

Abstract
Objective(s):
Targeted next-generation sequencing (NGS) provides a consequential opportunity to elucidate genetic factors in known diseases, particularly in profoundly heterogeneous disorders such as non-syndromic hearing loss (NSHL). Hearing impairments could be classified into syndromic and non-syndromic types. This study intended to assess the significance of mutations in these genes to the autosomal recessive/dominant non-syndromic genetic load among Iranian families.
Materials and Methods:
Two families were involved in this research and two patients were examined by targeted next-generation sequencing. Here we report two novel mutations in the MYO7A and EYA1 genes in two patients detected by targeted NGS. They were confirmed by Sanger sequencing and quantitative real-time PCR techniques.
Results:
In this investigation, we identified a novel mutation in MYO7A, c.3751G>C, p.A1251P, along with another previously identified mutation (c.1708C>T) in one of the cases. This mutation is located in the MYTH4 protein domain which is a pivotal domain for the myosin function. Another finding in this research was a novel de-novo deletion which deletes the entire EYA1 coding region (EX1-18 DEL). Mutations in EYA1 gene have been found in branchiootorenal (BOR) syndrome. Interestingly the patient with EYA1 deletion did not show any other additional clinical implications apart from HL. This finding might argue for the sole involvement of EYA1 function in the mechanism of hearing.
Conclusion:
This investigation exhibited that the novel mutations in MYO7A, c.3751G>C, p.A1251P, and EYA1, EX1-18 DEL, were associated with NSHL. Our research increased the mutation spectrum of hearing loss in the Iranian population.
Keywords: EYA1, Mutation, MYO7A, MYTH4, NSHL
Introduction
Sensorineural hearing loss (SNHL) is a multifaceted disease with profoundly medical, social, and educational consequences. The term “hearing impaired” inter-changeably refers to people with any degree of SNHL and “Deaf” is a conversational term that implies hearing thresholds in the audiometry severe to profound span.
The diagnosis of SNHL relies on the verification of reduced hearing acuity by auditory testing. Hearing is measured in decibels (dB) and a child’s hearing acuity is deemed as normal if it is within 20 dB of defined thresholds. The intensity of hearing loss is classified into five categories: mild (20–40 dB), moderate (41–55 dB), moderately severe (56–70 dB), severe (71–90 dB), or profound (90 dB)(1). The severity of hearing loss is graded as low (500 Hz), middle (501–2000 Hz) and high (2000 Hz) (2). Moreover, hearing loss is categorized by type, onset, severity, and frequency.
In most cases, type of loss is classified as sensori-neural, conductive, mixture of both, and either pro-gressive or stable types. Time of onset is arranged as either hereditary or late-onset (acquired). Causality is categorized as either hereditary (genetic) or environ-mental (non-genetic) (3). All types of the Mendelian inheritances in hearing loss such as autosomal recessive or dominant and X-linked and mitochondrial manner have been identified (4).
Hearing impairments could be categorized into syndromic and non-syndromic types. Nonsyndromic HL can further be classified by its mode of inheritance. Approximately one-fifth of nonsyndromic sensorineural hearing loss (NSSHL) is inherited as autosomal domi-nant which is implied to as DFNA. Autosomal recessive (DFNB), non-syndromic hearing loss (ARNSHL) causes around 80% of all cases of hearing loss. The inheritance of X-linked and mitochondrial modes are less than 1 percent. To date, 142 loci have been mapped for NSHL (5).
Autosomal dominant non-syndromic hearing loss (NSHL) is usually first detected in school-aged children during common audio-logical screening (6). Some types of syndromic SNHL are also first recognized at this time for the diagnosis of associated comorbidity that was not beforehand noted. Most known examples include Pendred and Usher syndromes; they are both inherited in an autosomal recessive manner. So a family history is contributory for diagnosis and risk assessment (1).
Hearing impairment results from etiologically heterogeneous genetic and environmental factors such as prenatal TORCH infections and postnatal secondary to bacterial meningitis by Neisseria meningitidis, Haemophilus influenzae, and Streptococcus pneumoniae (2, 7). Approximately, 50% of hearing loss has genetic causes (Table 1).
Table 1.
Distribution of causes for hearing loss in infancy
| Environmental-50% | TORCH infection, ototoxicity, drug consumption, prematurity, other infections | |
|---|---|---|
| Genetic 50% | ||
| Syndromic 30% | Alport, Pendred, Usher, Wardenberg, KSS1, MERRF2, CHARGE, Norrie, Stickler, Treacher Collins, Perrault syndrome, BORS3, Jervell & Lange-Nielsen and so on | |
| Non-syndromic 70% | Autosomal recessive (70-80%) Autosomal dominant (20-25%) X chromosomal/mitochondrial (1.5%) | |
TORCH=Toxoplasmosis, other (syphilis, varicella-zoster, parvovirus B19), rubella, cytomegalovirus (CMV), and herpes infections
Kearns-Sayre syndrome
Myoclonic epilepsy with ragged-red fibers
branchio-oto-renalsyndrome
The etiology of minor degrees of HL in the newborn period is not well perceived
In this report, we identified 2 novel mutations in two Iranian families. In the AT1220 case, we identified a novel variant c.3751G>C, p.A1251P along with a previously identified mutation (c.1708C>T; p.Arg570 Ter) as compound heterozygote mutations in MYO7A (NM_000260). The p.A1251P mutation has a delete-rious impression on Myosin protein by interference in protein folding. Besides, this mutation is located at a highly conserved region of ELEA. The pathogenicity of the mutation has been confirmed by in-silico predictions. In the second family, a de novo and novel deletion (EX1_18DEL) in the EYA1 gene was found in the YK1132 patient. Haploinsufficiency of the trans-cription co-activator EYA1 causes the branchio–oto–renal syndrome (BOR) that is related to profound hearing loss (8); however, in this family, we could not find any symptoms of the BOR syndrome and it is the first case of NSHL influenced by EYA1 deletion.
