

Abstract
A versatile, bottom‐up approach allows the controlled fabrication of polydopamine (PD) nanostructures on DNA origami. PD is a biosynthetic polymer that has been investigated as an adhesive and promising surface coating material. However, the control of dopamine polymerization is challenged by the multistage‐mediated reaction mechanism and diverse chemical structures in PD. DNA origami decorated with multiple horseradish peroxidase‐mimicking DNAzyme motifs was used to control the shape and size of PD formation with nanometer resolution. These fabricated PD nanostructures can serve as “supramolecular glue” for controlling DNA origami conformations. Facile liberation of the PD nanostructures from the DNA origami templates has been achieved in acidic medium. This presented DNA origami‐controlled polymerization of a highly crosslinked polymer provides a unique access towards anisotropic PD architectures with distinct shapes that were retained even in the absence of the DNA origami template.
Keywords: DNA origami, DNAzyme, nanostructures, polydopamine
Nanofabrication of polymer materials with high precision and resolution is in high demand for numerous modern technologies in the information era. From biochips for controlling cell adhesion and behavior, micro/nanofluidic systems with highly integrated functions, to photonic materials with quantum optical properties, these state‐of‐the‐art devices contain immense information given their small footprint.1 Polymers, which are quintessential in many of these devices, would naturally be required to break its limits on their fabrication. However, with current available techniques mainly based on top‐down strategies, such as lithography, high instrumental costs and long operation times as well as its resolution of control limit wider use. On the other hand, although restricted by the scale of operation, bottom‐up approaches have recently seen development with the assistance of DNA origami nanotechnology.2 DNA origami is a powerful DNA nanotechnology that offers systematic design of a large variety of defined DNA nanostructures by self‐assembling scaffold DNA and staple DNA sequences.3 Computer‐assisted design provides almost limitless access to any defined nanostructure in 2D and 3D4 with each DNA staple sequence encoding a specific position that can be precisely modified. This concept has been utilized to arrange metal nanoparticles,5 proteins,6 and fluorescent molecules7 with nanometer resolution. For higher complexity, DNA origami has been employed as a versatile framework to cast metal nanoparticles8 and liposomes9 in their confined 3D space. By integrating DNA origami with polymer chemistry, we have previously established this platform for spatially controlling “grafting from” polymer growth using atom transfer radical polymerization.10 In this manuscript, the DNA templated strategy was applied for the first time to control the polymerization of PD and the formation of anisotropic PD nanostructures with distinct shapes. PD is a bioinspired polymer from invertebrate mussels known for its highly uncontrolled self‐polymerization and its adhesive properties.11 A variety of functional groups available on PD provide this adhesive property as well as an avenue for post‐functionalization with polymers, biomolecules, and metal nanoparticles.11, 12 Due to its natural biocompatibility, it immediately attracted very broad attention in the field of biomaterials and nanomedicine.13 Micropatterned PD surfaces efficiently captured cells, proteins, chemicals, and metals14 for advanced biochips and sensors. Hence, there is great interest to fabricate PD with defined nanoscale structures/patterns, which has so far remained elusive due to 1) the molecular mechanisms of PD formation, that are still not fully understood since many intermediate and short‐lived chemical species are involved,15 2) the rapid polymerization kinetics of dopamine, and 3) its adhesiveness that often causes aggregation or uncontrolled adsorption to any surface.
Herein, a G‐quadruplex (G4)/hemin‐based DNA enzyme (DNAzyme) possessing a horseradish peroxidase (HRP)‐mimicking H2O2‐mediated oxidation ability was employed as redox catalyst (Scheme 1). This strategy has been reported for catalyzing the oxidative polymerization of polyaniline,16 but has never been explored for controlling dopamine polymerization. In detail, the control of dopamine polymerization centers on the DNAzyme, which is known to oxidize dopamine to dopaminochrome. This oxidized state of dopamine is one of the intermediates for PD formation17 and Gao et al has separately found that the HRP could accelerate the polymerization of dopamine.18 In combining both concepts, we speculate that organizing multiple DNAzyme moieties at distinct nanodomains on DNA origami provides the local environment allowing a controlled dopamine polymerization.
