

Abstract
Objective(s):
Therapeutic effect of many selectable methods applied in clinical practice for treating hypertrophic scar (HS) is not still so satisfactory. Meanwhile, a few medicines may lead to several undesirable complications. The traditional Chinese medicine, Rg3, has been reported for multiple antitumor effects previously. We have conducted series of animal experiments and confirmed the inhibitory effect of Rg3 in HS before. The aim of this study was to further verify the conclusions of previous studies and reveal the specific functional mechanisms of Rg3.
Materials and Methods:
The HS specimens were obtained from the patients aged from 15 to 36 years without systemic diseases and the primary cultured cells were isolated from the scar tissue and expanded in vitro. In every experiment, hypertrophic scar fibroblasts (HSFs) were divided into three groups and respectively cultured in medium with or without different Rg3 concentrations (50, 100 μg/ml). Cell viability assay, flow cytometry analysis (FCM), quantitative PCR, cell migration assay, immunofluorescence staining, western blot and ELISA were employed.
Results:
The outcomes demonstrated that Rg3 could suppress cell proliferation, vascularization and extracellular matrix (ECM) deposition of HSFs in vitro by TGF-β/SMAD and Erk signaling pathways. Significant statistical differences were between control group and Rg3-treated groups (P<0.05).
Conclusion:
This study provides sufficient in vitro evidences for Rg3 as a promising drug in the treatment of human HS.
Keywords: Fibroblast, Ginsenoside Rg(3), Hypertrophic scar, Signal transduction, Wound healing
Introduction
Hypertrophic scar (HS) is one type of the pathologic scars, which responds to the process of wound healing of dermis after skin trauma or serious burns (1). In addition to keloids, HS is characterized by disordered fibrotic proliferation and excessive accumulation of extracellular matrix (ECM) (2). However, they do not invade beyond the boundaries of the original lesions and constantly recrudesce after surgical operation, which serve as the characteristics of keloids (3).
Although the pathogenesis of HS is still ambiguous and indeterminate, the aberrant inflammation, superabundant ECM amassing, expansive neovascula-rization, lessened apoptosis and atypical ECM remodeling have been reported as the main features and mechanisms of HS formation process (4). Thus, treatments should be targeted at preventing or inhibiting these probable pathological processes. Nowadays, many selectable methods such as pressure therapy, adhesive gel sheet, corticosteroids, etc. are
applied in clinical practice and several new treat-ments such as interferon, calcium channel blocker, etc. have been reported (5, 6). Although these methods have their own superiorities, they can only function in certain specific stages and have no effect in other courses of HS formation. Even if they are combined to deal with HS, the therapeutic effect is not still so satisfactory. In addition, a few medicines may bring the risk of systemic diseases, local erythema, molting, pigmentation, wound ulcers and other undesirable complications (7). Therefore, it urged us to seek novel and better treatment strategies or drugs for HS patients.
The time length of wound closure is one of the important factors affecting the occurrence probability of HS (8). Hence, the ideal drugs or other treatments are better to function in the inflammatory phase, which is the first stage of the wound healing process to shorten the healing time of the lesion. Meanwhile, because of the numerous mechanisms during the whole formation process of HS, the treatments should aim at the multiple steps of the entire course.
Ginseng has been one of the most commonly used herbal medicines in the Orient. Ginsenosides are a group of glycosylated triterpenes, also known as saponins, which represent the pharmacologically active compounds found in the Panax root. Ginsenoside Rg3 is the most active one among ginseng saponins, which includes two different optical isomers named 20(R)-G. Rg3 and 20(S)-G. Rg3. There have been reported that G. Rg3 had vigorous biological activity and wide ranges of clinical and pharmacological effects, especially the 20(R)-G. Rg3 (9). Many studies have demonstrated that Rg3 can inhibit cell proliferation and decrease the angiogenesis of various kinds of tumors (10).
Our previous studies demonstrated that Rg3 could restrict the excessive inflammation in vivo, but do not delay the wound healing process, indicating that Rg3 could serve as an early intervention to treat HS (11). The previous published researches mainly focused on animal experiments and the effect of implantable G. Rg3-loaded electrospun fibrous membranes on HS in vivo. To further verify the conclusions of previous studies and reveal the specific functional mechanisms of Rg3, we carry out a series of related molecular and cellular experiments in vitro, which may consummate the exploration of treatment on HS by G. Rg3.
Materials and Methods
Chemicals
G. Rg3 (98.6% purity, Fusheng Pharmaceutical Ltd. Dalian, China) was dissolved in dimethyl sulfoxide (DMSO) and diluted with DMEM solution containing 10% fetal bovine serum (FBS) (HyClone, South Logan, UT) to form the final concentrations gradient (50, 100 μg/ml), as Kim et al. described (12). The final concentrations of DMSO in the culture medium were < 0.1%.
Patients
Eighteen Chinese HS patients were recruited in our research. The HS specimens were obtained from these patients aged from 15 to 36 years without systemic diseases. The selected scars were in the active stage and none of them had undergone any treatment before. All the patients were informed of the purpose and procedure of this research and agreed to provide their scars before operations. The HS tissues were completely removed from the patients’ skin of face, neck and abdomen. Prior written informed consent was obtained from all participants and every procedure had received the approval of the Ethic Committee.
Culture of hypertrophic scar fibroblasts
The dermis was cut into 1×1 mm pieces after removing the adipose tissues and epidermis. Then, these pieces were digested in 0.25% collagenase for about three hours. After centrifugation and discarding the supernatant, the remaining precipitate including abundant hypertrophic scar fibroblasts (HSFs) and some small pieces of scar tissues were cultured simultaneously in high glucose DMEM (Gibco BRL, Grand Island, NY) supplemented with 10% FBS, penicillin (100 U ml-1) and streptomycin (100 μg ml-1) (Sigma-Aldrich, St Louis, MO) in the 100 mm diameter dishes. They were put into humidified incubator at 37°C with 5% CO2. Culture media were replaced every two days. The HSFs gradually attached the bottom surface of the dish or migrated out of the tiny pieces in 3 to 5 days. The second to forth passage of HSFs were used in this research.