The aim of the present investigation was to determine the spectrum of the deleterious mutations related to NSHL in the Iranian population. Our results increased the mutational spectrum of MYO7A and EYA1 genes. The proteins encoded by these genes are involved in embryonic development, gene regulation, ionic and osmotic homeostasis and are required for normal function of cochlear hair cells.
Materials and Methods
Patients
The study was approved by the local ethics committee of Tarbiat Modares University. Informed consents were obtained from all patients and their healthy relatives. The patients in the YK1132 and AT12120 families were 33 and 30 year old males, respectively. All of the patient’s clinical information was collected at DeNA Laboratory, Tehran, Iran and the medical histories were obtained using a special ques-tionnaire that included following issues: type of HL (syndromic or non-syndromic), age of onset, bilateral or unilateral HL, presence of thyroid problems, extraordinary skin pigmentation, tinnitus and vertigo, exposure to teratogenic infections like TORCH in infancy or consuming special drugs during pregnancy, pathological changes in the ear and other related manifestations (9). Audiological evaluations of the patients exhibited symmetrical and bilateral sensori-neural hearing loss. According to audiological assess-ments, the severity of hearing loss varied among affected individuals, ranging from mild to profound. None of the patients displayed any additional symptoms apart from hearing loss, so they were deemed as NSHL patients and the NSHL panel was utilized to survey the pathogenic mutation(s). (The patient’s clinical features are described in Table 2).
Table 2.
Clinical features of patients and genes/variants that have been found in our study
| Patient ID | Phenotype | Gene Exon/CDS | Protein | N.A/pro. alteration | Zygosity | SIFT | Polyphen | Mutation taster | phastCons | EXAC/1K /10K genome frequency |
|---|---|---|---|---|---|---|---|---|---|---|
| AT1220 | MYO7A EX30/CDS29 | Myosin 7a | c.3751G>C* p.Ala1251Pro | Het | Damaging | Probably damaging (0.998) | Disease causing | 1 | Not reported | |
| Mild hearing loss | MYO7A EX15/CDS14 | Myosin 7a | c.1708C>T p.Arg570Ter | Het | Damaging | Damaging | Disease causing | 1 | Not reported | |
| WFS1 EX8/CDS7 | Wolfamin | c.2452C>T p.Arg818Cys | Het | Damaging | Damaging | Disease causing | 1 | Reported | ||
| YK1132 | EYA1 EX1-18 | EYES ABSENT 1 | C.Ex1_18DEL* | Het | - | - | Disease causing | 1 | - | |
| Severe hearing loss | WFS1 EX5/CD4 | Wolfamin | c.577A>C p.Lys193Gln | Het | Benign | Possibly damaging | Disease causing | 1 | Reported |
N.A: Nucleic acid, Pro.: Protein, phastCons: is a program for identifying evolutionarily conserved elements in a multiple alignment, for highly conserved sequences it is 1.
Novel mutations; EX: Exon; Het: Heterozygote
DNA extraction and Sequencing
Genomic DNA was extracted from peripheral blood samples using the Roche DNA Extraction Kit (Product No. 11814770001). There are about 127 genes involved in HL based on the reports of deafness variation database (DVD) (http://deafness-variationdatabase.org/letter). About 50% of the auto-somal recessive types of HL are due to mutations in GJB2/6 genes. GJB2 is also a major cause of hearing loss in Iranian patients (10-12). Therefore, at the first imperative step, these two cases were screened for connexin 26 mutations by Sanger sequencing. After ruling out GJB2 mutations all the contributed genes in HL were studied by utilizing a commercially available targeted NGS panel for hereditary NSHL from BGI, HongKong. In general, point mutations, micro-insertions, deletions and duplications (<20 bp) can be simultaneously detected by this targeted NGS panel with over 90% sensitivity (13). List of genes included in this panel is provided in appendix Table 1.
A filtering pipeline was established to recognize supposedly disease-causing variants. Due to the rarity of HL causing mutations, only variants with a frequency below 0.01 were selected (14). Frequencies of identified variants were also checked in ExAC (Exome Aggregation Consortium) (15), DVD (deafness variants database), and N.A: Nucleic acid, Pro.: Protein, phastCons: is a program for identifying evolutionarily conserved elements in a multiple alignment, for highly conserved sequences it is 1. * Novel mutations; EX: Exon; Het: Heterozygote ESP (Exon Sequencing Projects) publicly available databases. Mutations that have been described in HGMD (Human Gene Mutation Database) (16) were given the highest supremacy. For variants leading to missense mutations, pathogenicity predictions from at least 3 online databases namely SIFT, Polyphen2, and Mutation Taster (17) were compared. To predict the effect of c.3751G>C mutation on splicing, the HSF (Human Splicing Finder V 3.0) software was utilized. Also, ConSurf (http://www.consurf.tau.ac.il) was applied to provide evolutionary conservation profile for the myosin VII protein (Figure 2).
Figure 1.