Scheme 1.
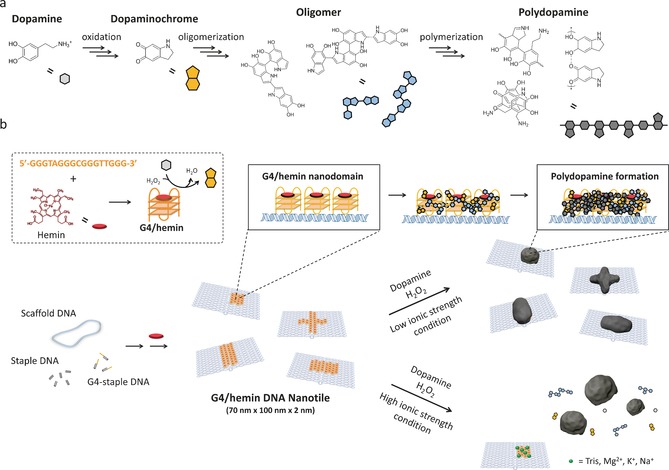
a) Proposed mechanism of PD formation (also see Figure S1) and b) Illustration of fabricating defined PD nanostructure on the DNA nanotile (light blue) carrying G4 (orange)/ hemin (red) DNAzyme domain. The DNAzyme domain locally oxidizes dopamine to dopaminochrome. And further processes yield PD. On the other hand, no PD formation on DNA nanotile occurs under the high ionic strength conditions (also see Figure S8).
To introduce the DNAzyme reaction center at distinct locations on DNA origami, 20 staple DNA sequences were modified with additional G‐rich sequences (TTGGGTAGG GCGGGTTGGG) at their 3′ ends (see Figure S2, S3 and Table S1 in the Supporting Information). The introduced G4 moieties were designed to appear as a 20 nm square domain at the surface of the Rothemund's DNA origami rectangle sheet (Figure S3a, 70 nm×100 nm dimensions),3 denoted as DNA nanotile. AFM measurements proved the correct formation of the DNA nanotile with desired dimensions and the G4 domain with an approximate 2 nm height increase at the designated positions (Figure S3c).
In the next step, DNAzyme on DNA nanotile was activated by incorporating its cofactor, hemin, resulting in no morphological differences by AFM (Figure S3c), but a characteristic shift of the hemin Soret band was detected in the UV/Vis spectra (Figure S4) that corresponds to existing literature.19 The catalytic activity of DNAzyme domain on DNA nanotile was evaluated by tracking the oxidation of 2,2′‐azino‐bis(3‐ethylbenzothiazoline‐6‐sulphonic acid (ABTS) in the presence of H2O2. It was observed that the activity of G4/hemin DNA nanotile was relatively low in TAE/ Mg buffer (20 mm Tris, 10 mm acetic acid, 1 mm EDTA, 12 mm MgCl2, pH 7.8), which is commonly used for DNA origami storage and functionalization (Figure S5). The DNAzyme activity was improved by supplementing 10 mm K+, which is known to stabilize G4.20 Additionally, lowering pH to acidic conditions (pH 5.3) significantly improved the activity profile while preventing oxidation of dopamine by dissolved oxygen. It should be also noted that G4/hemin domain on DNA nanotile showed an almost five fold faster reaction kinetic compared to free G4/hemin molecules with the same concentration, which was possibly due to a cooperative effect as a consequence of the locally concentrated G4/hemin reaction centers (Figure 1 a). Fabrication of a PD nanostructure on G4/hemin DNA nanotile was initially pursued in the optimized buffer as discussed above. However, no changes were observed from the G4/hemin DNA nanotile in AFM measurements, but many small particles were found in solution (Figure S6). Most likely, the high ionic strength and cation concentrations (tris‐ethylenediamine and Mg2+) could shield the negative charges of the DNA backbone.16 In this way, dopamine molecules that were oxidized to dopaminochrome on the DNA nanotile would rapidly diffuse back into the bulk solution and spontaneously self‐polymerize in solution.
Figure 1.