Cell viability assay
The viability of HSFs treated with or without Rg3 was tested by CCK-8 assay (Cell counting kit-8, Dojindo, Kumamoto, Japan). The HSFs were cultured in serum-free medium for 24 hr for synchronization and then seeded in 96-well plates at a density of 1×104 cells/ml. After incubating overnight for 24 hr at 37°C under 5% CO2, the medium with different Rg3 concentrations (50, 100 μg/ml) was added into the wells. Ten microliter CCK-8 was added into each well after one, two, three, four and five days of cultivation, respectively. After 2.5 hr of incubation, 100 μl aliquot of incubated medium was pipetted into another 96-well plate, and the colorimetric absorbance was recorded by a microplate reader (Thermo labsystems, Helsinki, Finland) at 450 nm.
Flow cytometric analysis of annexin V-FITC staining
Flow cytometry method (FCM) was used to detect the rate of apoptosis by the annexin V-FITC kit (Miltenyi Biotec, Teterow, Germany). The fibroblasts were incubated in the 6-well plates with the medium containing different Rg3 concentrations (50, 100 μg/ml). After incubation for 72 hr, the HSFs in each well were collected by centrifugation and resuspended in 500 μl of binding buffer. Ten microliter of annexin V-FITC was added into the HSFs suspension and the mixture was incubated for 10 min at room temperature in the dark. After that, another 10 μl propidium iodide (PI) was added and performed like annexin V-FITC. Finally, the reaction was prevented with ice-bath. We conducted the quantitative analysis by the flowcytometer (BD Bioscience, San Jose, CA). Over 10,000 cells from each well were counted and the apoptotic percentage was quantitatively analyzed by Cell Quest software (BD Bioscience, San Jose, CA).
RNA isolation and quantitative PCR (Q-PCR)
HSFs suspension was added into 10 cm culture dish with a density of 5×104 cells/ml. After 48 hr, the medium was changed for new with or without different Rg3 concentrations (50, 100 μg/ml) and continued to culture for 72 hr. Afterwards, fibroblasts were harvested and total RNA was extracted by Trizol Reagent (Invitrogen, Carlsbad, CA). cDNA was converted from 2 μg of total RNA with Avian Myeloblastosis Virus (AMV) reverse transcriptase (Promega, Madison, WI) in a 20 μl of reaction system (4 μl of 5×buffer, 1 μl of oligo-(dT), 2 μl of dNTP, 0.5 μl of RNase inhibitor, 0.5 μl of AMV reverse transcriptase, 12 μl of RNA and ddH2O). The 20 μl of reaction system was incubated at 30°C for 10 min, 42°C for 60 min, 95°C for 5 min, and 5°C for 5 min. Q-PCR was operated by the Power SYBRGreen PCR master mix (2×) (Applied Biosystems, Foster City, CA) in a real-time thermal cycler (Stratagene Mx3000PTM Q-PCR System, La Jolla, CA). The amplified products were normalized against the internal reference of GAPDH. The primers for Q-PCR analysis are listed in Table 1.
Table 1.
Primer pairs used in Q-PCR analysis are listed as follows
| Gene | Primer sequence (5’-3’) | Product size (bp) |
|---|---|---|
| Type I Collagen | Forward: GGCGGCCAGGGCTCCGACCC Reverse: AATTCCTGGTCTGGGGCACC |
319 |
| Type III Collagen | Forward: TGGTGTTGGAGCCGCTGCCA Reverse: CTCAGCACTAGAATCTGTCC |
346 |
| Fibronectin | Forward: GCCACTGGAGTCTTTACCACA Reverse: CCTCGGTGTTGTAAGGTGGA |
61 |
| α-SMA | Forward: CATCATGCGTCTGGATCTGG Reverse: GGACAATCTCACGCTCAGCA |
107 |
| CTGF | Forward: ACAAGGGCCTCTTCTGTGACTT Reverse: GGTACACCGTACCACCGAAGAT |
102 |
| PCNA | Forward: GTGATTCCACCACCATGTTC Reverse: TGAGACGAGTCCATGCTCTG |
145 |
| TGF-β1 | Forward: GAAGTGGATCCACGAGCCCAAG Reverse: GCTGCACTTGCAGGAGCGCAC |
227 |
| TGF-β3 | Forward: GGTTTTCCGCTTCAATGTGT Reverse: GCTCGATCCTCTGCTCATTC |
119 |
| VEGF | Forward: ACGAAGTGGTGAAGTTCATGGAA Reverse: AAGATGTCCACCAAGGTCTCGAT |
73 |
| PAI-1 | Forward: TCATCATCAATGACTGGGTGAAGAC Reverse: TTCCACTGGCCGTTGAAGTAGAG |
127 |
| SMAD-7 | Forward: GTGGCATACTGGGAGGAGAA Reverse: GATGGAGAAACCAGGGAACA |
309 |
| MMP2 | Forward: CGTCTGTCCCAGGATGACATC Reverse: ATGTCAGGAGAGGCCCCATA |
59 |
| MMP9 | Forward: TTCCAGTACCGAGAGAAAGCCTAT Reverse: GGTCACGTAGCCCACTTGGT |
102 |
| MMP13 | Forward: ACTGAGAGGCTCCGAGAAATG Reverse: GAACCCCGCATCTTGGCTT |
103 |
| IL-6 | Forward: TGTAGCCGCCCCACACA Reverse: GGATGTACCGAATTTGTTTGTCAA |
68 |
| MCP-1 | Forward: CAAACTGAAGCTCGCACTCTCGCC Reverse: ATTCTTGGGTTGTGGAGTGAGTGTTCA |
354 |
| α-TNF | Forward: TCTTCTCGAACCCCGAGTGA Reverse: GGAGCTGCCCCTCAGCTT |
70 |
| TIMP-1 | Forward: CGCTGACATCCGGTTCGT Reverse: TGTGGAAGTATCCGCAGACACT |
59 |
| VEGFR | Forward: GAAGGAGAGGACCTGAAACTGTC Reverse: ACTCTTTCAATAAACAGCGTGCTG |
458 |
| PDGF | Forward: AGTGACCACTCGATCCGCTCCT Reverse: TTTGGGGCGTTTTGGCTCGCTG |
517 |
| GAPDH | Forward: TCACCATCTTCCAGGAGCG Reverse: CTGCTTCACCACCTTCTTGA |
572 |
α-SMA, alpha-smooth muscle actin; CTGF, connective tissue growth factor; PCNA, proliferating cell nuclear antigen; TGF-β, transforming growth factor-β; VEGF, vascular endothelial growth factor; PAI-1, plasminogen activator inhibitor-1; Smads, drosophila mothers against decapentaplegic protein; MMPs, matrix metalloproteinases; IL-6, interleukin 6; MCP-1, monocyte chemoattractant protein-1; α-TNF, tumor necrosis factor alpha; TIMP-1, tissue inhibitor of metalloproteinase-1; VEGFR, vascular endothelial growth factor receptor; PDGF, platelet derived growth factor
Fibroblast migration assay
For the scratch wound assay, 2×105 HSFs were seeded in each well of 6-well culture plates and incubated until reached almost 100% confluence. Then, a scratch wound was created on cells by a sterile 200 μl pipette tip, which was perpendicular to the bottom of the dish. The cell cultures were changed for serum-free DMEM with or without different Rg3 concentrations (50, 100 μg/ml). Photographs of each wound were taken in five random views under a microscope (Nikon, Tokyo, Japan) immediately or at assigned time point (24 hr or 48 hr) after scratch. The results were quantified and analyzed by the commercial software Image pro-plus version 6.0 (Media Cybernetics, Silver Spring, MD) and public domain image processing program. Data (mean±SD, n=3) were expressed as the percentage of the scratched cell-free zone filled by HSFs.
