

Abstract
Multiple epigenetic mechanisms, including histone acetylation and nucleosome remodeling, are known to be involved in long-term memory formation. Enhancing histone acetylation by deleting histone deacetylases, like HDAC3, typically enhances long-term memory formation. In contrast, disrupting nucleosome remodeling by blocking the neuron-specific chromatin remodeling subunit BAF53b impairs long-term memory. Here, we show that deleting HDAC3 can ameliorate the impairments in both long-term memory and synaptic plasticity caused by BAF53b mutation. This suggests a dynamic interplay exists between histone acetylation/deacetylation and nucleosome remodeling mechanisms in the regulation of memory formation.
Recent work has shown that chromatin modifying and remodeling mechanisms play an important role in long-term memory formation (Kwapis and Wood 2014). Chromatin modification refers to mechanisms that modify histone proteins via post-translational modifications (e.g., acetylation). Chromatin remodeling refers to mechanisms involving ATP-dependent protein complexes that reposition, remove, and exchange nucleosomes. Chromatin modification and remodeling mechanisms are believed to actively restrict or provide access to specific genes in response to a learning event, allowing for dynamic and precise production of mRNA necessary for long-term memory formation. Although both processes are important for long-term memory formation (Alarcón et al. 2004; Korzus et al. 2004; Levenson et al. 2004; Wood et al. 2005; Vecsey et al. 2007; McQuown et al. 2011; Vogel-Ciernia et al. 2013, 2017; Kwapis et al. 2017), little is known about how these major epigenetic mechanisms interact.
Nucleosome remodeling can alter gene expression by shifting nucleosomes along the DNA strand, removing nucleosomes, or exchanging nucleosomes. Understanding how nucleosome remodeling complexes contribute to memory formation is critical, as mutations in multiple different subunits of the nBAF (neuron-specific Brg1-associated factor) nucleosome remodeling complex have been implicated in a variety of human intellectual disabilities, including Coffin-Siris (Santen et al. 2012; Tsurusaki et al. 2012) and Nicolaides–Baraitser syndromes (Van Houdt et al. 2012), sporadic mental retardation (Halgren et al. 2012; Hoyer et al. 2012; Santen et al. 2012; Tsurusaki et al. 2012), and even autism (Neale et al. 2012; O'Roak et al. 2012; Krupp et al. 2017). Previous work from our laboratory has demonstrated that mutating a neuron-specific subunit of nBAF, BAF53b (also known as Actl6b), disrupts both long-term memory and synaptic plasticity (Vogel-Ciernia et al. 2013, 2017). This suggests that functional nBAF-mediated nucleosome remodeling is critical for the formation of long-lasting forms of synaptic plasticity and memory.
A second major epigenetic mechanism linked to memory formation is histone acetylation, which is modified by two classes of enzymes: histone acetyltransferases (HATs) and histone deacetylases (HDACs). HATs, which add acetyl groups to histones, typically loosen chromatin structure to facilitate transcription. In contrast, HDACs remove acetyl groups from histones, condensing chromatin to restrict transcription. In general, facilitating histone acetylation by enhancing HAT activity or reducing HDAC activity improves long-term memory formation (Kwapis and Wood 2014). There is a powerful opposition of activity between HATs and HDACs, revealed by HDAC inhibition, which leads to significantly increased histone acetylation (Levenson et al. 2004; Vecsey et al. 2007; McQuown et al. 2011; Kwapis et al. 2017). One HDAC in particular, HDAC3, has been shown to be a powerful negative regulator of long-term memory formation. Using both genetic deletion and pharmacological inhibition of HDAC3, our laboratory has shown that disruption of HDAC3 can transform a subthreshold learning event into one that produces robust and persistent long-term memory (McQuown et al. 2011; Malvaez et al. 2013; Rogge et al. 2013; Kwapis et al. 2017). Similarly, pharmacological disruption of HDAC3 can improve associative hippocampal long-term potentiation (LTP) in slices from aging rats (Sharma et al. 2015). Together, these studies demonstrate that HDAC3 negatively regulates both long-term memory and synaptic plasticity.
Although it is clear that BAF53b disruption impairs long-term memory formation whereas disruption of HDAC3 enhances memory formation, little is understood about how these major epigenetic mechanisms interact. As mutations in the nBAF complex are linked to human intellectual disability disorders, it is critical to determine whether these deficits can be overcome by promoting access to chromatin through other epigenetic mechanisms, including HDAC3 deletion. If deletion of HDAC3 can ameliorate the memory disruption caused by BAF53b mutation, this represents a potential therapeutic avenue for improving cognition in individuals with disorders stemming from impaired nucleosome remodeling mechanisms.
All animals were between 8 and 20-wk old at the time of behavioral testing. Mice had free access to food and water and were maintained on a 12:12h light–dark cycle, with all behavior performed during the light portion of the cycle. Animals were backcrossed at least five generations to C57BL/6J mice (Jackson Labs). All experiments were conducted according to NIH guidelines for animal care and use and were approved by the Institutional Animal Care and Use Committee of the University of California, Irvine.
