

Abstract
The population dynamics of pathogens within hosts result from interactions between host immune responses and mechanisms of the pathogen to evade or resist immune responses. Vertebrate hosts have evolved adaptive immune responses to eliminate the infection, while many pathogens evade immune clearance through altering surface antigens. Such interactions can result in a characteristic pattern of pathogen population dynamics within hosts consisting of population growth after infection, rapid population decline following specific immune responses, followed by persistence at low densities during a chronic infection stage. Despite the medical importance of chronic infections, little is known about the conditions of the interactions between variable antigens and the adaptive immune system that cause the characteristic pathogen population dynamics. Using the vls antigenic variation system of the Lyme disease pathogen, Borrelia burgdorferi, as a model system, we investigated conditions of the interaction between the antigenic variation system and the adaptive immune response that can explain the within-host population dynamics of B. burgdorferi using mathematical modelling. This characteristic population dynamic pattern can be explained by models that assume a variable immune removal rate of antibody-bound B. burgdorferi. However, models with a constant immune removal rate could reproduce the rapid population decline of B. burgdorferi populations but not their long-term persistence within hosts using parameter values determined by fitting empirical data. The model predictions, along with the assumptions about the interactions between B. burgdorferi and the immune response, can be tested experimentally to estimate the likelihood that each mechanism affects B. burgdorferi population dynamics in real infections.
Keywords: antigenic variation, VlsE, Borrelia burgdorferi, within-host dynamics
Introduction
The evolutionary fitness of many microbial pathogens is a function of the duration of infection, which increases opportunities for transmission to naïve hosts, and the population size of the pathogen within the host, which increases the transmission rate per contact with naïve hosts [1–3]. However, pathogen persistence within hosts, as well as large pathogen population sizes, is correlated with symptom severity and decreases in host fitness [4–6]. Vertebrate hosts have evolved to respond to pathogen infections through an adaptive immune response aimed at reducing infection duration and pathogen population sizes. In turn, microbial pathogens have evolved antigenic variation systems to evade adaptive immune responses in order to persist within hosts at elevated population sizes [1–3, 5, 7]. Antigenic variation systems alter the antigens on the surface of pathogens, giving rise to pathogen subpopulations with distinct antigenic variants that are not recognized by antibodies targeting previously expressed antigens [8, 9]. The interaction between the host immune response and microbial antigenic variation controls the population dynamics of pathogens within the host, often resulting in a characteristic population dynamic pattern for chronic pathogens in which the pathogen population size increases rapidly in recently infected hosts, declines rapidly after the proliferation of specific antibodies, and then persists at low densities [10–13]. It has been hypothesized that this characteristic pattern is important for pathogenesis and transmission of the pathogens [12, 13]. It remains unclear, however, what conditions in the host immune system or in antigenic variation systems of chronic pathogens are sufficient to explain the population dynamics pattern for chronic pathogens. Here we use the well-characterized vls antigenic variation system of Borrelia burgdorferi to explore the interactions between variable antigens and host immune system that result in this characteristic pathogen population dynamic pattern by using mathematical modelling.
The population dynamics of B. burgdorferi within hosts resemble those of many infectious diseases. Multiple investigations of the population dynamics within experimentally infected mice have shown that B. burgdorferi density rapidly increases during the acute phase, declines following the activation of an adaptive immune response against surface exposed proteins including the VlsE antigen [14], and is followed by a low-level, chronic infection [10, 11]. Long-term persistence within hosts relies on continuous alteration of the immunodominant VlsE surface antigen in order to evade the antibody response [15–19]. Novel VlsE variants are generated by unidirectional recombination of a random segment from one of several unexpressed, paralogous vls cassettes into the vlsE expression site [19, 20], creating subpopulations of B. burgdorferi with novel VlsE sequences [21]. The magnitude of diversity among VlsE variants generated by the vls antigenic variation system is correlated with the ability to evade antibody recognition and permit chronic infections [16–19, 22–26].
In this study, we investigated conditions under which the interaction between adaptive immunity and the vls antigenic variation system can explain the within-host population dynamic pattern that is characteristic of B. burgdorferi and other chronic pathogens. A previous modelling study suggested that vlsE antigenic variation could impact B. burgdorferi densities within hosts, although specific interactions between the antigenic variation system and host immune response that could result in the characteristic dynamics pattern and the persistent infection of B. burgdorferi were not investigated [27]. In the present study, we investigated B. burgdorferi population dynamics during early stage infections by explicitly incorporating the well-characterized dynamics of VlsE antigenic variation and different assumptions about the functioning of the adaptive immune response. The models were parameterized using independent experimental data and the results were compared to B. burgdorferi population dynamics patterns observed in experimental infections [10, 23]. These models identify conditions under which the characteristic population dynamic pattern can be explained and describe the impact of multiple aspects of the host immune system – including antibody binding and removal of antibody-bound pathogen cells – on B. burgdorferi population dynamics. The models also make qualitative predictions about B. burgdorferi population dynamics that can be experimentally tested to estimate the likelihood that specific immune mechanisms control the persistence and population abundances of B. burgdorferi during infections.
