

Abstract
Tumor metabolism is a thrilling discipline that focuses on mechanisms used by cancer cells to earn crucial building blocks and energy to preserve growth and overcome resistance to various treatment modalities. At the same time, therapies directed specifically against aberrant signalling pathways driven by protein tyrosine kinases (TKs) involved in proliferation, metastasis and growth count for several years to promising anti-cancer approaches. In this respect, small molecule inhibitors are the most widely used clinically relevant means for targeted therapy, with a rising number of approvals for TKs inhibitors. In this review, we discuss recent observations related to TKs-associated metabolism and to metabolic feedback that is initialized as cellular response to particular TK-targeted therapies. These observations provide collective evidence that therapeutic responses are primarily linked to such pathways as regulation of lipid and amino acid metabolism, TCA cycle and glycolysis, advocating therefore the development of further effective targeted therapies against a broader spectrum of TKs to treat patients whose tumors display deregulated signalling driven by these proteins.
Keywords: Tyrosine kinase inhibitors, Metabolomics, Targeted therapies, Glycolysis, Glucose, TCA cycle, Energy metabolism, Amino acids, Lipid metabolism
Background
The switch from normal tissue to malignancy is a result of oncogenes-driven biochemical processes aimed at sustaining an accelerated rate of proliferation and growth [1]. Otto Warburg in 1956 described for the first time a specific metabolic characteristic of neoplasms by demonstrating that a cancer cell, unlike an untransformed cell, relies mainly on a higher glycolytic flux without a change in oxidative phosphorylation even in the presence of oxygen [2]. The so-called Warburg effect is nowadays considered a major hallmark of cancer and numerous studies have been repeatedly reporting that various metabolic pathways appear to be distinctive in individual tumour cells [3, 4]. Many of these alterations emerge as a consequence of the gain of mutations accumulated during oncogenesis, providing proliferative advantage for cancer cells in their microenvironment.
In recent years, in addition to investigating the role of cell metabolism in tumor cell development, particular attention has been devoted to metabolic changes occurring as a response to targeted treatments [5–7]. In view of the role that TKs seem to play in the regulation of cellular metabolism [8–11], it is crucial to determine whether the antitumor activity of particular tyrosine kinase inhibitors (TKIs) is related to their effect at a given metabolic level. Such insights may subsequently serve as an important ground for novel personalized therapeutic options and combination treatments. Assessment of biological conformity in changes in metabolites following administration of a particular TKI has already shown to provide important translational observations as to particular sensitive metabolic pathways [12]. Consequently, metabolomics has the potential to identify subgroups of patients that are likely to profit from given targeted perturbations and, of a similar importance, determine subgroups that may encounter toxicity or resistance.
Protein kinases constitute an immense enzyme family that emerges as a strikingly valuable set of targets in therapy of various tumors considering their high sensitivity to specific kinase inhibitors, which are often relatively well tolerated by normal cells. Development of TKIs created a therapeutic window for selective diminishing of malignancies with constitutively active kinase. The majority of these compounds share a common mechanism of action – they competitively inhibit adenosine triphosphate (ATP) at the catalytic binding site of the targeted protein [13]. As aforementioned, accumulating evidence suggests that key oncogenic pathways program the adaptation of metabolism with explicit changes for the selective advantage of tumor cells, many of them regulated by tyrosine kinase activity [14–16]. In this review, we summarize and discuss principal metabolic changes following administration of particular kinase inhibitors on different levels of cellular metabolism (key metabolites and molecules affected by TKIs in cancer are summarized in Table 1).
Table 1.
