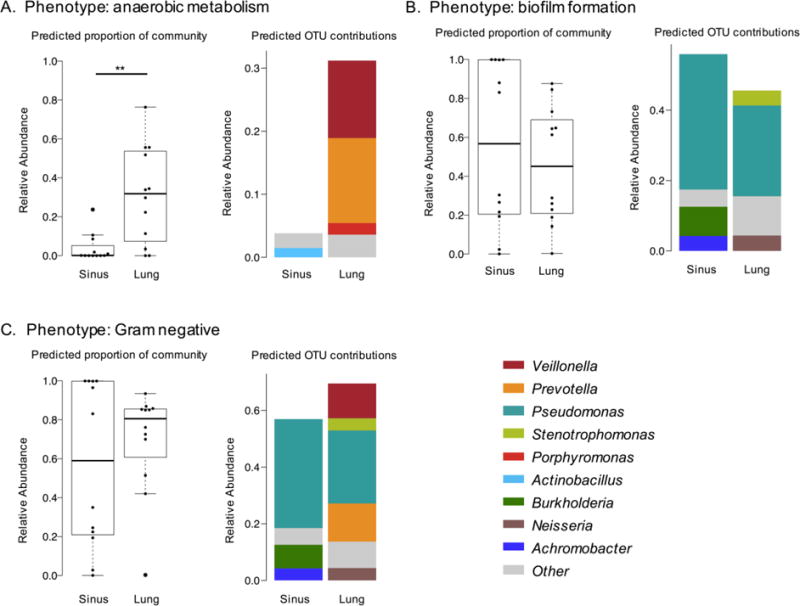
A. Anaerobic metabolism is significantly enriched in lung samples (Wilcoxon signed-rank test, P=0.01). B. Biofilm formation does not differ significantly with sample type (Wilcoxon signed-rank test, P=0.57). C. Gram-negative phenotype is driven by presence of Veillonella, Prevotella and Pseudomonas in lung samples (Wilcoxon signed-rank test, P=0.47). (* P ≤ 0.05, ** P ≤ 0.01, *** P ≤ 0.001)
DISCUSSION
It is poorly understood how complex bacterial communities promote the development of sinus disease. It is also not known how upper airway microbiota contributes to lower airway infections that are a primary cause of CF patient morbidity. The sinuses have been proposed as a reservoir in which individual pathogens adapt to the host prior to lung colonization, but it is unclear whether polymicrobial community composition of the CF lung can be a reasonable surrogate for upper airway microbiota and treatment of CF-CRS. We therefore took an ecological approach towards exploring bacterial diversity throughout the airways.
Our cohort showed striking variability in bacterial community membership between subjects. Despite this variability, we found that lung samples harbored an increased diversity (evenness) relative to sinuses which were dominated by either Staphylococcus or Pseudomonas. The abundances of these two canonical pathogens are commonly revealed by sinus culture [9], though many other taxa identified in our study are not. For example, when compared to summarized culture data comparing the upper and lower CF airways [8], the most striking difference between analyses was the identification of obligate anaerobes. These differences highlight the utility of molecular-based methods in detecting fastidious and anaerobic bacterial taxa. Given the emerging evidence of anaerobes being causative agents of pulmonary disease [29], increased use of culture-independent diagnostics to detect these bacteria throughout the airways is warranted.
Our most intriguing data were that several sinus samples harbored canonical CF pathogens, yet these bacteria were absent in the paired lung samples. Conversely, the lungs of several subjects were colonized by a dominant pathogen that went undetected in the upper airways. This study outcome has significant clinical implications; respiratory cultures and sequence data from the two sites are not necessarily interchangeable for the determination of antibiotic therapy. These findings are consistent with those of Muhlebach [30] who showed that bronchoalveolar lavage and oropharyngeal cultures are poor predictors (40–50% accuracy) of sinus bacteriology in children. Together, these observations may partially explain why patient response to therapy is poor, and suggest that the expanded use of non-invasive, sequence-based sinonasal sampling measures could be beneficial in steering patient therapies.