Methods
Modelling B. burgdorferi population dynamics
We formulated a model that describes the abundance of B. burgdorferi subpopulations expressing different VlsE variants as well as the specificity, reactivity and abundances of antibody subpopulations. The model extends a prior model [27] by explicitly describing sequence changes in the VlsE antigen that can result in antibody evasion. Each B. burgdorferi subpopulation grows logistically and shrinks either due to antibody-mediated death or by altering the VlsE variant expressed. We let Bi represent the size of the subpopulation expressing VlsE type i that is not bound by antibodies, and Bi* corresponds to the size of the antibody-bound B. burgdorferi subpopulation. B. burgdorferi population dynamics – including growth, recombination and immune clearance – are described by:
| (1) |
| (2) |
such that the B. burgdorferi subpopulation expressing VlsE variant type i (Bi) grows logistically with an intrinsic growth rate β. The within-host carrying capacity, K, which applies to the entire B. burgdorferi population within the host, is derived from bacterial growth rate data [27]. Antibody-mediated removal of B. burgdorferi is determined by the strength of the interaction between expressed VlsE variants and all contemporary antibodies () and the removal rate of antibody-bound cells (ρ). The strength of the interaction between VlsE variant type i and all antibodies present within the host () is a function of the concentration of each antibody type and the reactivity of that antibody for VlsE variant type i, summed across all antibodies present in the host (details below). B. burgdorferi expressing VlsE variant type i (Bi) transitions to the susceptible state (Bi*) when bound by contemporary antibodies at rate μ and can be removed by the immune system at rate ρ or the bound antibodies can disassociate at rate ν. B. burgdorferi cells in subpopulation i can found a new subpopulation by altering their expressed VlsE variant through recombination. Recombination between an unexpressed vls cassette and the vlsE expression site occurs at rate r. All model parameters are presented in Table 1.
Table 1. Parameters used in the numerical simulations.
| Parameter | Unit | Value | Derived from | Description | |
|---|---|---|---|---|---|
| Parameter values derived from previous studies |
β | h−1 | 0.06 | [42] | Bacterial growth rate |
| K | Cells | 150 000 | [10] | Bacterial carrying capacity | |
| Amax | µg ml−1 | 8.5 | [30] | Maximum total antibody concentration | |
| n | – | 4 | [27, 29] | Hill coefficient in antibody response function | |
| r | h−1 | 0.0059 | [23] | Recombination rate | |
| μ | ml µg−1 h−1 | 0.0085 | [27] | Antibody association rate | |
| ν | h−1 | 1 | [27] | Antibody dissociation rate | |
| tlag | h | 72 | [29] | Time until antibody response starts | |
| θ | h−1 | 0.08 | [43] | Immune effector cells death rate | |
| h | 100 | [27, 29] | Time until antibody reaches half maximal concentration | ||
| Parameter values estimated for Model 1 |
x | – | 1.000 [0.379, 1.621]* | Estimated | Antibody specificity |
| ρ | h−1 | 0.829 [0.605, 1.052]* | Estimated | Immune removal rate of antibody-bound cells | |
| Parameter values estimated for Model 2 (1 antibody) |
x | – | 1.008 [0.664, 1.352]* | Estimated | Antibody specificity |
| ρ | h−1 | 0.801 [0.533, 1.068]* | Estimated | Immune removal rate of antibody-bound cells | |
| Td | h | 652 [590,714] * | Estimated | Duration of each dominant antibody | |
| Parameter values estimated for Model 2 (10 antibody) |
x | – | 1.108 [0.657, 1.558]* |
Estimated | Antibody specificity |
| ρ | h−1 | 0.822 [0.525, 1.120]* | Estimated | Immune removal rate of antibody-bound cells | |
| Td | h | 939 [803, 1075]* | Estimated | Duration of each dominant antibody | |
| Parameter values estimated for Model 3 |
x | – | 1.208 [1.164, 1.252]* | Estimated | Antibody specificity |
| φ | Cells h−1 | 0.010 [0.006, 0.014]* | Estimated | Intrinsic recruitment rate of immune effector cells | |
| ψ | h−1 | 0.141 [0.083, 0.198]* | Estimated | Pathogen-dependent recruitment rate of immune effector cells | |
| C | Cells | 24 969 [18764, 31 175]* | Estimated | Pathogen population size that yields half-maximal recruitment of immune effector cells | |
| ρ | h−1 | 0.092 [0.051, 0.133]* | Estimated | Immune removal rate of antibody-bound cells |
*95 % confidence intervals for the estimated parameters were shown below the best-fit parameter values.
Antigenic variation
Novel VlsE variants are generated by recombination between the vlsE expression site and the vls cassettes [19, 20]. The difference between any two VlsE variants within a host is a function of the number of recombination events that have occurred since the variants shared a common ancestor and the number of amino acids altered per recombination event. The number of amino acids altered per recombination event depends upon the sequence diversity among the vls cassettes that recombine into vlsE [24]. The distributions of sequence similarities between pairs of VlsE variants were estimated by explicitly simulating recombination between unexpressed vls cassettes and the expression site. First, 1000 parental VlsE variants (, ) were generated by simulating 1000 sequential recombination events starting from the vlsE sequence of B. burgdorferi clone 5A3 [19] as previously described [26]. From each of the 1000 parental VlsEn sequences, we simulated 20 daughter sequences (–) that experienced nr=1, 2, 3…20 recombination events in order to estimate sequence similarity between VlsE variants as a function of the number of recombination events that have occurred since the variants shared a common ancestor (nr). Pairwise sequence similarity between each parental VlsEn and each of its daughter sequences (–) was calculated as amino acid sequence identity. We assumed that every recombination event results in a novel VlsE variant, similar to an assumption of infinite alleles [28]. The sequence similarity between VlsE variant type i and j [S(i,j)] that have experienced nr recombination events since sharing a common ancestor was determined by randomly sampling from the sequence similarity values between VlsEn and (). We set nr=20 for pairs of variants that have nr>20 because sequence differences saturated at nr values greater than 20 (Fig. S1, available with the online Supplementary Material).