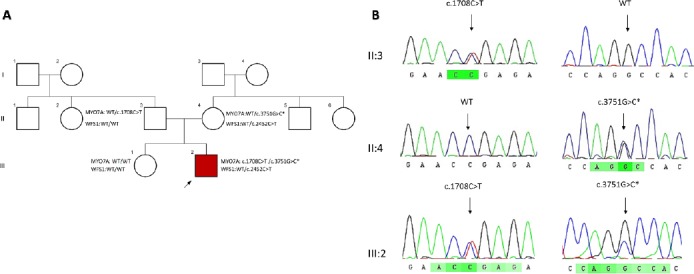
Identification of a novel missense mutation in the MYO7A gene
(A)Pedigree of the AT1220 family is comprised of three generations, squares and circles indicate females and males, respectively and the arrow appoints the proband of the family. The MYO7A mutations are co-segregating with the disease in this family as compound heterozygote. The star exhibits the novel mutation (B) Chromatograms showing nucleotide sequences of MYO7A in the regions of c.3751G>C and c.1708C>T mutations found in AT1220 family. Black arrows are indicating the heterozygous nucleotide substitution
Figure 2.
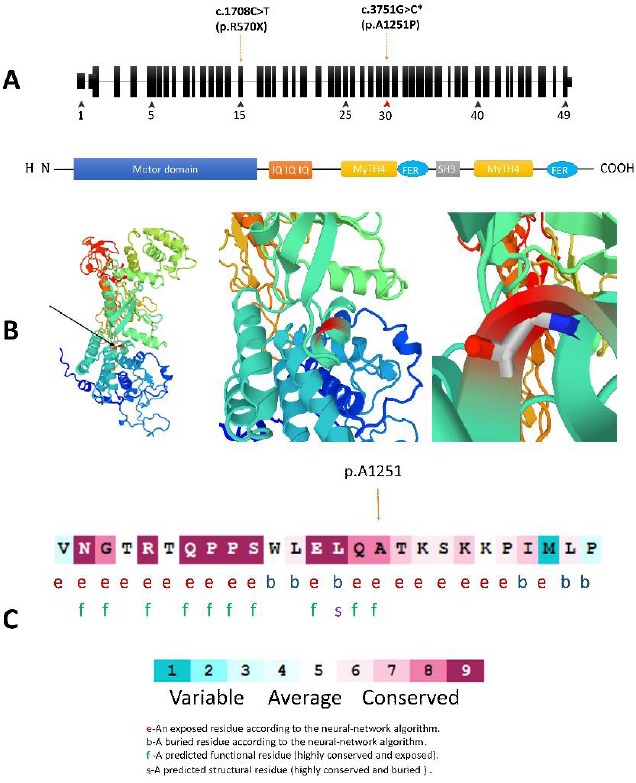
(A) Schematic structure of MYO7A and its encoded protein domains. The c.3735C>T located in the myosin tail homology 4 (MYTH4) domain that is coded by Exon 30. (B) A prediction of structural alternation resulted from MYO7A c. 3751G>C mutation designed by SWISS-MODEL and PMP online tools. This substitution is not conserved because the substituted amino acid, Proline, has poor helix-forming propensity and it can stir the secondary structure in this protein. (C) Multiple alignments of the MYO7A c.3751G homologous sequences of eight different vertebrates based on UCSC Multiz alignments tool. The amino acid substituted by the missense mutation p.A1251P (the red types) is highly conserved among the different vertebrate species. (D) The amino acid sequence MYO7A (p.Ala1251) colored based on conservation scores by ConSurf database. ConSurf demonstrates evolutionary conservation profiles for proteins of known structure in the PDB according to the phylogenetic relations between homologous sequences as well as amino acid’s structural and functional importance
Segregation study for variants: validation by Sanger sequencing
Appropriate primers were designed using primer3 plus a web-based tool (http://www.bioinformatics.nl/cgi-bin/primer3plus/primer3plus.cgi/) for the regions of identified variants (Table 3). Primer specificity was checked by in-silico-PCR and blat tools of UCSC genome browser and lack of SNPs in the genomic region corresponding to the 3′ ends of primers were checked by looking through the dbSNP database. PCR reactions were done according to the standard protocols and followed by Sanger sequencing. Sequence chromate-graphs were analyzed by Codon code aligner V.6.0.2 and DNA Baser Assembler V.4.36 to identify the variants. After confirming the mutation by Sanger sequencing in the studied sample by NGS, the co-segregation in the parents and healthy available members of the family were checked by Sanger sequencing. Additionally, the frequency of the variants was confirmed by utilizing at least 100 unrelated normal controls in the Iranian genome project database (https://www.irangenes.com/).
Table 3.