For the transwell assay, the transwell chambers with 8 μm membrane pore size were put into 24-well plates. After cultivation overnight in serum-free medium, the cell suspensions (1×105 cells/well) were added into the upper chambers and incubated with or without different Rg3 concentrations (50, 100 μg/ml) for 24 hr and 48 hr. Medium with 10% FBS was put into the lower chamber. After incubation for specific time period, cells remaining on the upper surface of the membrane were completely removed away by a cotton swab. Afterwards, fibroblasts that migrated to the bottom surface of the membrane were fixed with 4 % paraformaldehyde and stained with DAPI dye. The number of migrated cells was counted in five random fields under the fluorescence microscope (Olympus, Tokyo, Japan).
Immunofluorescence staining for type I collagen, α-SMA expression and ki-67
HSFs were seeded in the 6-well plates at a density of 2×104 cells/ml. After 2-3 days of regular cultivation, the medium was changed for new, which contained different Rg3 concentrations (50, 100 μg/ml) and the cultivation was continued for another 72 hr. Afterwards, HSFs were fixed in paraformaldehyde and the cell membranes were permeabilized by Triton X-100. After incubated overnight at 4°C with primary antibodies against type I collagen, alpha-smooth muscle actin (α-SMA) and ki-67 (rabbit anti-human, Abcam, Cambridge, MA), the cells were cultivated with appropriate fluorescent secondary antibodies (goat anti-rabbit, Jackson Immunoresearch Laboratories, West Grove, PA). The nuclei were stained with DAPI. The positive cells (green) and nuclear DAPI dye (blue) were photographed under a fluorescence microscope (Olympus, Tokyo, Japan). The staining of each section that represented expression level was compared between different groups. The incorporation ratio of ki-67-positive cells was calculated by dividing the number of ki-67-positive cells to total cells. The outcomes were counted in five randomly selected fields.
Western blot analysis
Western blot analysis was performed essentially as described previously (13), with primary antibodies specific for type I collagen, type III collagen, fibronectin, p-SMAD2, p-SMAD3, total-SMAD2/3, SMAD7, p-Erk1/2, and total-Erk1/2 (Cell Signaling Technology, Beverly, MA). The Western blots were developed using a Super-GL ECL reagent (Novland, Shanghai, China) and exposed onto a KODAK X-Omat BT Film (Kodak, New York). The outcomes were analyzed by digital imaging system equipped with AlphaEaseFC software (Alpha Imager 2000, Alpha Innotech).
ELISA assay
HSFs suspension was added into 6 cm culture dish with a density of 4×104 cells/ml. After 48 hr, the culture medium was replaced with serum-free DMEM with or without different Rg3 concentrations (50, 100 μg/ml) and continued to culture for another 72 hr. Afterwards, the culture media that contained the released transforming growth factor-beta 1 (TGF-β1) and vascular endothelial growth factor (VEGF) proteins from HSFs were collected and measured by ELISA Development Kits (R&D Systems, Minneapolis, MN). The absorbance measurement was implemented at a wavelength of 450 nm by a microreader (Thermo Fisher Scientific, Waltham, MA).
Statistical analysis
All assays were performed in triplicate and repeated in three samples. The data were presented as mean±standard deviation (SD). The differences among groups were measured by one-way ANOVA test and differences between two groups were tested with a post-hoc statistical method. SPSS 21.0 software (Chicago, IL) was used in statistical analysis. P<0.05 is considered as statistically significance.
Results
Ginsenoside Rg3 inhibited HSFs proliferation and induced apoptosis at high concentration
CCK-8 assay was used to detect the effect of Rg3 on HSFs proliferation. As shown in Figure 1(a), both of the two Rg3-treated groups could obviously inhibit HSFs proliferation after 48 hr of incubation compared with the control group (*P<0.05). However, the apparent difference between 50 μg/ml and 100 μg/ml Rg3-treated groups was detected from day 3 (&P<0.05). There also was apparent difference between DMSO group and Rg3-treated groups (#P<0.05), but no obvious difference was found between control group and DMSO group.
Figure 1.