Antibody kinetics
The expression of antibodies specifically targeting VlsE variant type i is triggered by the first presence of Bi. The concentration of antibodies specific to VlsE type i () was determined using a time-dependent function as previously described [27]:
| (3) |
where: tinit is the time when the antibody response targeting VlsE variant type i is initiated; tlag is the time in hours following initiation that antibody production begins; n is the Hill coefficient for the antibody response; t½ is the time to half the maximal antibody concentration; Amax is the experimentally observed maximum antibody concentration [29, 30]; and k is the total number of antibodies in the host at time t. The concentration of each of the k antibody types was normalized to the observed total antibody concentration at time t ().
Antibody effects on B. burgdorferi dynamics
Specific antibodies, as well as non-specific but cross-reactive antibodies, can bind and eliminate B. burgdorferi (equation 1). The quantitative impact of antibodies specific to VlsE variant j on the subpopulation of B. burgdorferi expressing VlsE variant i is a function of the abundance of antibodies specific to variant j () and the degree of cross-reactivity between these antibodies and subpopulation Bi. Therefore the strength of the interaction between VlsE variant i and all contemporary antibodies can be expressed as:
| (4) |
where f(S(i,j)) defines the relationship between antibody reactivity and sequence similarity between VlsE variants i and j. All antigenic components other than VlsE were assumed to be identical among the B. burgdorferi subpopulations such that the degree of cross reactivity between antibody j and Bi is a function of only the similarity between VlsE types i and j. This function should satisfy f(0)=1 where antibody reactivity is identical when variants have identical sequences. Here we modelled cross-reactivity using a power function:
| (5) |
where larger values of x correspond to higher antibody specificity (i.e. lower cross-reactivity).
Additional mechanisms of the immune system
We analyse three versions of the model that differ in their assumptions of the functioning of the host immune system.
Model 1
Host antibodies are generated against all existing VlsE variants and all antibody-bound variants are eliminated at a constant rate ρ.
Model 2 (antibody immunodominance)
Host antibodies are generated against only a subset of VlsE variants and all antibody-bound variants are eliminated at a constant rate ρ. Model 2 considers a well-characterized phenomenon known as immunodominance, where only a small subset of B cell clones proliferate in response to the presence of antigens even though the naïve B cell repertoire is highly heterogeneous [31, 32]. The effect of antibody immunodominance on the population dynamics of B. burgdorferi was investigated by allowing only 1 or 10 immunodominant antibody types to be actively produced per unit time against the most abundant 1 or 10 VlsE variants. Immunodominant antibodies proliferate for Tdhours prior to the initiation of the next set of dominant antibodies, which then replace the previously dominant antibodies.
Model 3 (immune effector cell dynamics)
Host antibodies are generated against all existing VlsE variants and antibody-bound variants are eliminated by immune effector cells with explicitly modelled dynamics. In model 3, the rate at which antibody-bound B. burgdorferi cells are eliminated depends upon the concentration of immune effector cells, such as phagocytes near the site of infection [27, 33]. Immune effector cell dynamics were modelled as previously described [27], where immune effector cells have an intrinsic migration rate (φ), a recruitment rate in response to the presence of pathogens (ψ) and an intrinsic death rate (θ):
| (6) |
where C is the B. burgdorferi population size that corresponds to half the maximum recruitment rate of immune effector cells. For model 3, dynamics of antibody-bound B. burgdorferi cells becomes:
| (7) |
where the rate of removal of the antibody-bound B. burgdorferi follows the law of mass action.
Fitting empirical data
Although the aim of the study is to investigate theoretical conditions that qualitatively explain the characteristic dynamic pattern of infectious pathogens, we approached this aim quantitatively by fitting models to experimental data, which constrains the parameter space to explore when analysing the behaviours of the models under different parameter values. The mathematical models were fitted to two types of empirical data: (1) within-mice (C3H) B. burgdorferi population density measured overtime using qPCR [10]; and, (2) the relative abundance of B. burgdorferi expressing wild-type VlsE measured over time by sequencing isolates cultured from experimentally infected C3H mice [23]. Model fitting was conducted by minimizing the sum of the squared residuals (SSR). We used the constrained optimization by linear approximation algorithm – a derivative-free method implemented in the Nlopt package – with the constraint ρ<1. AIC was estimated for each of the models as:
| (8) |
where q is the total number of parameters, N is the total number of datapoints and SSR is the sum of squared residuals. For parameter values estimated by fitting to data, 95 % confidence intervals were determined as:
| (9) |
where Best fit (pi) is the best fit value for the ith parameter, tN=q (0.05) is the value from the t-distribution for 95 % confidence at N-q degrees of freedom, cov(i,i) is the ith diagonal element of the covariance matrix of the estimated parameters. The first 10 states (burn-in period) from the fitting iterations were not used for confidence interval calculations. Parameter values estimated by fitting to empirical data, as well as parameter values derived from previous studies, are summarized in Table 1.