Sequences of the primers used to validate the mutations by Sanger sequencing
| Patient ID | Gene and Variants | Primers |
|---|---|---|
| MYO7A | F5’-TGTGGTGGAACTAGGTGGAT-3’ | |
| c.1708C>T | R5’-TTCCCGAACAACAGAAACCG-3’ | |
| MYO7A | F5’-AGAGAGCCAAAGTCCAGAGG-3’ | |
| AT1220 | c.3751G>C* | F5’-ACAGGGCAATGTAGAGGGAG-3’ |
| WFS1 | F5’-TTGAGATTACCGTGGGCATG-3’ | |
| c.2452C>T | R5’-CTGGTGGGTGAGAGCTGG-3’ | |
| YK1132 | EYA1* (Real-Time primer) | F 5’-AGTGGTTGCTGAAATGTTGCT-3’ (INTRON 3) R 5’-TGCCTAACCAAATTTGAAGACCT-3’ |
| WFS1 | F5’-CGAGACCGACCTGGAGAG-3’ | |
| c.577A>C | R5’-TGACCTGGCCGACATTCT-3’ |
Structural-based analysis and modeling
Wild-type and mutant protein comparison based on 3D (three dimensional) for a p.A1251P mutation in MYO7A was performed using SWISS-MODEL (18) and protein model portal (PMP) (19), automated homo-logy, comparative modeling programs, and automatic modeling approaches were used to apply the complete protein sequence of human MYO7A. The protein structure was fetched from the Protein Data Bank with the accession code 3PVL (20). The target sequences were uploaded in DeepView (21) and mapped on to wild-type template and then sequence alignment optimization was performed. Afterward, the complete project file was submitted to SWISS-MODEL. The effects of amino acid substitution on MYO7A were analyzed and predicted by in-silico tools such as I-mutant 2.0 (22) and site-directed mutator instruments (SDM) (23). The whole protein aggre-gation propensities in its folded states were predicted by Aggrescan3D (24). In order to verify the obtained 3D structure, ProSA (https://prosa.services.came.sbg.ac.at/prosa.php) was used, which analyzes the protein structure and matches with X-ray and NMR calculated structures.
Results
In this study, we identified two NSHL affected families and utilized the targeted next-generation sequencing for recognizing the pathogenic variants. After applying different filtering steps in case AT1220, one variant in WFS1 and two variants in MYO7A genes, and in case YK1132 a deletion in EYA1 gene and another variant in WFS1 remained as candidates (Table 2). The co-segregation study was performed for all of these variants in each family.
In AT1220 a novel variant (c.3751G>C, p.A1251P) and a previously identified mutation c.1708C>T; p.R570Ter in MYO7A (NM_000260) were identified as compound heterozygotes. The AT1220, was a 33-year old male with mild hearing loss. In addition to this mutation, another known mutation in WFS1 (NM_006005, c.2452C>T, p.R818C) was also found in this patient. The novel c.3751G>C variant affects the exon 30 of MYO7A coding the myosin tail homology 4 (MyTH4) domain, which is known to play a role in the connection between actin-based motor proteins and the microtubule cytoskeleton (Figure 2) (25). The MyTH4 domain comprises four highly conserved regions designated as MG or MGDhP, LRDE, RGW, and ELEA. Associations between this gene and Usher syndrome type 1B and autosomal dominant deafness type 11 have been previously reported (OMIM: 276903, OMIM: 600060). The novel p.A1251P is located in the conserved region of ELEA. Co-segregation study in this family showed that the MYO7A variants, c.1708C>T, and c.3751G>C, co-segregated with the disease as a compound hetero-zygote but the variant found in WFS1 (c.2452C>T) did not co-segregate with the disease (Figure 1A).
In the YK1132 patient also after applying different filtering steps, only two mutations remained as candidates: a heterozygote c.577A>C, p.K193Q mutation in WFS1 (NM_006005) and a heterozygote EX1-18 DEL dele-tion in the EYA1 (NM_000503) gene.
Segregation study in the family showed that the c.577A>C variant in the WFS1 gene cannot be the cause of disease in this family as the healthy father was also a carrier for this variant. On the other hand, the EX1_18 DEL deletion was a de-novo mutation that occurred in the patient only and none of the parents was the carrier for this deletion; therefore, this variant can be considered as the cause of disease in this patient. This is a de-novo interstitial deletion with the size of ~165 Kb on the long arm of chromosome 8 (8q13.3). This region contains the entire EYA1 gene. It is known that haploinsufficiency of EYA1 gene is responsible for the clinical features of patients with branchiootorenal (BOR) syndrome. According to the deafness variation database (DVD) to date, this deletion has not been reported for nonsyndromic hearing loss. Apart from hearing loss, any other obvious clinical features were not observed in this patient so it can be considered as NSHL. Quantitative real-time PCR was applied to confirm the deletion (Figure 3B).
Figure 3.
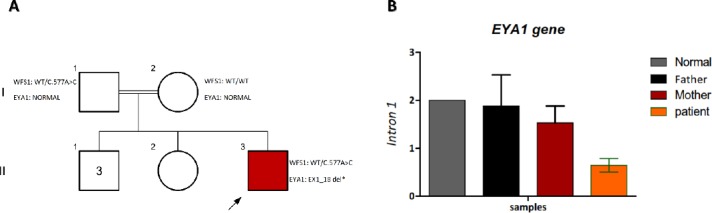
A) mutations identified in family YF-KHZ with hearing loss.
A) Pedigree of the YK1132 family, presenting the segregation of the c.577A>C mutation. B) Validation of EYA1 deletion by Real-time CQ-PCR. The relative quantity of Introns 1 in the Patient was almost half of the patient’s father and mother. Values are expressed as the mean ± standard error
Discussion
Hereditary hearing loss is strongly heterogeneous in different populations. To date, 142 loci have been mapped and over 127 genes have been found for HL. Despite such heterogeneity, mutations in some of the genes play important roles in the nonsyndromic hearing loss. These genes belong to the wide variety of the protein classes such as Myosins and related cytoskeletal proteins, transcription factors, proteins that belong to channels and gap junction structures, extracellular proteins and other genes with unknown functions (26).