To determine whether deletion of HDAC3 ameliorates memory impairments in BAF53b mutant mice, we generated a double mutant by crossing two mouse lines previously used by our laboratory: HDAC3flox/flox mice (McQuown et al. 2011) and BAF53bΔSB2 mice (Vogel Ciernia et al. 2017). HDAC3flox/flox mice carry LoxP sites flanking exons four through seven in the Hdac3 gene, so that local infusion of AAV-CaMKII-Cre creates a site-specific deletion of Hdac3 in excitatory neurons (McQuown et al. 2011). BAF53bΔSB2 mice express a transgene with a mutant form of BAF53b with a deletion of subdomain two, the region of BAF53b that is most distinct from BAF53a, its non-neuronal homolog (Vogel Ciernia et al. 2017). This mutant transgene is expressed under the CaMKIIα promoter, restricting expression to forebrain excitatory neurons during post-natal development. Crossing these mice produced four different genotypes: 1. HDAC3+/+::BAF53bWT, 2. HDAC3+/+::BAF53bΔSB2, 3. HDAC3flox/flox::BAF53bWT, and 4. HDAC3flox/flox::BAF53bΔSB2.
To delete HDAC3 from the dorsal hippocampus of HDAC3flox/flox mice, all animals were anesthetized with 2%–4% isoflurane in 100% O2 and locally infused with AAV2.1-CaMKII-Cre via an infusion needle positioned in the dorsal CA1 area of the hippocampus (AP −2.0 mm; ML ±1.5 mm; DV −1.5 mm). One microliter of virus was infused into each hemisphere at a rate of 6 µL/h as previously described (McQuown et al. 2011; Kwapis et al. 2017). Two weeks later (to allow for optimal gene deletion (McQuown et al. 2011)), mice were either handled and trained in the object location memory (OLM) task (Experiment 1) or were sacrificed for LTP (Experiment 2).
Following behavior, mice were euthanized by cervical dislocation and their brains were removed and flash-frozen in ice-cold isopentane. To verify the deletion of HDAC3 protein in HDAC3flox/flox mice, 20 µm slices were collected throughout the dorsal hippocampus and immunofluorescence was performed and quantified as previously described (McQuown et al. 2011; Kwapis et al. 2017). To verify the presence of the mutant transgene in BAF53bΔSB2 mice, punches (1.0 µm diameter) were also collected from area CA1 of the dorsal hippocampus and RT-qPCR was performed to measure expression of the BAF53bΔSB2 transgene as previously described (White et al. 2016; Vogel Ciernia et al. 2017).
First, we verified both genetic manipulations in our double mutant mice. To confirm that AAV-CaMKII-Cre infusion produced a complete focal deletion of HDAC3 in HDAC3flox/flox::BAF53bWT and HDAC3flox/flox::BAF53bΔSB2 mice, we measured immunoreactivity to HDAC3 in slices from each of our four genotypes. HDAC3+/+ animals showed robust HDAC3 immunoreactivity (green) throughout the dorsal hippocampus (Fig. 1A). HDAC3flox/flox animals, in comparison, showed a near complete loss of HDAC3 immunoreactivity throughout region CA1 of the dorsal hippocampus. General hippocampal morphology appeared normal in all four groups, as determined by the fluorescent nissl stain NeuroTrace (NT; red). Quantification of this staining confirmed that HDAC3flox/flox animals had significantly reduced levels of HDAC3 protein compared to HDAC3+/+ animals, regardless of whether they carried the BAF53bΔSB2 transgene (Fig. 1B). We next confirmed that the BAF53bΔSB2 transgene expressed normally in both HDAC3+/+::BAF53bΔSB2 and HDAC3flox/flox::BAF53bΔSB2 mice using RT-qPCR. We observed significantly higher expression of the BAF53bΔSB2 transgene in the dorsal hippocampus of BAF53bΔSB2 animals than in BAF53bWT controls (Fig. 1C). Further, both HDAC3+/+ and HDAC3flox/flox animals showed similar expression levels of the BAF53bΔSB2 transgene, indicating that the presence or absence of HDAC3 did not alter transgene expression.
Figure 1.