Results
B. burgdorferi population dynamic patterns
Experimental mouse infections in multiple studies show that B. burgdorferi increases in density after infection, followed by a rapid decrease in density [10, 11]. Yet the immune system is rarely capable of eliminating the pathogen population, allowing the pathogen to establish chronic infection. This empirical pattern cannot be qualitatively reproduced by model 1 or model 2 (models with constant ρ and restricted or unrestricted antibody diversity), both of which predict elimination of the B. burgdorferi population within the first month of the infection (Fig. 1). Conversely, the model with unrestricted antibody diversity and variable ρ (model 3) qualitatively reproduces the rapid increase and decrease in B. burgdorferi density after infection and allows the B. burgdorferi population to establish a chronic infection (Fig. 1). Additionally, model 3 fits to the empirical data with lower SSR than model 1 and model 2 (SSR=286.9 for model 1, SSR=237.5 for model 2 with one antibody, SSR=227.8 for model 2 with 10 antibodies, and SSR=123.7 for model 3). Model 1, which has the smallest number of parameters, has the highest quality based on AIC (AIC=33.81 for model 1, AIC=38.96 for model 2 with one antibody, AIC=38.54 for model 2 with 10 antibodies, and AIC=61.74 for model 3).
Fig. 1.

The characteristic pathogen population dynamic pattern (a), as well as the dynamics of VlsE variants (b), are predicted by model 3 but do not from model 1 nor model 2. (a) All models can fit the characteristic population growth after infection and rapid population declines following specific immune responses. However, models 1 and 2 predict that B. burgdorferi populations are eliminated within hosts while model 3 predicts the long-term persistence of B. burgdorferi observed in natural infections. Open circles represent B. burgdorferi levels experimentally observed by Pahl et al. [10]. (b) The proportion of B. burgdorferi cells retaining the parental VlsE sequences predicted by the numerical simulation from all models was similar to the proportion retaining the parental sequence in experimental infections observed by Coutte et al. [23] in C3H mice (open circles).
Analyses of model features that result in different predictions
The three models predict different trajectories and fates of B. burgdorferi within hosts due to qualitatively and quantitatively different interactions between the host immune response and subpopulations of B. burgdorferi producing different VlsE variants. Specifically, the models differ in assumptions about two processes that control the ability of the immune response to constrain B. burgdorferi: (1) activation of specific host antibodies and binding of B. burgdorferi cells; and, (2) the rate at which the immune system removes antibody-bound B. burgdorferi. The empirical pattern of B. burgdorferi population dynamics suggests that the probability of antibody binding or the rate of removing antibody-bound cells is greater at high B. burgdorferi density and lower at low B. burgdorferi density. The subsequent sections describe the features of each model that result in differences in model predictions in the fate of B. burgdorferi population during infection.
B. burgdorferi bound by antibodies
Antibody-mediated suppression of a B. burgdorferi infection is a function of the probability that a B. burgdorferi cell is bound by antibodies, which can be reflected by the fraction of B. burgdorferi cells that are bound by antibodies at a given time. The proportion of antibody-bound B. burgdorferi cells in model 1, which does not account for antibody immunodominance, is determined by both the antigenic variation rate (r) and the specificity of the antibodies (x) (Fig. 2) and consistent decreases in the proportion of antibody-bound B. burgdorferi cells in the whole population are correlated with increases in either r or x (Fig. 2). However, using parameter values that closely fit to the B. burgdorferi population dynamic patterns (r=0.005 and x=1, Fig. 2c), the proportion of antibody-bound cells remains stable despite rapid population declines between days 7 and 20 post-infection (Fig. 1). Thus, the B. burgdorferi population decreases at a relatively constant rate until it is completely eliminated and will not establish persistent infection (Fig. 1).
Fig. 2.

The rate at which the immune system can suppress a B. burgdorferi infection is negatively correlated with both the VlsE antigenic variation rate (r) and the antibody specificity (x). Antigenic variation allows subpopulations of B. burgdorferi to evade immune detection, thus reducing the proportion of antibody-bound B. burgdorferi cells and reducing the rate at which B. burgdorferi are eliminated. Variant subpopulations are further protected from immune detection at high antibody specificity, as non-targeted VlsE variants are less likely recognized and bound by antibodies against previously expressed variants.
Models that restrict antibody diversity (model 2), where antibodies specifically target only one or a few dominant VlsE variants but can cross-react with other variants, permits the B. burgdorferi population to rapidly decrease in size due to the rapid elimination of the dominant subpopulations (Fig. 1). The variation in antibody pressure among the variants results in greater fractions of antibody-bound B. burgdorferi () in subpopulations that express the dominant VlsE variants than in subpopulations expressing variants that are only targeted by cross-reactive antibodies, depending on the cross-reactivity of the antibodies (Fig. 3a–c, grey dashed lines and Fig. 3d). Such variation in antibody pressure could potentially lead to rapid removal of the dominant subpopulation, while the non-dominant subpopulations are not eliminated, leading to persistent infection within the host.
Fig. 3.

Antibody immunodominance increases the variation in the fraction of antibody-bound cells among B. burgdorferi subpopulations but does not decrease the overall fraction antibody-bound cells. (a–c) The fraction of antibody-bound B. burgdorferi cells predicted by model 2 is similar to that predicted by model 1. The grey dashed lines represent the standard deviation in the fraction of antibody-bound cells among B. burgdorferi subpopulations expressing different VlsE variants (). (d) Variance in antibody-bound cells among B. burgdorferi subpopulations expressing different VlsE variants () predicted by model 2 is higher than that predicted by model 1. Numerical simulations were conducted with r=0.005 h−1 and x=0.5, 1.0 or 2.0. Here, the fraction of antibody-bound B. burgdorferi cells does not include newly generated VlsE variants that have not yet activated any antibodies.