Summary of key metabolites and molecules affected by TKIs in cancer. Up- or downregulation highly depends on the inhibitor and model of the study used
| Metabolite | Function in | Sense of Regulation | Rerence(s) |
|---|---|---|---|
| Fructose 1,6-bisphosphate | glycolysis | ↓ | [27, 34, 38, 41, 45, 48, 49, 58] |
| Dihydroxyacetone phosphate | |||
| 3-phosphoglycerate | |||
| Glucose (consumption) | |||
| Phosphoenolpyruvate | glycolysis and gluconeogenesis | ↓ | [22, 27, 38, 45] |
| Lactate | |||
| Glyceraldehyde 3-phosphate | |||
| Pyruvate | |||
| 6-phosphogluconate | pentose phosphate pathway | ↓ | [27, 58] |
| Ribulose-5-phosphate | |||
| Ribose-5-phosphate | |||
| Xylulose-5-phosphate | |||
| D-sedoheptulose 1,7-bisphosphate | pentose phosphate pathway | ↑ | [34] |
| Deoxyribose phosphate | |||
| Glucose-6-phosphate | glycolysis and PPP | ↓ | [27, 58] |
| Glutamate | amino acid metabolism | ↑ | [27, 30, 34, 45, 74] |
| Valine | |||
| Lysine | |||
| Tyrosine | |||
| Aspartate | |||
| Proline | |||
| Threonine | |||
| Histidine | |||
| Asparagine | |||
| Tryptophan | |||
| Alanine | |||
| NADPH | pentose poshosphate pathway | ↓ | [34, 51] |
| oxidation-reduction pathways | |||
| ATP, GTP, CTP, TTP | energy metabolism | ↑ | [32, 45, 58] |
| Fumarate | TCA cycle | ↓ | [27, 30] |
| Malate | |||
| Citrate | |||
| Arginine | amino acid metabolism | ↓ | [74, 76] |
| Citrate | TCA cycle | ↑ | [32] |
| ATP | energy metabolism | ↓ | [60, 62] |
| Phosphocholine | glycerophospholipid metabolism | ↓ | [45, 87–89] |
Abbreviations: ↑—Up-regulation; ↓—Down-regulation; TCA cycle Tricarboxylic acid cycle; NADPH Nicotinamide adenine dinucleotide phosphate; ATP Adenosine triphosphate; GTP Guanosine triphosphate; CTP Cytidine triphosphate; TTP Thymidine triphosphate
Impact of TKIs on Glycolysis and glucose-related pathways
As metabolic reprogramming towards aerobic glycolysis has been suggested as one of the hallmarks of cancer, considerable research efforts focused for over a decade on enzymes and metabolites of the glycolytic pathway following antineoplastic treatments. Glucose metabolism, a paramount energetic resource for the cell, is a very complex process regulated in neoplastic cells by different oncogenes on multiple levels, ranging from transcription to post-translation modifications [14]. In that respect, for example, c-MYC controls key metabolic enzymes including those that are involved in glucose metabolism such as hexokinase 2 (HK2), glucose transporter 1 (GLUT1), pyruvate kinase muscle isozyme 2 (PKM2) and lactate dehydrogenase A (LDHA) [17].
Oncogene-conducted activation of glycolytic pathway takes frequently place through hypoxia-inducible factor 1α (HIF-1α) [18, 19]. The already mentioned Warburg effect is a result of deregulated genes, leading to upregulation of glucose transporters 1 and 3, with resulting elevated glucose consumption [20, 21]. Glucose metabolism does not necessarily encompass glycolysis only. Indeed, other glucose-related metabolic pathways, as the pentose phosphate pathway (PPP), which provides nicotinamide adenine dinucleotide phosphate (NADPH), the hexosamine pathway, a minor branch of glycolysis needed for glycosylation of proteins, and glycogenesis that generates glycogen used as a glucose repository, are all critical branches of cellular glucose metabolism [22]. Since it has been shown that many RTKs inhibitors suppress among others also metabolic pathways as for example the PI3K/Akt pathway, it is expected that they would inhibit glucose metabolism in a similar manner [23, 24]. In this section we summarize how glycolysis and other glucose-related pathways are reprogrammed in malignant cells following particular TKI targeting (summarized in Fig. 1).
Fig. 1.
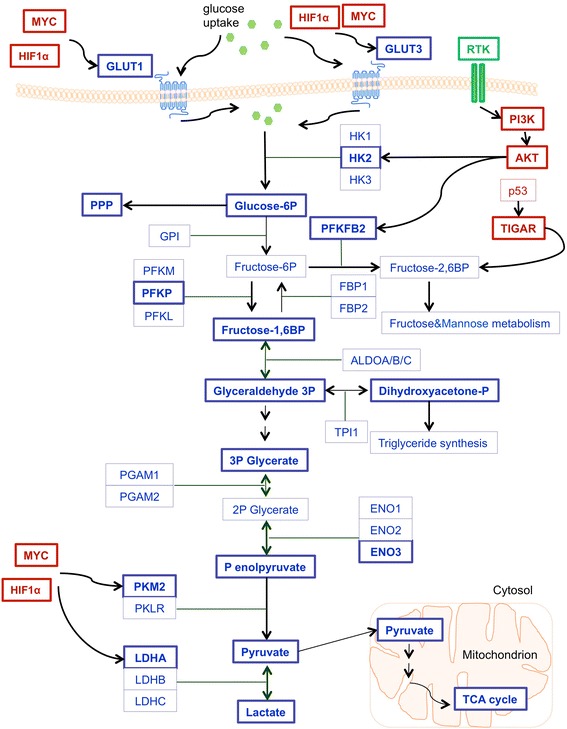
TKI-induced regulation of glycolytic pathway. Highlighted in bold are proteins and metabolites (blue) together with glycolytic regulators (red) that were shown to be affected by the inhibition of TKs. Abbreviations: GLUT1/3—glucose transporter 1/3; HK1/2/3—hexokinase 1/2/3; TIGAR—TP53-inducible glycolysis and apoptosis regulator; P—phosphate; BP—bisphosphate; PPP—pentose phosphate pathway; GPI—glucose-6-phosphate isomerase; PFKFB2— 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase 2; PFK—6- phosphofructokinase(three isoforms – muscle (PFKM), liver (PFKL) and platelet (PFKP)); FBP1/2—fructose-bisphosphatase 1/2; ALDOA/B/C—aldolase A/B/C; TPI1—triosephosphate isomerase; PGAM1/2—phosphoglycerate mutase 1/2; ENO1/2/3—enolase 1/2/3; PKM2—pyruvate kinase isozyme M2; PKLR—Pyruvate kinase isozymes L/R; LDHA/B/C—lactate dehydrogenase A/B/C; TCA cycle—tricarboxylic acid cycle
ErbB family
Epidermal growth factor receptor (EGFR)
EGFR, a broadly studied RTK system, is overexpressed, deregulated and mutated in a large number of malignancies. Specifically, EGFR protein overexpression was detected in tumors of breast, brain, cervix, ovary, colon, head and neck and lung [25, 26], creating a strong motivation to develop novel antitumor agents focused on EGFR.