Deleting HDAC3 ameliorates the hippocampal memory impairments induced by the BAF53bΔSB2 transgene. (A) Representative immunofluorescence image of HDAC3 expression (green) in each of the four genotypes following intrahippocampal injection of AAV-CaMKII-Cre. Cell bodies were counterstained with a fluorescent Nissl stain (NeuroTrace, NT, red). HDAC3 expression was deleted throughout area CA1 in both HDAC3flox/flox::BAF53bWT and HDAC3flox/flox::BAF53bΔSB2 mice. (B) Mean intensity of HDAC3 immunofluorescence sampled from CA1 (normalized to background). HDAC3 expression was significantly reduced in both groups of HDAC3flox/flox mice relative to HDAC3+/+ controls (two-way ANOVA: main effect of HDAC3 (F(1,31) = 85.53, P < 0.0001), no effect of BAF53b or interaction; Sidak's post hoc comparing HDAC3+/+ to HDAC3flox/flox: BAF53bWT, P < 0.0001; BAF53bΔSB2, P < 0.0001; n = 10 (5 female), 5 (0 female), 11 (6 female), 9 (6 female)). (C) RT-qPCR measuring BAF53bΔSB2 transgene expression. Both groups of BAF53bΔSB2 mice displayed significantly higher levels of BAF53bΔSB2 transgene expression than BAF53bWT mice (two-way ANOVA: main effect of BAF53b (F(1,26) = 67.83, P < 0.0001), no effect of HDAC3 or interaction; Sidak's post hoc comparing BAF53bWT to BAF53bΔSB2: HDAC3+/+, P < 0.0001; HDAC3flox/flox, P < 0.0001; n = 12 (5 female), 6 (0 female), 11 (7 female), 5 (2 female)). (D) Schematic of the OLM task. (E) Deletion of HDAC3 rescues the impairments in OLM induced by BAF53bΔSB2. For HDAC3+/+ mice, expression of the BAF53bΔSB2 transgene significantly impairs memory. HDAC3flox/flox mice, however, show robust memory for OLM regardless of whether the transgene is expressed (two-way ANOVA: main effects of HDAC3 (F(1,52) = 5.36, P < 0.05), BAF53b (F(1,52) = 6.41, P < 0.05), and significant interaction (F(1,52) = 4.22, P < 0.05; Sidak's post hoc comparing BAF53bWT to BAF53bΔSB2: HDAC3+/+, P < 0.05, HDAC3flox/flox P = 0.9; n = 14 (9 female), 7 (2 female), 19 (10 female), 16 (11 female)). (F) There was no significant difference in total exploration time between groups during testing (two-way ANOVA; no main effect or interaction).
For experiment 1, we tested whether HDAC3 deletion could rescue the OLM impairments induced by disruption of BAF53b (Vogel Ciernia et al. 2017). Following 4 d of handling (2-min per day), mice were habituated for six consecutive days (5-min per day), during which mice were exposed to the context without objects. The following day, mice were given a training trial (Fig. 1D), in which they were placed into the context with two identical objects for 10 min, a protocol that produces robust long-term memory in young wild-type mice (Vogel Ciernia et al. 2017). Twenty-four hours later, mice were given a 5-min retention test in which one object was moved to a new location in a counterbalanced manner (Fig. 1D). Videos were recorded using AnyMaze and exploration of objects during training and testing was hand-scored (Vogel-Ciernia and Wood 2014). Exploration times were used to calculate a discrimination index (DI): (tnovel − tfamiliar)/(tnovel + tfamiliar) × 100. Memory for OLM is indicated by increased exploration of the moved object relative to the unmoved object, reflected as a positive DI score.
For HDAC3+/+ animals, expression of the mutant BAF53bΔSB2 transgene significantly impaired memory for OLM, consistent with previous work from our laboratory (Vogel-Ciernia et al. 2013). HDAC3+/+::BAF53bWT mice showed a strong preference for the moved object, indicating robust long-term memory for the 10-min training session (Fig. 1E, black bar), whereas HDAC3+/+::BAF53bΔSB2 mice showed little preference for the moved object, indicating poor long-term memory (Fig. 1E, dark blue bar). When hippocampal HDAC3 was deleted, however (HDAC3flox/flox mice), expression of the BAF53bΔSB2 transgene had no effect on memory, with both HDAC3flox/flox::BAF53BWT and HDAC3flox/flox::BAF53bΔSB2 mice demonstrating robust and comparable preference for the moved object (light gray and blue bars). No difference in total exploration was observed between any of the groups during the test session (Fig. 1F). Together, these results suggest that deleting HDAC3 in the dorsal hippocampus can overcome memory impairments induced by disruption of BAF53b.
Next, for Experiment 2, we asked whether synaptic plasticity deficits induced by the BAF53bΔSB2 transgene could similarly be rescued by hippocampal HDAC3 deletion. Previous work from our laboratory has demonstrated that mutation of BAF53b disrupts the stabilization of LTP in the Schaffer-commissural pathway of the hippocampus (Vogel-Ciernia et al. 2013, 2017). To determine whether hippocampal deletion of HDAC3 could ameliorate this impairment, we examined LTP in acute hippocampal slices from each of our four experimental groups (HDAC3+/+::BAF53bWT, HDAC3+/+::BAF53bΔSB2, HDAC3flox/flox::BAF53bWT, and HDAC3flox/flox::BAF53bΔSB2, 3–6 mo of age).