The advent and development of the next-generation sequencing (NGS) technologies have tended to increase the rate of gene detection for genetically heterogeneous diseases such as hearing loss with reducing costs and producing massive sequencing data (27). By high-throughput sequencing technologies, it could be possible to sequence a large number of exons or other genomic sequences in a person to confirm or rule out a suspected genetic condition (28). As for a plurality of genetic factors that account for hearing loss, we utilized the NGS technologies to detect the pathogenic mutations in previously known causative genes.
In this study, we identified two novel variants in MYO7A and EYA1 that were associated with NSHL. Myosin VIIA belongs to the unconventional myosin superfamily of proteins that consists of over 35 classes (29). The unconventional myosin VIIa is required for normal auditory functions and its mutations are associated with NSHL. This gene is expressed in hair cell stereocilia. Several disruptive variants in this gene can result in hearing impairment associated with vestibular dysfunction and retinitis pigmentosa.
The most conserved sequence in these proteins is the N-terminal region with the characteristic head domain followed by a more variable neck region. The N-terminal domain binds to actin and ATP, and regulatory domain or neck domain consists of IQ domains that bind to calmodulin or calmodulin-like light chains. The C-terminal domain corresponds to the tail domain and it is the highly variable region between myosin classes (30). Mutations in MYO7A have been previously associated with the Usher Syndrome, especially Usher syndrome type IB, and autosomal dominant or recessive NSHL (31). MYO7A mutations have been shown to be the cause of hearing loss in a significant percentage of deaf Iranian families (32).
Myosins comprise a superfamily of ATP-dependent motor proteins and they are well-known for their function in muscle contraction, vesicles transportation, cell division, and so on (33). It is shown that myosin VIIa plays a critical role in transporting harmonin to the tip region of the stereocilium and has a significant role in shaping the sensory hair cell bundle (34).
Myosin VIIa has two MyTH4 domains and several mutations in this domain have been described previously to be the common causes of DFNB3 hearing loss (35). Two domains of MyTH4-FERM (MF; myosin tail homology 4; band 4.1, ezrin, radixin, moesin), which are separated by an SH3 domain, are located in the Myo7 tail region and essential for interaction with PDZ domain and microtubules (36, 37).
In this study, we report a novel mutation in ELEA conserved region of the MyTH4 domain in MYO7A. The alanine-to-proline substitution results in obstacles in α-helix formation due to poor helix-forming propensity of proline. This alteration is predicted to negatively effect the function of the myosin VIIa protein and it could be the cause of hearing loss. Based on I-mutant 2.0, this substitution can change the stability and flexibility of the protein because of proline’s unique propensities (38). According to Aggrescan3D, no certain alteration in protein folding is predicted. In a nutshell, additional studies, especially in animal models, must be applied to identify certain pathogenic role(s) of the mutation.
Meanwhile, we described another family with hearing loss phenotype with a mutation in WFS1 and a denovo mutation in EYA1 gene.
An interesting point is that in both families studied here, after various stringent filtering steps, we still ended up with previously reported mutations in WFS1, c.577A>C and c.2452C>T, and surprisingly in both cases, segregation study in the families could rule out these mutations as the pathogen, because they were present in the healthy members as well despite the fact that both of these mutations have been previously reported in several publications as pathogenic (39-41). This finding shows how much a study like this one is important to elucidate the repertoires of mutations in different populations. It is also highly needed to publish and submit these variants and their clinical outcomes in the public databases in order to avoid misinterpretation of the neutral variants.
EYA1 gene encodes the EYA1 protein which is identified as a transcription factor or transcription coactivator. This protein interacts with several other proteins, such as SIX proteins. In the organogenesis stage, these protein interactions are necessary for the normal formation of many tissues (42). It has been illustrated that Eya1 knocked out mice show several deficiencies in ears and kidneys as well as abnormal apoptosis of organ primordia (43). This protein plays important roles in the control of the cell polarity, cell fate, spindle orientation and Notch signaling in distal embryonic lung epithelium (44). It can explain the lung deficiencies in BOR patients (45). At least 160 mutations in the EYA1 gene have been reported in individuals with BOR syndrome so far (46-50). This syndrome is being identified by the famous manifestation of malformations in ears and kidneys. Several mutations in this gene are related to eye disorders such as clouding of the clear front surface of the eye (the cornea) and clouding of the lens (cataracts) (51). Mutations in EYA1 gene have been found in 40% of BOR patients (47). In patients with BOR syndrome, the malformations of the middle and inner ear structures have been revealed by analysis of computed tomography (CT) imaging of the temporal bone (52, 53).
Given the large number of EYA1 mutations iden-tified in various populations, in this study, we imply-cated pathogenic variants in EYA1 as a novel cause of NSHL. EYA1 encodes a member of the eyes absent (EYA) family of proteins. This protein has various important functions in developing several organs such as ears, branchial arches, eyes, and kidneys. Mutations in this gene are associated with the bran-chiootorenal dysplasia syndrome, branchiootic synd-rome, sporadic cases of congenital cataracts, and NSHL (54).
The YK1132 is a sporadic case and parents who were clinically healthy did not carry the EYA1 mutation, indicating a de-novo mutation in this case. After precise physical examinations we did not find any obvious expected symptoms of the BOR syndrome in this patient. Interestingly our findings suggest that the complete deletion of EYA1 leads to non-syndrome hearing loss without any other additional clinical implications. This finding might argue for the sole involvement of EYA1 function in the mechanism of hearing.
