

Abstract
Tick-borne encephalitis virus (TBEV) is a member of the genus Flavivirus. It can cause serious infections in humans that may result in encephalitis/meningoencephalitis. Although several studies have described the involvement of specific genes in the host response to TBEV infection in the central nervous system (CNS), the overall network remains poorly characterized. Therefore, we investigated the response of DAOY cells (human medulloblastoma cells derived from cerebellar neurons) to TBEV (Neudoerfl strain, Western subtype) infection to characterize differentially expressed genes by transcriptome analysis. Our results revealed a wide panel of interferon-stimulated genes (ISGs) and pro-inflammatory cytokines, including type III but not type I (or II) interferons (IFNs), which are activated upon TBEV infection, as well as a number of non-coding RNAs, including long non-coding RNAs. To obtain a broader view of the pathways responsible for eliciting an antiviral state in DAOY cells we examined the effect of type I and III IFNs and found that only type I IFN pre-treatment inhibited TBEV production. The cellular response to TBEV showed only partial overlap with gene expression changes induced by IFN-β treatment – suggesting a virus-specific signature – and we identified a group of ISGs that were highly up-regulated following IFN-β treatment. Moreover, a high rate of down-regulation was observed for a wide panel of pro-inflammatory cytokines upon IFN-β treatment. These data can serve as the basis for further studies of host–TBEV interactions and the identification of ISGs and/or lncRNAs with potent antiviral effects in cases of TBEV infection in human neuronal cells.
Keywords: tick-borne encephalitis virus, neuronal cells, transcriptome analysis, host response, interferon
Introduction
Tick-borne encephalitis virus (TBEV) is a medically important tick-borne flavivirus and is the causative agent of tick-borne encephalitis (TBE). TBE is widespread in Europe and North Asia, and more than 10 000 cases per year are reported [1]. The Czech Republic has the second highest incidence of TBE in Europe after Russia [2]. The clinical outcome of TBE can vary from sub-clinical cases to severe encephalitis/meningoencephalitis. The European subtype of TBEV is associated with a high ratio of sub-clinical or asymptomatic cases (estimated 70–95 %). Neurologic sequelae were reported in up to 30 % of the patients and the case fatality in adult patients is <2 % [3].
The mechanism(s) by which TBEV crosses the blood–brain barrier (BBB) and enters the CNS are still not clear. Several routes have been suggested: (i) direct infection of epithelial cells and transport of viruses across basolateral membranes, (ii) cytokine-mediated breakdown of BBB, or (iii) a ‘Trojan horse’ pathway in which TBEV-infected leukocytes can migrate across the BBB [4, 5]. Once TBEV enters the CNS, neurons are the predominantly infected cell type [6]. Astrocytes were recently also shown to be susceptible to TBEV infection [7]. Immunocytochemistry analysis of brain autopsies from fatal TBE cases detected localization of viral antigens in the spinal cord, brainstem, cerebellum and basal ganglia. Labelling was consistently found in the perikarya and processes of Purkinje cells, large neurons of the dentate nucleus, inferior olives and anterior horns [6]. The suggested mechanism of neural tissue damage during TBEV infection is virus-associated cell death combined with an immunopathogenic role of the cellular/humoral responses of the host immune system, especially CD8+ granzyme B–releasing cytotoxic T cells and macrophages/microglia [8, 9].
The interferon (IFN) response is part of the innate immune system. IFNs activate the expression of hundreds of genes, known as IFN-stimulated genes (ISGs), which elicite the antiviral state [10–12]. In most cell types, type I IFNs (IFN-α and IFN-β), which signal through the IFNAR1/IFNAR2 receptors, are the primary IFNs produced. With regard to the production of type I IFNs in CNS, murine astrocytes and microglia were observed to be the main IFN producers following La Crosse virus (LACV) infection [13]. However, a study by Delhaye et al. showed that 16 % of IFN-producing cells in the CNS of mice infected with either Theiler's encephalomyelitis virus (TMEV) or LACV corresponded to neurons [14]. The importance of the type I IFN system in preventing CNS infection in mice was also characterized for West Nile virus (WNV) [15]. Furthermore, the role of IFN-β in preventing viral infection in neuronal cells was shown for human granule cell neurons and cortical neurons when IFN-β pre-treatment resulted in the inhibition of WNV and Saint Louis encephalitis (SLEV) flaviviruses [16]. Recently, type III IFNs were found to play an important role in the immune response to neurotropic viruses. IFN-λ1/2 pre-treatment of human neurons and astrocytes resulted in inhibition of herpes simplex virus 1 (HSV1) [17] and IFN-λ2 pre-treatment reduced WNV infection in murine CNS by decreasing BBB permeability [18]. Type III IFNs bind to IFNLR1/IL10β, which signals through a similar pathway to the type I IFN receptor complex and induces many of the same ISGs [19, 20].
To date, only the type I IFN system has been shown to be essential for control of TBEV and related Langat virus (LGTV) systemic infection of the murine CNS [21, 22]. Moreover, type I IFN responses have been shown to protect murine astrocytes – a CNS cell type – from tick-borne flavivirus infection [23]. IFN-ɑ pre-treatment of murine neuroblastoma cells resulted in a decrease in the production of LGTV [24]. However, to date no study has described the host response of human neuronal cells upon TBEV infection. Here we investigated the responses to TBEV infection and type I IFNs in DAOY cells (human medulloblastoma cells derived from cerebellar neurons) by transcriptome analysis. We previously used this cell line to investigate morphological changes post-TBEV infection [25], and here expanded our study of virus–cell interactions. Our results show that in response to TBEV infection DAOY cells modulate the expression of ISGs, type III IFNs and pro-inflammatory cytokines. We found that the virus-induced responses differed from those induced by IFN-ß, with partial overlap. We examined the protective effect of type I and III IFNs on TBEV infection to assess pathways capable of eliciting an antiviral state in DAOY cells. Host responses mediated by type I but not type III IFNs mediated antiviral protection. Virus-specific host response signatures may be relevant for understanding TBEV pathogenesis.
Results
Human DAOY medulloblastoma cell line expresses markers typical for neural precursor cells
As TBEV infection can result in CNS damage, we studied the antiviral host response against TBEV strain Neudoerfl (Western subtype) in vitro in the human medulloblastoma-derived neuronal cell line, DAOY HTB-186. These cells are derived from the cerebellum [26], one of the brain areas affected most during TBE infection [6], and were shown to be susceptible to TBEV strain Hypr [25]. In order to determine the infection rate of TBEV Neudoerfl, DAOY cells were infected at a multiplicity of infection (m.o.i.) of 0.1, 1, and 5, respectively, and levels of viral NS3 protein were analysed at 24 h post infection (h p.i.) using an immunofluorescence assay. The infection rates for an m.o.i. 0.1, 1 or 5 at 24 h p.i. were 1.5 % (sd ±0.44), 5.0 % (sd ±0.93) and 19.6 % (sd ±2.25), respectively (Fig. 1a). The m.o.i. refers to the TBEV titre in PS cells (see below); infection rates may therefore vary in other cell lines.
Fig. 1.
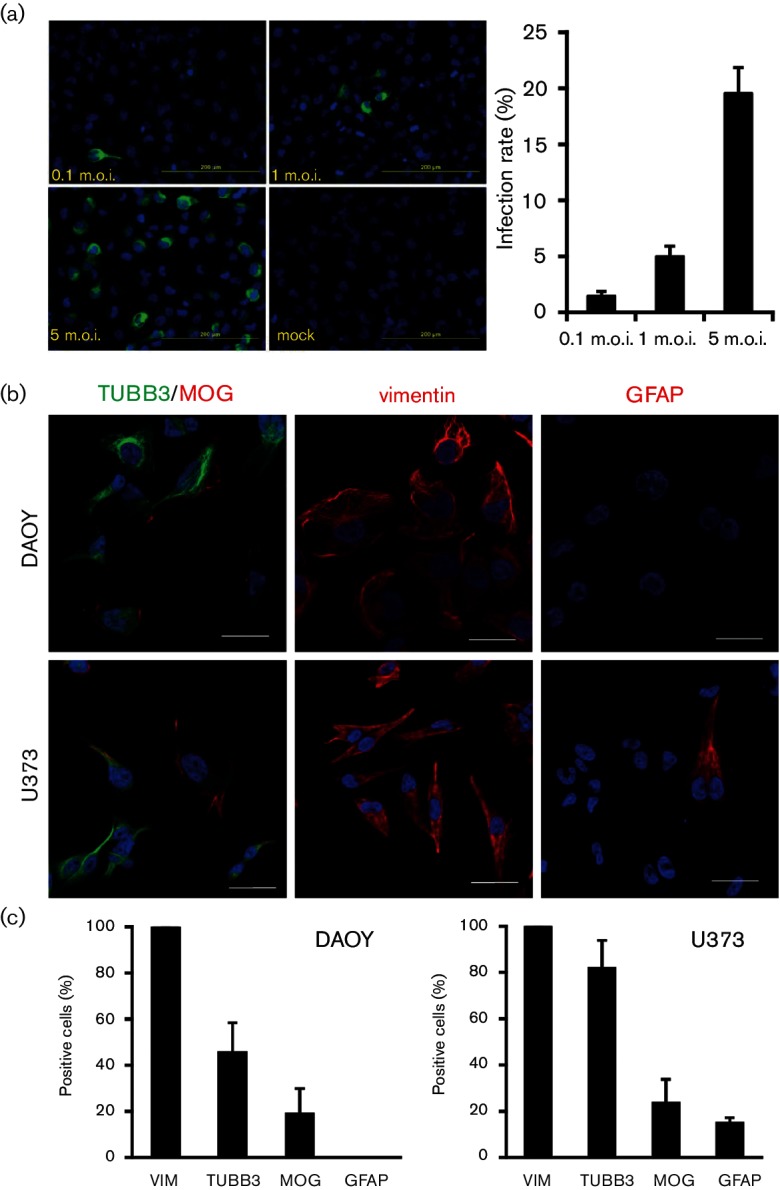
DAOY cells were susceptible to TBEV infection and expressed both neuronal and glial markers. (a) DAOY cells were seeded on a chamber slide and infected at an m.o.i. of 0.1, 1 and 5 with TBEV Neudoerfl strain. Detection of viral NS3 protein via immunofluorescence was carried out at 24 h p.i. using anti-NS3 antibodies. Representative pictures from two independent experiments (in triplicate per experiment) are shown. The infection rate was calculated as a ratio of the total number of infected cells (positive signal for NS3) to the total number of cells. The average with standard deviation is shown. (b) DAOY and U373 cells were seeded on a chamber slide and incubated 24 h. Cells were stained with antibodies for TUBB3, MOG, VIM and GFAP. Representative pictures from two independent experiments (in triplicate per experiment) are shown. Scale bar represents 20 µm. (c) Cell counts for each of the markers are presented. The average with standard deviation from two independent experiments (in triplicate per experiment) is shown.