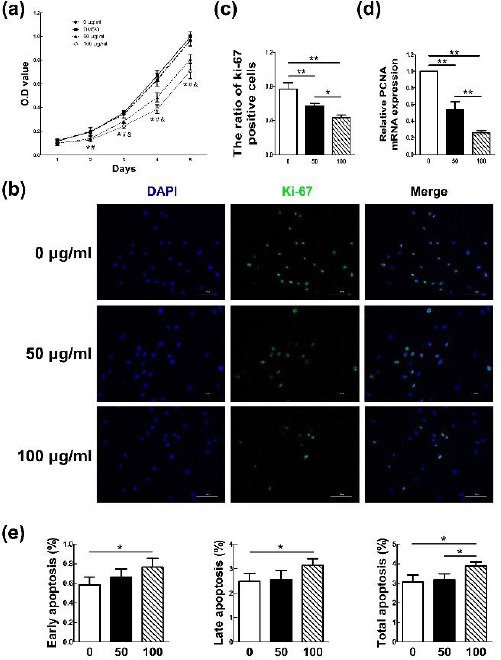
Hypertrophic scar fibroblasts (HSFs) proliferation could be effectively inhibited by Rg3 and the apoptosis could be induced at high Rg3 concentration. The proliferation rates in the HSFs treated with or without different Rg3 concentrations (50, 100 μg/ml) at day 1, 2, 3, 4, and 5 are showed in Figure 1(a). Both of the two Rg3-treated groups could obviously inhibit HSFs proliferation after 48 hr of incubation (*P<0.05). However, the apparent difference between 50 μg/ml and 100 μg/ml Rg3-treated group was detected from day 3 (&P<0.05). Apparent difference also existed between DMSO group and Rg3-treated groups (#P<0.05). As shown in Figure 1(b) and 1(c), the expression of the proliferation marker Ki-67 in Rg3-treated groups was clearly decreased as fewer HSFs were able to merge Ki-67 (*P<0.05, **P<0.01). The level differences of the proliferating cell nuclear antigen (PCNA) proliferation gene between experimental and control groups were showed in Figure 1(d). Evident difference existed between the two groups (**P<0.01). The results of flowcytometry analysis in Figure 1(e) showed that obvious difference existed not only between control group and 100 μg/ml-Rg3-treated group (**P<0.01), but also between the two experimental groups. However, there was no obvious difference between the control group and 50 μg/ml-Rg3-treated group in each apoptosis phase
Then, the Ki-67 immunofluorescence in combi-nation with DAPI nuclear staining was performed. As shown in Figure 1(b), the expression of the proliferation marker Ki-67 in Rg3-treated groups was clearly decreased as fewer HSFs were able to merge Ki-67 compared with that in control group, and the difference of percentages of Ki-67-positive cells between non-treated and Rg3-treated groups was demonstrated in Figure 1(c) (*P<0.05, **P<0.01).
Meanwhile, the difference of the proliferating cell nuclear antigen (PCNA) proliferation gene between experimental and control groups was detected by Q-PCR. There was significant difference between each two groups (**P<0.01) as shown in Figure 1(d). The mRNA expression of PCNA was obviously decreased as the Rg3 concentration increased.
The results of FCM in Figure 1(e) showed that there only was statistical difference between control group and 100 μg/ml-Rg3-treated group in separate early apoptosis phase and late phase (*P<0.05). Statistical difference is not only between control group and 100 μg/ml-Rg3-treated group (**P<0.01), but also there is between the two experimental groups. However, there was no obvious difference between the control group and 50 μg/ml-Rg3-treated group in each apoptosis phase.
G. Rg3 reduced collagen production and ECM accumulation
The mRNA or protein levels of collagen I, and III, fibronectin, connective tissue growth factor (CTGF), Interleukin 6 (IL-6), monocyte chemoattractant protein 1 (MCP-1), tumor necrosis factor alpha (α-TNF), α-SMA, TGF-β3, the ratio of matrix metalloproteinase-2/tissue inhibitor of matrix metalloproteinase 1 (MMP2/TIMP1), MMP9/TIMP1 and MMP13/TIMP1 quantified in HSFs by Q-PCR and Western blot were showed in Figure 2. Treatment of Rg3 could visibly suppress both mRNA and protein levels of collagen I, and III and fibronectin as shown in Figure 2(a) and (e). Moreover, Rg3 apparently reduced the levels of other pro-fibrogenic genes such as CTGF, IL-6, MCP-1, α-TNF, and α-SMA (Figure 2(b)), and simultaneously strengthen the level of anti-fibrosis gene such as TGF-β3 (Figure 2(d)) in HSFs. There is difference between the two groups (*P<0.05, **P<0.01). Furthermore, the incubation of Rg3 could distinctly raise the ratio of MMP2/TIMP1, MMP9/TIMP1 and MMP13/TIMP1. There were also evident differences between the two groups (*P<0.05, **P<0.01), which are demonstrated in Figure 2(c).
Figure 2.