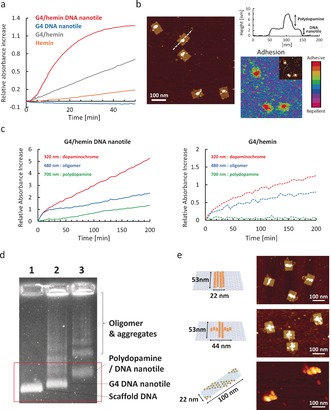
a) ABTS assay of G4/hemin DNA nanotile (3.5 nm containing 70 nm G4/hemin) and G4/hemin only (70 nm). b) AFM image of DNA nanotile after PD formation and adhesion mapping (inset: corresponding height image). c) Reaction kinetics of PD formation on G4/hemin DNA nanotile (3.5 nm, left) and G4/hemin only (70 nm, right). d) AGE of 1: M13MP18 DNA, 2: G4 DNA nanotile, and 3: PD/ DNA nanotile. e) Illustrations of DNA nanotile with different G4/hemin (yellow circle) positions (vertical line [top], cross patterning [middle], striped tube [bottom]) and corresponding AFM images.
This self‐polymerization in solution was suppressed efficiently, when a buffer containing low concentrations of TAE/Mg/K (0.3 mm Tris, 0.2 mm acetic acid, 0.06 mm EDTA, 0.6 mm MgCl2, 10 mm KCl, pH 5.3) was applied (Figure S5). Only under these conditions, new objects appeared on the DNAzyme domain (Figure 1 b). The AFM image showed the height increase of the DNAzyme domain from 2 nm to about 4–6 nm after the reaction. Additionally, mechanical property mapping revealed an increase in adhesion force by 73±16 pn from the DNA nanotile (Figure 1 b and S7), which taken together, indicated the successful PD formation. It is hypothesized that the electrostatic interaction between positively charged dopaminochrome/dopamine and negatively charged DNA nanotile coupled with multiple DNAzyme molecules in close proximity facilitated a high local concentration of PD intermediates including oligomers, leading to the preferential polymerization directly at the DNAzyme domain (Figure S8).
To further characterize PD formation, the reaction kinetics were monitored by UV/Vis spectroscopy (Figure 1 c and S9). The early oxidation (dopaminochrome, 320 nm) and polymerization products (oligomer, 480 nm) formed immediately after initiating the reaction, followed by the occurrence of PD (700 nm), after 50 min.21 A control reaction conducted with free DNAzyme molecules in solution could only produce dopaminochrome and oligomers, but no PD (Figure 1 c and S9), clearly indicating that multiple DNAzymes in the immediate vicinity at low ionic strength were essential for producing PD locally and directly on the DNAzyme domain. Additionally, an experiment replacing dopamine with its neutral analog l‐Dopa (at pH 5.3) revealed no polymerization (Figure S10), further strengthening the importance of the association of dopamine and its intermediates with the DNAzyme domain. As a control without hemin, PD was not formed at all due to a lack of DNAzyme activity (Figure S11). The quantity of PD and therefore the height increase was controlled and tuned with the amount of H2O2 added to the reaction (Figure S12). Anchoring of the PD to G4 locally limits the growth providing control over the final shape and size of the nanostructures. PD nanostructures grown on the DNA nanotile were purified by size exclusion chromatography for agarose gel electrophoresis (AGE) (Figure 1 d).
Next, we applied these optimized reaction conditions to fabricate different PD nanopatterns since the framework of DNA origami allows the positioning of every single DNAzyme moiety on the DNA nanotile with molecular precision. Vertical line, horizontal line, cross and striped patterning of DNAzyme domains were designed on DNA nanotile and PD nanostructures adopting similar shapes were achieved (Figure 1 e, S2, S13, and S14). And under these optimized reaction conditions, there was no polydopamine induced aggregation of DNA origami observed via AFM.
One of the major features of PD is related to its high adhesiveness, serving as glue in many applications. We have exploited the precise positioning of the adhesive PD nanostructures to induce and control DNA origami folding. By growing PD from the DNA tile with horizontal line patterning at the edge or in the middle, partially or fully folded DNA nanotile structures were observed (Figure 2, S15 and S16). This unique feature is likely due to the secondary interaction between the functional groups on PD surfaces (Mg2+ bridging, hydrogen bonding, π–π stacking).