The 2014 study from Makinoshima and collaborators [27] provided one of the first comprehensive analyses of EGFR TKI-mediated modulations of metabolism. The presence of EGFR TKIs erlotinib (Tarceva®) and gefitinib (Iressa®) repressed lactate production and glucose consumption in three distinct lung adenocarcinoma (LAD) cell lines, HCC827, NCI-H1975 and PC-9 [27]. Importantly, HCC827 and PC-9 both carry the EGFR exon 19 delE746-A750 mutation and are sensitive to EGFR TKIs whereas H1975 harbors the EGFR L858R + T790 M mutation, which causes resistance to both gefitinib and erlotinib [28]. The authors hypothesized that lactate production is regulated by MYC via transcriptional regulation, since MYC is decreased at both protein and mRNA levels following treatment by EGFR TKIs. Interestingly, western blot analysis showed that MYC-regulated proteins HK2 and GLUT3, but not GLUT1, were reduced in EGFR TKI-sensitive cell lines upon treatment [27]. Metabolome analysis using Capillary Electrophoresis Time of Flight Mass Spectrometer (CE-TOFMS) exposed intermediate key metabolites in glucose metabolism that were altered following erlotinib treatment in both EGFR TKI-sensitive cell lines HCC827 and PC-9. Specifically, fructose 1,6-bisphosphate (FBP), dihydroxyacetone phosphate (DHAP), 3-phosphoglycerate (3PG), phosphoenolpyruvate (PEP), lactate (LA), and 6-phosphogluconate (6PG) were all decreased in TKI-sensitive HCC827 and PC9 cells after 6 h of erlotinib treatment, but not in TKI-resistant NCI-H1975 cells [27]. Furthermore, PPP metabolites, glucose 6-phosphate (G6P), glyceraldehyde 3-phosphate (G3P), pyruvate (PA), ribulose 5-phosphate (Ribu5P), and ribose 5-phosphate (R5P) were significantly reduced in both HCC827 and PC9 cells [27]. Measuring the extracellular acidification rate (ECAR), an indirect readout of the glycolytic rate, Lim et al. reported an attenuation of ECAR by the co-treatment with EGF stimulation together with gefitinib in an EGFR-overexpressing breast cancer cell line MDA-MB-468 [29]. Moreover, they showed that EGFR binds, phosphorylates and inhibits PKM2, a rate-limiting glycolytic enzyme that catalyses the last glycolysis step [29]. On the contrary, ECAR was increased in triple-negative breast cancer (TNBC) mesenchymal-like cell lines MDA-MB-231 and Hs578T upon treatment by erlotinib or the MET inhibitor capmatinib (INC280) [30]. The impact of EGFRi on glycolysis was further confirmed by the Heath group in 2015, who reported, as assessed by the 18F–FDG radioassay, a reduction of consumption of glucose and hexokinase activity following erlotinib treatment in patient-derived glioblastoma (GBM) neurosphere tumor cells (GBM39) that express EGFR [31]. Outlining similarities with other authors’ models, further recent report conducted by De Rosa et al., where one EGFR inhibition-sensitive cell line (HCC827) and two EGFR inhibition-resistant cell lines (H1975 and H1993 (both bearing MET gene amplification)) were exposed to WZ4002 (a specific EGFRT790M inhibitor), erlotinib or PHA665752 (a first generation MET inhibitor) and their impacts on glycolytic enzymes and transporters were investigated [32]. Although protein levels of HKI, PKM1/2 and GLUT1 remained consistent across all cell lines, all three studied inhibitors led to a concentration-dependent downregulation of HKII and to upregulation of levels of GLUT3 with efficient inhibitors of the corresponding cell line (curiously, the levels of GLUT3 were upregulated after 72 h treatment of H1975 with WZ4002 or following treatment of H1993 cells with PHA665752) [32]. Moreover, a reduction of pPKM2 was observed in HCC827 and H1993 treated with erlotinib and PHA665752, respectively [32]. The in vitro observations were further substantiated in vivo by using H1975 and H1993 cells injected into female BALB/c (nu/nu) mice treated with WZ4002 and crizotinib (Xalkori® a MET inhibitor), respectively [32]. This differential regulation of glycolysis brings a rationale for a potential combination therapy targeting both the EGFR pathway and