Two weeks after infusion of AAV-CaMKII-Cre, mice were sacrificed and hippocampal slices were collected for LTP. Transverse hippocampal slices (300 µm) were prepared as previously described (Vogel Ciernia et al. 2017). Following a 2-h incubation period, a single glass pipette (2–3 MΩ) filled with 1 M NaCl was placed in CA1b stratum radiatum to record fEPSPs evoked by bipolar stainless steel stimulation electrodes placed at sites CA1a and CA1c. Orthodromic stimulation (CA1c) was used to induce LTP whereas antidromic stimulation (CA1a) was used to monitor slice viability and baseline stability. After a 20-min stable baseline recording, five θ bursts (each burst consists of four pulses at 100 Hz; each burst separated by 200 msec) were delivered to elicit LTP.
As previously reported (Vogel Ciernia et al. 2017), we found that expression of the mutant BAF53bΔSB2 transgene impaired the stabilization of LTP in slices from HDAC3+/+ mice. Slices from HDAC3+/+::BAF53bWT mice showed stable LTP following a single train of five theta bursts (Fig. 2A, black symbols). In contrast, slices from HDAC3+/+::BAF53bΔSB2 mice, showed comparable short-term potentiation (STP) that failed to stabilize and ultimately decayed to a significantly lower level than in control mice (Fig. 2A, blue symbols). When hippocampal HDAC3 was deleted, however, expression of the BAF53bΔSB2 transgene did not disrupt LTP. Slices from both HDAC3flox/flox::BAF53bWT and HDAC3flox/flox::BAF53bΔSB2 mice produced stable, persistent potentiation following the same stimulation protocol (Fig. 2B). There was no significant difference in the level of potentiation 60 m post-TBS between slices from HDAC3flox/flox::BAF53bWT and HDAC3flox/flox::BAF53bΔSB2 mice (Fig. 2C), indicating that the presence of the transgene had little effect on LTP stabilization in the absence of hippocampal HDAC3. Finally, in an effort to determine whether these genetic deletions interfered with synaptic events used to induce LTP (e.g., NMDA receptor function), we measured STP immediately following induction (1–2 min post-TBs) and to what extent the burst response facilitated over the course of a theta train (Larson and Lynch 1988; Arai and Lynch 1992). There were no notable differences between groups in either STP (Fig. 2A,B) or burst area (Fig. 2D). Together, these results suggest that genetic deletion of HDAC3 ameliorated the impairments in LTP stabilization induced by disruption of the BAF53b subunit of the nBAF complex by engaging a mechanism that follows the triggering events for LTP.
Figure 2.

Deleting HDAC3 can also overcome the deficits in hippocampal LTP induced by the BAF53bΔSB2 transgene. (A) fEPSP slope measurement after five theta bursts in hippocampal slices from HDAC3+/+::BAF53bWT and HDAC3+/+::BAF53bΔSB2 mice. Following the application of TBS, STP (1–2 min post-TBS) was indistinguishable between groups (two-way ANOVA: no main effects (F(1,10) = 2.19, P = 0.17), and no significant interaction (F(5,50) = 0.71, P = 0.61). Stable potentiation was observed in slices from HDAC3+/+::BAF53bWT mice 50–60 min post-TBS, but the same stimulation protocol produced only weak potentiation in slices from HDAC3+/+::BAF53bΔSB2 mice, consistent with our previous work showing the BAF53bΔSB2 transgene impairs stabilization of LTP. (Top) representative fEPSP traces collected during baseline (solid line) and 1 h after stimulation (dashed line). Scale bar, 1 mV per 5 msec. (B) fEPSP slope measurement after five theta bursts in HDAC3flox/flox::BAF53bWT and HDAC3flox/flox::BAF53bΔSB2 slices. STP was again not significantly different between groups (two-way ANOVA: no main effects (F(1,11) = 1.49, P = 0.25), and no interaction (F(5,55) = 0.32, P = 0.90). Following induction, hippocampal slices from both HDAC3flox/flox groups showed stable LTP 50–60 min post-TBS, indicating that the BAF53bΔSB2 transgene failed to disrupt LTP in the absence of HDAC3. (Top) representative fEPSP traces collected during baseline (solid line) and 1 h after stimulation (dashed line). Scale bar, 1 mV per 5 msec. (C) Summary graph showing the mean fEPSP slope for each group 50–60 m after stimulation. The BAF53bΔSB2 transgene caused a significant impairment in the level of LTP in HDAC3+/+ mice, as previously reported (Vogel Ciernia et al. 2017). Deleting HDAC3 in mice carrying the BAF53bΔSB2 transgene (HDAC3flox/flox::BAF53bΔSB2), however, produced LTP comparable to that of control (HDAC3+/+::BAF53bWT) mice (two-way ANOVA: main effects of HDAC3 (F(1,21) = 22.02, P < 0.001), BAF53b (F(1,21) = 19.51, P < 0.001), but no significant interaction (F(1,21) = 0.38, P > 0.05); Sidak's post hoc tests: HDAC3+/+::BAF53bWT versus HDAC3+/+::BAF53bΔSB2, P < 0.01; HDAC3+/+::BAF53bΔSB2 versus HDAC3flox/flox::BAF53bWT, P < 0.0001; HDAC3+/+::BAF53bΔSB2 versus HDAC3flox/flox::BAF53bWT, P < 0.01; all other post hoc comparisons not significant (P > 0.05); n = 6 slices (from 2 male, 1 female), 6 slices (from 1 male, 2 female), 7 slices (from 2 male, 3 female), 6 slices (from 1 male, 2 female)). (D) No significant differences in burst area were observed between the four groups (two-way ANOVA: main effect of burst number (F(4,84) = 43.40, P < 0.001) and significant interaction (F(12,84) = 2.61, P < 0.01) but no main effect of genotype (F(3,21) = 0.13, P > 0.05); all Sidak's post hoc tests, P > 0.05).