Figure 2.
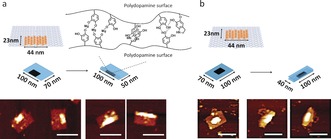
PD‐induced folding of DNA nanotile with horizontal line‐shape DNAzyme domain. Depending on the position of DNAzyme domain (a: side, and b: middle), DNA nanotile was folded either partially (a) or completely (b). Scale bar: 100 nm.
By adjusting the position of the PD “nanoglue” in the center or closer to the outer edge of the origami nanotile, different folded origami shapes could be imaged by AFM (Figure 2 a and b). On the other hand, when the line DNAzyme domain was designed along the short axis (70 nm), most of the DNA nanotile structures remained as non‐folded states (Figure S17). We believe that the rigid dsDNA parallel to the long axis most likely inhibited folding along its short axis where the energy required for the curvature cannot be compensated by the non‐covalent interactions. This observation shows that PD could serve as “supramolecular glue” for controlling different DNA origami conformations with low nanometer resolution, which outperforms other, less precise methods, for example, by using DNA intercalators.22 The formation of precise polymeric architectures with nanoscale precision similar to biomolecules such as proteins, tRNA has always been a holy grail in polymer chemistry. Dopamine is unique as it serves as monomer as well as cross‐linker thereby leading to rigid and highly crosslinked materials after polymerization. Therefore, PD nanostructures formed on DNA origami preserved their engineered size and geometry after removal of the template given the innate stability differences between PD and DNA. DNA origami is unstable in acidic pH and quickly degrades due to the hydrolysis of the phosphodiester backbone, bases, and glycosidic bonds.22 Hence, the liberation of PD nanostructures was achieved by treating the deposited PD/DNA on mica surface with 1 m HCl for 10 seconds (Figure 3 and S18). Subsequent replacement of the buffer and re‐engagement of AFM imaging revealed the structural preservation of the PD architecture (Figure 3).
Figure 3.
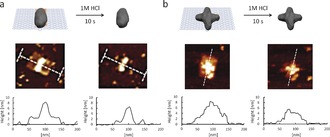
Extraction of PD nanostructure (a: vertical line, b: cross) from DNA nanotile via HCl treatment.
In summary, we have presented the first strategy for creating defined anisotropic PD nanostructures on DNA origami. The most valuable features of this approach are the precise localization of dopamine polymerization areas in nanoscale by using precisely patterned multiple DNAzyme as reaction centers. In the method reported herein, dopamine molecules are oxidized locally at DNAzyme domains on DNA origami resulting in the formation of PD structures with nanoscale precision. Also, the well‐known random and uncontrolled self‐polymerization behavior of dopamine in solution was efficiently subverted by conducting the reaction in acidic pH. The kinetics of PD growth and the control over the height of the architecture was achieved by adjusting the H2O2 concentration. Coupled with the spatial programmability of functionalities on DNA origami, one could envision creating various desired shapes of PD nanostructures in 2D and 3D with unprecedented resolution.
Based on the unique secondary adhesive properties of PD, conformational changes of the DNA origami nanostructures were induced. Here, PD served as “supramolecular glue”, connecting different areas of the DNA origami in a fast and controlled fashion, which serves as a versatile tool to manipulate the post‐modified conformation of DNA origami. The robust and tightly crosslinked PD features allow programmed PD nanostructures to retain their shapes after removal of the template. In this way, the organization of nanostructured PD over large surface areas24 as a nanoarray or the application of distinct PD shapes to probe and optimize cell interactions could be envisaged. Collectively, the strategy reported herein provides an integrated methodology for designing functional DNA nanodevices and for fabricating PD with spatial control, which will create new frontiers in DNA nanotechnology and the various fields of material science and nanomedicine.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We acknowledge the support by the European Research Council (ERC) under the program Synergy Grant 319130‐BioQ and the BMBF project “Selekomm” within the Biotechnologie 2020+ initiative. C.C. thanks for support from the Promotionskolleg Pharmaceutical Biotechnology. We thank Dr. Rüdiger Berger for critical reading of the manuscript and for many valuable comments.