Conclusion
We have utilized the targeted NGS panel for the discovery and diagnosis of the new variants and causative mutations in syndromic hearing loss. In this report, we described two novel mutations in the highly conserved regions of MYO7A (c.3751G>C) and EYA1 (EX1_18 deletion) genes, suggesting the potentially conserved role of these genes. Although in-silico predictions have shown that the novel mutation in MYO7A (c.3751G>C) is a pathogenic variant, more in vivo studies or animal models are necessary to conclude the pathogenic impact. Furthermore, this study illustrates for the first time the impression of EYA1 gene deletion in a patient with NSHL. Besides, these novel mutations expand the mutational spectrum of MYO7A and EYA1 in the Iranian population, which will contribute to the clinical understanding of hearing loss, caused by mutations in these genes and will help physicians in better understanding the hearing loss etiology.
Acknowledgment
We would like to appreciate our patients and their families for participating in this study. We thank the staff of DeNA Laboratory, Tehran, Iran, for helping us in this research. This research received no specific grant from any funding agency, commercial or not-for-profit sectors.
Conflicts of interest
The authors declare that they have no conflicts of interest.
References
- 1.Smith RJ, Bale JF, Jr, White KR. Sensorineural hearing loss in children. Lancet. 2005;365:879–890. doi: 10.1016/S0140-6736(05)71047-3. [DOI] [PubMed] [Google Scholar]
- 2.Kochhar A, Hildebrand MS, Smith RJ. Clinical aspects of hereditary hearing loss. Genet Med. 2007;9:393–408. doi: 10.1097/gim.0b013e3180980bd0. [DOI] [PubMed] [Google Scholar]
- 3.Mattox DE, Simmons FB. Natural history of sudden sensorineural hearing loss. Ann Otol Rhinol Laryngol. 1977;86:463–480. doi: 10.1177/000348947708600406. [DOI] [PubMed] [Google Scholar]
- 4.Pandya A. Nonsyndromic hearing loss and deafness, mitochondrial. Washington: Gene Reviews; 1993. [Google Scholar]
- 5.Van Camp G, Smith RJ. Hereditary hearing loss homepage. London: Whurr Publisher; 2006. [Google Scholar]
- 6.Cremers CW, Smith R. Genetic hearing impairment:its clinical presentations. Switzerland: Karger Medical and Scientific Publishers; 2002. [Google Scholar]
- 7.Nance WE. The genetics of deafness. Ment Retard Dev Disabil Res Rev. 2003;9:109–119. doi: 10.1002/mrdd.10067. [DOI] [PubMed] [Google Scholar]
- 8.Zou D, Erickson C, Kim EH, Jin D, Fritzsch B, Xu PX. Eya1 gene dosage critically affects the development of sensory epithelia in the mammalian inner ear. Hum Mol Genet. 2008;17:3340–3356. doi: 10.1093/hmg/ddn229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.ACMG. Genetics evaluation guidelines for the etiologic diagnosis of congenital hearing loss. Genetic Evaluation of Congenital Hearing Loss Expert Panel. ACMG statement. Genet Med. 2002;4:162–171. doi: 10.1097/00125817-200205000-00011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tang HY, Fang P, Ward PA, Schmitt E, Darilek S, Manolidis S, et al. DNA sequence analysis of GJB2, encoding connexin 26:observations from a population of hearing impaired cases and variable carrier rates, complex genotypes, and ethnic stratification of alleles among controls. Am J Med Genet A. 2006;140:2401–2415. doi: 10.1002/ajmg.a.31525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tabatabaiefar MA, Montazer ZM, Shariati L, Safari CJ, Ashrafi K, Gholami A, et al. Mutation analysis of GJB2 and GJB6 genes and the genetic linkage analysis of five common DFNB loci in the Iranian families with autosomal recessive non-syndromic hearing loss. J Sci Islamic Republic Iran. 2010;21:105–112. [Google Scholar]
- 12.Chaleshtori MH, Farhud DD, Taylor R, Hadavi V, Patton MA, Afzal AR. Deafness–associated connexin 26 gene (GJB2) mutations in Iranian population. Iran J Public Health. 2002;31:75–79. [Google Scholar]
- 13.Liu Y, Wei X, Kong X, Guo X, Sun Y, Man J, et al. Targeted Next-Generation Sequencing for Clinical Diagnosis of 561 Mendelian Diseases. PLoS One. 2015;10:e0133636. doi: 10.1371/journal.pone.0133636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stitziel NO, Kiezun A, Sunyaev S. Computational and statistical approaches to analyzing variants identified by exome sequencing. Genome Biol. 2011;12:227. doi: 10.1186/gb-2011-12-9-227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Browser E. Exome aggregation consortium website. New York: ExAC Browser; 2015. [Google Scholar]
- 16.Stenson PD, Ball EV, Mort M, Phillips AD, Shiel JA, Thomas NS, et al. Human gene mutation database (HGMD®):2003 update. Hum Mutat. 2003;21:577–581. doi: 10.1002/humu.10212. [DOI] [PubMed] [Google Scholar]