To summarize, we found that focal deletion of the histone deacetylase HDAC3 in the dorsal hippocampus can ameliorate the impairments in both long-term memory and synaptic plasticity caused by mutation of the BAF53b subunit of the nBAF nucleosome remodeling complex. As previously observed (Vogel Ciernia et al. 2017), both OLM and theta burst-induced LTP were impaired by the presence of the mutant BAF53bΔSB2 transgene. In the absence of HDAC3, on the other hand, the BAF53bΔSB2 transgene had no effect on either long-term memory (Fig. 1) or LTP (Fig. 2). Importantly, focal deletion of HDAC3 did not affect transgene expression in the BAF53bΔSB2 mice (Fig. 1C). Broadly, these findings suggest that disrupting nucleosome remodeling impairs memory formation, but this impairment can be overcome by facilitating local transcription through other epigenetic mechanisms, including histone acetylation.
In the current study, although it is not clear how HDAC3 deletion restored long-term memory in BAF53bΔSB2 mice, one possibility is that deleting HDAC3 allowed for enhanced histone acetylation at memory-relevant genes (McQuown et al. 2011; Kwapis et al. 2017), promoting a permissive chromatin structure even in the absence of normal nucleosome remodeling (Fig. 3A–C). Thus, even though the learning event may have failed to stimulate normal nucleosome remodeling in BAF53bΔSB2 mice, the enhanced histone acetylation following HDAC3 deletion was sufficient to overcome the deficits in both long-term memory and synaptic plasticity, possibly by promoting an open chromatin structure at key plasticity-related genes (Fig. 3D).
Figure 3.
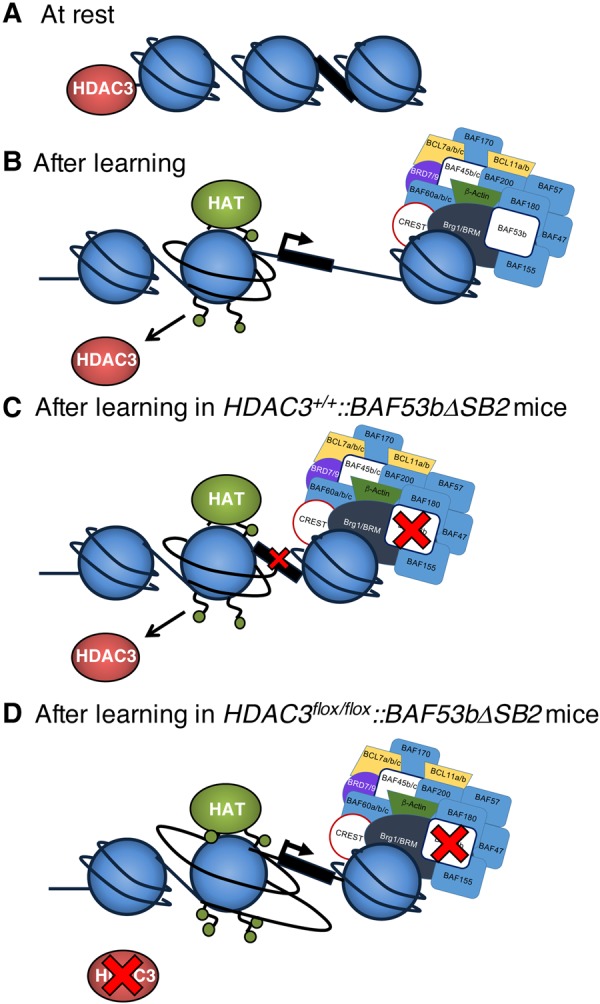
Simplified illustration of hypothesized transcriptional regulation of key memory-relevant genes in HDAC3flox/flox::BAF53bΔSB2 mice (drawing not to scale). (A) In the absence of a learning event, HDAC3 is tightly bound to the chromatin, functioning as a “molecular brake pad” to maintain memory-relevant genes in a silent state (McQuown and Wood 2011). (B) Following a significant learning event, HDAC3 and its associated corepressors are removed and the nBAF complex shifts, deletes, or exchanges nucleosomes, allowing for an open chromatin state around key memory-relevant genes that permits transcription. (C) In the presence of the BAF53bΔSB2 transgene, nBAF-mediated nucleosome remodeling is impaired, restricting transcriptional access to key genes. (D) When HDAC3 is deleted, the hyperacetylated nucleosome promotes an open chromatin structure at key genes, providing transcriptional access even in the context of disrupted nucleosome remodeling in mice carrying the BAF53bΔSB2 transgene.