In order to verify the neuronal origin of DAOY cells, we analysed them for the presence of CNS biomarkers – tubulin beta 3 class III (TUBB3), vimentin (VIM) and myelin oligodendrocyte glycoprotein (MOG) [27]. These three CNS biomarkers were found to be among the genes with the highest expression according to transcriptomic analysis [see Tables S1 and S2 (available with the online Supplementary Material) for lists of glial and neuronal markers identified in DAOY cells]. In addition, we characterized the presence of glial fibrillary acidic protein (GFAP) in DAOY cells, which is a commonly used marker for glial cells [28]. The human glioblastoma cell line U-373 MG (Uppsala) was used as a control for glial origin [29]. The expression of GFAP was only detected in U373 cells (14.6 %, sd ±2.5), whereas MOG was detected in both DAOY (18.9 %, sd ±11.0) and U373 (23.4 %, sd ±10.4). Both cell lines were also positive for TUBB3 (45.4 %, sd ±13.1, and 81.7 %, sd ±12.2 for DAOY and U373, respectively) as well as VIM (100 %, sd ±0, and 100 %, sd ±0 for DAOY and U373, respectively), as shown in Fig. 1(b, c) (see also Fig. S1 for separate TUBB3/MOG staining).
Furthermore we analysed the expression of selected glial/neuronal markers in DAOY cells upon TBEV infection, and whether TBEV preferentially targets certain cells (m.o.i. 5; analysis at 24 h p.i.). As shown in Fig. S2(a), the expression rates of TUBB3, MOG and VIM were not significantly changed upon TBEV infection in comparison to control cells (Student‘s t-test; P=0.9679, P=0.9249 and P=0.2244, respectively). No signal was detected for GFAP, which correlates with the data described in Fig. 1. In order to determine whether the presence of a particular marker affects the ability of DAOY cells to be infected with TBEV, we also quantified infected cells positive for TUBB3, MOG or VIM (30.4 %, sd ±5.1; 7.9 %, sd ±2.5; 100.0 %, sd ±0, respectively) (Fig. S2b). These numbers largely correlated with CNS marker expression levels in infected and uninfected cells, as shown in Fig. S2(a), suggesting that TBEV did not preferentially infect cells expressing a specific marker.
IFN-β pre-treatment of DAOY cells resulted in reduced production of TBEV
It was previously shown that in vitro type I IFN pre-treatment of neuronal cells resulted in decreased production of several neurotropic RNA viruses, including LGTV, WNV and SLEV [16, 30]. To analyse whether IFN-β pre-treatment can impair TBEV infection, DAOY and A549 cells were pre-treated with human recombinant IFN-β (10, 100, and 1000 ng ml−1) and infected with TBEV 12 h later at an m.o.i. of 5. A549 cells were used as controls, given their wide use in type I IFN studies (for example [31–34]). Cells were incubated for 5 days until a virus-induced cytopathic effect (CPE) was observed in the control wells. Viability assays using MTT were subsequently performed (see the Methods section). We analysed the rescue of cell viability in the presence of recombinant IFN-β compared to uninfected samples. In both cases, rescue effects were observed at IFN concentrations of 10 ng ml−1, culminating at 100 ng ml−1 in DAOY cells (Fig. 2a). To further verify the antiviral effect of IFN-β and determine the kinetics of virus production, DAOY cells were treated with recombinant IFN-β (10 ng ml−1) and subsequently infected with TBEV at an m.o.i. of 5 at 12 h post-IFN-β treatment. Samples were harvested at 12, 24, 48 and 72 h p.i. An over 10000-fold decrease in viral titres was observed starting at 24 h p.i. in IFN-β-pre-treated cells (Fig. 2b). Moreover, a decrease of viral NS3 protein levels in IFN-β-pre-treated cells was also observed, as shown in Fig. 2(c).
Fig. 2.

IFN-β pre-treatment resulted in decreased virus production. (a) DAOY and A549 cells were treated with human recombinant IFN-β at concentrations of 10, 100 and 1000 ng ml−1, and after 12 h infected with TBEV (m.o.i. 5). MTT viability assays were performed at 5 d p.i., when CPE was observed in infected cells without IFN pre-treatment. The numbers represents the percentage of living cells normalized to the untreated control. The average with standard deviation from three independent experiments (in triplicate per experiment) is shown. *Represent a significant difference from the TBEV-infected control as calculated by Student’s t-test (*P<0.05; **P<0.01). (b) IFN-β- (10 ng ml−1) or mock (no IFN-β)-treated DAOY cells were infected (m.o.i. 5) at 12 h post-treatment and viral titres (at 12, 24, 48 and 72 h p.i.) were determined by plaque assay. Time 0 stands for the initial infection input (6×105 p.f.u.). The average with standard deviation from three independent experiments is shown. Significant difference from the control was calculated by Student’s t-test (****P<0.0001). (c) Cell lysates from (b) were used for the detection of viral NS3 levels by Western blot. GAPDH was used as a loading control. (d) DAOY and A549 cells were first co-transfected with p125Luc IFN-β promoter reporter expressing Firefly luciferase (500 ng well−1) and pRL-CMV internal control expressing Renilla luciferase (2 ng well−1). IFN-β promoter was stimulated following a secondary transfection of poly I:C (either 0, 1 or 10 µg well−1) at 24 h post-first transfection. Relative luciferase activity was analysed at 24 h post-second transfection. The mean with standard error is shown for three independent experiments performed in triplicate. Data were normalized to cells co-transfected with p125Luc and pRL-CMV without poly I:C treatment. Significant differences from the control were calculated by Student’s t-test (**P<0.01).
In order to analyse the activity of viral pattern-recognition/signalling pathways leading to IFN-β expression, DAOY cells were co-transfected with the p125Luc plasmid encoding the Firefly luciferase reporter gene under the control of IFN-β promoter [35] and the pRL-CMV plasmid encoding Renilla luciferase as internal control. Cells were stimulated with poly I:C (1 or 10 µg ml−1) at 24 h post-first transfection and luciferase actvity was determined 24 h post-second transfection. Again, A549 was used for positive controls [36]. No activation of IFN-β promoter was observed in DAOY cells upon poly I:C treatment (Fig. 2d).
IFN-β treatment and TBEV infection induce characteristic transcriptome changes in DAOY cells
To characterize the cellular response to TBEV infection and identify the differentially expressed genes responsible for the inhibition of TBEV replication after IFN-β treatment, an unbiased transcriptome analysis was performed. Infected (m.o.i. 5) and mock-infected DAOY cells at 24 h p.i. in the presence or absence of IFN-β pre-treatment (carried out 12 h prior to infection) were utilized for this analysis. Three biological replicates for each of the four combinations were prepared and successful infection was confirmed by plaque titration assays; again, a decrease in viral titre was observed in IFN-β-pre-treated cells (Fig. 3a). On average, ~48 million reads/sample were generated (Phred quality >30), and these were assembled against the Homo sapiens genome using TopHat2 [37]. In total, 94.3 % of the sequence reads were assembled to the reference genome. Differentially expressed genes (Benjamini Hochberg P-value≤0.05 and fold change >1.5 or <−1.5) in comparison to mock-treated cells were identified using Cuffdiff2 [38]. The analysis showed that TBEV infection resulted in the differential expression of 498 genes (Fig. 3b; see Table S3 for a comprehensive list of differentially expressed genes). Moreover, either 155 or 778 genes were found to be differentially expressed in mock- or TBEV-infected cells pre-treated with IFN-β, respectively, thus confirming the high sensitivity of DAOY cells to IFN-β treatment (Fig. 3 and Table S3). Interestingly, IFN-β pre-treatment resulted in the altered expression of a rather unique set of genes: only 12.3 and 29.2 % of the differentially expressed genes identified in mock- and TBEV-infected cells that had been pre-treated with IFN-β were also identified in TBEV-infected cells. The differential expression analysis was further validated by the relative quantification of eight selected genes using qRT-PCR (Fig. 3c). Significantly decreased numbers of reads mapped to the TBEV genome in IFN-β-pre-treated cells infected with TBEV (613 reads; sd ±61) compared to TBEV-infected cells without IFN-β pre-treatment (176 000 reads; sd ±15733) were observed (Student’s t-test; P<0.0001). In addition, the Kraken tool was used to verify any contamination present in the samples. As shown in Table S4, the DAOY cells were free of bacterial or viral contamination, including Mycoplasma spp. or human cytomegalovirus, which might have interfered with host responses.
Fig. 3.

Analysis of differentially expressed genes in DAOY cells upon TBEV infection and/or IFN-β treatment. (a) IFN-β- or mock-treated DAOY cells were infected (at 12 h post-treatment) with TBEV (m.o.i. 5) and viral titres were determined by plaque assay at 24 h p.i.; the mean of three biological replicates with standard deviation is shown. Significant difference from the control as calculated by Student’s t-test (****P<0.0001). (b) Venn diagrams of either down-regulated (left) or up-regulated (right) transcripts in comparison to untreated mock-infected cells. In both diagrams, differentially expressed genes with a greater than 1.5-fold change as a cut-off condition are shown. (c) qRT-PCR validation of transcript level changes detected in transcriptome analysis for selected genes; the ΔΔ-ct method using HPRT as housekeeping gene was employed for relative fold-change calculation (control: mock-infected cells). The mean of three biological replicates with standard deviation is shown. Significant difference from control, as calculated by Student’s t-test (*P<0.05).