Y. Tokura, S. Harvey, C. Chen, Y. Wu, D. Y. W. Ng, T. Weil, Angew. Chem. Int. Ed. 2018, 57, 1587.
Contributor Information
Prof. Dr. Yuzhou Wu, Email: wuyuzhou@hust.edu.cn
Dr. David Y. W. Ng, Email: david.ng@mpip-mainz.mpg.de.
Prof. Dr. Tanja Weil, Email: tanja.weil@mpip-mainz.mpg.de.
References
- 1.
- 1a. Geissler M., Xia Y. N., Adv. Mater. 2004, 16, 1249–1269; [Google Scholar]
- 1b. Hammond P. T., Adv. Mater. 2004, 16, 1271–1293; [Google Scholar]
- 1c. Kim P., Lee S. E., Jung H. S., Lee H. Y., Kawai T., Suh K. Y., Lab Chip 2006, 6, 54–59. [DOI] [PubMed] [Google Scholar]
- 2.
- 2a. Endo M., Yang Y., Sugiyama H., Biomater. Sci. 2013, 1, 347–360; [DOI] [PubMed] [Google Scholar]
- 2b. Okholm A. H., Kjems J., Adv. Drug Delivery Rev. 2016, 106, 183–191; [DOI] [PubMed] [Google Scholar]
- 2c. Komiyama M., Yoshimoto K., Sisido M., Ariga K., Bull. Chem. Soc. Jpn. 2017, 90, 967–1004. [Google Scholar]
- 3. Rothemund P. W. K., Nature 2006, 440, 297–302. [DOI] [PubMed] [Google Scholar]
- 4. Veneziano R., Ratanalert S., Zhang K. M., Zhang F., Yan H., Chiu W., Bathe M., Science 2016, 352, 1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.
- 5a. Schreiber R., Do J., Roller E. M., Zhang T., Schuller V. J., Nickels P. C., Feldmann J., Liedl T., Nat. Nanotechnol. 2014, 9, 74–78; [DOI] [PubMed] [Google Scholar]
- 5b. Zhang T., Neumann A., Lindlau J., Wu Y., Pramanik G., Naydenov B., Jelezko F., Schüder F., Huber S., Huber M., Stehr F., Högele A., Weil T., Liedl T., J. Am. Chem. Soc. 2015, 137, 9776–9779. [DOI] [PubMed] [Google Scholar]
- 6.
- 6a. Saccà B., Meyer R., Erkelenz M., Kiko K., Arndt A., Schroeder H., Rabe K. S., Niemeyer C. M., Angew. Chem. Int. Ed. 2010, 49, 9378–9383; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 9568–9573; [Google Scholar]
- 6b. Fu J. L., Yang Y. R., Johnson-Buck A., Liu M. H., Liu Y., Walter N. G., Woodbury N. W., Yan H., Nat. Nanotechnol. 2014, 9, 531–536. [DOI] [PubMed] [Google Scholar]
- 7.
- 7a. Dutta P. K., Varghese R., Nangreave J., Lin S., Yan H., Liu Y., J. Am. Chem. Soc. 2011, 133, 11985–11993; [DOI] [PubMed] [Google Scholar]
- 7b. Lin C. X., Jungmann R., Leifer A. M., Li C., Levner D., Church G. M., Shih W. M., Yin P., Nat. Chem. 2012, 4, 832–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.
- 8a. Helmi S., Ziegler C., Kauert D. J., Seidel R., Nano Lett. 2014, 14, 6693–6698; [DOI] [PubMed] [Google Scholar]
- 8b. Sun W., Boulais E., Hakobyan Y., Wang W. L., Guan A., Bathe M., Yin P., Science 2014, 346, 717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.