- 17.Schwarz JM, Rödelsperger C, Schuelke M, Seelow D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat Methods. 2010;7:575–576. doi: 10.1038/nmeth0810-575. [DOI] [PubMed] [Google Scholar]
- 18.Guex N, Peitsch MC. SWISS-MODEL and the Swiss-Pdb Viewer:an environment for comparative protein modeling. Electrophoresis. 1997;18:2714–2723. doi: 10.1002/elps.1150181505. [DOI] [PubMed] [Google Scholar]
- 19.Haas J, Roth S, Arnold K, Kiefer F, Schmidt T, Bordoli L, et al. The protein model portal-a comprehensive resource for protein structure and model information. Database. 2013;2013:31. doi: 10.1093/database/bat031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wu L, Pan L, Wei Z, Zhang M. Structure of MyTH4-FERM domains in myosin VIIa tail bound to cargo. Science. 2011;331:757–760. doi: 10.1126/science.1198848. [DOI] [PubMed] [Google Scholar]
- 21.Kaplan W, Littlejohn TG. Swiss-PDB viewer (deep view) Brief Bioinform. 2001;2:195–197. doi: 10.1093/bib/2.2.195. [DOI] [PubMed] [Google Scholar]
- 22.Capriotti E, Fariselli P, Casadio R. I-Mutant2.0:predicting stability changes upon mutation from the protein sequence or structure. Nucleic Acids Res. 2005;33:W306–W310. doi: 10.1093/nar/gki375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Worth CL, Preissner R, Blundell TL. SDM--a server for predicting effects of mutations on protein stability and malfunction. Nucleic Acids Res. 2011;39:W215–W222. doi: 10.1093/nar/gkr363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zambrano R, Jamroz M, Szczasiuk A, Pujols J, Kmiecik S, Ventura S. AGGRESCAN3D (A3D):server for prediction of aggregation properties of protein structures. Nucleic Acids Res. 2015;43:W306–W313. doi: 10.1093/nar/gkv359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Weber KL, Sokac AM, Berg JS, Cheney RE, Bement WM. A microtubule-binding myosin required for nuclear anchoring and spindle assembly. Nature. 2004;431:325–329. doi: 10.1038/nature02834. [DOI] [PubMed] [Google Scholar]
- 26.Van Laer L, Cryns K, Smith RJ, Van Camp G. Nonsyndromic hearing loss. Ear Hear. 2003;24:275–288. doi: 10.1097/01.AUD.0000079805.04016.03. [DOI] [PubMed] [Google Scholar]
- 27.Ari Ş, Arikan M. Next-generation sequencing:advantages, disadvantages, and future. Plant Omics: Trends and Applications; 2016. pp. 109–135. [Google Scholar]
- 28.Yan D, Tekin M, Blanton SH, Liu XZ. Next-generation sequencing in genetic hearing loss. Gen Test Mol Biom. 2013;17:581–587. doi: 10.1089/gtmb.2012.0464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Well D, Blanchard S, Kaplan J, Guilford P, Gibson F, Walsh J, et al. Defective myosin VIIA gene responsible for Usher syndrome type IB. Nature. 1995;374:60–61. doi: 10.1038/374060a0. [DOI] [PubMed] [Google Scholar]
- 30.Hartman MA, Spudich JA. The myosin superfamily at a glance. J Cell Sci. 2012;125:1627–1632. doi: 10.1242/jcs.094300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gerber S, Bonneau D, Gilbert B, Munnich A, Dufier JL, Rozet JM, et al. USH1A:chronicle of a slow death. Am J Hum Genet. 2006;78:357–359. doi: 10.1086/500275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Asgharzade S, Reiisi S, Tabatabaiefar MA, Chaleshtori MH. Screening of Myo7A mutations in Iranian patients with autosomal recessive hearing loss from west of Iran. Iran J Public Health. 2017;46:76–82. [PMC free article] [PubMed] [Google Scholar]
- 33.Mermall V, Post PL, Mooseker MS. Unconventional myosins in cell movement, membrane traffic, and signal transduction. Science. 1998;279:527–533. doi: 10.1126/science.279.5350.527. [DOI] [PubMed] [Google Scholar]
- 34.Boëda B, El-Amraoui A, Bahloul A, Goodyear R, Daviet L, Blanchard S, et al. Myosin VIIa, harmonin and cadherin 23, three Usher I gene products that cooperate to shape the sensory hair cell bundle. EMBO J. 2002;21:6689–6699. doi: 10.1093/emboj/cdf689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Reiisi S, Tabatabaiefar MA, Sanati MH, Chaleshtori MH. Screening of DFNB3 in Iranian families with autosomal recessive non-syndromic hearing loss reveals a novel pathogenic mutation in the MyTh4 domain of the MYO15A gene in a linked family. Iran J Basic Med Sci. 2016;19:772–778. [PMC free article] [PubMed] [Google Scholar]
- 36.Planelles-Herrero VJ, Blanc F, Sirigu S, Sirkia H, Clause J, Sourigues Y, et al. Myosin MyTH4-FERM structures highlight important principles of convergent evolution. Proc Natl Acad Sci U S Am. 2016;113:E2906–E2915. doi: 10.1073/pnas.1600736113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yu IM, Planelles-Herrero VJ, Sourigues Y, Moussaoui D, Sirkia H, Kikuti C, et al. Myosin 7 and its adaptors link cadherins to actin. Nat Commun. 2017;8:15864. doi: 10.1038/ncomms15864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cordes FS, Bright JN, Sansom MS. Proline-induced distortions of transmembrane helices. J Mol Biol. 2002;323:951–960. doi: 10.1016/s0022-2836(02)01006-9. [DOI] [PubMed] [Google Scholar]