Host response-associated genes, including type III IFNs, are activated upon TBEV infection of DAOY cells
It was recently shown that TBEV infection of mouse brain and human astrocytes resulted in inflammatory responses, which included elevated production of cytokines (IL-1α, IL-1β, IL-6, IL-8, IFN-α and IFN-γ) and chemokines (CCL2/MCP1, CCL3/MIP12α, CCL4/MIP1β, CCL5/RANTES and CXCL10/IP-10) [7, 39]. As shown in Fig. 4(a), DAOY cells activated a similar panel of cytokine-coding genes upon TBEV infection (CCL3/MIP1ɑ, CCL4/MIP1β, CCL5/RANTES, CXCL10/IP-10, IL-6 and TNF-ɑ). In addition, five new cytokine-coding genes were identified as being significantly up-regulated, (CXCL2/MIP2ɑ, CXCL11/IP-9, IL-12ɑ, IL-15 and IL-23ɑ), together with the IL-18 receptor accessory protein (IL18RAP). Hundreds of ISGs have been identified as being induced following viral infection (reviewed in [10–12]). Transcriptome analysis of TBEV-infected DAOY cells revealed significant induction of a number of ISGs (Table S3), including IFIT1, IFIT2, RSAD2, OASL, IFIT3, OAS2, ISG15 and ISG20 amongst the most up-regulated (fold-change >2.5; Fig. 4a and Table S5). RIG-I/DDX58 and MDA5/IFIH1 of the retinoic acid-inducible gene I-like receptor family, which are responsible for sensing viral RNA [40], were also significantly up-regulated. A TBEV-directed decrease in IL-2 and IL-4 mRNA levels was documented in the murine spleen [41]. A panel of 277 significantly down-regulated genes in TBEV-infected DAOY cells (fold-change >1.5; Fig. 3b and Table S3) was also observed. RNA28S5, RN7SL2, NOTCH3, COL1A1, BCL9L, BCORL1, POLR2A, FAM71D, IGF2, RN7SL3 and HSPG2 were found to be the most strongly down-regulated genes (fold-change >2.5; Fig. 4 and Table S5). Other than protein-coding genes, a number of non-coding RNAs were also identified as being differentially expressed upon TBEV infection, as shown in Fig. 4(b). However, of these, RN7SL2, RN7SL3 and RNA28S5 are the only RNA genes with known functions. The remaining differentially expressed RNAs were long non-coding RNAs (lncRNAs) with unknown functions. The observed pattern of general activation of host responses upon TBEV infection was also confirmed by Ingenuity Pathway Analysis (IPA) software. Table 1 shows the 10 most significantly affected canonical pathways that include IFN signalling. Furthermore, the unfolded protein response and endoplasmic reticulum stress pathways were also identified as being significantly affected. The TBEV-induced expression changes for genes that are functionally involved in these two pathways correlates with recent findings of TBEV-driven reorganization of the ER structure in infected cells [25, 42].
Fig. 4.

Overview of selected differentially expressed genes. DAOY cells were pre-treated with IFN-β (10 ng ml−1) and/or infected with TBEV (m.o.i. 5) after 12 h. Three independent biological replicates were included for each of the combinations [untreated mock cells (control); IFN-β-treated mock cells; untreated cells infected with TBEV; IFN-β-pre-treated cells infected with TBEV]. Total cellular RNA was isolated at 24 h p.i. and used for transcriptome analysis. (a) List of selected protein-coding genes identified to be differentially expressed in at least one of the combinations over control (Benjamini Hochberg P-value ≤0.05 and fold change >1.5 or <−1.5; down-regulated in red and up-regulated in green). To emphasize the up-regulation of IFN-λ1, information for transcripts of IFN-ɑ, IFN-β and IFN-γ was also included. (b) List of selected non-coding genes identified to be differentially expressed in at least one of the combinations over control (Benjamini Hochberg P-value ≤0.05 and fold change >1.5 or <−1.5; down-regulated in red and up-regulated in green).
Table 1. List of top 10 predicted canonical pathways affected by TBEV infection or IFN-β pre-treatment according to IPA software analysis.
| Untreated mock cells versus TBEV-infected cells | ||
| Canonical pathway | P-value | Pathway-associated genes found to be differentially expressed |
| Unfolded protein response | 1,10E-13 | DDIT3, DNAJB9, DNAJC3, EIF2AK3, ERO1LB, HSP90B1, HSPA1A, PPP1R15A, SEL1L, XBP1 |
| Role of hypercytokinemia/hyperchemokinemia in the pathogenesis of influenza | 5,09E-12 | CCL3, CCL4, CCL5, CXCL10, IFNL1, IL6, IL15, IL12A, TNF |
| Pathogenesis of multiple sclerosis | 5,93E-10 | CCL3, CCL4, CCL5, CXCL10, CXCL11 |
| Activation of IRF by cytosolic pattern recognition receptors | 8,51E-10 | CREBBP, DDX58, DHX58, IFIH1, IFIT2, IL6, ISG15, LTA, PPIB, TNF |
| VDR/RXR activation | 2,39E-09 | CCL5, CXCL10, EP300, IGFBP5, IGFBP6, IL12A, KLF4, LRP5, MXD1, NCOR2, RXRG |
| Differential regulation of cytokine production in macrophages and T helper cells by IL-17A and IL-17F | 3,15E-09 | CCL3, CCL4, CCL5, IL6, IL12A, TNF |
| Endoplasmic reticulum stress pathway | 5,06E-09 | DDIT3, DNAJC3, EIF2AK3, HSP90B1, HSPA5, XPB1 |
| Role of macrophages, fibroblasts and endothelial cells in rheumatoid arthritis | 5,98E-09 | CCL5, CREBBP, EP300, FGF2, IL6, IL15, IL18RAP, JAK2, LRP5, LTA, MMP13, NFATC1, PLCD4, PLCG2, SFRP2, TCF7L2, TNF, TRAF1, WNT7B, WNT9A |
| Interferon signalling | 7,53E-09 | IFI6, IFIT1, IFIT3, IRF1, ISG15, JAK2, MX1 |
| Granulocyte adhesion and diapedesis | 9,68E-09 | CCL3, CCL4, CCL5, CCL3L1, CCL4L2, CLDN1, CXCL2, CXCL10, CXCL11, IL18RAP, MMP13, MMP15, SDC4, TNF |
| Untreated mock cells vs IFN-β-treated cells | ||
| Canonical pathway | P-value | Pathway-associated genes found to be differentially expressed |
| Granulocyte adhesion and diapedesis | 1,43E-07 | CCL3, CCL4, CCL5, CCL3L1, CCL4L2, CLDN1, CXCL2, CXCL10, CXCL11, IL18RAP, MMP13, MMP15, SDC4, TNF |
| Agranulocyte Adhesion and Diapedesis | 2,00E-07 | CCL3, CCL20, CLDN1, CLDN14, CXCL1, CXCL2, CXCL3, CXCL5, CXCL8, IL1A, IL1B, IL1RN, IL36RN, MMP1 |
| Differential regulation of cytokine production in macrophages and T helper cells by IL-17A and IL-17F | 4,45E-07 | CCL3, CCL4, CCL5, IL6, IL12A, TNF |
| Role of cytokines in mediating communication between immune cells | 5,53E-07 | CSF2, CSF3, CXCL8, IL6, IL1A, IL1B, IL1RN, IL36RN |
| Communication between innate and adaptive immune cells | 6,11E-07 | CCL3, CSF2, CXCL8, HLA-B, IL6, IL1A, IL1B, IL1RN, IL36RN |
| Differential regulation of cytokine production in intestinal epithelial cells by IL-17A and IL-17F | 1,17E-06 | CCL3, CSF2, CSF3, CXCL1, IL1A, IL1B |
| Role of hypercytokinemia/hyperchemokinemia in the pathogenesis of influenza | 3,24E-06 | CCL3, IL8, IL6, IL1A, IL1B, IL1RN, IL36RN |
| Role of IL-17F in allergic inflammatory airway diseases | 4,30E-06 | CSF2, CXCL1, CXCL5, CXCL8, IL6, IL11, IL1B |
| Role of IL-17A in psoriasis | 7,89E-06 | CCL20, CXCL1, CXCL3, CXCL5, CXCL8 |
| Hematopoesis from pluripotent stem cells | 2,10E-05 | CSF2, CSF3, CXCL8, IL6, IL11, IL1A, LIF |
| Untreated mock cells versus IFN-β-treated cells infected with TBEV | ||
| Canonical pathway | P-value | Pathway-associated genes found to be differentially expressed |
| Hepatic fibrosis/hepatic stellate cell activation | 6,85E-11 | BCL2, COL13A1, COL1A1, COL1A2, COL3A1, COL4A2, COL5A1, COL5A3, COL6A1, CXCL3, CXCL8, ECE1, EGFR, IGF2, IGFBP3, IL6, IL1A, IL1B, MMP1, MMP9, MYH9, MYH14, PDGFA, PDGFRB, SERPINE1, TIMP2 |
| Role of osteoblasts, osteoclasts and chondrocytes in rheumatoid arthritis | 6,79E-08 | BCL2, BMP5, CBL, COL1A1, FZD7, IL6, IL11, IL1A, IL1B, IL1RN, IL36RN, ITGA5, ITGB3, LRP1, MAP2K3, MMP1, MMP14, NFATC1, PIK3CD, SFRP2, SPP1, TCF7L1, WNT7B, WNT9A |
| Agranulocyte adhesion and diapedesis | 8,17E-08 | CCL3, CCL20, CLDN1, CLDN14, CXCL1, CXCL2, CXCL3, CXCL5, CXCL8, IL1A, IL1B, IL1RN, IL36RN, MMP1 |
| Granulocyte adhesion and diapedesis | 1,16E-07 | CCL3, CCL4, CCL5, CCL3L1, CCL4L2, CLDN1, CXCL2, CXCL10, CXCL11, IL18RAP, MMP13, MMP15, SDC4, TNF |
| Atherosclerosis signalling | 2,32E-07 | APOC1, COL1A1, COL1A2, COL3A1, COL5A3, CXCL8, IL6, IL1A, IL1B, IL1RN, IL36RN, MMP1, MMP9, PDGFA, RARRES3, SELPLG, TNFSF14 |
| Communication between innate and adaptive immune cells | 2,95E-06 | CCL3, CSF2, CXCL8, HLA-B, IL6, IL1A, IL1B, IL1RN, IL36RN |
| Role of IL-17F in allergic inflammatory airway diseases | 5,92E-06 | CSF2, CXCL1, CXCL5, CXCL8, IL6, IL11, IL1B |
| Graft versus host disease signalling | 1,26E-05 | HLA-B, HLA-C, HLA-DRA, HLA-F, IL6, IL1A, IL1B, IL1RN, IL36RN |
| Inhibition of matrix metalloproteases | 1,92E-05 | HSPG2, LRP1, MMP1, MMP9, MMP14, MMP15, THBS2, TIMP2 |
| Role of IL-17A in psoriasis | 2,74E-05 | CCL20, CXCL1, CXCL3, CXCL5, CXCL8 |
Basal expression of IFN-λ1 and its receptor, IFNLR1/IL10Rβ, was documented in human brain tissue and a set of human neuronal cells including primary human neurons, NT2-N neurons and neuroblastoma cell lines [43]. Our transcriptome data, together with qRT-PCR analysis, demonstrated that IFN-λ1 was expressed in non-infected DAOY cells and highly up-regulated upon TBEV infection. Surprisingly, type I IFNs (IFN-α and IFN-β) as well as type II IFNs (IFN-γ) were not found to be up-regulated in response to TBEV infection in DAOY cells (Fig. 5a, b). In addition, the basal levels of IFN-ɑ and IFN-β, but not INF-γ, were found to be significantly lower than the basal levels of IFN-λ1. IPA software analysis confirmed that a wide spectrum of genes involved in the IFN-λ signalling pathway were differentially expressed upon TBEV infection, as shown in Fig. 5(c). In order to assess the potential antiviral effect of IFN-λ1 on TBEV infection, we performed CPE inhibition in pre-treated DAOY and A549 cells. The cells were pre-treated with human recombinant IFN-λ1 (10, 100 and 1000 ng ml−1) and then the analysis was carried out 12 h p.i. with TBEV at m.o.i. 5. Cells were incubated for 5 days until a virus-induced CPE was observed in non-treated control cells and then viability assays using MTT were performed. A significant rescue effect by type III IFNs in A549 cells was only seen when 100 ng IFN-λ1/ml was applied. No significant rescue of cell viability was observed in the case of DAOY cells, as shown in Fig. 6(a). The potential antiviral effects of IFN-λ1 pre-treatment were further validated via plaque assays and the detection of TBEV NS3 protein by Western blot. Again, no significant changes were observed in the viral titres and protein levels of NS3 in DAOY cells pre-treated with either 10 or 100 ng ml−1 of IFN-λ1, as shown in Fig. 6(b, c). In order to verify whether these concentrations of IFN-λ1 are sufficient to trigger the expression of ISGs, we analysed the mRNA expression of six genes that were found to be up-regulated most after TBEV infection. This suggested that the activation of their expression could be influenced by elevated levels of endogenous IFN-λ. No ISG expression was observed in DAOY cells treated with 10 ng ml−1 of IFN-λ1, but in cells treated with 100 ng ml−1 of IFN-λ a strong induction of RSAD2 (viperin) gene expression was detected (Fig. 6d). Moreover, high basal mRNA expression of receptor subunits for both type I and type III IFNs was documented in DAOY cells (Fig. 6e).