However, persistent infections cannot be achieved using parameter values estimated by fitting to empirical data. Specifically, the antibody specificity value (x) leading to predictions that most closely resemble empirical data suggests that the host antibodies are highly cross-reactive (x=1.0) such that all B. burgdorferi subpopulations, whether or not specifically targeted, are efficiently eliminated at rates that are uncorrelated with population density (Fig. 1). Thus, the overall fraction of antibody-bound B. burgdorferi at x=1.0 decreases only by less than 10 % (Fig. 3b) when B. burgdorferi density decreases between day 7 and day 20 post-infection (Fig. 1). This high cross-reactivity causes the B. burgdorferi population size to constantly decrease until completely eliminated, a trajectory similar to model 1. Further, the overall fraction of antibody-bound B. burgdorferi predicted by model 2 is not lower than in model 1 (Fig. 3a–c), suggesting that restricting antibody diversity does not decrease the probability that a VlsE variant is bound by antibodies. This is because antibody reactivity to a given VlsE variant is a function of the genetic distance as measured by the number of recombination events (nr) between the VlsE variant and the variant to which the antibody specifically targets. In model 1, antibodies are activated by all circulating VlsE variants, while antibodies are only activated by a few ancestral VlsE variants that have high relative abundance in model 2. The average distance (nr) between all pairs of coexisting variants is larger than the average distance between the parental VlsE and coexisting variants (Fig. 4). Thus, the average antibody reactivity in model 2 is greater than in model 1, which contributes to the rapid elimination of B. burgdorferi predicted by model 2.
Fig. 4.
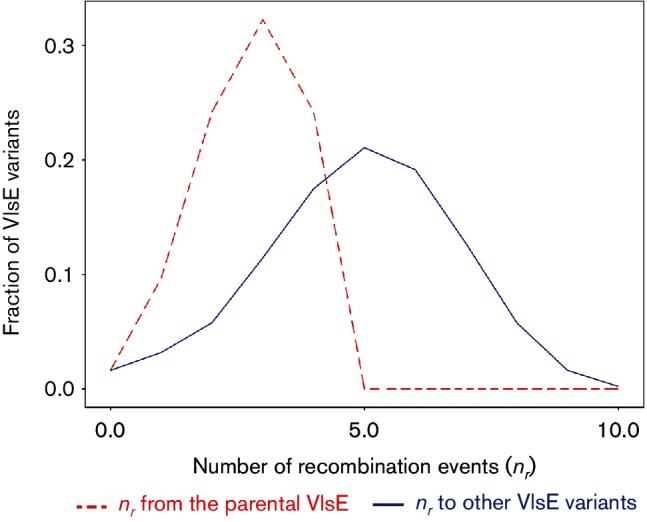
The average antibody reactivity in model 2 is greater than in model 1 because antigenic distances between pairs of coexisting variants is larger on average than the difference between variants and the parental VlsE that was targeted by the immunodominant antibody. The figure describes the frequency distribution of the number of recombination events (nr) between pairs of VlsE variants (solid line) and between variants and the parental VlsE variant (dashed line). Numerical simulations were conducted with r=0.005 h−1; x=1.0; 7 days post-infection.
Immune removal of antibody-bound B. burgdorferi cells
Model 3 differs from models 1 and 2 in that the immune killing rate of antibody-bound B. burgdorferi cells is dynamic. Model 3 describes predator–prey-like interactions between the immune effector cells and B. burgdorferi, observed as an increase in the immune killing rate following an increase in B. burgdorferi density (Fig. 5). Thus, model 3 predicts that the immune response removes B. burgdorferi efficiently at high pathogen density and less efficiently at low density, which is consistent to the observed dynamic pattern in experimental infections. This population dynamic pattern is predicted by model 3 under a wide range of parameter values, although the quantitative estimates of both B. burgdorferi and immune effector cells population sizes are sensitive to parameter specifications (Fig. 5). In contrast to models 1 and 2, which remove B. burgdorferi at a constant rate and result in pathogen elimination, model 3 allows both the sharp drop in population density and the persistent infection of the B. burgdorferi population, reproducing the experimentally observed population dynamics both qualitatively and quantitatively.
Fig. 5.

Model 3 describes predator–prey-like dynamics where the immune killing rate increases following an increase in B. burgdorferi density. Contemporaneous B. burgdorferi population densities (left axis, red) and immune killing rates (ρM) normalized to their maximum value (right axis, blue) demonstrate that high efficiency of the immune response at high B. burgdorferi densities and low efficiency at low pathogen density, consistent with the observed dynamic patterns in experimental infections. Numerical simulations were conducted with r=0.005 h−1, x=1.0, and multiple values for parameters controlling the dynamics of immune effector cells (φ, ψ and C).