Often in microbiome surveys, bacterial membership may differ dramatically between samples, but functional capabilities remain conserved [31]. Here, we highlight this relationship in the airways. Contrasting differences in bacterial diversity and community composition between sinuses and lungs, we found that the predicted functional capacity in both niches was similar. In the context of bacterial contribution to CF disease, it is plausible that there are many similarities in the microenvironments of the upper and lower airways that may contribute, or even result from these conserved functions.
This exploratory study reveals many opportunities for future research. Notably, this work focuses on a small adult cohort with lung function lower than 70% predicted (mean). While the inter-individual variability reported is not unfounded, it highlights the need for increased enrollment for assessing clinical and sequencing variables. Moreover, future work should extend to both pediatric and adult subjects across the spectrum of disease severity, as our conclusions may not be generalizable to all subjects. For example, it is possible that in patients where lung-adapted strains differ in prominence, the influence of bacterial trafficking from the upper airways on bacterial structure also varies. Likewise, longitudinal studies are warranted to investigate microbiota trafficking dynamics over time as patients age. Our work also does not account for airway microenvironments harboring distinct microbiomes. For example, a single maxillary sinus may not capture an infectious agent present in another sinus, whereas sputum may reflect multiple lower airway niches. Prior studies have shown that the middle meatus is a valid representation of the sinus cavity [33], though multiple samples that define the biogeography of the upper airways will be informative for infection management. Finally, a common concern in sputum-based studies is the potential for salivary contamination. Though we cannot rule out the contribution of oral flora to our analyses, samples from lung explants and bronchoalveolar lavage have clearly demonstrated the presence of oral-associated anaerobes in the lung [33,34]. Moreover, others suggest that contamination of sputum is limited during transit through the oral cavity [33,35]. Moving forward, oropharyngeal swabs should be included in analyses to help define the site of sequence origin, particularly for diagnostic purposes.
Despite these limitations, our data shed important light on microbial community relationships throughout the CF airways. Though airway niche spaces differ in diversity, shared taxa between the sample pairs reflects the interconnectedness of the airway and does not discount the sinuses as a source of lower airway colonization. However, the distinct bacterial community structures suggest sinus and lung microenvironments play a critical role in governing the prevalence and abundance of canonical CF pathogens. These data can be translated to the clinic by informing caregivers to utilize respiratory specimens originating from the site of infection. In the context of our findings, we advocate for the use of 16S rRNA gene sequences in the clinical setting to supplement culture data in the assessment and informed therapeutic approach towards the management of CF-associated CRS infections.
Supplementary Material
Highlights.
16S rRNA sequencing reveals a complex CF-associated sinus microbiota
Comparison of upper and lower airway communities demonstrates a discordance between sites
Community composition clusters more closely by sampling site than by patient.
It is not known whether this relationship holds across the spectrum of disease severity or patient age.
Metagenome prediction reveals that anaerobiosis is more strongly associated with the lung.
Acknowledgments
This work was supported by a Pathway to Independence Award from the National Heart Lung and Blood Institute to RCH (R00HL114862); an Investigator-Sponsored Research Grant from Gilead Sciences to RCH, JD, and HCB; a Lions 5M International award to RY; a National Institutes of Health T32 Fellowship (#T90 DE 0227232) awarded through the National Institute of Dental and Craniofacial Research to SKL; and the University of Minnesota Medical School. We acknowledge the Minnesota Supercomputing Institute (MSI), Daryl Gohl and John Garbe at the UMN Genomics Center for sequencing assistance. We thank Abayo Itabiyi, Ali Stockness and Rebecca Dove of the Department of Otolaryngology, and the members of BioNet at the University of Minnesota for facilitating sample collection. Our thanks go out to Tonya Ward, Dan Knights, and the Knights Lab at UMN for their assistance with the BugBase analysis.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Robertson JM, Friedman EM, Rubin BK. Nasal and sinus disease in cystic fibrosis. Paediatr Respir Rev. 2008;9:213–9. doi: 10.1016/j.prrv.2008.04.003. [DOI] [PubMed] [Google Scholar]
- 2.Becker KA, Reithmuller J, Zhang Y, Gulbins E. The role of sphingolipids and ceramide in pulmonary fibrosis. Open Resp Med J. 2010;4:39–47. doi: 10.2174/1874306401004010039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Babinski D, Trawinska-Bartincka M. Rhinosinusitis in cystic fibrosis: not a simple story. Int J Pediatr Otorhinolaryngol. 2008;72:619–624. doi: 10.1016/j.ijporl.2008.01.010. [DOI] [PubMed] [Google Scholar]
- 4.Benninger MS, Ferguson BJ, Hadley JA, Hamilos DL, Jacobs M, Kennedy DW, Lanza DC, Marple BF, Osguthorpe JD, Stankiewicz JA, Anon J, Denneny J, Emanuel I, Levine H. Adult chronic rhinosinusitis: definitions, diagnosis, epidemiology, and pathophysiology. Otolaryngol Head Neck Surg. 2003;129:S1–32. doi: 10.1016/S0194-5998(03)01397-4. [DOI] [PubMed] [Google Scholar]
- 5.Roby BB, McNamara J, Finkelstein M, Sidman J. Sinus surgery in cystic fibrosis patients: comparison of sinus and lower airway cultures. Int J Pediatr Otorhinolaryngol. 2008;72:1365–1369. doi: 10.1016/j.ijporl.2008.05.011. [DOI] [PubMed] [Google Scholar]
- 6.Cox MJ, Allgaier M, Taylor B, Baek MS, Huang YJ, Daly RA, Karaoz U, Anderson GL, Brown R, Fujimura KE, Wu B, Tran D, Koff J, Kleinhenz M, Nielson D, Brodie EL, Lynch SV. Airway microbiota and pathogen abundance in age-stratified cystic fibrosis patients. PLoS One. 2010;5:e11044. doi: 10.1371/journal.pone.0011044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bonestroo HJC, de Winter-de Groot KM, van der Ent CK, Arets HGM. Upper and lower airway cultures in children with cystic fibrosis: do not neglect the upper airways. J Cyst Fibros. 2010;9:130–134. doi: 10.1016/j.jcf.2010.01.001. [DOI] [PubMed] [Google Scholar]
- 8.Fischer N, Hentschel J, Markert UR, Keller PM, Pletz MW, Mainz JG. Non-invasive assessment of upper and lower airway infections and inflammation in cystic fibrosis patients. Pediatr Pulmonol. 2014;49:1065–1075. doi: 10.1002/ppul.22982. [DOI] [PubMed] [Google Scholar]
- 9.Mainz JG, Naehrlich L, Schien M, Käding M, Schiller I, Mayr S, Schneider G, Wiedemann B, Wiehlmann L, Cramer N, Pfister W, Kahl BC, Beck JF, Tümmler B. Concordant genotype of upper and lower airways P aeruginosa and S aureus isolates in cystic fibrosis. Thorax. 2009;64:535–540. doi: 10.1136/thx.2008.104711. [DOI] [PubMed] [Google Scholar]
- 10.Johansen H, Aanaes K, Pressler T, Nielsen K, Fisker J, Skov M, Hoiby N, Buchwald C. Colonisation and infection of the paranasal sinuses in cystic fibrosis patients is accompanied by a reduced PMN response. J Cyst Fibros. 2012;11:525–31. doi: 10.1016/j.jcf.2012.04.011. [DOI] [PubMed] [Google Scholar]
- 11.Ciofu O, Johansen H, Aanaes K, Wassermann T, Alhede M, Buchwald C, Iby N. Paeruginosa in the paranasal sinuses and transplanted lungs have similar adaptive mutations as isolates from chronically infected CF lungs. J Cyst Fibros. 2013;12:729–36. doi: 10.1016/j.jcf.2013.02.004. [DOI] [PubMed] [Google Scholar]
- 12.Syed SA, Whelan FJ, Waddell B, Rabin HR, Parkins MD, Surette MG. Reemergence of lower-airway microbiota in lung transplant patients with cystic fibrosis. Ann Am Thorac Soc. 2016;13:2132–2142. doi: 10.1513/AnnalsATS.201606-431OC. [DOI] [PubMed] [Google Scholar]
- 13.Rudkjøbing V, Aanaes K, Wolff T, Buchwald C, Johansen H, Thomsen T. An exploratory study of microbial diversity in sinus infections of cystic fibrosis patients by molecular methods. J Cyst Fibros. 2014;13:645–652. doi: 10.1016/j.jcf.2014.02.008. [DOI] [PubMed] [Google Scholar]
- 14.Hansen SK, Rau MH, Johansen HK, Ciofu O, Jelsbak L, Yang L, Folkesson A, Jarmer HO, Aaneas K, von Buchwald C, Hoiby N, Molin S. Evolution and diversification of Pseudomonas aeruginosa in the paranasal sinuses of cystic fibrosis children have implications for chronic lung infection. ISME J. 2012;6:31–45. doi: 10.1038/ismej.2011.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mainz JG, Hentschel J, Schien C, Cramer N. Sinonasal persistence of Pseudomonas aeruginosa after lung transplantation. J Cyst Fibros. 2012;11:158–161. doi: 10.1016/j.jcf.2011.10.009. [DOI] [PubMed] [Google Scholar]
- 16.Coburn B, Wang P, Caballero J, Clark S, Brahma V, Donaldson S, Zhang Y, Surendra A, Gong Y, Tullis E, Yau Y, Waters V, Hwang D, Guttman D. Lung microbiota across age and disease stage in cystic fibrosis. Sci Rep. 2015;5:10241. doi: 10.1038/srep10241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Boutin S, Graeber SY, Weitnauer M, Panitz J, Stahl M, Clausznitzer D, Kaderali L, Einarsson G, Tunney MM, Elborn JS, Mall MA, Dalpke AH. Comparison of microbiomes from different niches of upper and lower airways in children and adolescents with cystic fibrosis. PLoS One. 2015;10:e0116029. doi: 10.1371/journal.pone.0116029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rivas Caldas R, Boisrame S. Upper aero-digestive contamination by Pseudomonas aeruginosa and implications for cystic fibrosis. J Cyst Fibros. 2015;14:6–15. doi: 10.1016/j.jcf.2014.04.008. [DOI] [PubMed] [Google Scholar]
- 19.Lavin J, Bhushan B, Schroeder JW. Correlation between respiratory cultures and sinus cultures in children with cystic fibrosis. Int J Pediatr Otorhinolaryngol. 2013;77:686–689. doi: 10.1016/j.ijporl.2013.01.018. [DOI] [PubMed] [Google Scholar]
- 20.Osborn AJ, Leung R, Ratjen F, James AL. Effect of endoscopic sinus surgery on pulmonary function and microbial pathogens in a pediatric population with cystic fibrosis. Arch Otolaryngol Head Neck Surg. 2011;137:542–7. doi: 10.1001/archoto.2011.68. [DOI] [PubMed] [Google Scholar]
- 21.Mainz JG, Koitschev A. Pathogenesis and management of nasal polyposis in cystic fibrosis. Curr Allerg Asthma Rep. 2012;12:163–174. doi: 10.1007/s11882-012-0250-y. [DOI] [PubMed] [Google Scholar]
- 22.Abreu N, Nagalingam N, Song Y, Roediger F, Pletcher S, Goldberg A, Lynch S. Sinus Microbiome Diversity Depletion and Corynebacterium tuberculostearicum Enrichment Mediates Rhinosinusitis. Sci Transl Medicine. 2012;4:151ra124–151ra124. doi: 10.1126/scitranslmed.3003783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gohl D, Vangay P, Garbe J, MacLean A, Hauge A, Becker A, Gould T, Clayton J, Johnson T, Hunter R, Knights D, Beckman KB. Systematic improvement of amplicon marker gene methods for increased accuracy in microbiome studies. Nat Biotechnol. 2016;34:942–9. doi: 10.1038/nbt.3601. [DOI] [PubMed] [Google Scholar]
- 24.Langille MG, Zaneveld J, Caporaso JG, McDonald D, Knights D, Reyes JA, Clemente JC, Burkepile DE, Vega Thurber RL, Knight R, Beiko RG, Huttenhower C. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat Biotechnol. 2013;31:814–21. doi: 10.1038/nbt.2676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ward T, Larson J, Meulemans J, Hillmann B, Lynch J, Sidiropoulos D, Spear J, Caporaso G, Blekhman R, Knight R, Fink R, Knights D. BugBase predicts organism level microbiome phenotypes. BioRxiv. 2017;133462 doi: 10.1101/133462. [DOI] [Google Scholar]
- 26.Hill MO. Diversity and Evenness: A Unifying Notation and Its Consequences. Ecology. 1973;54:427–432. doi: 10.2307/1934352. [DOI] [Google Scholar]
- 27.Charlson ES, Bittinger K, Haas AR, Fitzgerald AS, Frank I, Yadav A, Bushman FD, Collman RG. Topographical Continuity of Bacterial Populations in the Healthy Human Respiratory Tract. Am J Respir Crit Care Med. 2011;184:957–963. doi: 10.1164/rccm.201104-0655OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lozupone C, Knight R. UniFrac : a New Phylogenetic Method for Comparing Microbial Communities. Appl Environ Microbiol. 2005;71:8228–8235. doi: 10.1128/AEM.71.12.8228-8235.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Flynn JM, Niccum D, Dunitz JM, Hunter RC. Evidence and role for bacterial mucin degradation in cystic fibrosis airway disease. PLoS Pathogens. 2016:e1005846. doi: 10.1371/journal.ppat.1005846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Muhlebach MS, Miller MB, Moore C, Wedd JP, Drake AF, Leigh MW. Are lower airway or throat cultures predictive of sinus bacteriology in cystic fibrosis? Pediatr Pulmonol. 2006;41:445–451. doi: 10.1002/ppul.20396. [DOI] [PubMed] [Google Scholar]
- 31.Human Microbiome Project Consortium. Structure, function and diversity of the healthy human microbiome. Nature. 2012;486:207–214. doi: 10.1038/nature11234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ramakrishnan VR, Gitomer S, Kofonow JM, Robertson CE, Frank DN. Investigation of sinonasal microbiome spatial organization in chronic rhinosinusitis. Int Forum Allergy Rhinol. 2016;7:16–23. doi: 10.1002/alr.21854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Blainey PC, Milla CE, Cornfield DN, Quake SR. Quantitative analysis of the human airway microbial ecology reveals a pervasive signature for cystic fibrosis. Sci Transl Med. 2012;4:153ra130. doi: 10.1126/scitranslmed.3004458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hogan DA, Willger SD, Dolben EL, Hampton TH, Stanton BA, Morrison HG, Sogin ML, Czum J, Ashare A. Analysis of lung microbiota in bronchoalveolar lavage, protected brush and sputum samples from subjects with mild-to-moderate cystic fibrosis lung disease. PLoS One. 2016;11:e0149998. doi: 10.1371/journal.pone.0149998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Charlson ES, Bittinger K, Haas AR, Fitzgerald AS, Frank I, Yadav A, Bushman FD, Collman RG. Topographical continuity of bacterial populations in the health human respiratory tract. Am J Resp Crit Care Med. 2011;184:957–963. doi: 10.1164/rccm.201104-0655OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.