Fig. 5.

Expression of type III IFN (IFN-λ1) is induced in DAOY cells following TBEV infection. DAOY cells were treated with IFN-β (10 ng ml−1) and, where indicated, infected with TBEV after 12 h (m.o.i. 5), or only infected with TBEV, or mock-treated. Three independent biological replicates were included for each of the combinations (untreated mock cells; IFN-β-treated mock cells; untreated cells infected with TBEV; IFN-β-pre-treated cells infected with TBEV). Total cellular RNA was isolated at 24 h p.i. and further processed for transcriptome analysis. (a) Relative quantification of type I, II and III IFN mRNA levels in DAOY cells. The ΔΔ-ct method, using HPRT as a housekeeping gene, was employed for relative fold-change calculation; the mean of three biological replicates with standard deviation is shown. Significant differences to the control (mock-infected cells) were calculated by Student’s t-test (**P<0.01). (b) -Δ ct values of type I, II and III IFNs normalized to the HPRT gene; the mean of three biological replicates with standard deviation is shown. The dotted line represents the sensitivity of the qPCR. Significant differences were calculated by Student’s t-test (***P<0.001, ****P<0.0001). (c) Schematic overview of the IFN-λ (IFNL1) signalling network (as identified by IPA software). Identified up-regulated (red) transcripts in TBEV-infected DAOY cells are displayed.
Fig. 6.

IFN-λ1 pre-treatment results in non-altered virus production. (a) DAOY and A549 cells were first treated with human recombinant IFN-λ1 at concentrations of 10, 100 and 1000 ng ml−1, and then after 12 h infected with TBEV (m.o.i. 5). MTT viability assays were performed at 5 d p.i. when the CPE was observed in infected cells in the absence of IFN pre-treatment. Numbers represents the percentage of living cells normalized to the untreated control. Averages with standard deviation from three independent experiments performed in triplicates are shown. Significant differences to control were calculated by Student’s t-test (*P<0.05). (b, c) IFN-λ1 (10 and 100 ng ml−1, respectively) or mock-treated DAOY cells were infected (m.o.i. 5) at 12 h post-treatment and viral titres (at 12, 24, 48 and 72 h p.i.) were determined by plaque assay. The time 0 value stands for the initial infection input (6×105 p.f.u.). Averages with standard deviation from three independent experiments are shown. Cell lysates were further used for detection of the TBEV protein NS3 levels by Western blot. GAPDH was used as a loading control. A representative blot of three independent experiments is shown. (d) Total RNA isolated from DAOY cells 12 h post-IFN-λ1 treatment (10 ng ml−1; 100 ng ml−1) was used for relative quantification of the indicated ISG mRNAs. The ΔΔ-ct method, using HPRT as a housekeeping gene, was used for relative fold-change calculation; the mean of three independent experiments with standard deviation is shown. Significant difference from control was calculated by Student’s t-test (****P<0.0001). (e) -Δ ct values of type I and III IFN receptor subunits normalized to the HPRT gene; the mean of three biological replicates with standard deviation is shown. The dotted line indicates the sensitivity of the assay.
IFN-β pre-treatment results in up-regulation of ISGs and down-regulation of pro-inflammatory cytokines
Transcriptome analysis showed that IFN-β treatment of DAOY resulted in the altered expression of 155 genes, as shown in Fig. 3(b), thus confirming the high sensitivity of human neuronal cells to type I IFNs. Based on the inhibitory effects observed following IFN-β pre-treatment in TBEV infected-DAOY cells, we speculated that IFN-β activates the expression of the genes responsible for this antiviral effect. We searched for genes that were highly up-regulated upon IFN-β pre-treatment in both mock- and TBEV-infected DAOY cells; 41 genes were found to be up-regulated in both data sets (Fig. 3b). Analysis of the literature showed that five genes (encoding BST2, IFI27, IFITM3, ISG15 and RSAD2) were also expressed in IFN-β-treated human granule and cortical neurons [16]. Out of the 41 identified genes, IFI27, IFI6, RMRP, RN7SK, IFITM1, BST2, EPSTI1, IFITM3 and CRABP2 were up-regulated most (fold-change >2.5; Fig. 4a Tables S6 and S7). IFI6 and IFI27 were both characterized as being most highly expressed upon IFN-β treatment in mock- and TBEV-infected cells (Fig. 4a, Tables S6 and S7). Interestingly, a panel of genes down-regulated following IFN-β treatment consisted mainly of cytokine coding genes (Fig. 4a and Table S3). The same rate of down-regulation for CCL3/MIP1α, CSF3, CCL20/MIP3α, IL36RN, CXCL1/KC, CXCL2/MIP2α, CXCL3/MIP2β, CXCL5/ENA78, IL1α, IL1β, IL6, IL8 and IL11 was observed in both mock- and TBEV-infected DAOY cells pre-treated with IFN-β, but not in TBEV-infected cells (Fig. 4a). These data suggest a unique response of DAOY cells to type I IFN treatment in terms of the decreased mRNA levels of pro-inflammatory cytokines. The down-regulation of cytokine expression was by far the most significant expression pattern observed after IFN-β treatment. These findings were also supported by IPA analysis, which identified that for the most part cytokine-related canonical pathways were most significantly affected (Table 1). Our transcriptome analysis also revealed altered IFN-β-driven expression of a wide panel of non-coding RNAs (Fig. 3b). RNA5-8SP6, RMRP and RN7SK were the most up-regulated non-coding RNAs in this condition. Their expression may, however, be negatively influenced by TBEV, since a lower rate of up-regulation was seen for all three genes in IFN-β-treated cells subsequently infected with TBEV (Fig. 4b; Tables S6 and S7).
Discussion
Here we established a model for studying the interactions between TBEV and cells of neural origin. Human DAOY medulloblastoma cells are derived from cerebellum and their neuronal origin was analysed using TUBB3, MOG, VIM and GFAP biomarkers. TUBB3 and VIM expression is typical for neural precursor cells; TUBB3 expression was documented in neuronal precursor cells [44], whereas vimentin is typical for radial glia, a primary progenitor cells capable of both neuro- and gliogenesis [27]. Therefore, our findings support the neuronal origin of DAOY cells, as well as their dedifferentiated state, which is typical for cancer cells. In the case of U373 cells, TUBB3 and VIM expression also points to a dedifferentiated state (especially TUBB3 for an ascending histological grade of malignancy). However, the presence of GFAP supports their glial origin. Despite its expression in radial glia, the presence of VIM in both cell lines may also serve as a cancer marker, since the high expression of VIM and CD44 results in an epithelial– mesenchymal transition that is typical for metastasis [45]. Relatively low expression of MOG, a minor component of myelin (0.05 %) in both cell lines, could point to an ascending histological grade of malignancy, since this protein is located in oligodendrocytes (a fully differentiated type of glia) [46, 47]. Importantly, to the best of our knowledge, our data, as described below, also describe the innate immune properties of this cell line for the first time in the context of virus infection, and provide extensive transcriptome information. This is important information for others in the field who wish to make use of this particular cell line. It is often desirable in virology to use such cell lines before primary lines, which can be difficult to obtain, grow and infect. The data supplied here provide information on the nature of these cells, for example on how they compare to healthy neurons for experiments in virus–host interaction studies, drug screens, virus entry studies, etc.
Based on our transcriptome analysis, up-regulation of cytokines/chemokines as described here would mostly result in the activation/stimulation and chemotaxis of effector immune cells. This correlates with TBEV-associated immunopathogenesis in the brain [8, 9]. Transcriptome analysis of LGTV-infected HEK 293 T cells revealed enhanced expression of CCL5/RANTES, CXCL10/IP-10 and TNF-ɑ cytokines [48]. CCR5 (specifically the CCR5Delta32 allele) has been associated with the severity of TBEV-induced disease, suggesting that differential regulation of CCL5/RANTES, etc. may be clinically relevant [49]. We did not observe up-regulation of IFN-α, which is intriguing and may point to a defect in its regulation. In addition, we identified that RIG-I/DDX58 and MDA5/IFIH1 (RIG-I like receptors, RLRs) are up-regulated upon TBEV infection. The involvement of RLRs in sensing TBEV RNA was documented previously [50]. Viral dsRNA ‘hides’ in endoplasmic reticulum-derived vesicle packets and thus prevents the activation of host receptors and subsequent IFN-mediated antiviral response [51]. Moreover, a recent study showed enhanced mortality rates of IPS-1/MAVS (a downstream factor involved in RIG-I and MDA5 signalling cascade) knockout mice infected with TBEV or LGTV [22]. In addition, RLR signalling can induce type III IFN expression [52]. Therefore, up-regulation of RIG-I and MDA5 in the case of TBEV-infected DAOY cells may also contribute to the observed induction of IFN-λ1.