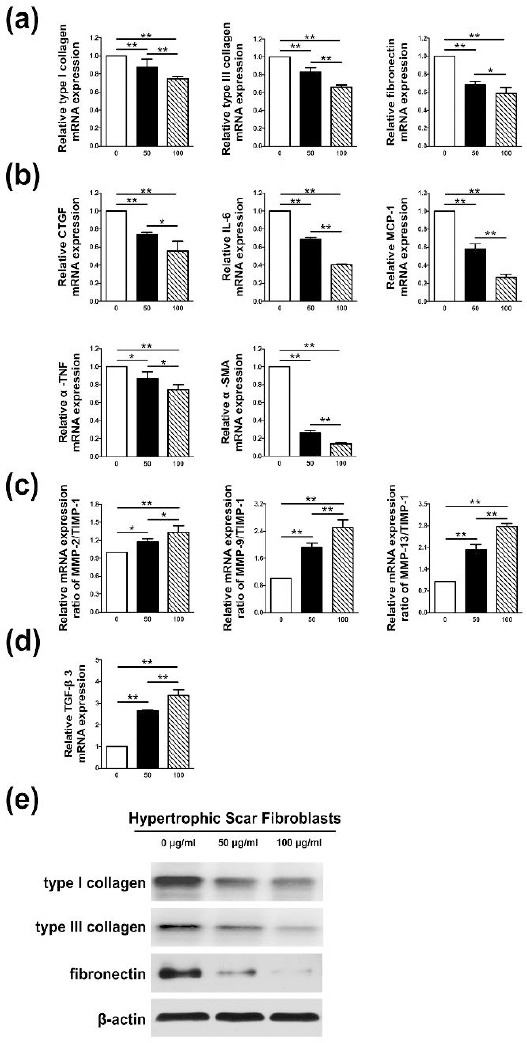
Collagen production and extracellular matrix (ECM) accumulation could be reduced by Rg3. As shown in Figure 2(a) and (e), Rg3 could visibly suppress both mRNA and protein levels of collagen I, and III and fibronectin. It could be observed in Figure 2(b) and (d) that Rg3 apparently reduced the levels of pro-fibrogenic genes such as connective tissue growth factor (CTGF), Interleukin 6 (IL-6), monocyte chemoattractant protein 1 (MCP-1), tumor necrosis factor alpha (α-TNF), and alpha-smooth muscle actin (α-SMA), and simultaneously strengthen the level of anti-fibrosis gene such as transforming growth factor-beta 3 (TGF-β3). The difference existed between the two groups (*P<0.05, **P<0.01). As shown in Figure 2(c), Rg3 could distinctly raise the ratio of matrix metalloproteinase-2/tissue inhibitor of matrix metalloproteinase 1 (MMP2/TIMP1), MMP9/TIMP1 and MMP13/TIMP1 (*P<0.05, **P<0.01), which signified the reduction of fibrotic ECM in hypertrophic scar fibroblasts
G. Rg3 restrained cell migration
We applied the scratch wound assay and a transwell system to assess the migration ability of HSFs under the treatment of Rg3. As shown in Figure 3(a), the HSFs in control group migrated 50.84±2.78% (mean±SD) of the scratched area, which showed blurry wound boundary after 24 hr of cultivation. The two sides of the wound that almost converged as 87.78±5.14% (mean±SD) of the scratched area were filled after 48 hr of cultivation. In comparison with the control group, the migration ability of HSFs in treated groups was remarkably weakened (*P<0.05, **P<0.01). The results were further confirmed by a transwell assay. As shown in Figure 3(b), Rg3 diminished the migration capability of HSFs by reducing cell numbers across the chamber membrane in a concentration-dependent manner. There was distinct difference between the two groups at both time points (*P<0.05, **P<0.01).
Figure 3.
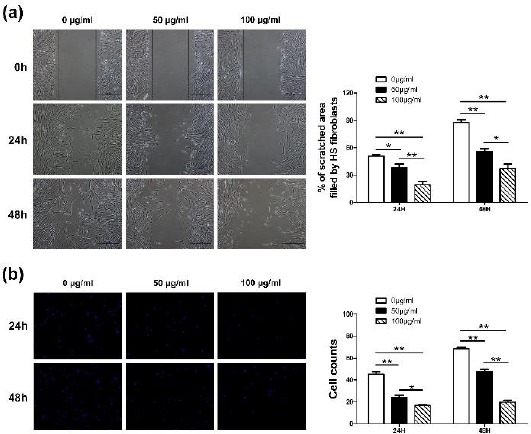
Cell migration was restrained by Rg3. As shown in Figure 3(a), the proportion of hypertrophic scar fibroblasts (HSFs)’ migrated area in control group was distinctly larger than that in treated groups after both time points. The migration ability of HSFs in treated groups was remarkably weakened (*P<0.05, **P<0.01). As shown in Figure 3(b), Rg3 diminished the migration capability of HSFs by reducing cell numbers across the chamber membrane in a concentration-dependent manner (*P<0.05, **P<0.01)
G. Rg3 affected the biological state of HSFs through TGF-β/SMAD and Erk signaling pathways
Several researches have proved that TGF-β1/SMAD and Erk pathways play crucial roles in fibrotic pathogenesis (14). As shown in Figure 4(a) and (b), both mRNA and protein expression levels of TGF-β1 and VEGF as well as the mRNA levels of VEGF receptor (VEGFR) and platelet-derived growth factor (PDGF) were remarkably decreased by Rg3. The statistical differences existed between control group and treated groups (*P<0.05, **P<0.01). Meanwhile, as demonstrated in Figure 4(c), the protein level of SMAD-7 was distinctly increased in Rg3-treated groups. Furthermore, the remedy of Rg3 obviously reduced the protein levels of phosphorylated SMAD2 and phosphorylated SMAD3. Simultaneously, the phosphorylated Erk1/2 was also overtly reduced in Rg3-treated groups in comparison with the control group in HSFs as demonstrated in the results of western blot. The protein levels of total SMAD2/3 and total Erk1/2 remained nearly the same in the three groups.
Figure 4.
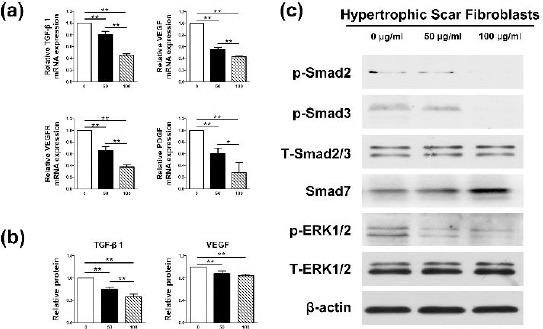
The biological states of hypertrophic scar fibroblasts (HSFs) were changed by Rg3 via TGF-β/SMAD and Erk signaling pathways. As shown in Figure 4(a) and (b), Rg3 apparently reduced both mRNA and protein expression levels of transforming growth factor-beta 1 (TGF-β1) and vascular endothelial growth factor (VEGF) as well as the mRNA levels of VEGF receptor (VEGFR) and platelet-derived growth factor (PDGF) (*P<0.05, **P<0.01). Meanwhile, Rg3 elevated the protein level of SMAD-7, which served as the negative feedback regulator in TGF-β1/SMAD pathway as shown in Figure 4(c). Furthermore, Rg3 obviously reduced the protein levels of phosphorylated SMAD2 and phosphorylated SMAD3, which acted as the downstream signal molecules after TGF-β1. The phosphorylated Erk1/2 was also overtly reduced in experimental groups as demonstrated in the results of western blot.