- 9a. Yang Y., Wang J., Shigematsu H., Xu W. M., Shih W. M., Rothman J. E., Lin C. X., Nat. Chem. 2016, 8, 476–483; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9b. Zhang Z., Yang Y., Pincet F., Llaguno M. C., Lin C. X., Nat. Chem. 2017, 9, 653–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Tokura Y., Jiang Y. Y., Welle A., Stenzel M. H., Krzemien K. M., Michaelis J., Berger R., Barner-Kowollik C., Wu Y. Z., Weil T., Angew. Chem. Int. Ed. 2016, 55, 5692–5697; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 5786–5791. [Google Scholar]
- 11. Lee H., Dellatore S. M., Miller W. M., Messersmith P. B., Science 2007, 318, 426–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.
- 12a. Lee H., Rho J., Messersmith P. B., Adv. Mater. 2009, 21, 431–434; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12b. Liu R., Guo Y. L., Odusote G., Qu F. L., Priestley R. D., ACS Appl. Mater. Interfaces 2013, 5, 9167–9171. [DOI] [PubMed] [Google Scholar]
- 13.
- 13a. Lynge M. E., van der Westen R., Postma A., Städler B., Nanoscale 2011, 3, 4916–4928; [DOI] [PubMed] [Google Scholar]
- 13b. Ding Y. H., Floren M., Tan W., Biosurface and Biotribology 2016, 2, 121–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chien H. W., Kuo W. H., Wang M. J., Tsai S. W., Tsai W. B., Langmuir 2012, 28, 5775–5782. [DOI] [PubMed] [Google Scholar]
- 15.
- 15a. Della Vecchia N. F., Avolio R., Alfe M., Errico M. E., Napolitano A., d'Ischia M., Adv. Funct. Mater. 2013, 23, 1331–1340; [Google Scholar]
- 15b. Liebscher J., Mrowczynski R., Scheidt H. A., Filip C., Hadade N. D., Turcu R., Bende A., Beck S., Langmuir 2013, 29, 10539–10548; [DOI] [PubMed] [Google Scholar]
- 15c. Liu Y. L., Ai K. L., Lu L. H., Chem. Rev. 2014, 114, 5057–5115. [DOI] [PubMed] [Google Scholar]
- 16. Wang Z. G., Liu Q., Ding B. Q., Chem. Mater. 2014, 26, 3364–3367. [Google Scholar]
- 17.
- 17a. Albada H. B., Golub E., Willner I., Chem. Sci. 2016, 7, 3092–3101; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17b. Golub E., Albada H. B., Liao W. C., Biniuri Y., Willner I., J. Am. Chem. Soc. 2016, 138, 164–172. [DOI] [PubMed] [Google Scholar]
- 18. Li J., Baird M. A., Davis M. A., Tai W., Zweifel L. S., Waldorf K. M. A., M. Gale, Jr. , Rajagopal L., Pierce R. H., Gao X., Nat. Biomed. Eng. 2017, 1, 0082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yang X., Fang C., Mei H., Chang T., Cao Z., Shangguan D., Chem. Eur. J. 2011, 17, 14475–14484. [DOI] [PubMed] [Google Scholar]
- 20. Travascio P., Li Y. F., Sen D., Chem. Biol. 1998, 5, 505–517. [DOI] [PubMed] [Google Scholar]
- 21. Bisaglia M., Mammi S., Bubacco L., J. Biol. Chem. 2007, 282, 15597–15605. [DOI] [PubMed] [Google Scholar]
- 22. Chen H., Zhang H., Ping J., Cha T., Li S., Andreasson J., Choi J. H., ACS Nano 2016, 10, 4989–4996. [DOI] [PubMed] [Google Scholar]
- 23. Kim H., Surwade S. P., Powell A., O'Donnell C., Liu H. T., Chem. Mater. 2014, 26, 5265–5273. [Google Scholar]
- 24.
- 24a. Hung A. M., Micheel C. M., Bozano L. D., Osterbur L. W., Wallraff G. M., Cha J. N., Nat. Nanotechnol. 2010, 5, 121–126; [DOI] [PubMed] [Google Scholar]
- 24b. Gopinath A., Rothemund P. W. K., ACS Nano 2014, 8, 12030–12040; [DOI] [PubMed] [Google Scholar]
- 24c. Aghebat Rafat A., Pirzer T., Scheible M. B., Kostina A., Simmel F. C., Angew. Chem. Int. Ed. 2014, 53, 7665–7668; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 7797–7801. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary