

Abstract
Objective(s):
Inflammation is involved in various forms of pulmonary arterial hypertension (PAH). Although the pathophysiology of PAH remains uncertain, NF-κB and p38 mitogen-activated protein kinase (p38 MAPK) has been reported to be associated with many inflammatory mediators of PAH. This study aimed to evaluate the effect of chronic intermittent hypobaric hypoxia (CIHH) on pulmonary inflammation and remodeling in monocrotaline (MCT) induced PAH in rats.
Materials and Methods:
An in vivo model of PAH induced by MCT was employed. Statistical analyses were done using one-way analysis of variance (ANOVA) or Fisher’s LSD test for multiple comparisons.
Results:
Four weeks of CIHH exposure following MCT injection resulted in significant reduction of mean pulmonary artery pressure (mPAP) level and improvement of right ventricular hypertrophy (RVH). Morphometric analyses showed decreased wall thickness of pulmonary arterioles in MCT+CIHH treated rats. These findings are consistent with the decrease in Ki-67 immunostaining. Following CIHH treatments, apoptotic analysis showed a consistent decrease in T lymphocytes together with lower levels of CD4+ T cell subset as measured in spleen and blood samples. Furthermore, CIHH treatment resulted in markedly reduced expression of TNF-α and IL-6 via the inhibition of NF-κB and p38 MAPK activity in rat lungs.
Conclusion:
Altogether, these results provide new evidence relating to the mode of action of CIHH in the prevention of PAH induced by MCT.
Keywords: Chronic intermittent -hypobaric hypoxia, Inflammation Monocrotaline, NF-κB, Pulmonary arterial -hypertension, p38, MAPK
Introduction
Pulmonary arterial hypertension (PAH) is a chronic and progressive clinical picture characterized by sustained increase of PA pressure and vascular remodeling (1), which ultimately leads to severe right ventricular hypertrophy (RVH) and failure or even fatal consequences. There can be many different underlying causes of PAH, but its exact pathophysiology still remains unclear. There is evidence in the literature that inflammation plays a key role in the genesis or progression of PAH (2). Recent studies have revealed the presence of inflammatory cells, including macrophages, mast cells, lymphocytes, and dendritic cells in the vicinity of vessels and plexiform lesions of PAH (3, 4). Moreover, proinflammatory cytokines such as inter-leukin (IL)-1β, IL-6, IL-8, and tumor necrosis factor-α (TNF-α) are excessively produced in patients with PAH (4, 5), which may correlate with high morbidity. In addition, experimental (6) and clinical (7) evidence demonstrated the key role of inflammation in PAH pathobiology, as well as the therapeutic potential of steroid or immunosuppressive agents in PAH (8-10). However, the benefit of anti-inflammatory therapy in the treatment of PAH and the molecular mechanisms still remain to be further investigated.
Nuclear factor-κB (NF-κB) is a key transcription factor in many cardiac disorders (11, 12), but the function of NF-κB was limited in MCT-induced pulmonary vascular remodeling. Recent reports reveal the critical role of NF-κB in the progression of PAH (13-15), however, the mechanism underlying PAH and its progression to the development of RVH still remains unclear. p38 MAPK is a key intermediary pathway in inflammatory pathological states and plays an important role in mediating cellular responses to proinflammatory cytokines. Alpha (α)-isoform of p38 MAPK is most often associated with inflammation (16-18). p38 MAPK was proved to be constitutively expressed in fibroblasts explanted from the pulmonary artery in a PAH rat model induced by chronic hypoxia(19-21). In addition, p38 MAPK has been implicated in the development of PAH in other animal models, although no clear mechanism has been suggested.
The present study aims to evaluate the cardiac and pulmonary effects of CIHH on an experimental PAH model and to determine the involved intracellular signaling pathways (22). A PAH rat model induced by monocrotaline (MCT) was used as the experimental setting. PAH induced by MCT (a plant alkaloid contained in the seeds of Crotalaria spectalalia and its metabolite) is an established experimental model that is similar to human PAH featured by vascular remodeling, pro-liferation of pulmonary artery smooth muscle cells, endothelial dysfunction, upregulation of inflammatory cytokines, and leukocyte infiltration (8). It is suggested that MCT treatment can cause direct damage to ECs, increase alveolar-capillary permeability, and induce inflammation that triggers the development of severe PAH (22-24). Furthermore, strong proinflammatory cytokines such as IL-1, IL-6, and TNF-α, are enhanced in animals treated with MCT (6). Herein, we try to evaluate the effect of CIHH on pulmonary inflammation and remodeling in MCT induced PAH in rats.
Materials and Methods
Animals
Forty male Sprague-Dawley (SD) rats (200–250 g) were obtained from Shanghai SLAC Laboratory Animal Company. Rats were housed with free access to food and water under a natural day/night cycle. Rats were acclimated for 7 days before any experimental procedures. All experiments were approved by the Institutional Animal Care and Use Committee of the Hebei Medical University.
Experimental design
The rats were randomly divided into four groups (n=10 rats for each group): CON (control, saline-treated), MCT (monocrotaline-treated), CIHH (chronic inter-mittent hypobaric hypoxia-treated), and MCT+CIHH (monocrotaline and chronic intermittent hypobaric hypoxia-treated). Rats from the MCT and MCT+CIHH groups were induced to pulmonary hypertension through a single subcutaneous injection of MCT (60 mg/kg body weight, Sigma, USA). Control rats received an equal volume of saline. Two weeks later, animals treated with the entire 4 weeks of nomoxia or hypoxia exposure. The rats designated for exposure to chronic hypoxia were housed intermittently in a hypobaric hypoxia chamber for 6 hr/day for 4 weeks. The hypoxic chamber was flushed with room air and 100% N2 to maintain 10% O2 concentration. The normoxic rats were housed at room air.