Discussion
The specialists have been familiar with the common approaches dealing with HS. However, the treatment for HS is still a thorny problem, which is embodied in several aspects as follows: 1) The treatment time is too long. 2) Lack of specific remedies. Various different treatments must join to apply, which are still not so much effective for the scar disease. 3) Many drugs have side-effects, leading to the limit of dosage and cure time. 4) The lack of early interventions may deprive the optimal time for treating. In light of the points listed above, we attempted to seek for a new drug, which may serve as the early intervention to treat HS more effectively or intensify the function of corticosteroids and chemotherapeutic agents.
As a kind of traditional Chinese medicine, Rg3 has been widely studied in many fields especially in tumor therapy (15). It has been confirmed in our previous researches that Rg3-loaded electrospun fibrous membranes could significantly inhibit HS formation on rabbit ears, with decreased production of collagen fibers and microvessels (11). In the present study, we further probed into the specific functional mechanisms of Rg3 and carried out related molecular and cellular experiments in vitro. The results demonstrated that the proliferation, angiogenesis and collagen synthesis of HSFs could be inhibited by Rg3 through TGF-β/SMAD and Erk signal pathways.
G. Rg3 has been revealed to decrease various tumor cells proliferation and promote apoptosis (16-18). Li Y et al. showed that Rg3 could effectively inhibit cell proliferation and induced apoptosis of multiple myeloma cells (16). In another study, it was reported that Rg3 both in vitro and in vivo had anti-proliferative function against melanoma by decreasing histone deacetylase 3 (HDAC3) and increasing acetylation of p53 (17). Other findings demonstrated that Rg3 could inhibit cell proliferation and promote apoptosis by increasing mitochondrial reactive oxygen species in human leukemia Jurkat cells (18). In our study, it could be confirmed that Rg3 has anti-proliferative function in HSFs. The cell proliferation assay, immuno-fluorescence (Ki-67) and decreased expression of PCNA in experimental groups indicated that Rg3 could suppress the growth of HS effectually. Meanwhile, the FCM outcome showed that only the high concen-tration of Rg3 could induce cell apoptosis.
As stated before, the excess collagen deposition and aberrant accumulation of ECM are the main features of HS. The levels of collagen I, collagen III, elastin, fibronectin, etc. all visibly elevate in HS and they function crucially in the formation of fibrotic ECM environments and matrix network (19). In our work, Rg3 treatment down-regulate both mRNA and protein levels of collagen I, collagen III and fibronectin. Numerous pro-fibrogenic molecules such as CTGF, IL-6, MCP-1, α-TNF, α-SMA, etc. play important roles in the process of HS formation, and higher levels of them have been found in HS versus normal dermis (20-23). In our study, these pro-fibrogenic molecules were all obviously declined after Rg3 treatment. Additionally, the balance between MMPs and their corresponding inhibitors TIMPs is pivotal in the normal wound healing process (24). The disproportionality of them could impact on natural wound healing and lead to the formation of pathological scars. It has been reported that MMP-2, MMP-9 and MMP-13 contributed mainly to degrade collagen I and collagen III in HS or keloid (25, 26). In our research, the ratios of MMP2/TIMP1, MMP9/ TIMP1 and MMP13/TIMP1 were adjusted to signi-ficantly higher levels in Rg3-treated groups, which implied the inhibition or reversal of fibrosis in HS after Rg3 treatment. All the results above indicated that Rg3 could reduce collagen production and ECM accumulation in HS.
It is accepted that a wide variety of profibrotic and anti-fibrotic cytokines are involved in the normal process of wound healing, keeping dynamic equilibrium with each other (19). Among these cytokines, TGF-β family are the extremely pivotal ones associated with many biologic behaviors such as cell proliferation, migration, ECM remodeling, inducing fibrosis, modulating other signal pathways, etc. (27). Once injury occurred, TGF-β is released in the site of damage and attracts multiple inflammatory cells and fibroblasts to the wound (28). TGF-β family mainly has three highly conserved subtypes in the human body named TGF-β1, TGF-β2 and TGF-β3. The former two isoforms are related to fibration, which can not only stimulate collagen synthesis but also prevent ECM disintegration (29). On the contrary, TGF-β3 mainly acts to reduce fibrosis and scarring. In the process, the TGF-β receptors (TGF-βRs) receive the stimulus from corresponding antigens and, serving as a downstream mediator of TGF-β action, transmit signals via SMAD signal-transduction pathway (30). Finally, the fibrosis-related genes are activated, leading to collagen production and ECM deposition. In the SMAD family, SMAD7 provides negative feedback to the pathway by preventing the phosphorylation of SMAD2 and SMAD3, which acted as the downstream signal molecules after TGF-β1, thus blocking the actions of TGF-β1 or TGF-β2. In our research, Rg3 evidently reduced the levels of TGF-β1, phosphor-rylated SMAD2 and phosphorylated SMAD3, yet increased the level of SMAD7, implying favorable inhibiting effect of Rg3 in the TGF-β/SMAD signal pathway.
Once the injury occurs, abundant nutrition supply and substance metabolism that come from neovascularization are needed to facilitate the renovation of damaged tissues. VEGF and PDGF play pivotal roles during the process. Therefore, the expression levels of VEGF and PDGF in HS are much higher than that in normal skin (31). After the binding reaction between antigen and antibody, VEGF and PDGF can all activate ERK1/2 signaling pathway and finally trigger the transcription of angiogenesis-related genes (32, 33). Numerous studies showed that Rg3 had the treatment efficacy against tumor angio-genesis. It is reported that Rg3 has anti-leukemia effect in some degree due to its anti-angiogenic action via inhibiting PI3K/Akt and ERK1/2 signal pathways (34). In addition, it is suggested that Rg3 targeted hypoxia-induced multiple signal pathways to down-regulate the level of VEGF in tumor cells (35). In our research, the levels of VEGF/VEGFR, PDGF and activated phosphorylated ERK1/2 were all down-regulated in experimental groups, suggesting the reductive vascularization in HS after Rg3 treatment.