Our study identified a wide panel of ISGs that were up-regulated in response to TBEV infection. These ISGs were found to inhibit a broad spectra of viruses [11, 53–57]. Viperin, encoded by the RSAD2 gene, was shown to inhibit TBEV replication in infected HEK293T cells [58]. Although various ISGs were up-regulated, high TBEV titres were observed in DAOY cells. This suggests the presence of counteracting measures by TBEV against host immune responses, at least in the infected cells. TBEV antagonizes type I IFN signalling in infected cells and NS5 protein inhibits JAK-STAT signalling [24, 59]. Although type I and type III IFNs signal through different receptors, downstream signalling pathways converge and lead to the formation of the ISGF3 transcription complex and subsequent expression of ISGs [60]. Whether IFN-λ1 signalling is antagonized via TBEV protein(s) is not known. Strong up-regulation of IFIT1, IFIT2 and RSAD2 transcripts in comparison to other ISGs was detected. This could be a result of IFN-independent transcriptional induction of either IRF1 or IRF3, as IFN-independent ISG induction pathways were characterized for all three genes [61, 62].
We also identified a panel of genes that were down-regulated upon TBEV infection in DAOY cells. These genes are mostly involved in transcription and translation processes, as well as the regulation of cell proliferation. Down-regulation of effectors involved in either transcription (POLR2A) or translation (RNA28S5, RN7SL2, RN7SL3) suggests a possible TBEV-driven transcriptional or translational shut off in host cells. Both transcriptional and translational shut off are well-documented phenomena [63, 64]. Similar rates of RNA28S5, NOTCH3, COL1A1, BCL9L, BCOR1, POLR2A, FAM71D, IGF2 and HSPG2 down-regulation were also evident in IFN-β-pre-treated cells infected with TBEV, where significantly lower viral titres were determined. Intriguingly, down-regulation of these genes was not documented in only IFN-β-pre-treated cells. Therefore, the down-regulation observed for these genes could be considered to be a hallmark of TBEV infection in DAOY cells.
Altered expression of host lncRNAs was described in the case of influenza A virus and severe acute respiratory syndrome coronavirus (SARS-CoV) infection [65]. Furthermore, a wide panel of lncRNAs was found to be regulated by type I IFNs [66]. Therefore it may be that lncRNAs play an important role in IFN-stimulated host immune responses to viral infection. This hypothesis was recently supported by the identification of an IFN-λ3 up-regulated lncBST2/BISPR that positively regulates expression of BST2/tetherin [67], an ISG with antiviral effect in murine neurons against measles virus [68]. Further studies are required to elucidate the possible involvement of host lncRNAs in response to TBEV infection. The possible function of RNA5-8SP6 remains unclear, since it is classified as a 5.8S ribosomal RNA 6 pseudogene. RN7SK is involved in the regulation of transcription by RNA polymerase II. Its enhanced expression upon IFN-β treatment may increase the expression of other ISGs. RMRP was shown to interact with TERT, forming a complex with RNA-dependent RNA polymerase activity. This complex produces dsRNA that is processed into siRNAs in a Dicer-dependent manner [69], suggesting a possible role for RMRP in the decrease of TBEV levels by recruiting the RNA interference pathway. The phenomenon of type I IFN-dependent expression regulation of lncRNAs was recently described [66, 70]. We also report that TBEV infection results in the differential expression of genes coding for lncRNAs (Fig. 3b). No change in RN7SL3 expression was observed in IFN-β-pre-treated cells infected with TBEV, although down-regulation of RN7SL3 took place in TBEV-infected cells. This observation indicates a negative effect of TBEV on RN7SL3 expression, however this effect seems to be dose-dependent, since lowered titres of virus (as IFN pre-treatment resulted in lower TBEV production) did not affect the RN7SL3 expression at all. The biological relevance of these data, however, needs to be investigated.
With regard to IFNs, based on our data, DAOY cells seem to exclusively activate the IFN-λ1 (type III IFN) pathway in response to TBEV infection; our data suggest a defect in the recognition of viral RNA, given the inability the of the RIG-I/MDA-5 ligand poly I:C to induce a type I IFN reporter gene while type I IFN signalling itself is functional. This is likely to account at least in part for these observations. The unique pattern of IFN signalling in DAOY cells might be virus- and cell-type-specific, since previous work characterized IFN-λ as an inducer of IFN-ɑ expression in HSV1-infected human neurons [17]. Furthermore, high up-regulation of IFN-β in LGTV-infected HEK 293T cells was also described [48]. However, DAOY cells express the type I IFN receptor and are responsive to IFN-β treatment. Furthermore, only IFN-β treatment resulted in significant inhibition of TBEV production. These results suggest that DAOY cells express IFN-λ1 in response to TBEV infection, however this endogenous response does not restrict TBEV production. However, it may be that TBEV does not inhibit the type III IFN pathway, because it does not affect virus replication, despite sharing elements of the signalling cascade [19, 20]. A comparable phenomenon was indeed observed for epithelial cell infection with human rotavirus [71].
We identified two genes that were highly up-regulated in IFN-β-treated DAOY cells, in the absence or presence of TBEV, IFI6 and IFI27. They belong to the FAM14 family of ISGs [72] and were documented as mitochondrial proteins involved in apoptosis regulation [73–75]. Over-expression of IFI6 inhibited DENV-induced apoptosis of endothelial cells [74, 75] and restricted HCV replication in hepatocarcinoma cells [76]. In addition, IFI6 was shown to block HCV entry into hepatocarcinoma cells [77]. IFI27 overexpression in human neurons resulted in decreased production of WNV, SLEV and MHV [16], while over-expression of murine IFI27 delayed Sindbis virus-induced encephalitis and death in neonatal mice [78]. This suggests that IFI6 and IFI27 are promising candidate proteinsthat may be responsible for the inhibition of TBEV infection in DAOY cells. Both IFITM1 and IFITM3 were documented to inhibit HCV entry into hepatocarcinoma cells [79, 80]. However, the antiviral effects of IFITMs seem to be RNA virus-specific [81]. An antiviral role in the case of HCV infection was also reported for EPSTI1, whose expression was induced upon IFN-λ2 treatment and resulted in a decreased rate of viral replication, assembly and release [82]. Moreover, BST2/tetherin inhibits HCV and DENV in hepatocarcinoma cells [83, 84], as well as measles virus in murine neurons [68].
In summary, our results provide novel insights into the response of neuronal cells to TBEV infection and the antiviral effects of type I and III IFN. Importantly, we found a partial overlap of host-induced genes for TBEV and type I IFN. Whether genes induced by both pathways are particularly important in restricting infection, or whether virus-specific responses may have unique roles in pathogenesis, remains to be investigated. Our findings should influence and encourage further studies into the pathogenic effects of infection, as well as inhibitors of TBEV that can be further investigated and targeted.
Methods
Cells, viruses and IFN pre-treatment
Human medulloblastoma (ATCC; DAOY HTB-186) and human lung adenocarcinoma (A549; available at the Institute of Parasitology, Biology Centre of the Academy of Sciences of the Czech Republic, Branišovská) lines were grown in low-glucose DMEM medium supplemented with 10 % foetal bovine serum (FBS), 1 % antibiotic/antimycotic (amphotericin B 0.25 µg ml−1, penicillin G 100 units/ml and streptomycin 100 µg ml−1) and 1 % l-glutamine. The human glioblastoma line (U373 MG Uppsala; kindly provided by T. Eckschlager, Charles University in Prague) was grown in IMDM medium supplemented with 10 % FBS, 1 % antibiotic/-antimycotic (amphotericin B 0.25 µg ml−1, penicillin G 100 units/ml, and streptomycin 100 µg ml−1) and 1 % l-glutamine. The DAOY medulloblastoma cell line was derived from desmoplastic cerebellar medulloblastoma [26], and the U373 MG Uppsala glioblastoma cell line was derived from malignant glioma/astrocytoma [29]. Porcine kidney stable (PS; cell line as in [85]; available at the Institute of Parasitology, Biology Centre of the Academy of Sciences of the Czech Republic, Branišovská) cells were grown in L15 medium with 3 % newborn calf serum (NCS), 1 % antibiotic/antimycotic and 1 % l-glutamine. All cell lines were grown at 37 °C and 5 % CO2 (PS cells at 37 °C without additional CO2).
The low-passage TBEV strain Neudoerfl (fourth passage in suckling mice brains; GenBank accession no. U27495) was provided by Professor F. X. Heinz (Medical University of Vienna, Austria) [86]. TBEV in growth medium was added to the cells 1 day post-seeding. Cells were incubated with the virus for 2 h, washed with PBS and then fresh pre-warmed medium was added. Human INF-β 1a (RayBiotech) or IFN-λ1 (Sigma Aldrich) were added to the A549 or DAOY cells 12 h prior to infection.
Virus titration
Viral titres were determined by plaque assay [87], with minor modifications. Briefly, PS cell monolayers (9×104 cells per well) were grown in 24-well plates and incubated with 10× serial dilutions of viral samples for 4 h at 37 °C. The samples were then covered by a 1 : 1 (v/v) overlay mixture of carboxy-methyl cellulose and 2× L15 medium including 6 % PTS, 2 % antibiotics and 2 % glutamine. After 5 days, the medium with overlay was removed, and the cells washed and subsequently fixed and stained with 0.1 % naphthalene black in 6 % acetic acid solution for 45 min. Virus-induced plaques were counted and the titres are stated as p.f.u./ml; it should be noted that infection rates can be different for other cell types.
IFN and antiviral activity assays
DAOY and A549 cells (1×104 cells well−1 and 2×104 cells well−1, respectively) were seeded in 96-well plates 12 h prior pre-treatment with recombinant IFN-β and IFN-λ using concentrations of 10, 100 and 1000 ng ml−1. Cells were infected at 12 h post-treatment with TBEV strain Neudoerfl at an m.o.i. of 5 and incubated at 37 °C and 5 % CO2 for 5 days until virus-induced the CPE was observed in control wells. Subsequently, an MTT assay with minor modifications was performed for the determination of cell viability [88]. Briefly, after removal of the medium, the cells were washed with PBS and 100 µl of fresh medium containing MTT (3-[4.5-dimethylthiazol-2-yl]−2,5-diphenyl tetrazolium bromide; Sigma Aldrich; 0,5 mg ml−1) was added to each well. After incubation at 37 °C for 2 h, the medium with MTT was removed and 100 µl of DMSO was added to each well. After shaking for 15 min at room temperature, the absorbance at 570 nm was determined using the microplate reader Synergy H1 (BioTek).