Discussion
Within-host pathogen population dynamics are functions of the interactions between the dynamically specific adaptive immune response and the continuously changing antigens on the surface of pathogens. These interactions often result in a characteristic population dynamic pattern in which the pathogen population size increases in recently infected hosts, declines rapidly due to proliferation of the initial adaptive antibody response, and then persists as a chronic infection [10–13]. We analysed the conditions under which within-host dynamics of B. burgdorferi can be explained by interactions between the host immune system and the VlsE antigens by comparing mathematical models to empirical data of B. burgdorferi population dynamics and VlsE antigenic variation [10, 23]. Simple models that account for B. burgdorferi population growth rate, antigenic variation and simple adaptive immune responses (model 1) cannot explain the persistence of B. burgdorferi due to the high antibody pressure across all VlsE variants required to reproduce the rapid population declines observed in empirical data. Thus, additional mechanisms such as antibody immunodominance or immune effector cell dynamics [27, 31] are necessary to account for the B. burgdorferi population dynamics observed in experimental infections.
Including antibody immunodominance into the B. burgdorferi population dynamic model (model 2) cannot reproduce the characteristic pathogen population dynamics using realistic parameter values for the rate of antigenic variation (r) and B. burgdorferi growth rate (β). In model 2, the dominant B. burgdorferi subpopulations each experience strong antibody pressure by one of the few proliferating antibody types, but the rapid population decline observed in experimental infections also requires immune-mediated suppression of the subpopulations expressing non-dominant VlsE variants (generated at rate r and growing at rate β) that are only affected by cross-reactive antibodies. Thus, the rate of population decline of B. burgdorferi within a host is contingent on the specificity of the immunodominant antibodies (x). If host antibodies are highly specific (large x), B. burgdorferi subpopulations expressing non-dominant VlsE variants will experience limited immune pressure from the antibodies targeting the dominant VlsE variants, leading to slower decreases in population densities than is characteristic in empirical pathogen population dynamics. Models assuming high cross-reactivity (small x) fit the characteristic rapid population declines as subpopulations expressing non-dominant VlsE variants experience substantial immune pressure. However, this cross-reactive immune pressure leads to extinction of the pathogen population during the early stages of the infection, which is rarely observed in experimental infections. The results of these models suggest the importance of cross-reactive immunity in controlling pathogens expressing variable antigens especially when the diversity of host antibodies is constrained. The influence of cross-reactive immunity on pathogen dynamics has also been emphasized in mathematical studies of other parasite systems such as trypanosome infections [8] although many earlier mathematical models of similar systems do not consider cross-reactivity [34] or restrict cross-reactivity a priori [35].
The population dynamics that result from models that assume variable immune killing rates (model 3) can recreate the characteristic population growth, decline and persistence within a host. The population dynamics described by model 3 result from predator–prey-like interactions between immune effector cells and the B. burgdorferi population. Here, the recruitment rate of predator-like immune effector cells is a function of the abundance of prey-like B. burgdorferi, resulting in rapid recruitment of immune effector cells and removal of B. burgdorferi when B. burgdorferi population sizes are large, followed by fewer immune effector cells and reduced killing when B. burgdorferi is rare [33]. The recruitment and depletion of immune effector cells such as neutrophils, macrophages and eosinophils have been empirically shown to impact on B. burgdorferi infections within hosts [36–38]. Model 3 also predicts continuous fluctuations in B. burgdorferi populations throughout the early stages of infection (Fig. 1) – a typical pattern observed in predator–prey systems that has not been empirically investigated in B. burgdorferi dynamics. The predicted pattern can be validated by more compact experimental time series of B. burgdorferi and immune cell densities within hosts.
In principle, the dynamics pattern observed in experimental infections could also be explained by immune or pathogen mechanisms that were not explored in these models. Due to the sparse time series in both the B. burgdorferi density and the VlsE sequence variation, we constrained our study to only the immune mechanisms with relatively simple mathematical representations, the early stage of the infection and qualitative predictions of the dynamics. Given the assumptions of the models explored, only when including mechanisms in addition to antibody–antigen interaction, such as immune effector cell dynamics, can the population dynamics of B. burgdorferi be qualitatively explained. A more compact time series will allow modelling attempts involving additional mechanisms such as variable immune intensity at different dissemination sites [39], difference in antibody reactivity towards protective or non-protective epitopes [40], difference in epitope efficacy among different residue types or positions [41] and potential variability in the recombination rates among B. burgdorferi clones.
Funding information
This research was supported by Burroughs Wellcome Fund (1012376), an NIH training grant (T32 AI055400) and grants from the National Institute of Allergy and Infectious Diseases (AI076342 and AI097137).
Acknowledgements
We are very thankful to Erol Akçay, Paul Sniegowski, Rahul Kohli and Timothy Linksvayer for helpful suggestions.
Conflicts of interest
The authors declare that there are no conflicts of interest.
Supplementary Data
Footnotes
Abbreviations: AIC, Akaike information criterion; SSR, sum of squared residuals; vls, VMP-like sequence; vlsE, expressed VMP-like sequence gene.
One supplementary figure is available with the online Supplementary Material.
Edited by: S. P. Diggle and M. Whiteley
References
- 1.Deitsch KW, Moxon ER, Wellems TE. Shared themes of antigenic variation and virulence in bacterial, protozoal, and fungal infections. Microbiol Mol Biol Rev. 1997;61:281–293. doi: 10.1128/mmbr.61.3.281-293.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Moxon ER, Rainey PB, Nowak MA, Lenski RE. Adaptive evolution of highly mutable loci in pathogenic bacteria. Curr Biol. 1994;4:24–33. doi: 10.1016/S0960-9822(00)00005-1. [DOI] [PubMed] [Google Scholar]
- 3.Schmid-Hempel P. Immune defence, parasite evasion strategies and their relevance for 'macroscopic phenomena' such as virulence. Philos Trans R Soc Lond B Biol Sci. 2009;364:85–98. doi: 10.1098/rstb.2008.0157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Frank SA. Models of parasite virulence. Q Rev Biol. 1996;71:37–78. doi: 10.1086/419267. [DOI] [PubMed] [Google Scholar]
- 5.Frank SA, Schmid-Hempel P. Mechanisms of pathogenesis and the evolution of parasite virulence. J Evol Biol. 2008;21:396–404. doi: 10.1111/j.1420-9101.2007.01480.x. [DOI] [PubMed] [Google Scholar]
- 6.Levin BR, Bull JJ. Short-sighted evolution and the virulence of pathogenic microorganisms. Trends Microbiol. 1994;2:76–81. doi: 10.1016/0966-842X(94)90538-X. [DOI] [PubMed] [Google Scholar]
- 7.Brunham RC, Plummer FA, Stephens RS. Bacterial antigenic variation, host immune response, and pathogen-host coevolution. Infect Immun. 1993;61:2273–2276. doi: 10.1128/iai.61.6.2273-2276.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Antia R, Nowak MA, Anderson RM. Antigenic variation and the within-host dynamics of parasites. Proc Natl Acad Sci USA. 1996;93:985–989. doi: 10.1073/pnas.93.3.985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Van der Woude MW, Bäumler AJ. Phase and antigenic variation in bacteria. Clin Microbiol Rev. 2004;17:581–611. doi: 10.1128/CMR.17.3.581-611.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pahl A, Kühlbrandt U, Brune K, Röllinghoff M, Gessner A. Quantitative detection of Borrelia burgdorferi by real-time PCR. J Clin Microbiol. 1999;37:1958–1963. doi: 10.1128/jcm.37.6.1958-1963.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hodzic E, Feng S, Freet KJ, Barthold SW. Borrelia burgdorferi population dynamics and prototype gene expression during infection of immunocompetent and immunodeficient mice. Infect Immun. 2003;71:5042–5055. doi: 10.1128/IAI.71.9.5042-5055.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Turner CM. Antigenic variation in Trypanosoma brucei infections: an holistic view. J Cell Sci. 1999;112:3187–3192. doi: 10.1242/jcs.112.19.3187. [DOI] [PubMed] [Google Scholar]
- 13.Matthews KR, Mcculloch R, Morrison LJ. The within-host dynamics of African trypanosome infections. Philos Trans R Soc Lond B Biol Sci. 2015;370:20140288. doi: 10.1098/rstb.2014.0288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wooten RM, Ma Y, Yoder RA, Brown JP, Weis JH, et al. Toll-like receptor 2 is required for innate, but not acquired, host defense to Borrelia burgdorferi. J Immunol. 2002;168:348–355. doi: 10.4049/jimmunol.168.1.348. [DOI] [PubMed] [Google Scholar]
- 15.Bankhead T, Chaconas G. The role of VlsE antigenic variation in the Lyme disease spirochete: persistence through a mechanism that differs from other pathogens. Mol Microbiol. 2007;65:1547–1558. doi: 10.1111/j.1365-2958.2007.05895.x. [DOI] [PubMed] [Google Scholar]
- 16.Labandeira-Rey M, Skare JT. Decreased infectivity in Borrelia burgdorferi strain B31 is associated with loss of linear plasmid 25 or 28-1. Infect Immun. 2001;69:446–455. doi: 10.1128/IAI.69.1.446-455.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Purser JE, Norris SJ. Correlation between plasmid content and infectivity in Borrelia burgdorferi. Proc Natl Acad Sci USA. 2000;97:13865–13870. doi: 10.1073/pnas.97.25.13865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rogovskyy AS, Bankhead T. Variable VlsE is critical for host reinfection by the Lyme disease spirochete. PLoS One. 2013;8:e61226. doi: 10.1371/journal.pone.0061226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang JR, Hardham JM, Barbour AG, Norris SJ. Antigenic variation in Lyme disease borreliae by promiscuous recombination of VMP-like sequence cassettes. Cell. 1997;89:275–285. doi: 10.1016/S0092-8674(00)80206-8. [DOI] [PubMed] [Google Scholar]
- 20.Zhang JR, Norris SJ. Genetic variation of the Borrelia burgdorferi gene vlsE involves cassette-specific, segmental gene conversion. Infect Immun. 1998;66:3698–3704. doi: 10.1128/iai.66.8.3698-3704.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rogovskyy AS, Casselli T, Tourand Y, Jones CR, Owen JP, et al. Evaluation of the importance of VlsE antigenic variation for the enzootic cycle of Borrelia burgdorferi. PLoS One. 2015;10:e0124268. doi: 10.1371/journal.pone.0124268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bykowski T, Babb K, Von Lackum K, Riley SP, Norris SJ, et al. Transcriptional regulation of the Borrelia burgdorferi antigenically variable VlsE surface protein. J Bacteriol. 2006;188:4879–4889. doi: 10.1128/JB.00229-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Coutte L, Botkin DJ, Gao L, Norris SJ. Detailed analysis of sequence changes occurring during vlsE antigenic variation in the mouse model of Borrelia burgdorferi infection. PLoS Pathog. 2009;5:e1000293. doi: 10.1371/journal.ppat.1000293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Graves CJ, Ros VI, Stevenson B, Sniegowski PD, Brisson D. Natural selection promotes antigenic evolvability. PLoS Pathog. 2013;9:e1003766. doi: 10.1371/journal.ppat.1003766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mcdowell JV, Sung SY, Hu LT, Marconi RT. Evidence that the variable regions of the central domain of VlsE are antigenic during infection with lyme disease spirochetes. Infect Immun. 2002;70:4196–4203. doi: 10.1128/IAI.70.8.4196-4203.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhou W, Brisson D. Potentially conflicting selective forces that shape the vls antigenic variation system in Borrelia burgdorferi. Infect Genet Evol. 2014;27:559–565. doi: 10.1016/j.meegid.2014.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Binder SC, Telschow A, Meyer-Hermann M. Population dynamics of Borrelia burgdorferi in Lyme disease. Front Microbiol. 2012;3:104. doi: 10.3389/fmicb.2012.00104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kimura M, Crow JF. The number of alleles that can be maintained in a finite population. Genetics. 1964;49:725–738. doi: 10.1093/genetics/49.4.725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Barthold SW, Persing DH, Armstrong AL, Peeples RA. Kinetics of Borrelia burgdorferi dissemination and evolution of disease after intradermal inoculation of mice. Am J Pathol. 1991;139:263–273. [PMC free article] [PubMed] [Google Scholar]
- 30.Wooten RM, Ma Y, Yoder RA, Brown JP, Weis JH, et al. Toll-like receptor 2 plays a pivotal role in host defense and inflammatory response to Borrelia burgdorferi. Vector Borne Zoonotic Dis. 2002;2:275–278. doi: 10.1089/153036602321653860. [DOI] [PubMed] [Google Scholar]
- 31.Frank SA. Immunology and Evolution of Infectious Disease. Princeton, New Jersey: Princeton University Press; 2002. [PubMed] [Google Scholar]
- 32.Rao KV. Selection in a T-dependent primary humoral response: new insights from polypeptide models. APMIS. 1999;107:807–818. doi: 10.1111/j.1699-0463.1999.tb01476.x. [DOI] [PubMed] [Google Scholar]
- 33.Hirschfeld M, Kirschning CJ, Schwandner R, Wesche H, Weis JH, et al. Cutting edge: inflammatory signaling by Borrelia burgdorferi lipoproteins is mediated by toll-like receptor 2. J Immunol. 1999;163:2382–2386. [PubMed] [Google Scholar]
- 34.Agur Z, Abiri D, Van der Ploeg LH. Ordered appearance of antigenic variants of African trypanosomes explained in a mathematical model based on a stochastic switch process and immune-selection against putative switch intermediates. Proc Natl Acad Sci USA. 1989;86:9626–9630. doi: 10.1073/pnas.86.23.9626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gatton ML, Cheng Q. Investigating antigenic variation and other parasite-host interactions in Plasmodium falciparum infections in naïve hosts. Parasitology. 2004;128:367–376. doi: 10.1017/S0031182003004608. [DOI] [PubMed] [Google Scholar]
- 36.Brown CR, Blaho VA, Loiacono CM. Treatment of mice with the neutrophil-depleting antibody RB6-8C5 results in early development of experimental Lyme arthritis via the recruitment of Gr-1- polymorphonuclear leukocyte-like cells. Infect Immun. 2004;72:4956–4965. doi: 10.1128/IAI.72.9.4956-4965.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Menten-Dedoyart C, Faccinetto C, Golovchenko M, Dupiereux I, van Lerberghe PB, et al. Neutrophil extracellular traps entrap and kill Borrelia burgdorferi sensu stricto spirochetes and are not affected by Ixodes ricinus tick saliva. J Immunol. 2012;189:5393–5401. doi: 10.4049/jimmunol.1103771. [DOI] [PubMed] [Google Scholar]
- 38.Xu Q, Seemanapalli SV, Reif KE, Brown CR, Liang FT. Increasing the recruitment of neutrophils to the site of infection dramatically attenuates Borrelia burgdorferi infectivity. J Immunol. 2007;178:5109–5115. doi: 10.4049/jimmunol.178.8.5109. [DOI] [PubMed] [Google Scholar]
- 39.Hyde JA. Borrelia burgdorferi keeps moving and carries on: a review of borrelial dissemination and invasion. Front Immunol. 2017;8:1–16. doi: 10.3389/fimmu.2017.00114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liang FT, Jacobs MB, Philipp MT. C-terminal invariable domain of VlsE may not serve as target for protective immune response against Borrelia burgdorferi. Infect Immun. 2001;69:1337–1343. doi: 10.1128/IAI.69.3.1337-1343.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kringelum JV, Lundegaard C, Lund O, Nielsen M. Reliable B cell epitope predictions: impacts of method development and improved benchmarking. PLoS Comput Biol. 2012;8:e1002829. doi: 10.1371/journal.pcbi.1002829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Barbour AG. Isolation and cultivation of Lyme disease spirochetes. Yale J Biol Med. 1983;57:521–525. [PMC free article] [PubMed] [Google Scholar]
- 43.van Furth R, Diesselhoff-den Dulk MM. The kinetics of promonocytes and monocytes in the bone marrow. J Exp Med. 1970;132:813–828. doi: 10.1084/jem.132.4.813. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.