Conclusion
Overall, our research demonstrated that Rg3 could suppress HSFs proliferation, vascularization and ECM deposition in vitro. The related TGF-β⁄SMAD and ERK1/2 signal pathways were proved to be involved in Rg3 action mechanisms. All of our studies that included the previous animal experiments offer the evidences that Rg3 may serve as a promising medicine in the treatment of human HS. The further clinical tests may need to be conducted in the future to roundly verify the Rg3 treatment effect.
Acknowledgment
The results described in this paper were part of student thesis. The authors are grateful to Shanghai Key Laboratory of Tissue Engineering for technical assistance. This study was supported in part by the National Natural Science Foundation of China (grant no. 81372073 and 81772099).
Conflict of Interest
The authors declare no financial or commercial conflict of interest.
References
- 1.Lorden ER, Miller KJ, Ibrahim MM, Bashirov L, Hammett E, Chakraborty S, et al. Biostable electrospun microfibrous scaffolds mitigate hypertrophic scar contraction in an immune-competent murine model. Acta Biomater. 2016;32:100–109. doi: 10.1016/j.actbio.2015.12.025. [DOI] [PubMed] [Google Scholar]
- 2.Guadanhim LR, Gonçalves RG, Bagatin E. Observational retrospective study evaluating the effects of oral isotretinoin in keloids and hypertrophic scars. Int J Dermatol. 2016;55:1255–1258. doi: 10.1111/ijd.13317. [DOI] [PubMed] [Google Scholar]
- 3.Ud-Din S, Bayat A. New insights on keloids, hypertrophic scars, and striae. Dermatol Clin. 2014;32:193–209. doi: 10.1016/j.det.2013.11.002. [DOI] [PubMed] [Google Scholar]
- 4.Van der Veer WM, Bloemen MC, Ulrich MM, Molema G, van Zuijlen PP, Middelkoop E, et al. Potential cellular and molecular causes of hypertrophic scar formation. Burns. 2009;35:15–29. doi: 10.1016/j.burns.2008.06.020. [DOI] [PubMed] [Google Scholar]
- 5.Hayashi T, Furukawa H, Oyama A, Funayama E, Saito A, Murao N, et al. A new uniform protocol of combined corticosteroid injections and ointment application reduces recurrence rates after surgical keloid/hypertrophic scar excision. Dermatol Surg. 2012;38:893–897. doi: 10.1111/j.1524-4725.2012.02345.x. [DOI] [PubMed] [Google Scholar]
- 6.Yang SY, Yang JY, Hsiao YC. Comparison of combination therapy (steroid, calcium channel blocker, and interferon) with steroid monotherapy for treating human hypertrophic scars in an animal model. Ann Plast Surg. 2015;74(Suppl 2):S162–167. doi: 10.1097/SAP.0000000000000470. [DOI] [PubMed] [Google Scholar]
- 7.Wang J, Jiao H, Stewart TL, Shankowsky HA, Scott PG, Tredget EE. Improvement in postburn hypertrophic scar after treatment with IFN-alpha2b is associated with decreased fibrocytes. J Interferon Cytokine Res. 2007;27:921–930. doi: 10.1089/jir.2007.0008. [DOI] [PubMed] [Google Scholar]
- 8.Berman B, Flores F. The treatment of hypertrophic scars and keloids. Eur J Dermatol. 1998;8:591–595. [PubMed] [Google Scholar]
- 9.He B, Chen P, Xie Y, Li S, Zhang X, Yang R, et al. 20(R)-Ginsenoside Rg3 protects SH-SY5Y cells against apoptosis induced by oxygen and glucose deprivation/reperfusion. Bioorg Med Chem Lett. 2017;27:3867–3871. doi: 10.1016/j.bmcl.2017.06.045. [DOI] [PubMed] [Google Scholar]
- 10.Keung MH, Chan LS, Kwok HH, Wong RN, Yue PY. Role of microRNA-520h in 20(R)-ginsenoside-Rg3-mediated angiosuppression. J Ginseng Res. 2016;40:151–159. doi: 10.1016/j.jgr.2015.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cheng L, Sun X, Hu C, Jin R, Sun B, Shi Y, et al. In vivo early intervention and the therapeutic effects of 20(S)-ginsenoside Rg3 on hypertrophic scar formation. PLoS One. 2014;9:e113640. doi: 10.1371/journal.pone.0113640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim SM, Lee SY, Cho JS, Son SM, Choi SS, Yun YP, et al. Combination of ginsenoside Rg3 with docetaxel enhances the susceptibility of prostate cancer cells via inhibition of NF-Kb. Eur J Pharmacol. 2010;10:1–9. doi: 10.1016/j.ejphar.2009.12.018. [DOI] [PubMed] [Google Scholar]
- 13.Shin JU, Lee WJ, Tran TN, Jung I, Lee JH. Hsp70 knockdown by siRNA decreased collagen production in keloid fibroblasts. Yonsei Med J. 2015;56:1619–1626. doi: 10.3349/ymj.2015.56.6.1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen QJ, Zhang MZ, Wang LX. Gensenoside Rg3 inhibits hypoxia-induced VEGF expression in human cancer cells. Cell Physiol Biochem. 2010;26:849–858. doi: 10.1159/000323994. [DOI] [PubMed] [Google Scholar]
- 15.Shin YM, Jung HJ, Choi WY, Lim CJ. Antioxidative, anti-inflammatory, and matrix metalloproteinase inhibitory activities of 20(S)-ginsenoside Rg3 in cultured mammalian cell lines. Mol Biol Rep. 2013;40:269–279. doi: 10.1007/s11033-012-2058-1. [DOI] [PubMed] [Google Scholar]
- 16.Li Y, Yang T, Li J, Hao HL, Wang SY, Yang J, et al. Inhibition of multiple myeloma cell proliferation by ginsenoside Rg3 via reduction in the secretion of IGF-1. Mol Med Rep. 2016;14:2222–2230. doi: 10.3892/mmr.2016.5475. [DOI] [PubMed] [Google Scholar]