IFN-β promoter activity assay
The in vitro activity of the IFN-β promoter was analysed in DAOY and A549 cells using p125Luc reporter vector expressing Firefly luciferase under the control of IFN-β promoter [35] and pRL-CMV vector expressing Renilla luciferase as an internal control. Transfections were carried out using the PolyJet transfection reagent (SignaGen) according to the manufacturer’s protocol. Briefly, cells (1.2×105 well−1 and 1.6×105 well−1 for DAOY and A549 cells, respectively) were seeded in 24-well plates 1 day prior to transfection. The first co-transfection of p125Luc (500 ng) and pRL-CMV (2 ng) was followed by a second transfection of poly I:C (1 or 10 µg well−1) after 24 h. Cells were lysed after a further 24 h in passive lysis buffer (Promega). The Firefly and Renilla luciferase activities were determined using a Dual Luciferase assay kit (Promega) in an H1 Synergy luminometer (BioTek).
RNA isolation
For transcriptome analysis, RNA from DAOY cells was extracted by using Trizol (Life Technologies). Briefly, cells were washed with phosphate buffer saline (PBS) and lysed in 1 ml Trizol. Chloroform (0.2 ml) was added, and the samples were mixed intensively and incubated for 5 min at room temperature. The upper aqueous phase was transferred to a new tube after centrifugation (12 000 g, 15 min, 4 °C) and mixed with 0.5 ml of isopropanol. After incubation at 4 °C for 10 min, the precipitated RNA was pelleted by centrifugation (12 000 g/15 min/4 °C) and washed with 75 % ethanol. The RNA pellet was dissolved in 20 µl of RNase-free water. The RNA was stored at −80 °C until further use.
For qRT-PCR analysis, total cellular RNA was isolated using the NucleoSpin RNA Plus kit (Macherey-Nagel).
Transcriptome analysis
RNA integrity was checked before sequencing using a 2200 TapeStation (Agilent). Five-hundred ng of total RNA from each sample was enriched for poly(A) RNA, and then fragmented and prepared for sequencing using a TruSeq stranded mRNA preparation kit (Illumina). Index-tagged libraries were pooled and single-end datasets with a read length of 76 nucleotides were generated on a NextSeq500 sequencer (Illumina). On average, 48 million reads were acquired for each sample.
FastQC software (http://www.bioinformatics.babraham.ac.uk/projects/fastqc) was used to check the RNA-Seq read quality. In order to check for possible contamination in the analysed samples we employed Kraken [89]. It is a system for assigning taxonomic labels to short DNA sequences, usually obtained through metagenomic studies. We mapped k-mers to a pre-built 4 GB database constructed from complete bacterial, archaeal and viral genomes in RefSeq. On average, only ~0.5 % reads aligned to the MiniKraken database constructed from bacterial, archaeal and viral genomes in RefSeq. TopHat2 [37], a fast splice junction mapper for RNA-Seq reads, aligns RNA-Seq reads to mammalian-sized genomes using the ultra high-throughput short-read aligner Bowtie2, and then analyses the alignment results to identify splice junctions between exons. In the present research, we aligned the short reads to the Homo sapiens genome (GRCh37) downloaded via the Ensembl genome browser.
Cuffdiff was used to identify differentially expressed genes [38]. Cuffdiff is a program in the Cufflinks package (version 2.2.1). It adopts an algorithm that controls cross-replicate variability and read-alignment ambiguity by using a model for fragment counts based on a beta negative binomial distribution. It can identify differentially expressed (DE) transcripts and genes, differential splicing and promoter preference changes, and returns far more statistically significant differentially expressed genes than microarray analysis.
After identifying the DE genes, the software IPA was applied for the function annotation and pathway analysis. Sequencing data were deposited in EBI (study accession number: PRJEB14767).
Real-time qPCR
For qPCR validation of gene expression from samples used for transcriptome analysis, 1 µg of total RNA was first treated with DNase using the TURBO DNA-free kit (Life Technologies) and then reverse-transcribed by SuperScript III reverse transcriptase (Life Technologies) with 500 ng of oligo d(T)15 primer according to the manufacturer's protocol. For qPCR reactions, 2 µl of 5× diluted cDNA reaction was used for the detection and amplification of selected genes; Fast SYBR Green Master Mix (Life Technologies) was used according to manufacturer's protocol.
For qPCR analysis of the IFN-λ1 treatment effect on DAOY cells, total RNA was first treated with dsDNase (Life Technologies) and 80 ng per reaction was used for RT-qPCR using the FAST Universal One-Step qRT-PCR kit (Kapa Biosystems) according to the manufacturer's protocol.
All data were analysed using the relative quantification ΔΔ Ct method and HPRT as the reference gene. A full list of primers is outlined in Table S8.
Western blotting
Cells were washed with PBS and subsequently lysed on ice for 15 min in 1× cell lysis buffer (Cell Signalling Technology) including protease and phosphatase inhibitors (Life Technologies). Lysate was sonicated and centrifuged at 4 °C for 15 min at 14 000 g to eliminate cellular debris, and then analysed by BCA assay for protein concentration quantification. Using SDS-PAGE, 8–12 µg of protein extract per well was separated. The proteins were then subsequently transferred to a PVDF membrane (GE Healthcare). For TBEV NS3 detection, chicken polyclonal primary antibodies in 1 : 5000 dilution ratio were used (kindly provided by Dr M. Bloom, National Institute of Allergy and Infectious Diseases, USA). Goat polyclonal antibodies (Abcam) for the detection of GAPDH were used at 1 : 500 dilution. For NS3/GAPDH detection, anti-goat and anti-chicken alkaline phosphatase-conjugated secondary antibodies (both 1 : 1000 dilution ratio; Vector Laboratories) were used and immuno-labelled proteins were visualized by chemiluminescence assay using Novex AP chemiluminescent substrate (CDP-Star) reagent (Life Technologies).
Immunofluorescence analysis
DAOY and U373 cells were seeded on a chamber slide (0.3 cm2 well−1; 1×104 cells well−1). For the detection of CNS markers, cells were fixed after 24 h post-seeding, and in the case of TBEV NS3 detection, cells were infected with TBEV at an m.o.i. of 0.1, 1 or 5, and fixed at 24 h p.i. Fixation was carried out by using 4 % paraformaldehyde for 15 min; cells were subsequently rinsed in PBS and permeabilized with 0.1 % Triton X-100 for 15 min. Cells were also treated with 50 mM NH4Cl in 1 % BSA in PBS to block formaldehyde auto-fluorescence. Following this, cells were blocked in 3 % BSA in PBS and incubated with chicken polyclonal anti-NS3, goat polyclonal anti-MOG (Abcam), rabbit polyclonal anti-TUBB3 (Abcam), rabbit polyclonal anti-VIM (Abcam), or rabbit polyclonal anti-GFAP (Dako) antibodies at 1 : 5000, 1 : 200, 1 : 200, 1 : 1000 and 1 : 500 dilutions, respectively. After washing with PBS, primary antibodies were labelled using DyLight488/594-conjugated secondary antibodies (Vector Laboratories) at a 1 : 1000 dilution. For MOG immunodetection, the Tyramide amplification signal kit (Life Technologies) was used according to the manufacturer’s instructions. Subsequently, the cells were mounted in Vectashield (Vector Laboratories). Analysis of NS3-labelling was carried out on an Olympus BX-51 fluorescence microscope equipped with an Olympus DP-70 CCD camera. For CNS marker expression imaging, an Olympus Fluoview FV10i confocal microscope was used. In order to analyse the numbers of cells expressing NS3 or one of the CNS markers, four–nine images (100× magnification) were taken for NS3 or markers in two independent experiments. Subsequently, the total number of NS3 or CNS marker expressing cells (as indicated in figures) were counted and transformed to percentages relating to the total number of cells. Average values and standard deviations were calculated from two independent experiments.
Funding information
A. K. and E. S. were funded by the UK MRC (MC_UU_12014). M. S. and L. G. were funded by the Czech Science Foundation (GACR) (15-03044S) and Czech Research Infrastructure for Systems Biology (C4SYS) (LM2015055).
Acknowledgements
We thank M. Bloom (NIAID, USA) for NS3 antibodies; F. X. Heinz (Medical University of Vienna, Austria) for providing TBEV; and T. Eckschlager (Charles University Prague, Czech Republic) for U373 MG Uppsala cells.
Conflicts of interest
The authors declare that there are no conflicts of interest.
Supplementary Data
Footnotes
Abbreviations: BBB, blood-brain-barrier; CNS, central nervous system; CPE, cytopathic effect; ISG, interferon-stimulated gene; TBEV, tick-borne encephalitis virus.
Eight supplementary tables and two supplementary figures are available with the online Supplementary Material.