An additional possibility is that disruption of BAF53b impairs the formation or gene-specific targeting of the nBAF complex, ultimately disrupting interactions between nBAF and other regulatory elements required to control expression of target genes. Specifically, it is possible that the BAF53bΔSB2 mutant prevents the formation of a functional nBAF-based enhanceosome, a protein complex that assembles at gene enhancer regions to regulate transcription through recruitment of modifying enzymes, like HATs (Thanos and Maniatis 1995; Merika and Thanos 2001; Panne 2008; López and Wood 2015). Consistent with this, loss of BAF53b disrupts the targeting of nBAF and its calcium-responsive subunit CREST (calcium-responsive transactivator, also known as SS18L1) to specific gene promoters (Wu et al. 2007). CREST is known to recruit CBP (Aizawa et al. 2004; Qiu and Ghosh 2008), a HAT known to facilitate long-term memory formation (Barrett and Wood 2008). Therefore, disruption of BAF53b might impair memory by preventing the nBAF-CREST complex from localizing to memory-relevant genes, ultimately preventing the recruitment of HATs, like CBP, and other elements that enhance transcriptional accessibility at these genes. In this case, HDAC3 deletion might improve memory formation in BAF53bΔSB2 mutants by restoring acetylation in the absence of the nBAF-CREST complex. More work will be required to determine precisely how the learning-induced gene expression profile is affected by HDAC3 deletion in BAF53bΔSB2 mice.
Recent studies have identified numerous mutations in chromatin remodeling complexes in patients with both intellectual disability and autism spectrum disorders (Neale et al. 2012), suggesting that disruption in nucleosome remodeling may be a key contributing factor to these disorders. Consistent with this, the current study and previous work from our laboratory (White et al. 2016; Vogel Ciernia et al. 2017) have found that disruption of the BAF53b subunit of the neuron-specific nBAF nucleosome remodeling complex severely impairs both long-term memory and synaptic plasticity in mice. Here, we show that deletion of the repressive histone deacetylase HDAC3 can ameliorate these impairments, suggesting that enhancing histone acetylation may be sufficient to overcome impairments in memory and plasticity induced by disruption of nucleosome remodeling. As both broad-spectrum pharmacological HDAC inhibitors (Khan and La Thangue 2012; Ganai et al. 2016) and HDAC3-specific inhibitors (Malvaez et al. 2013; Franklin et al. 2014; Bieszczad et al. 2015; Rumbaugh et al. 2015; Krishna et al. 2016; Phan et al. 2017; Suelves et al. 2017) already exist, this represents a potential novel therapeutic avenue to improve cognition in patients with disorders linked to impaired nucleosome remodeling.
This study demonstrates, for the first time, that deleting HDAC3 is sufficient to ameliorate the long-term memory and synaptic plasticity impairments caused by disruption of the BAF53b subunit of the nBAF nucleosome remodeling complex. Although there is much work to be done to determine how these and other epigenetic mechanisms interact during memory formation, this study confirms that histone acetylation and nucleosome remodeling cooperate to regulate long-term memory formation. Understanding how these major epigenetic mechanisms interact to shape chromatin structure during memory formation is critical to understanding how long-term memory formation normally occurs and how this process is disrupted in intellectual disability and autism spectrum disorders.
Acknowledgments
We would like to thank all members of the Wood laboratory for scientific discussions and technical assistance. This work was supported by grants from NIMH (MH101491) to M.A.W.; NIA (AG051807 and AG050787) to M.A.W.; and NIA (F32 AG052303 and K99 AG056596) to J.L.K.
Footnotes
Article is online at http://www.learnmem.org/cgi/doi/10.1101/lm.046920.117.