- 17.Shan X, Fu YS, Aziz F, Wang XQ, Yan Q, Liu JW. Ginsenoside Rg3 inhibits melanoma cell proliferation through down-regulation of histone deacetylase 3 (HDAC3) and increase of p53 acetylation. PLoS One. 2014;9:e115401. doi: 10.1371/journal.pone.0115401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xia T, Wang YN, Zhou CX, Wu LM, Liu Y, Zeng QH, et al. Ginsenoside Rh2 and Rg3 inhibit cell proliferation and induce apoptosis by increasing mitochondrial reactive oxygen species in human leukemia Jurkat cells. Mol Med Rep. 2017;15:3591–3598. doi: 10.3892/mmr.2017.6459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Berman B, Maderal A, Raphael B. Keloids and Hypertrophic Scars:pathophysiology, classification, and treatment. Dermatol Surg. 2017;43(Suppl 1):S3–S18. doi: 10.1097/DSS.0000000000000819. [DOI] [PubMed] [Google Scholar]
- 20.Ehrlich HP, Desmoulière A, Diegelmann RF, Cohen IK, Compton CC, Garner WL, et al. Morphological and immunochemical differences between keloid and hypertrophic scar. Am J Pathol. 1994;145:105–113. [PMC free article] [PubMed] [Google Scholar]
- 21.Mu S, Kang B, Zenfresultg W, Sun Y, Yang F. MicroRNA-143-3p inhibits hyperplastic scar formation by targeting connective tissue growth factor CTGF/CCN2 via the Akt/mTOR pathway. Mol Cell Biochem. 2016;416:99–108. doi: 10.1007/s11010-016-2699-9. [DOI] [PubMed] [Google Scholar]
- 22.Yamamoto T, Hartmann K, Eckes B, Krieg T. Role of stem cell factor and monocyte chemoattractant protein-1 in the interaction between fibroblasts and mast cells in fibrosis. J Dermatol Sci. 2001;26:106–111. doi: 10.1016/s0923-1811(00)00164-x. [DOI] [PubMed] [Google Scholar]
- 23.Salgado RM, Alcántara L, Mendoza-Rodríguez CA, Cerbón M, Hidalgo-González C, Mercadillo P, et al. Post-burn hypertrophic scars are characterized by high levels of IL-1βmRNA and protein and TNF-αtype I receptors. Burns. 2012;38:668–676. doi: 10.1016/j.burns.2011.12.012. [DOI] [PubMed] [Google Scholar]
- 24.Visse R, Nagase H. Matrix metalloproteinases and tissue inhibitors of metalloproteinases:structure, function, and biochemistry. Circ Res. 2003;92:827–839. doi: 10.1161/01.RES.0000070112.80711.3D. [DOI] [PubMed] [Google Scholar]
- 25.Uchida G, Yoshimura K, Kitano Y, Okazaki M, Harii K. Tretinoin reverses upregulation of matrix metalloproteinase-13 in human keloid-derived fibroblasts. Exp Dermatol. 2003;12(Suppl 2):35–42. doi: 10.1034/j.1600-0625.12.s2.6.x. [DOI] [PubMed] [Google Scholar]
- 26.Huang D, Liu Y, Huang Y, Xie Y, Shen K, Zhang D, et al. Mechanical compression upregulates MMP9 through SMAD3 but not SMAD2 modulation in hypertrophic scar fibroblasts. Connect Tissue Res. 2014;55:391–396. doi: 10.3109/03008207.2014.959118. [DOI] [PubMed] [Google Scholar]
- 27.Cheng F, Shen Y, Mohanasundaram P, Lindström M3, Ivaska J4, Ny T, et al. Vimentin coordinates fibroblast proliferation and keratinocyte differentiation in wound healing via TGF-β-Slug signaling. Proc Natl Acad Sci U S A. 2016;113:E4320–7. doi: 10.1073/pnas.1519197113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bullard KM, Longaker MT, Lorenz HP. Fetal wound healing:current biology. World J Surg. 2003;27:54–61. doi: 10.1007/s00268-002-6737-2. [DOI] [PubMed] [Google Scholar]
- 29.Kose O, Waseem A. Keloids and hypertrophic scars:are they two different sides of the same coin? Dermatol Surg. 2003;34:336–346. doi: 10.1111/j.1524-4725.2007.34067.x. [DOI] [PubMed] [Google Scholar]
- 30.Branton MH, Kopp JB. TGF-βand fibrosis. Microbes Infect. 1999;1:1349–1365. doi: 10.1016/s1286-4579(99)00250-6. [DOI] [PubMed] [Google Scholar]
- 31.Chun Q, ZhiYong W, Fei S, XiQiao W. Dynamic biological changes in fibroblasts during hypertrophic scar formation and regression. Int Wound J. 2016;13:257–262. doi: 10.1111/iwj.12283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wu Y, Zhang Q, Ann DK, Akhondzadeh A, Duong HS, Messadi DV, et al. Increased vascular endothelial growth factor may account for elevated level of plasminogen activator inhibitor-1 via activating ERK1/2 in keloid fibroblasts. Am J Physiol Cell Physiol. 2004;286:C905–912. doi: 10.1152/ajpcell.00200.2003. [DOI] [PubMed] [Google Scholar]
- 33.Li J, Zhang M, Ma J. Myricitrin inhibits PDGF-BB-stimulated vascular smooth muscle cell proliferation and migration through suppressing PDGFRβ/Akt/Erk signaling. Int J Clin Exp Med. 2015;8:21715–21723. [PMC free article] [PubMed] [Google Scholar]
- 34.Zeng D, Wang J, Kong P, Chang C, Li J, Li J. Ginsenoside Rg3 inhibits HIF-1αand VEGF expression in patient with acute leukemia via inhibiting the activation of PI3K/Akt and ERK1/2 pathways. Int J Clin Exp Pathol. 2014;7:2172–2178. [PMC free article] [PubMed] [Google Scholar]
- 35.Chen QJ, Zhang MZ, Wang LX. Gensenoside Rg3 inhibits hypoxia-induced VEGF expression in human cancer cells. Cell Physiol Biochem. 2010;26:849–858. doi: 10.1159/000323994. [DOI] [PubMed] [Google Scholar]