References
- 1.Kunz C, Heinz FX. Tick-borne encephalitis. Vaccine. 2003;21:S1–S2. doi: 10.1016/S0264-410X(02)00810-1. [DOI] [PubMed] [Google Scholar]
- 2.Süss J. Tick-borne encephalitis 2010: epidemiology, risk areas, and virus strains in Europe and Asia-an overview. Ticks Tick Borne Dis. 2011;2:2–15. doi: 10.1016/j.ttbdis.2010.10.007. [DOI] [PubMed] [Google Scholar]
- 3.Kaiser R. The clinical and epidemiological profile of tick-borne encephalitis in southern Germany 1994–98: a prospective study of 656 patients. Brain. 1999;122:2067–2078. doi: 10.1093/brain/122.11.2067. [DOI] [PubMed] [Google Scholar]
- 4.Dörrbecker B, Dobler G, Spiegel M, Hufert FT. Tick-borne encephalitis virus and the immune response of the mammalian host. Travel Med Infect Dis. 2010;8:213–222. doi: 10.1016/j.tmaid.2010.05.010. [DOI] [PubMed] [Google Scholar]
- 5.Miner JJ, Diamond MS. Mechanisms of restriction of viral neuroinvasion at the blood-brain barrier. Curr Opin Immunol. 2016;38:18–23. doi: 10.1016/j.coi.2015.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gelpi E, Preusser M, Garzuly F, Holzmann H, Heinz FX, et al. Visualization of Central European tick-borne encephalitis infection in fatal human cases. J Neuropathol Exp Neurol. 2005;64:506–512. doi: 10.1093/jnen/64.6.506. [DOI] [PubMed] [Google Scholar]
- 7.Palus M, Bílý T, Elsterová J, Langhansová H, Salát J, et al. Infection and injury of human astrocytes by tick-borne encephalitis virus. J Gen Virol. 2014;95:2411–2426. doi: 10.1099/vir.0.068411-0. [DOI] [PubMed] [Google Scholar]
- 8.Gelpi E, Preusser M, Laggner U, Garzuly F, Holzmann H, et al. Inflammatory response in human tick-borne encephalitis: analysis of postmortem brain tissue. J Neurovirol. 2006;12:322–327. doi: 10.1080/13550280600848746. [DOI] [PubMed] [Google Scholar]
- 9.Růzek D, Salát J, Palus M, Gritsun TS, Gould EA, et al. CD8+ T-cells mediate immunopathology in tick-borne encephalitis. Virology. 2009;384:1–6. doi: 10.1016/j.virol.2008.11.023. [DOI] [PubMed] [Google Scholar]
- 10.Melchjorsen J. Learning from the messengers: innate sensing of viruses and cytokine regulation of immunity – clues for treatments and vaccines. Viruses. 2013;5:470–527. doi: 10.3390/v5020470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ivashkiv LB, Donlin LT. Regulation of type I interferon responses. Nat Rev Immunol. 2014;14:36–49. doi: 10.1038/nri3581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schneider WM, Chevillotte MD, Rice CM. Interferon-stimulated genes: a complex web of host defenses. Annu Rev Immunol. 2014;32:513–545. doi: 10.1146/annurev-immunol-032713-120231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kallfass C, Ackerman A, Lienenklaus S, Weiss S, Heimrich B, et al. Visualizing production of beta interferon by astrocytes and microglia in brain of La Crosse virus-infected mice. J Virol. 2012;86:11223–11230. doi: 10.1128/JVI.01093-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Delhaye S, Paul S, Blakqori G, Minet M, Weber F, et al. Neurons produce type I interferon during viral encephalitis. Proc Natl Acad Sci USA. 2006;103:7835–7840. doi: 10.1073/pnas.0602460103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Samuel MA, Diamond MS. Alpha/beta interferon protects against lethal West Nile virus infection by restricting cellular tropism and enhancing neuronal survival. J Virol. 2005;79:13350–13361. doi: 10.1128/JVI.79.21.13350-13361.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cho H, Proll SC, Szretter KJ, Katze MG, Gale M, et al. Differential innate immune response programs in neuronal subtypes determine susceptibility to infection in the brain by positive-stranded RNA viruses. Nat Med. 2013;19:458–464. doi: 10.1038/nm.3108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li J, Hu S, Zhou L, Ye L, Wang X, et al. Interferon lambda inhibits herpes simplex virus type I infection of human astrocytes and neurons. Glia. 2011;59:58–67. doi: 10.1002/glia.21076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lazear HM, Daniels BP, Pinto AK, Huang AC, Vick SC, et al. Interferon-λ restricts West Nile virus neuroinvasion by tightening the blood-brain barrier. Sci Transl Med. 2015;7:284ra59. doi: 10.1126/scitranslmed.aaa4304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Marcello T, Grakoui A, Barba-Spaeth G, Machlin ES, Kotenko SV, et al. Interferons α and λ inhibit hepatitis C virus replication with distinct signal transduction and gene regulation kinetics. Gastroenterology. 2006;131:1887–1898. doi: 10.1053/j.gastro.2006.09.052. [DOI] [PubMed] [Google Scholar]
- 20.Bolen CR, Ding S, Robek MD, Kleinstein SH. Dynamic expression profiling of type I and type III interferon-stimulated hepatocytes reveals a stable hierarchy of gene expression. Hepatology. 2014;59:1262–1272. doi: 10.1002/hep.26657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Weber E, Finsterbusch K, Lindquist R, Nair S, Lienenklaus S, et al. Type I interferon protects mice from fatal neurotropic infection with Langat virus by systemic and local antiviral responses. J Virol. 2014;88:12202–12212. doi: 10.1128/JVI.01215-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kurhade C, Zegenhagen L, Weber E, Nair S, Michaelsen-Preusse K, et al. Type I interferon response in olfactory bulb, the site of tick-borne flavivirus accumulation, is primarily regulated by IPS-1. J Neuroinflammation. 2016;13:22. doi: 10.1186/s12974-016-0487-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lindqvist R, Mundt F, Gilthorpe JD, Wölfel S, Gekara NO, et al. Fast type I interferon response protects astrocytes from flavivirus infection and virus-induced cytopathic effects. J Neuroinflammation. 2016;13:277. doi: 10.1186/s12974-016-0748-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Best SM, Morris KL, Shannon JG, Robertson SJ, Mitzel DN, et al. Inhibition of interferon-stimulated JAK-STAT signaling by a tick-borne flavivirus and identification of NS5 as an interferon antagonist. J Virol. 2005;79:12828–12839. doi: 10.1128/JVI.79.20.12828-12839.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Růzek D, Vancová M, Tesarová M, Ahantarig A, Kopecký J, et al. Morphological changes in human neural cells following tick-borne encephalitis virus infection. J Gen Virol. 2009;90:1649–1658. doi: 10.1099/vir.0.010058-0. [DOI] [PubMed] [Google Scholar]
- 26.Jacobsen PF, Jenkyn DJ, Papadimitriou JM. Establishment of a human medulloblastoma cell line and its heterotransplantation into nude mice. J Neuropathol Exp Neurol. 1985;44:472–485. doi: 10.1097/00005072-198509000-00003. [DOI] [PubMed] [Google Scholar]
- 27.Howard BM, Zhicheng Mo Z, Filipovic R, Moore AR, Antic SD, et al. Radial glia cells in the developing human brain. Neuroscientist. 2008;14:459–473. doi: 10.1177/1073858407313512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Eng LF, Ghirnikar RS, Lee YL. Glial fibrillary acidic protein: GFAP-thirty-one years (1969–2000) Neurochem Res. 2000;25:1439–1451. doi: 10.1023/A:1007677003387. [DOI] [PubMed] [Google Scholar]
- 29.Pontén J, Macintyre EH. Long term culture of normal and neoplastic human glia. Acta Pathol Microbiol Scand. 1968;74:465–486. doi: 10.1111/j.1699-0463.1968.tb03502.x. [DOI] [PubMed] [Google Scholar]
- 30.Best SM, Morris KL, Shannon JG, Robertson SJ, Mitzel DN, et al. Inhibition of interferon-stimulated JAK-STAT signaling by a tick-borne flavivirus and identification of NS5 as an interferon antagonist. J Virol. 2005;79:12828–12839. doi: 10.1128/JVI.79.20.12828-12839.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bosworth A, Dowall SD, Garcia-Dorival I, Rickett NY, Bruce CB, et al. A comparison of host gene expression signatures associated with infection in vitro by the Makona and Ecran (Mayinga) variants of Ebola virus. Sci Rep. 2017;7:43144. doi: 10.1038/srep43144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Voigt EA, Swick A, Yin J. Rapid induction and persistence of paracrine-induced cellular antiviral states arrest viral infection spread in A549 cells. Virology. 2016;496:59–66. doi: 10.1016/j.virol.2016.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang W, Wang WH, Azadzoi KM, Su N, Dai P, et al. Activation of innate antiviral immune response via double-stranded RNA-dependent RLR receptor-mediated necroptosis. Sci Rep. 2016;6:22550. doi: 10.1038/srep22550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chiang C, Beljanski V, Yin K, Olagnier D, Ben Yebdri F, et al. Sequence-specific modifications enhance the broad-spectrum antiviral response activated by RIG-I agonists. J Virol. 2015;89:8011–8025. doi: 10.1128/JVI.00845-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yoneyama M, Suhara W, Fukuhara Y, Sato M, Ozato K, et al. Autocrine amplification of type I interferon gene expression mediated by interferon stimulated gene factor 3 (ISGF3) J Biochem. 1996;120:160–169. doi: 10.1093/oxfordjournals.jbchem.a021379. [DOI] [PubMed] [Google Scholar]
- 36.Donald CL, Brennan B, Cumberworth SL, Rezelj VV, Clark JJ, et al. Full Genome sequence and sfRNA interferon antagonist activity of Zika virus from Recife, Brazil. PLoS Negl Trop Dis. 2016;10:e0005048. doi: 10.1371/journal.pntd.0005048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, et al. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013;14:R36. doi: 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Trapnell C, Hendrickson DG, Sauvageau M, Goff L, Rinn JL, et al. Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat Biotechnol. 2013;31:46–53. doi: 10.1038/nbt.2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Palus M, Vojtíšková J, Salát J, Kopecký J, Grubhoffer L, et al. Mice with different susceptibility to tick-borne encephalitis virus infection show selective neutralizing antibody response and inflammatory reaction in the central nervous system. J Neuroinflammation. 2013;10:77. doi: 10.1186/1742-2094-10-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yoneyama M, Onomoto K, Jogi M, Akaboshi T, Fujita T, et al. Viral RNA detection by RIG-I-like receptors. Curr Opin Immunol. 2015;32:48–53. doi: 10.1016/j.coi.2014.12.012. [DOI] [PubMed] [Google Scholar]
- 41.Tun MM, Aoki K, Senba M, Buerano CC, Shirai K, et al. Protective role of TNF-α, IL-10 and IL-2 in mice infected with the Oshima strain of Tick-borne encephalitis virus. Sci Rep. 2014;4:5344. doi: 10.1038/srep05344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Miorin L, Romero-Brey I, Maiuri P, Hoppe S, Krijnse-Locker J, et al. Three-dimensional architecture of tick-borne encephalitis virus replication sites and trafficking of the replicated RNA. J Virol. 2013;87:6469–6481. doi: 10.1128/JVI.03456-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhou L, Wang X, Wang YJ, Zhou Y, Hu S, et al. Activation of toll-like receptor-3 induces interferon-λ expression in human neuronal cells. Neuroscience. 2009;159:629–637. doi: 10.1016/j.neuroscience.2008.12.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Katsetos CD, Herman MM, Mörk SJ. Class III β-tubulin in human development and cancer. Cell Motil Cytoskeleton. 2003;55:77–96. doi: 10.1002/cm.10116. [DOI] [PubMed] [Google Scholar]