Hemodynamic measurements
After 4 weeks hypoxia exposure, the rats were anesthetized with isoflurane 1–2.5% at 6 mL/min and the right external jugular vein was exposed by blunt dissection. A polyethylene catheter full with heparin-saline solution (125 U/ml) linked to a miniature liquid pressure transducer (RT14M2, 971004, Japan) was gently inserted into the right ventricle through the jugular vein to measure right ventricular systolic pressure (RVSP). After 20 min stabilization, the catheter was advanced into the pulmonary artery to measure mPAP. Experimental data of RVSP and mPAP were then recorded using the Power Lab Software (ADI Instruments). Cardiovascular recordings were performed with the animals under conscious, free-moving conditions, and all data were obtained from steady-state waveform for 5 min.
Afterward, all rats were euthanized and blood samples were taken from the heart for serum preparation. Then, lungs and heart were excised and weighted for histological evaluation or frozen in liquid nitrogen for further analysis. The right ventricular (RV) free wall and left ventricle plus septum (LV+S) were collected, and the weight ratio of (RV/LV+S) was measured and calculated.
Morphology analysis
After overnight fixation in 4% polyformaldehyde solution, the right lung was embedded in paraffin, and multiple 4 μm-thick sections were stained with hema-toxylin and eosin as previously reported (25). Morphologic changes in the small pulmonary artery (50–200 μm) were detected using a Zeiss microscope digital camera. The external diameter (ED), medial wall thickness (MT), vessel lumen transverse area (VA) and total vessel area (TVA) of pulmonary arteries were measured. The percent medial wall thickness (MT %) and percent vessel lumen transverse area (VA %) were calculated to present pulmonary vascular structure remodeling. MT%=2×MT/ED×100%, VA% = VA/ TVA× 100%.
Enzyme-linked immunosorbent assay (ELISA)
Serum samples obtained from the heart were rapidly centrifuged at 3000 rpm for 20 min, and the serum concentrations of TNF-α and IL-6 were measured with rat TNF-α and IL-6 ELISA kits (R&D systems, Minneapolis, USA) according to the manufacturer’ instructions. The levels of TNF-α and IL-6 were expressed as pg/ml protein.
Immunohistochemical staining
Five μm lung sections were deparaffinized, rehydrated, the antigens retrieved, and then incubated with 1% H2O2 in methanol for 15 min to block endogenous peroxidase. Once the antigen retrieval was performed, the sections were blocked for 1 hr with normal goat serum, and then incubated overnight with anti- TNF-α antibody (1:500 dilution) or anti- IL-6 antibody (1:500 dilution) at 4 °C. After sections were washed in phosphate buffered saline (PBS) and incubated with the corresponding secondary antibody and streptavidin-horseradish peroxidases (Zymed Laboratories, South San Fran-cisco, CA) 3, 3’-diaminobenzidine was applied as a chromogen. Immunoreactivity was visualized using diaminobenzidine. Then a light hematoxylin counter-stain was applied. For negative control purposes, the same procedure was followed except that the primary antibody was replaced by PBS. The level of TNF-α or IL-6 expression was calculated by combining an estimate of the percentage of positive cells with an estimate of the staining intensity as follows. No cell with positive staining was scored as 0, 1-10% as 1, 10-50% as 2, 50-80% as 3, and 80-100% as 4. Staining intensity was rated as follows: negative (no color) as 0; weak (weak yellow) as 1, moderate (yellow) as 2, and strong (brown) as 3. Immuno-histochemical score (IHS) was calculated by multiplying the quantity and staining intensity scores. The IHS score >3 was considered as positive expression (26).
Immunofluorescence staining
Double immunofluorescence staining for Ki67 was performed on sections. Sections were deparaffinized, rehydrated, the antigens retrieved and endogenous peroxidase blocked. After blocking with normal goat serum for 1 hr, sections were incubated overnight with the anti-Ki67 antibody (1:1000 dilution) at 4 °C. Then, Alexa Fluor 488 (green) goat anti-rabbit IgG conjugated antibody was incubated on the sections for 1 hr at room temperature. Cell nuclei were stained with DAPI. Images were taken by fluorescence microscope (Olympus BX51; Olympus, Tokyo, Japan), Olympus FV100i (Olympus), or Leica SP-8 (Leica, Mannheim, Germany) confocal microscope.
Apoptosis assays
The apoptotic T cells were detected by staining with the CD3 antibody, followed by Annexin-V Apoptosis Detection Kit I (BD PharMingen). Splenocytes were harvested from rats and labeled with PE anti-rat CD3 antibody for 20 min at room temperature followed by added erythrolysin and continued incubation at room temperature for an additional 20 min. The cells were then washed twice with PBS and resuspended with 100 μl/ml of annexin binding buffer and 5 µl of annexin V-FITC working solution was added. The cells were incubated at room temperature, protected from light, for 15 min. After the incubation period, we added 400 μl annexin-binding buffer, mixed gently and kept the samples on ice until the fluorescence of cells was determined using a FACS Calibur flow cytometer (BD Biosciences). Results were obtained by analyzing data with FlowJo version 7.6.1 software (TreeStar). All assays were repeated 3 times.
Flow cytometry
Whole peripheral blood was stained with PE anti-rat CD4, and FITC anti-rat CD8 antibodies for 30 min on ice followed by added erythrolysin and incubation was continued at room temperature for an additional 20 min. The cells were then washed twice with PBS and resuspended in staining buffer, mixed gently and the samples kept on ice. All samples were analyzed with a FACS Calibur flow cytometer (BD Bioscience).
Western blot
Pulmonary artery samples were homogenized on ice in RIPA lysis buffer (Beyotime Inc., Jiangsu, China). The protease inhibitor of phenylmethylsulfonyl fluoride (PMSF, 1 mM) was added to the buffer in advance. Protein concentrations of samples were determined by the Bradford method (Bio-Rad Protein Assay). Equivalent amounts of protein (50 μg) from each sample were separated on 12% SDS-polyacrylamide gels and then transferred onto 0.22 μM nitrocellulose filter membranes (Millipore, Billerica, MA, USA). The membranes were blocked for 1 hr at room temperature with 5% nonfat milk in Tris-buffered saline solution (TBS) containing 3% bovine serum albumin (Sigma) and 0.05% Tween 20 (Bio-Rad), and then incubated with specific antibodies against p38, phospho-p38, and NF-κB p65 (1:1000 dilution) (Cell Signaling Technology, Inc., Danvers, MA) at 4 °C overnight and then incubated with the corresponding secondary antibody for 1 hr at room temperature. The signals were developed using Super-Signal West Pico chemiluminescent substrate (Pierce, Rockford, USA) and visualized with Quantity One software 4.6.2.