References
- Aizawa H, Hu SC, Bobb K, Balakrishnan K, Ince G, Gurevich I, Cowan M, Ghosh A. 2004. Dendrite development regulated by CREST, a calcium-regulated transcriptional activator. Science 303: 197–202. [DOI] [PubMed] [Google Scholar]
- Alarcón JM, Malleret G, Touzani K, Vronskaya S, Ishii S, Kandel ER, Barco A. 2004. Chromatin acetylation, memory, and LTP are impaired in CBP+/− mice: a model for the cognitive deficit in Rubinstein-Taybi syndrome and its amelioration. Neuron 42: 947–959. [DOI] [PubMed] [Google Scholar]
- Arai A, Lynch G. 1992. Factors regulating the magnitude of long-term potentiation induced by θ pattern stimulation. Brain Res 598: 173–184. [DOI] [PubMed] [Google Scholar]
- Barrett RM, Wood MA. 2008. Beyond transcription factors: the role of chromatin modifying enzymes in regulating transcription required for memory. Learn Mem 15: 460–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bieszczad KM, Bechay K, Rusche JR, Jacques V, Kudugunti S, Miao W, Weinberger NM, McGaugh JL, Wood MA. 2015. Histone deacetylase inhibition via RGFP966 releases the brakes on sensory cortical plasticity and the specificity of memory formation. J Neurosci 35: 13124–13132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franklin AV, Rusche JR, McMahon LL. 2014. Increased long-term potentiation at medial-perforant path-dentate granule cell synapses induced by selective inhibition of histone deacetylase 3 requires Fragile X mental retardation protein. Neurobiol Learn Mem 114: 193–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganai SA, Ramadoss M, Mahadevan V. 2016. Histone deacetylase (HDAC) inhibitors - emerging roles in neuronal memory, learning, synaptic plasticity and neural regeneration. Curr Neuropharmacol 14: 55–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halgren C, Kjaergaard S, Bak M, Hansen C, El-Schich Z, Anderson CM, Henriksen KF, Hjalgrim H, Kirchhoff M, Bijlsma EK, et al. 2012. Corpus callosum abnormalities, intellectual disability, speech impairment, and autism in patients with haploinsufficiency of ARID1B. Clin Genet 82: 248–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoyer J, Ekici AB, Endele S, Popp B, Zweier C, Wiesener A, Wohlleber E, Dufke A, Rossier E, Petsch C, et al. 2012. Haploinsufficiency of ARID1B, a member of the SWI/SNF-A chromatin-remodeling complex, is a frequent cause of intellectual disability. Am J Hum Genet 90: 565–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan O, La Thangue NB. 2012. HDAC inhibitors in cancer biology: emerging mechanisms and clinical applications. Immunol Cell Biol 90: 85–94. [DOI] [PubMed] [Google Scholar]
- Korzus E, Rosenfeld MG, Mayford M. 2004. CBP histone acetyltransferase activity is a critical component of memory consolidation. Neuron 42: 961–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishna K, Behnisch T, Sajikumar S. 2016. Inhibition of histone deacetylase 3 restores amyloid-β oligomer-induced plasticity deficit in hippocampal CA1 pyramidal neurons. J Alzheimer's Dis 51: 783–791. [DOI] [PubMed] [Google Scholar]
- Krupp DR, Barnard RA, Duffourd Y, Evans SA, Mulqueen RM, Bernier R, Rivière JB, Fombonne E, O'Roak BJ. 2017. Exonic mosaic mutations contribute risk for autism spectrum disorder. Am J Hum Genet 101: 369–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwapis JL, Alaghband Y, López AJ, White AO, Campbell RR, Dang RT, Rhee D, Tran AV, Carl AE, Matheos DP, et al. 2017. Context and auditory fear are differentially regulated by HDAC3 activity in the lateral and basal subnuclei of the amygdala. Neuropsychopharmacology 42: 1284–1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwapis JL, Wood MA. 2014. Epigenetic mechanisms in fear conditioning: implications for treating post-traumatic stress disorder. Trends Neurosci 37: 706–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson J, Lynch G. 1988. Role of N-methyl-D-aspartate receptors in the induction of synaptic potentiation by burst stimulation patterned after the hippocampal 0-rhythm. Brain Res 441: 111–118. [DOI] [PubMed] [Google Scholar]
- Levenson JM, O'Riordan KJ, Brown KD, Trinh MA, Molfese DL, Sweatt JD. 2004. Regulation of histone acetylation during memory formation in the hippocampus. J Biol Chem 279: 40545–40559. [DOI] [PubMed] [Google Scholar]
- López AJ, Wood MA. 2015. Role of nucleosome remodeling in neurodevelopmental and intellectual disability disorders. Front Behav Neurosci 9: 100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malvaez M, McQuown SC, Rogge GA, Astarabadi M, Jacques V, Carreiro S, Rusche JR, Wood MA. 2013. HDAC3-selective inhibitor enhances extinction of cocaine-seeking behavior in a persistent manner. Proc Natl Acad Sci 110: 2647–2652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McQuown SC, Barrett RM, Matheos DP, Post RJ, Rogge GA, Alenghat T, Mullican SE, Jones S, Rusche JR, Lazar MA, et al. 2011. HDAC3 is a critical negative regulator of long-term memory formation. J Neurosci 31: 764–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McQuown SC, Wood MA. 2011. HDAC3 and the molecular brake pad hypothesis. Neurobiol Learn Mem 96: 27–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merika M, Thanos D. 2001. Enhanceosomes. Curr Opin Genet Dev 11: 205–208. [DOI] [PubMed] [Google Scholar]