- 45.Päll T, Pink A, Kasak L, Turkina M, Anderson W, et al. Soluble CD44 interacts with intermediate filament protein vimentin on endothelial cell surface. PLoS One. 2011;6:e29305. doi: 10.1371/journal.pone.0029305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Johns TG, Bernard CC. The structure and function of myelin oligodendrocyte glycoprotein. J Neurochem. 1999;72:1–9. doi: 10.1046/j.1471-4159.1999.0720001.x. [DOI] [PubMed] [Google Scholar]
- 47.Solly SK, Thomas JL, Monge M, Demerens C, Lubetzki C, et al. Myelin/oligodendrocyte glycoprotein (MOG) expression is associated with myelin deposition. Glia. 1996;18:39–48. doi: 10.1002/(SICI)1098-1136(199609)18:1<39::AID-GLIA4>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 48.Mlera L, Lam J, Offerdahl DK, Martens C, Sturdevant D, et al. Transcriptome analysis reveals a signature profile for tick-borne flavivirus persistence in HEK 293T cells. MBio. 2016;7:e00314-16. doi: 10.1128/mBio.00314-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kindberg E, Mickiene A, Ax C, Akerlind B, Vene S, et al. A deletion in the chemokine receptor 5 (CCR5) gene is associated with tickborne encephalitis. J Infect Dis. 2008;197:266–269. doi: 10.1086/524709. [DOI] [PubMed] [Google Scholar]
- 50.Overby AK, Popov VL, Niedrig M, Weber F. Tick-borne encephalitis virus delays interferon induction and hides its double-stranded RNA in intracellular membrane vesicles. J Virol. 2010;84:8470–8483. doi: 10.1128/JVI.00176-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Overby AK, Weber F. Hiding from intracellular pattern recognition receptors, a passive strategy of flavivirus immune evasion. Virulence. 2011;2:238–240. doi: 10.4161/viru.2.3.16162. [DOI] [PubMed] [Google Scholar]
- 52.Odendall C, Dixit E, Stavru F, Bierne H, Franz KM, et al. Diverse intracellular pathogens activate type III interferon expression from peroxisomes. Nat Immunol. 2014;15:717–726. doi: 10.1038/ni.2915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hornung V, Hartmann R, Ablasser A, Hopfner KP. OAS proteins and cGAS: unifying concepts in sensing and responding to cytosolic nucleic acids. Nat Rev Immunol. 2014;14:521–528. doi: 10.1038/nri3719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lenschow DJ. Antiviral properties of ISG15. Viruses. 2010;2:2154–2168. doi: 10.3390/v2102154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang W, Xu L, Su J, Peppelenbosch MP, Pan Q, et al. Transcriptional regulation of antiviral interferon-stimulated genes. Trends Microbiol. 2017 doi: 10.1016/j.tim.2017.01.001. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fitzgerald KA. The interferon inducible gene: viperin. J Interferon Cytokine Res. 2011;31:131–135. doi: 10.1089/jir.2010.0127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Liu SY, Sanchez DJ, Cheng G. New developments in the induction and antiviral effectors of type I interferon. Curr Opin Immunol. 2011;23:57–64. doi: 10.1016/j.coi.2010.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Upadhyay AS, Vonderstein K, Pichlmair A, Stehling O, Bennett KL, et al. Viperin is an iron-sulfur protein that inhibits genome synthesis of tick-borne encephalitis virus via radical SAM domain activity. Cell Microbiol. 2014;16:834–848. doi: 10.1111/cmi.12241. [DOI] [PubMed] [Google Scholar]
- 59.Werme K, Wigerius M, Johansson M. Tick-borne encephalitis virus NS5 associates with membrane protein scribble and impairs interferon-stimulated JAK-STAT signalling. Cell Microbiol. 2008;10:696–712. doi: 10.1111/j.1462-5822.2007.01076.x. [DOI] [PubMed] [Google Scholar]
- 60.Kotenko SV, Gallagher G, Baurin VV, Lewis-Antes A, Shen M, et al. IFN-λ s mediate antiviral protection through a distinct class II cytokine receptor complex. Nat Immunol. 2003;4:69–77. doi: 10.1038/ni875. [DOI] [PubMed] [Google Scholar]
- 61.Defilippis VR, Robinson B, Keck TM, Hansen SG, Nelson JA, et al. Interferon regulatory factor 3 is necessary for induction of antiviral genes during human cytomegalovirus infection. J Virol. 2006;80:1032–1037. doi: 10.1128/JVI.80.2.1032-1037.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dixit E, Boulant S, Zhang Y, Lee AS, Odendall C, et al. Peroxisomes are signaling platforms for antiviral innate immunity. Cell. 2010;141:668–681. doi: 10.1016/j.cell.2010.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rivas HG, Schmaling SK, Gaglia MM. Shutoff of host gene expression in influenza A virus and herpesviruses: similar mechanisms and common themes. Viruses. 2016;8:102. doi: 10.3390/v8040102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Walsh D, Mohr I. Viral subversion of the host protein synthesis machinery. Nat Rev Microbiol. 2011;9:860–875. doi: 10.1038/nrmicro2655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Josset L, Tchitchek N, Gralinski LE, Ferris MT, Eisfeld AJ, et al. Annotation of long non-coding RNAs expressed in collaborative cross founder mice in response to respiratory virus infection reveals a new class of interferon-stimulated transcripts. RNA Biol. 2014;11:875–890. doi: 10.4161/rna.29442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kambara H, Niazi F, Kostadinova L, Moonka DK, Siegel CT, et al. Negative regulation of the interferon response by an interferon-induced long non-coding RNA. Nucleic Acids Res. 2014;42:10668–10680. doi: 10.1093/nar/gku713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Barriocanal M, Carnero E, Segura V, Fortes P. Long non-coding RNA BST2/BISPR is induced by IFN and regulates the expression of the antiviral factor tetherin. Front Immunol. 2014;5:655. doi: 10.3389/fimmu.2014.00655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Holmgren AM, Miller KD, Cavanaugh SE, Rall GF. Bst2/Tetherin is induced in neurons by type I interferon and viral infection but is dispensable for protection against neurotropic viral challenge. J Virol. 2015;89:11011–11018. doi: 10.1128/JVI.01745-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Maida Y, Yasukawa M, Furuuchi M, Lassmann T, Possemato R, et al. An RNA-dependent RNA polymerase formed by TERT and the RMRP RNA. Nature. 2009;461:230–235. doi: 10.1038/nature08283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Carnero E, Barriocanal M, Segura V, Guruceaga E, Prior C, et al. Type I interferon regulates the expression of long non-coding RNAs. Front Immunol. 2014;5:548. doi: 10.3389/fimmu.2014.00548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Saxena K, Simon LM, Zeng XL, Blutt SE, Crawford SE, et al. A paradox of transcriptional and functional innate interferon responses of human intestinal enteroids to enteric virus infection. Proc Natl Acad Sci USA. 2017;114:E570. doi: 10.1073/pnas.1615422114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Parker N, Porter AC. Identification of a novel gene family that includes the interferon-inducible human genes 6-16 and ISG12. BMC Genomics. 2004;5:8. doi: 10.1186/1471-2164-5-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Rosebeck S, Leaman DW. Mitochondrial localization and pro-apoptotic effects of the interferon-inducible protein ISG12a. Apoptosis. 2008;13:562–572. doi: 10.1007/s10495-008-0190-0. [DOI] [PubMed] [Google Scholar]
- 74.Qi Y, Li Y, Zhang Y, Zhang L, Wang Z, et al. IFI6 inhibits apoptosis via mitochondrial-dependent pathway in Dengue virus 2 infected vascular endothelial cells. PLoS One. 2015;10:e0132743. doi: 10.1371/journal.pone.0132743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Huang J, Li Y, Qi Y, Zhang Y, Zhang L, et al. Coordinated regulation of autophagy and apoptosis determines endothelial cell fate during Dengue virus type 2 infection. Mol Cell Biochem. 2014;397:157–165. doi: 10.1007/s11010-014-2183-3. [DOI] [PubMed] [Google Scholar]
- 76.Itsui Y, Sakamoto N, Kurosaki M, Kanazawa N, Tanabe Y, et al. Expressional screening of interferon-stimulated genes for antiviral activity against hepatitis C virus replication. J Viral Hepat. 2006;13:690–700. doi: 10.1111/j.1365-2893.2006.00732.x. [DOI] [PubMed] [Google Scholar]
- 77.Meyer K, Kwon YC, Liu S, Hagedorn CH, Ray RB, et al. Interferon-α inducible protein 6 impairs EGFR activation by CD81 and inhibits hepatitis C virus infection. Sci Rep. 2015;5:9012. doi: 10.1038/srep09012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Labrada L, Liang XH, Zheng W, Johnston C, Levine B, et al. Age-dependent resistance to lethal alphavirus encephalitis in mice: analysis of gene expression in the central nervous system and identification of a novel interferon-inducible protective gene, mouse ISG12. J Virol. 2002;76:11688–11703. doi: 10.1128/JVI.76.22.11688-11703.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wilkins C, Woodward J, Lau DT, Barnes A, Joyce M, et al. IFITM1 is a tight junction protein that inhibits hepatitis C virus entry. Hepatology. 2013;57:461–469. doi: 10.1002/hep.26066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Narayana SK, Helbig KJ, Mccartney EM, Eyre NS, Bull RA, et al. The interferon-induced transmembrane proteins, IFITM1, IFITM2, and IFITM3 Inhibit hepatitis C virus entry. J Biol Chem. 2015;290:25946–25959. doi: 10.1074/jbc.M115.657346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Warren CJ, Griffin LM, Little AS, Huang IC, Farzan M, et al. The antiviral restriction factors IFITM1, 2 and 3 do not inhibit infection of human papillomavirus, cytomegalovirus and adenovirus. PLoS One. 2014;9:e96579. doi: 10.1371/journal.pone.0096579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Meng X, Yang D, Yu R, Zhu H. EPSTI1 is involved in IL-28A-mediated inhibition of HCV infection. Mediators Inflamm. 2015;2015:1–13. doi: 10.1155/2015/716315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Pan XB, Qu XW, Jiang D, Zhao XL, Han JC, et al. BST2/Tetherin inhibits hepatitis C virus production in human hepatoma cells. Antiviral Res. 2013;98:54–60. doi: 10.1016/j.antiviral.2013.01.009. [DOI] [PubMed] [Google Scholar]
- 84.Pan XB, Han JC, Cong X, Wei L. BST2/Tetherin inhibits dengue virus release from human hepatoma cells. PLoS One. 2012;7:e51033. doi: 10.1371/journal.pone.0051033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kozuch O, Mayer V. Pig kidney epithelial (PS) cells: a perfect tool for the study of flaviviruses and some other arboviruses. Acta Virol. 1975;19:498. [PubMed] [Google Scholar]
- 86.Heinz FX, Kunz C. Homogeneity of the structural glycoprotein from European isolates of tick-borne encephalitis virus: comparison with other flaviviruses. J Gen Virol. 1981;57:263–274. doi: 10.1099/0022-1317-57-2-263. [DOI] [PubMed] [Google Scholar]
- 87.de Madrid AT, Porterfield JS. A simple micro-culture method for the study of group B arboviruses. Bull World Health Organ. 1969;40:113–121. [PMC free article] [PubMed] [Google Scholar]
- 88.Zhang L, Li Y, Gu Z, Wang Y, Shi M, et al. Resveratrol inhibits enterovirus 71 replication and pro-inflammatory cytokine secretion in rhabdosarcoma cells through blocking IKKs/NF-κB signaling pathway. PLoS One. 2015;10:e0116879. doi: 10.1371/journal.pone.0116879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Wood DE, Salzberg SL. Kraken: ultrafast metagenomic sequence classification using exact alignments. Genome Biol. 2014;15:R46. doi: 10.1186/gb-2014-15-3-r46. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.