Statistical analysis
The statistical significance between groups was analyzed using GraphPad Prism v 4.0 Software (San Diego, CA, USA). Statistical analyses were performed with one-way analysis of variance (ANOVA) or Fisher’s LSD test for multiple comparisons. The data were shown as means±SD. A significant difference was accepted as significant if P<0.05.
Results
CIHH attenuated MCT-induced PAH and pulmonary artery remodeling in rats
After injection of MCT, the average mPAP of the MCT group was significantly increased compared with the control group. However, mPAP of the MCT+CIHH treated group was much lower than that of the MCT alone group (Figure 1A). The RV hypertrophy index (RVHI) (as measured by the weight ratio of RV/LV+S) showed a similar trend as mPAP, MCT-induced elevation of the ratio of RVHI was inhibited by the application of CIHH (Figure 1B). However, CIHH altered neither mPAP nor RVHI in the experimental rats treated with CIHH alone. To investigate the effect of CIHH on MCT-induced pulmonary artery remodeling, MT % and VA% of pulmo-nary arteries, which were stained with hematoxylin and eosin were evaluated (Figure 2A). As shown in Figure 2, MCT-treated rats had a significant increase in MT % and decreased VA% in pulmonary arteries compared to the control rats. The elevated medial wall thickness and reduced vessel lumen transverse area induced by MCT was substantially inhibited with CIHH treatment (Figures 2B and 2C).
Figure 1.
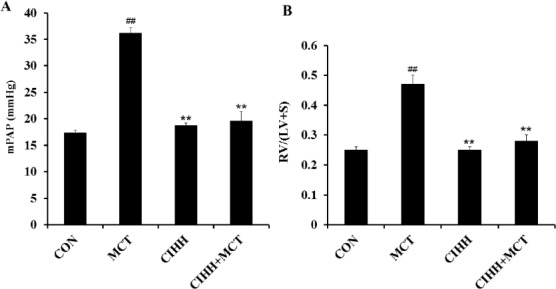
Effect of CIHH on mPAP (A) and RV/(LV+S) (B) in MCT-treated rats. mPAP and RV/(LV+S) were significantly higher in MCT-treated rats compared to the controls. CIHH did not affect mPAP or RV/(LV+S) compared to control animals, but significantly reduced the MCT-induced elevation of mPAP and RV/(LV+S). mPAP: mean pulmonary artery pressure, RV/(LV+S): weight ratio of the right ventricular wall (RV) and the left ventricular wall (LV) and septum (S). Values are means ± SD, n = 10 for each group. **P<0.01, compared with CON group; ##P<0.01, compared with PAH
Figure 2.
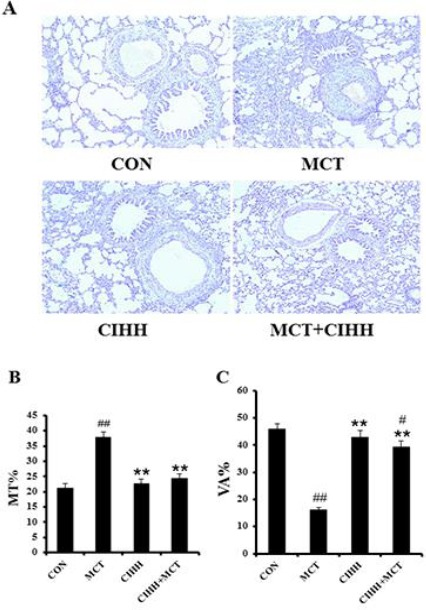
CIHH reverses MCT-induced increase of medial wall thickness in small pulmonary arteries. Representative photomicrographs of H/E stained lung sections, all images are shown with the same magnification (×200) (A). Percentage of medial wall thickness (MT %) (B). Percentage of the vessel lumen transverse area (VA %) (C). Values are means±SD, n = 10 for each group. **P<0.01, compared to MCT group; #P<0.05, ##P<0.01, compared to control group
CIHH reduced macrophage infiltration and proinflammatory cytokine expression
A number of studies revealed that inflammatory cells exist around pulmonary remodeled arteries. Macrophages in the lung tissue, particularly in the alveolar spaces and the surrounding pulmonary artery, increased in rats treated with MCT and MCT+CIHH compared with the control animals. CIHH treatment corresponded with decreased macrophage infiltration; however, this did not reach statistical significance (Figure 3A). TNFα and IL-6, also known to play pivotal roles in PAH and pulmonary vascular remodeling (4, 27), were evaluated with ELISA and immunohistochemical staining. ELISA data showed that CIHH prevented the MCT-induced increase in TNF-α and IL-6 levels when compared to the level detected in the MCT group. Moreover, no significant difference was observed between MCT+CIHH-treated and control animals (Figures 3B and 3C). IHC studies were performed to identify the pattern of cellular expression of TNF-α or IL-6 in the lung. Positive staining for TNF-α or IL-6 was significantly higher in lungs and pulmonary arteries from the MCT-treated group compared to those from the control group (Figures 3D and 3E). Taken together, our results demonstrate that CIHH treatment can reduce pulmonary inflammation induced by MCT in PAH rats.
Figure 3.

Effect of CIHH on perivascular inflammation and pro-inflammatory cytokines expression in MCT-treated rats. Peri-vascular inflammation score was increased by MCT injection and the effect of MCT was prevented by CIHH treatment (A). TNF-α (B) and IL-6 (C) levels in serum of the rats treated with MCT and CIHH. Immunohistochemical staining of pulmonary TNF-α (D) and IL-6 (E) protein expression in experimental rats (magnification ×200). MCT induced an increase of TNF-α and the IL-6 level was significantly antagonized by CIHH. Values are means±SD, n=10 for each group. **P<0.01, compared to MCT group; #P<0.05, ##P<0.01, compared to control group
CIHH inhibited pulmonary artery cell proliferation
In order to explore the mechanism underlying CIHH-associated reduction in vascular remodeling, immunostaining was performed to assess cell proliferation in lung tissues derived from control, MCT, CIHH, and MCT+CIHH-treated rats. Image analyses following Ki67 (green) and DAPI counters-taining (blue) revealed levels of cellular proliferation were increased in lung tissues derived from MCT-treated rats compared with the level found in lung tissues of the control rats (Figures 4A and 4B). On the other hand, CIHH treatment reduced proliferative cell numbers in pulmonary arteries following MCT exposure (Figure 4D). Quantitative analysis revealed 48.35 ± 1.31% of Ki67 positive cells per vessel in lung sections derived from MCT+CIHH treated rats compared with 72.54 ± 2.23% in lung sections derived from MCT-treated rats and 26.29 ± 1.50% in lung sections of control rats (Figure 4E).
Figure 4.
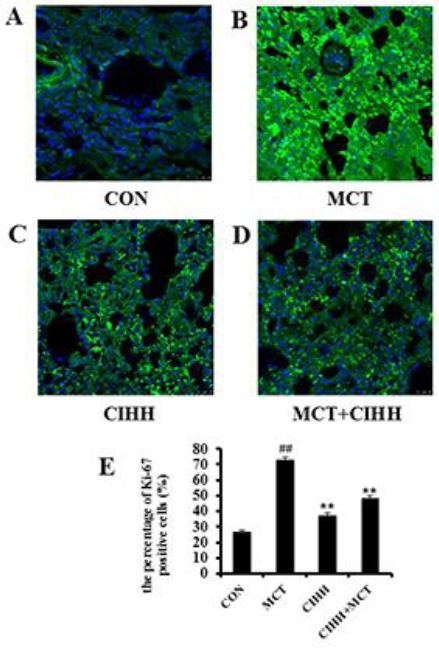
Analysis of the percentage of Ki67-positive cells in pulmonary arteries by immunofluorescence staining. Representative photomicrographs of Ki-67 protein expression (green) in pulmonary arteries (A). Scale bar: 25 μm. The bar graph shows the number of Ki67-positive cells relative to the total SMCs in pulmonary arteries (%) (B). Values are means±SD, n= 10 for each group. **P<0.01, compared to MCT group; ##P<0.01, compared to control group
CIHH promoted T lymphocytes apoptosis and reduced differential frequencies of lymphocyte subclasses
The apoptosis assay of total numbers T lymphocytes (CD3+ cells) from the spleen of rats in each group was performed in triplicate. The percentage of apoptotic CD3+ T lymphocytes found in the spleen of PAH rats were less than cells from controls (27.67±1.6% vs. 38.61±2.5%). Addition of CIHH treatment suppressed the reduced apoptosis of CD3+ T cells (36.95±2.3%) in spleens induced by MCT (Figure 5A). We next sought to determine which cellular subset of spleen cells used for CIHH was essential for the prevention of vascular injury and PAH. Within the T cell subset, we found a trend toward an increase in the proportion of CD4+ T cells in the peripheral blood of PAH versus controls (48.92±2.5% vs. 37.33±1.8%) at the expense of a significant decrease in CD8+ T cells (28.28±1.2% vs. 38.41±1.9%) (Figure 5B). The CD4/CD8 relation consecutively increased. With CIHH treatment, we found significantly less CD4+ T cells (39.23±1.9% vs. 48.92±2.5%) and more CD8+ T cells (37.29±1.7% vs. 28.28±1.2%) in MCT-treated rats compared with those of the MCT alone group and a significantly decreased ratio of CD4/CD8 T cells (Figure 5B). These studies demonstrated that both populations of T cells exhibited regulatory activity and were required to prevent PAH.
Figure 5.
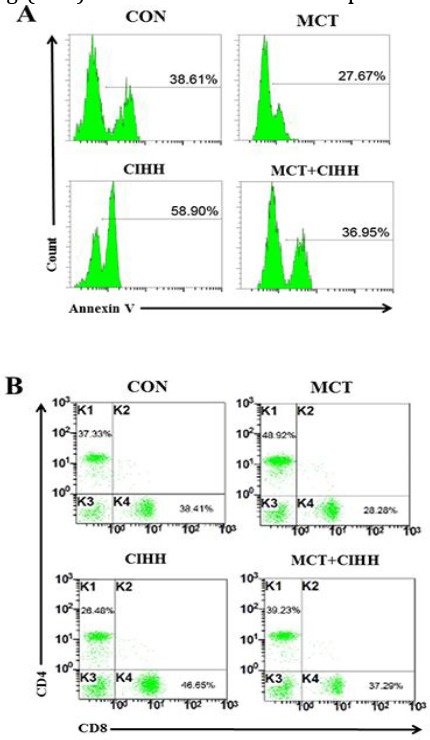
Effect of CIHH on T lymphocytes apoptosis and differential frequencies of lymphocyte subclasses in MCT-treated rats. Representative flow chart of spleen apoptotic CD3+T lymphocytes in rats of each group (A). Representative flow chart of CD4+ and CD8+T lymphocyte levels in peripheral blood of rats in each group (B)
Attenuated NF-kB/ p38 activation involved in the treatment of CIHH
In order to explore the molecular pathways that are involved in CIHH intervention in MCT-induced PAH, protein levels of NF-κB and p38 MAPK were analyzed. Western blot analysis revealed that MCT injection resulted in appearance of phosphorylated p65 subunit staining in lung fractions (Figure 6A). However, CIHH treatments suppressed the phosphor-rylation of p65 subunit of NF-κB in lung tissues compared to that found in the MCT-treated group (Figure 6A). A significant reduction of 77±4.2% was observed in CIHH-treated rats following comparative analysis of P-NF-κB/total NF-κB ratio (Figure 6B).
Figure 6.

CIHH treatment alters expression of p-p38 and NF-κB p-P65 protein in rats. Representative western blotting shows the levels of p-p38 (A) and NF-κB p65 (C) subunit in lung tissue. p38 and Lamin B1 served as internal controls. Normalized band intensity quantification shows the fold change of p-p38 (B) and NF-κB p65 (D) subunit in lung tissue. Data are expressed as means±SD from 4 independent mice. *P<0.05, **P<0.01, compared to MCT group; #P<0.05, ##P<0.01, compared to control group
Activation of MAPKs, especially p38 MAPK, plays a key role in vascular remodeling, vascular endothelial dysfunction and inflammation in hypertensive animal models (28-30). Thus, we examined the level of p38 MAPK phosphorylation in lung homogenate derived from control, MCT, CIHH, and MCT+CIHH treated rats. Phosphorylation of p38 MAPK was increased in preparations derived from MCT rats compared to that from control rats (Figure 6C). However, the level of p38 MAPK phosphorylation was decreased in preparation derived from the MCT+CIHH group when compared to the MCT-treated group (Figure 6C). A significant reduction of 81±3.7% was observed in CIHH treated rats following comparative analysis of P-p38 MAPK/ total p38 MAPK ratio (Figure 6D).
Discussion
In this study, the ability of CIHH to prevent pulmonary inflammation and remodeling in a rat model of PAH induced by MCT was investigated. The data proved that CIHH treatment can markedly reverse mPAP elevation and RV hypertrophy and reduce the pulmonary artery remodeling caused by MCT. This observation is suggested by the results obtained from histological findings as well. CIHH was also found to reduce the intensity of the inflammatory infiltrate in the arteries and proinflammatory cytokine expression, and increase the number of apoptotic T lymphocytes with a predominance of the CD8+ T cell subset. Moreover, our results suggest that CIHH may have a therapeutic function in MCT-induced PAH by modulating pulmonary artery inflammation, which is probably mediated via the suppression of NF-κB and p38 MAPK pathway.
Increasing evidence indicates that inflammation may play an important role in the pathophysiology of PAH (3, 4, 31-33). For example, IL-2, IL-6, TNF-α, soluble TNF-receptor type I, and interferon-γ are elevated in patients with the rare POEMS syndrome (plasma cell dyscrasia and polyneuropathy, organo-megaly, endocrinopathy, monoclonal gammopathy, and skin changes) in which PAH invariably occurs(34). Similarly, increased number of neutrophils, macro-phages, and T and B lymphocytes have been shown in animal lungs in response to chronic hypoxia and MCT (35). These findings implicate immune cell dysfunction and loss of immunologic self-tolerance in the pathogen-nesis of this disease (36). The potential roles of infla-mmatory mediators in the development of PAH have been definitively shown in the MCT model. These include IL-6 (6), IL-1(37), leukotrienes(38), and TNF-α (39). MCT is a pyrrolizidine alkaloid that can cause endothelial injury and PAH. Infiltration by mononuclear cells, especially macrophages, is a prominent pathologic feature of MCT induced PAH in the early phase (40). Our current findings also show the higher levels of TNF-α, IL-6, and inflammatory infiltration in MCT-induced PAH.
Lung histology of patients with PAH revealed perivascular infiltrates of inflammatory cells consis-ting of T and B cells and macrophages exclusively around plexiform lesions (41, 42). It is reported that the majority of CD3+ T lymphocytes in the walls of resistance arteries in PAH patients showed NFATc2 (a nuclear factor of activated T cell) activation (9). Some reports have described a decreased percentage of circulating CD8+ T cells in patients with PAH compared with controls, as well as an increased proportion of FoxP3+ regulatory cells within the CD4+ T cell population, presumably identifying an increase in circulating Tregs with suppressor activity (43). Prolonged and intermittent exposure to aerosolized OVA antigen in mice has been linked to the develop-ment of PAH, a response that is suppressed in CD4+ T-cell depleted mice (44). Thus, there is ample evidence for a link between lymphocytes and the patho-physiology of PAH. The present study demonstrates a decrease in apoptotic CD3+ lymphocytes in the spleen of rats with PAH compared with controls. Importantly, the increases in CD4+ T cells and a reduction in CD8+ T lymphocytes in the peripheral blood of PAH rats found in this study are consistent with the previous study conducted in circulating blood of PAH patients (43). Overall then, our evidence suggests that inflammation should be considered as a possible factor in the pathological underpinnings of PAH. The present data reveal that in animals developing MCT-induced PAH, CIHH treatment inhibits the inflammation and cell proliferation and results in reduction of RVH and vascular remodeling of pulmonary arteries.
In an attempt to clarify the molecular mechanisms underlying the anti-proliferative and anti-migratory effects of CIHH on MCT-induced PAH in rats, we examined NF-κB and p38 MAPK activation by analyzing lung homogenates derived from rats of different groups.
NF-κB and p38 MAPK are activated in response to pro-oxidants, inflammatory stimuli, various growth factors, and mechanical stress, and regulate the expression of genes associated with inflammation, growth, invasion, and angiogenesis (45, 46). Activated NF-κB and p38 MAPK pathways can be found in animal models of PAH and in clinical pathology (29, 30, 45, 46). Our data indicate an increase of NF-κB and p38 MAPK activity in MCT-treated rat lungs, and we prove that the generation and action of many potential inflammatory mediators of PAH are associated with activation of the NF-κB and p38 MAPK signaling pathway (47). Activated NF-κB and p38 MAPK induce the transcription of a range of proliferative and proinflammatory genes, including those that encode COX-2, intercellular adhesion molecule-1 (ICAM-1), vascular endothelial growth factor (VEGF), matrix metalloproteinases (MMPs), TNFα, IL-1β, IL-8, IL-6, and inducible nitric oxide synthase (45, 48). A recent study has examined the potential therapeutic effect of specific p38 MAPK inhibitors on inflammatory diseases (49). Inhibition of NF-κB by pharmacological inter-vention or nanoparticle (decoy of NF-κB) delivery or by genetic manipulation has been reported to attenuate the progression of PAH and RVH (13, 14, 50, 51). In the MCT model used herein, CIHH down-regulated the phosphorylated form of p65 NF-κB, thus decreasing p65 translocation to the nucleus. Moreover, CIHH treatments reduce the level of phosphorylated p38 MAPK in the PAH rat model used in our study.
Conclusion
In summary, we first report that CIHH can down-regulate the MCT-stimulated expression of TNFα and IL-6 in the PAH rat model of our study. Furthermore, our results clearly indicate that NF-κB and p38 MAPK may be important molecular mediators in the biological actions of CIHH in MCT-induced PAH.
Conflict of Interest
The authors declare that they have no conflict of interest.
References
- 1.Rubin LJ. Primary pulmonary hypertension. N Engl J Med. 1997;336:111–117. doi: 10.1056/NEJM199701093360207. [DOI] [PubMed] [Google Scholar]
- 2.Simonneau G, Galie N, Rubin LJ, Langleben D, Seeger W, Domenighetti G, et al. Clinical classification of pulmonary hypertension. J Am Coll Cardiol. 2004;43:S5–S12. doi: 10.1016/j.jacc.2004.02.037. [DOI] [PubMed] [Google Scholar]
- 3.Dorfmüller P, Perros F, Balabanian K, Humbert M. Inflammation in pulmonary arterial hypertension. Eur Respir J. 2003;22:358–363. doi: 10.1183/09031936.03.00038903. [DOI] [PubMed] [Google Scholar]
- 4.Humbert M, Monti G, Brenot F, Sitbon O, Portier A, Grangeot-Keros L, et al. Increased interleukin-1 and interleukin-6 serum concentrations in severe primary pulmonary hypertension. Am J Respir Crit Care Med. 1995;151:1628–1631. doi: 10.1164/ajrccm.151.5.7735624. [DOI] [PubMed] [Google Scholar]
- 5.Soon E, Holmes AM, Treacy CM, Doughty NJ, Southgate L, Machado RD, et al. Elevated levels of inflammatory cytokines predict survival in idiopathic and familial pulmonary arterial hypertension. Circulation. 2010;122:920–927. doi: 10.1161/CIRCULATIONAHA.109.933762. [DOI] [PubMed] [Google Scholar]
- 6.Bhargava A, Kumar A, Yuan N, Gewitz M, Mathew R. Monocrotaline induces interleukin-6 mRNA expression in rat lungs. Heart Dis (Hagerstown, Md) 1998;1:126–132. [PubMed] [Google Scholar]
- 7.Karmochkine M, Wechsler B, Godeau P, Brenot F, Jagot J, Simonneau G. Improvement of severe pulmonary hypertension in a patient with SLE. Ann Rheum Dis. 1996;55:561. doi: 10.1136/ard.55.8.561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Price L, Montani D, Tcherakian C, Dorfmüller P, Souza R, Gambaryan N, et al. Dexamethasone reverses monocrotaline-induced pulmonary arterial hypertension in rats. Eur Respir J. 2011;37:813–822. doi: 10.1183/09031936.00028310. [DOI] [PubMed] [Google Scholar]
- 9.Bonnet S, Rochefort G, Sutendra G, Archer SL, Haromy A, Webster L, et al. The nuclear factor of activated T cells in pulmonary arterial hypertension can be therapeutically targeted. Proc Natl Acad Sci. 2007;104:11418–11423. doi: 10.1073/pnas.0610467104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Suzuki C, Takahashi M, Morimoto H, Izawa A, Ise H, Hongo M, et al. Mycophenolate mofetil attenuates pulmonary arterial hypertension in rats. Biochem Biophys Res Commun. 2006;349:781–788. doi: 10.1016/j.bbrc.2006.08.109. [DOI] [PubMed] [Google Scholar]
- 11.Gordon JW, Shaw JA, Kirshenbaum LA. Multiple facets of NF-κB in the heart to be or not to NF-κB. Circ Res. 2011;108:1122–1132. doi: 10.1161/CIRCRESAHA.110.226928. [DOI] [PubMed] [Google Scholar]
- 12.Hall G, Hasday JD, Rogers TB. Regulating the regulator:NF-κB signaling in heart. J Mol Cell Cardiol. 2006;41:580–591. doi: 10.1016/j.yjmcc.2006.07.006. [DOI] [PubMed] [Google Scholar]
- 13.Chen L, Nakano K, Kimura S, Matoba T, Iwata E, Miyagawa M, et al. Nanoparticle-mediated delivery of pitavastatin into lungs ameliorates the development and induces regression of monocrotaline-induced pulmonary artery hypertension. Hypertension. 2011;57:343–350. doi: 10.1161/HYPERTENSIONAHA.110.157032. [DOI] [PubMed] [Google Scholar]
- 14.Kimura S, Egashira K, Chen L, Nakano K, Iwata E, Miyagawa M, et al. Nanoparticle-mediated delivery of nuclear factor κB decoy into lungs ameliorates monocrotaline-induced pulmonary arterial hypertension. Hypertension. 2009;53:877–883. doi: 10.1161/HYPERTENSIONAHA.108.121418. [DOI] [PubMed] [Google Scholar]
- 15.Hassoun PM, Mouthon L, Barberà JA, Eddahibi S, Flores SC, Grimminger F, et al. Inflammation, growth factors, and pulmonary vascular remodeling. J Am Coll Cardiol. 2009;54:S10–S19. doi: 10.1016/j.jacc.2009.04.006. [DOI] [PubMed] [Google Scholar]
- 16.Sugden PH, Clerk A. “Stress-responsive” mitogen-activated protein kinases (c-Jun N-terminal kinases and p38 mitogen-activated protein kinases) in the myocardium. Circ Res. 1998;83:345–352. doi: 10.1161/01.res.83.4.345. [DOI] [PubMed] [Google Scholar]
- 17.Rose F, Grimminger F, Appel J, Heller M, Pies V, Weissmann N, et al. Hypoxic pulmonary artery fibroblasts trigger proliferation of vascular smooth muscle cells:role of hypoxia-inducible transcription factors. FASEB J. 2002;16:1660–1661. doi: 10.1096/fj.02-0420fje. [DOI] [PubMed] [Google Scholar]
- 18.Welsh DJ, Scott PH, Peacock AJ. p38 MAP kinase isoform activity and cell cycle regulators in the proliferative response of pulmonary and systemic artery fibroblasts to acute hypoxia. Pulm Pharmacol Ther. 2006;19:128–138. doi: 10.1016/j.pupt.2005.04.008. [DOI] [PubMed] [Google Scholar]
- 19.Welsh DJ, Peacock AJ, MacLEAN M, Harnett M. Chronic hypoxia induces constitutive p38 mitogen-activated protein kinase activity that correlates with enhanced cellular proliferation in fibroblasts from rat pulmonary but not systemic arteries. Am J Respir Crit Care Med. 2001;164:282–289. doi: 10.1164/ajrccm.164.2.2008054. [DOI] [PubMed] [Google Scholar]
- 20.Jin N, Hatton N, Swartz DR, Xia X-l, Harrington MA, Larsen SH, et al. Hypoxia activates un-N-terminal kinase, extracellular Signal–regulated protein kinase, and p38 kinase in pulmonary arteries. Am J Respir Cell Mol Biol. 2000;23:593–601. doi: 10.1165/ajrcmb.23.5.3921. [DOI] [PubMed] [Google Scholar]
- 21.Scott PH, Paul A, Belham CM, Peacock AJ, Wadsworth RM, Gould GW, et al. Hypoxic stimulation of the stress-activated protein kinases in pulmonary artery fibroblasts. Am J Respir Crit Care Med. 1998;158:958–962. doi: 10.1164/ajrccm.158.3.9712130. [DOI] [PubMed] [Google Scholar]
- 22.Wang Q, Zuo X-r, Wang Y-y, Xie W-p, Wang H, Zhang M. Monocrotaline-induced pulmonary arterial hypertension is attenuated by TNF-αantagonists via the suppression of TNF-αexpression and NF-κB pathway in rats. Vascul Pharmacol. 2013;58:71–77. doi: 10.1016/j.vph.2012.07.006. [DOI] [PubMed] [Google Scholar]
- 23.Hardziyenka M, Campian ME, de Bruin-Bon HR, Michel MC, Tan HL. Sequence of echocardiographic changes during development of right ventricular failure in rat. J Am Soc Echocardiogr. 2006;19:1272–1279. doi: 10.1016/j.echo.2006.04.036. [DOI] [PubMed] [Google Scholar]
- 24.Stenmark KR, Meyrick B, Galie N, Mooi WJ, McMurtry IF. Animal models of pulmonary arterial hypertension:the hope for etiological discovery and pharmacological cure. Am J Physiol Lung Cell Mol Physiol. 2009;297:L1013–L1032. doi: 10.1152/ajplung.00217.2009. [DOI] [PubMed] [Google Scholar]
- 25.Yuan Y, Liao L, Tulis DA, Xu J. Steroid receptor coactivator-3 is required for inhibition of neointima formation by estrogen. Circulation. 2002;105:2653–2659. doi: 10.1161/01.cir.0000018947.95555.65. [DOI] [PubMed] [Google Scholar]
- 26.Friedrichs K, Gluba S, Eidtmann H, Jonat W. Overexpression of p53 and prognosis in breast cancer. Cancer. 1993;72:3641–3647. doi: 10.1002/1097-0142(19931215)72:12<3641::aid-cncr2820721215>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- 27.Fujita M, Shannon JM, Irvin CG, Fagan KA, Cool C, Augustin A, et al. Overexpression of tumor necrosis factor-αproduces an increase in lung volumes and pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol. 2001;280:L39–L49. doi: 10.1152/ajplung.2001.280.1.L39. [DOI] [PubMed] [Google Scholar]
- 28.Morin C, Hiram R, Rousseau E, Blier PU, Fortin S. Docosapentaenoic acid monoacylglyceride reduces inflammation and vascular remodeling in experimental pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol. 2014 doi: 10.1152/ajpheart.00814.2013. ajpheart. 00814.02013. [DOI] [PubMed] [Google Scholar]
- 29.Takahashi M, Okazaki H, Ogata Y, Takeuchi K, Ikeda U, Shimada K. Lysophosphatidylcholine induces apoptosis in human endothelial cells through a p38-mitogen-activated protein kinase-dependent mechanism. Atherosclerosis. 2002;161:387–394. doi: 10.1016/s0021-9150(01)00674-8. [DOI] [PubMed] [Google Scholar]
- 30.Weerackody RP, Welsh DJ, Wadsworth RM, Peacock AJ. Inhibition of p38 MAPK reverses hypoxia-induced pulmonary artery endothelial dysfunction. Am J Physiol Lung Cell Mol Physiol. 2009;296:H1312–H1320. doi: 10.1152/ajpheart.00977.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fartoukh M, Emilie D, Le Gall C, Monti G, Simonneau G, Humbert M. Chemokine macrophage inflammatory protein-1αmRNA expression in lung biopsy specimens of primary pulmonary hypertension. Chest J. 1998;114:50S–51S. doi: 10.1378/chest.114.1_supplement.50s. [DOI] [PubMed] [Google Scholar]
- 32.Cool CD, Kennedy D, Voelkel NF, Tuder RM. Pathogenesis and evolution of plexiform lesions in pulmonary hypertension associated with scleroderma and human immunodeficiency virus infection. Hum Pathol. 1997;28:434–442. doi: 10.1016/s0046-8177(97)90032-0. [DOI] [PubMed] [Google Scholar]