- Neale B, Kou Y, Liu L, Ma'ayan A, Samocha KE, Sabo A, Lin CF, Stevens C, Wang LS, Makarov V, et al. 2012. Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature 485: 242–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Roak BJO, Vives L, Girirajan S, Karakoc E, Krumm N, Coe P, Levy R, Ko A, Lee C, Smith JD, et al. 2012. Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature 485: 246–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panne D. 2008. The enhanceosome. Curr Opin Struct Biol 18: 236–242. [DOI] [PubMed] [Google Scholar]
- Phan ML, Gergues MM, Mahidadia S, Jimenez-Castillo J, Vicario DS, Bieszczad KM. 2017. HDAC3 inhibitor RGFP966 modulates neuronal memory for vocal communication signals in a songbird model. Front Syst Neurosci 11: 65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu Z, Ghosh A. 2008. A calcium-dependent switch in a CREST-BRG1 complex regulates activity-dependent gene expression. Neuron 60: 775–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogge GA, Singh H, Dang R, Wood MA. 2013. HDAC3 is a negative regulator of cocaine-context-associated memory formation. J Neurosci 33: 6623–6632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rumbaugh G, Sillivan SE, Ozkan ED, Rojas CS, Hubbs CR, Aceti M, Kilgore M, Kudugunti S, Puthanveettil SV, Sweatt JD, et al. 2015. Pharmacological selectivity within class i histone deacetylases predicts effects on synaptic function and memory rescue. Neuropsychopharmacology 40: 2307–2316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santen GWE, Aten E, Sun Y, Almomani R, Gilissen C, Nielsen M, Kant SG, Snoeck IN, Peeters EAJ, Hilhorst-Hofstee Y, et al. 2012. Mutations in SWI/SNF chromatin remodeling complex gene ARID1B cause Coffin-Siris syndrome. Nat Genet 44: 379–380. [DOI] [PubMed] [Google Scholar]
- Sharma M, Shivarama Shetty M, Arumugam TV, Sajikumar S. 2015. Histone deacetylase 3 inhibition re-establishes synaptic tagging and capture in aging through the activation of nuclear factor κ B. Sci Rep 5: 16616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suelves N, Kirkham-McCarthy L, Lahue RS, Ginés S. 2017. A selective inhibitor of histone deacetylase 3 prevents cognitive deficits and suppresses striatal CAG repeat expansions in Huntington's disease mice. Sci Rep 7: 6082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thanos D, Maniatis T. 1995. Virus induction of human IFN-b gene expression requires the assembly of an enhancosome. Cell 83: 1091–1100. [DOI] [PubMed] [Google Scholar]
- Tsurusaki Y, Okamoto N, Ohashi H, Kosho T, Imai Y, Hibi-Ko Y, Kaname T, Naritomi K, Kawame H, Wakui K, et al. 2012. Mutations affecting components of the SWI/SNF complex cause Coffin-Siris syndrome. Nat Genet 44: 376–378. [DOI] [PubMed] [Google Scholar]
- Van Houdt JKJ, Nowakowska BA, Sousa SB, van Schaik BDC, Seuntjens E, Avonce N, Sifrim A, Abdul-Rahman OA, van den Boogaard M-JH, Bottani A, et al. 2012. Heterozygous missense mutations in SMARCA2 cause Nicolaides-Baraitser syndrome. Nat Genet 44: 445–449. [DOI] [PubMed] [Google Scholar]
- Vecsey CG, Hawk JD, Lattal KM, Stein JM, Fabian SA, Attner MA, Cabrera SM, McDonough CB, Brindle PK, Abel T, et al. 2007. Histone deacetylase inhibitors enhance memory and synaptic plasticity via CREB: CBP-dependent transcriptional activation. J Neurosci 27: 6128–6140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel Ciernia A, Kramár EA, Matheos DP, Havekes R, Hemstedt TJ, Magnan CN, Sakata K, Tran A, Azzawi S, Lopez A, et al. 2017. Mutation of neuron-specific chromatin remodeling subunit BAF53b: rescue of plasticity and memory by manipulating actin remodeling. Learn Mem 24: 199–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel-Ciernia A, Matheos DP, Barrett RM, Kramár EA, Azzawi S, Chen Y, Magnan CN, Zeller M, Sylvain A, Haettig J, et al. 2013. The neuron-specific chromatin regulatory subunit BAF53b is necessary for synaptic plasticity and memory. Nat Neurosci 16: 552–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel-Ciernia A, Wood MA. 2014. Examining object location and object recognition memory in mice. Curr Protoc Neurosci 2014: 8.31.1–8.31.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White AO, Kramár EA, López AJ, Kwapis JL, Doan J, Saldana D, Davatolhagh MF, Alaghband Y, Blurton-Jones M, Matheos DP, et al. 2016. BDNF rescues BAF53b-dependent synaptic plasticity and cocaine-associated memory in the nucleus accumbens. Nat Commun 7: 11725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood MA, Kaplan MP, Park A, Blanchard EJ, Oliveira AMM, Lombardi TL, Abel T. 2005. Transgenic mice expressing a truncated form of CREB-binding protein (CBP) exhibit deficits in hippocampal synaptic plasticity and memory storage. Learn Mem 12: 111–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu JI, Lessard J, Olave IA, Qiu Z, Ghosh A, Graef IA, Crabtree GR. 2007. Regulation of dendritic development by neuron-specific chromatin remodeling complexes. Neuron 56: 94–108. [DOI] [PubMed] [Google Scholar]