- 39.Fukuoka H, Kanda Y, Ohta S, Usami S. Mutations in the WFS1 gene are a frequent cause of autosomal dominant nonsyndromic low-frequency hearing loss in Japanese. J Hum Genet. 2007;52:510–515. doi: 10.1007/s10038-007-0144-3. [DOI] [PubMed] [Google Scholar]
- 40.Goncalves AC, Matos TD, Simoes-Teixeira HR, Pimenta Machado M, Simao M, Dias OP, et al. WFS1 and non-syndromic low-frequency sensorineural hearing loss:a novel mutation in a Portuguese case. Gene. 2014;538:288–291. doi: 10.1016/j.gene.2014.01.040. [DOI] [PubMed] [Google Scholar]
- 41.Cryns K, Pfister M, Pennings RJ, Bom SJ, Flothmann K, Caethoven G, et al. Mutations in the WFS1 gene that cause low-frequency sensorineural hearing loss are small non-inactivating mutations. Hum Genet. 2002;110:389–394. doi: 10.1007/s00439-002-0719-1. [DOI] [PubMed] [Google Scholar]
- 42.Abdelhak S, Kalatzis V, Heilig R, Compain S, Samson D, Vincent C, et al. A human homologue of the Drosophila eyes absent gene underlies branchio-oto-renal (BOR) syndrome and identifies a novel gene family. Nat Genet. 1997;15:157–164. doi: 10.1038/ng0297-157. [DOI] [PubMed] [Google Scholar]
- 43.Xu PX, Adams J, Peters H, Brown MC, Heaney S, Maas R. Eya1-deficient mice lack ears and kidneys and show abnormal apoptosis of organ primordia. Nat Genet. 1999;23:113–117. doi: 10.1038/12722. [DOI] [PubMed] [Google Scholar]
- 44.Pourquié O. El-Hashash, editor. Publisher's Note:eya1 controls cell polarity, spindle orientation, cell fate and Notch signaling in distal embryonic lung epithelium. Development doi:10.1242/dev. 058479. Development. 2017;144:529. doi: 10.1242/dev.148718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Spahiu L, Merovci B, Ismaili Jaha V, Batalli Këpuska A, Jashari H. Case report of a novel mutation of the EYA1 gene in a patient with branchio-oto-renal syndrome. Balkan J Med Genet. 2016;19:91–94. doi: 10.1515/bjmg-2016-0042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Krug P, Morinière V, Marlin S, Koubi V, Gabriel HD, Colin E, et al. Mutation screening of the EYA1, SIX1, and SIX5 genes in a large cohort of patients harboring branchio-oto-renal syndrome calls into question the pathogenic role of SIX5 mutations. Hum Mutat. 2011;32:183–190. doi: 10.1002/humu.21402. [DOI] [PubMed] [Google Scholar]
- 47.Song MH, Kwon TJ, Kim HR, Jeon JH, Baek JI, Lee WS, et al. Mutational analysis of EYA1, SIX1 and SIX5 genes and strategies for management of hearing loss in patients with BOR/BO syndrome. PloS One. 2013;8:e67236. doi: 10.1371/journal.pone.0067236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sanggaard KM, Rendtorff ND, Kjaer KW, Eiberg H, Johnsen T, Gimsing S, et al. Branchio–oto–renal syndrome:detection of EYA1 and SIX1 mutations in five out of six Danish families by combining linkage, MLPA and sequencing analyses. Eur J Hum Genet. 2007;15:1121–1131. doi: 10.1038/sj.ejhg.5201900. [DOI] [PubMed] [Google Scholar]
- 49.Matsunaga T, Okada M, Usami S, Okuyama T. Phenotypic consequences in a Japanese family having branchio-oto-renal syndrome with a novel frameshift mutation in the gene EYA1. Acta Otolaryngol. 2007;127:98–104. doi: 10.1080/00016480500527185. [DOI] [PubMed] [Google Scholar]
- 50.Kwon MJ, Boo SH, Kwon MJ, Boo SH, Kim HJ, Cho YS, et al. A novel splice site mutation in the EYA1 gene in a Korean family with branchio-oto (BO) syndrome. Acta Otolaryngol. 2009;129:688–693. doi: 10.1080/00016480802342432. [DOI] [PubMed] [Google Scholar]
- 51.Washington NL, Haendel MA, Mungall CJ, Ashburner M, Westerfield M, Lewis SE. Linking human diseases to animal models using ontology-based phenotype annotation. PLoS Biol. 2009;7:e1000247. doi: 10.1371/journal.pbio.1000247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chen A, Francis M, Ni L, Cremers CW, Kimberling WJ, Sato Y, et al. Phenotypic manifestations of branchiootorenal syndrome. Am J Med Genet A. 1995;58:365–370. doi: 10.1002/ajmg.1320580413. [DOI] [PubMed] [Google Scholar]
- 53.Propst EJ, Blaser S, Gordon KA, Harrison RV, Papsin BC. Temporal bone findings on computed tomography imaging in branchio-oto-renal syndrome. Laryngoscope. 2005;115:1855–1862. doi: 10.1097/01.mlg.0000177032.98386.20. [DOI] [PubMed] [Google Scholar]
- 54.Ou Z, Martin DM, Bedoyan JK, Cooper ML, Chinault AC, Stankiewicz P, et al. Branchiootorenal syndrome and oculoauriculovertebral spectrum features associated with duplication of SIX1, SIX6, and OTX2 resulting from a complex chromosomal rearrangement. Am J Med Genet A. 2008;146:2480–2489. doi: 10.1002/ajmg.a.32398. [DOI] [PubMed] [Google Scholar]