- 33.Humbert M, Morrell NW, Archer SL, Stenmark KR, MacLean MR, Lang IM, et al. Cellular and molecular pathobiology of pulmonary arterial hypertension. J Am Coll Cardiol. 2004;43:S13–S24. doi: 10.1016/j.jacc.2004.02.029. [DOI] [PubMed] [Google Scholar]
- 34.Lesprit P, Godeau B, Authier F-J, Soubrier M, Zuber M, Larroche C, et al. Pulmonary hypertension in POEMS syndrome:a new feature mediated by cytokines. Am J Respir Crit Care Med. 1998;157:907–911. doi: 10.1164/ajrccm.157.3.9707095. [DOI] [PubMed] [Google Scholar]
- 35.Minamino T, Christou H, Hsieh C-M, Liu Y, Dhawan V, Abraham NG, et al. Targeted expression of heme oxygenase-1 prevents the pulmonary inflammatory and vascular responses to hypoxia. Proc Natl Acad Sci. 2001;98:8798–8803. doi: 10.1073/pnas.161272598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Barst RJ, Loyd JE. Genetics and immunogenetic aspects of primary pulmonary hypertension. Chest J. 1998;114:231S–236S. doi: 10.1378/chest.114.3_supplement.231s. [DOI] [PubMed] [Google Scholar]
- 37.Voelkel NF, Tuder R. Interleukin-1 receptor antagonist inhibits pulmonary hypertension induced by inflammationa. Ann N Y Acad Sci. 1994;725:104–109. doi: 10.1111/j.1749-6632.1994.tb39794.x. [DOI] [PubMed] [Google Scholar]
- 38.Tabata T, Ono S, Song C, Noda M, Suzuki S, Tanita T, et al. [Role of leukotriene B4 in monocrotaline-induced pulmonary hypertension] Nihon Kyobu Shikkan Gakkai Zasshi. 1997;35:160–166. [PubMed] [Google Scholar]
- 39.Miyata M, Ueno Y, Sekine H, Ito O, Sakuma F, Koike H, et al. Protective effect of beraprost sodium, a stable prostacyclin analogue, in development of monocrotaline-induced pulmonary hypertension. J Cardiovasc Pharmacol. 1996;27:20–26. doi: 10.1097/00005344-199601000-00004. [DOI] [PubMed] [Google Scholar]
- 40.Sugita T, Hyers TM, Dauber I, Wagner W, McMurtry I, Reeves J. Lung vessel leak precedes right ventricular hypertrophy in monocrotaline-treated rats. J Appl Physiol. 1983;54:371–374. doi: 10.1152/jappl.1983.54.2.371. [DOI] [PubMed] [Google Scholar]
- 41.Tuder RM, Groves B, Badesch DB, Voelkel NF. Exuberant endothelial cell growth and elements of inflammation are present in plexiform lesions of pulmonary hypertension. Am J Pathol. 1994;144:275. [PMC free article] [PubMed] [Google Scholar]
- 42.Alpert M, Goldberg S, Singsen B, Durham J, Sharp G, Ahmad M, et al. Cardiovascular manifestations of mixed connective tissue disease in adults. Circulation. 1983;68:1182–1193. doi: 10.1161/01.cir.68.6.1182. [DOI] [PubMed] [Google Scholar]
- 43.Ulrich S, Nicolls MR, Taraseviciene L, Speich R, Voelkel N. Increased regulatory and decreased CD8+cytotoxic T cells in the blood of patients with idiopathic pulmonary arterial hypertension. Respiration. 2007;75:272–280. doi: 10.1159/000111548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Daley E, Emson C, Guignabert C, de Waal Malefyt R, Louten J, Kurup VP, et al. Pulmonary arterial remodeling induced by a Th2 immune response. J Exp Med. 2008;205:361–372. doi: 10.1084/jem.20071008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Behr T, Berova M, Doe C, Ju H, Angermann CE, Boehm J, et al. p38 mitogen-activated protein kinase inhibitors for the treatment of chronic cardiovascular disease. Curr Opin Investig Drugs. 2003;4:1059–1064. [PubMed] [Google Scholar]
- 46.Karin M, Lin A. NF-κB at the crossroads of life and death. Nat Immunol. 2002;3:221–227. doi: 10.1038/ni0302-221. [DOI] [PubMed] [Google Scholar]
- 47.Lee JC, Laydon JT, McDonnell PC, Gallagher TF, Kumar S, Green D, et al. A protein kinase involved in the regulation of inflammatory cytokine biosynthesis. Nature. 1994;372:739–746. doi: 10.1038/372739a0. [DOI] [PubMed] [Google Scholar]
- 48.Matsumori A. Anti-inflammatory therapy for heart failure. Curr Opin Pharmacol. 2004;4:171–176. doi: 10.1016/j.coph.2003.11.003. [DOI] [PubMed] [Google Scholar]
- 49.Lee JC, Kumar S, Griswold DE, Underwood DC, Votta BJ, Adams JL. Inhibition of p38 MAP kinase as a therapeutic strategy. Immunopharmacology. 2000;47:185–201. doi: 10.1016/s0162-3109(00)00206-x. [DOI] [PubMed] [Google Scholar]
- 50.Hosokawa S, Haraguchi G, Sasaki A, Arai H, Muto S, Itai A, et al. Pathophysiological roles of NF-kB in pulmonary arterial hypertension:Effects of synthetic selective NF-kB inhibitor IMD-0354. Cardiovasc Res. 2013:cvt105. doi: 10.1093/cvr/cvt105. [DOI] [PubMed] [Google Scholar]
- 51.Kumar S, Wei C, Thomas CM, Kim I-K, Seqqat R, Kumar R, et al. Cardiac-specific genetic inhibition of nuclear factor-κB prevents right ventricular hypertrophy induced by monocrotaline. Am J Physiol Heart Circ Physiol. 2012;302:H1655–H1666. doi: 10.1152/ajpheart.00756.2011. [DOI] [PubMed] [Google Scholar]