glucose metabolism for enhanced therapeutic effect [32]. Interestingly, the impact of EGFR inhibition on glucose-related metabolism was recently substantiated both in cell culture and in vivo using HCC827 and H1650 (bearing E746-A750 deletion of exon 19) cell lines, where erlotinib therapy reduced expression of MYC and HIF1α and their downstream targets GLUT1, HKII, neutral amino acid transporter B(0) (SLC1A5) together with sodium-coupled neutral amino acid transporter 1 (SLC38A1) [33]. These results further correlated with decreased 18F–FDG and 11C–Gln uptake seen in HCC827 xenografts following the erlotinib treatment [33]. In addition, metabolic profiling of myeloma cancer cells LP-1 (no NRAS, KRAS or BRAF mutation), L-363 (harbouring NRAS mutation), RPMI-8226 (KRAS mutation), and U-266 (BRAF mutation) revealed that following the treatment with gefitinib, metabolites from the PPP such as ribose-phosphate, D-sedoheptulose-1,7-bisphosphate, O8P-O19 and deoxyribose phosphate were significantly increased in LP-1 cell line and unchanged in KRAS/NRAS/BRAF mutant myeloma cancer cells [34]. As PPP is a main source of NADPH supplying R5P for nucleotide synthesis, the authors hypothesized that the upregulation of these metabolites is a metabolic compensatory mechanism to prevent complete therapeutic response towards EGFR inhibition [34]. This hypothesis was experimentally confirmed by the use of the antimetabolite 6AN, a PPP inhibitor, together with gefitinib [34] The combinational therapy supressed the proliferation of LP-1 cells, which was recovered by supplementation of NADPH. Analogous results were reported using afatinib (Giotrif®), a dual EGFR and ERBB2 inhibitor [34] as well as in another study, where MET or EGFR inhibition both sensitized TNBC cell line MDA-MB-468 to nucleotide enzymes knockdown [30].
HER2
Similarly to EGFR, HER2, encoded by the ERBB2 gene, is also often overexpressed in cancer and its deregulation is associated with aggressive phenotype and shortened survival [35]. Targeting HER2 by the humanized murine monoclonal antibody trastuzumab (Herceptin®) leads to a 40% improved overall survival in patients with breast cancer that show approximately 15%–25% amplification or overexpression of HER2 [36, 37].
Zhao et al. reported that trastuzumab inhibits glucose uptake and lactate production in BT474 and ZR-7530 breast cancer cell lines without a change in cell growth inhibition, hypothesising that glycolysis inhibition is not a consequence of the cell growth inhibition [38]. Their previous study showed that the ErbB2-heat shock factor1 (HSF1)-lactate dehydrogenase A (LDHA) pathway has a main role in glucose regulation in breast cancer cells [39]. Therefore they suggested and subsequently also reported that trastuzumab inhibits glycolysis through downregulation of the HSF1-LDHA axis and, moreover, this axis contributes to the resistance of breast cancer cells to this monoclonal antibody [38]. Similar response on glycolysis was shown with lapatinib (Tykerb®), a dual inhibitor of EGFR and ErbB2/HER2 that is usually used in combination with capecitabine for the treatment of HER2-positive metastatic breast cancer [40]. Specifically, Komurov et al. reported that lapatinib treatment of ErbB2-positive SKBR3 breast cancer cells induced glucose deprivation, suggesting a blockage of glucose-dependent EGFR/HER2 signaling [41]. Additional study by Ruprecht et al. unveiled that phosphorylation of Ser466 of 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase 2 (PFKFB2) is inhibited following lapatinib treatment in lapatinib-sensitive BT-474 breast cancer cell line, however it recovers to its initial levels of phosphorylation in lapatinib-resistant BT-474 clone BT-474-J4 [42]. Phosphorylation of Ser466 was reported to trigger PFKFB2 kinase activity that activates the production of metabolite fructose-2,6-bisphosphate, pointing out a possible link between lapatinib therapeutic action and metabolic reprogramming in resistance [42].
The results of research efforts focusing on ErbB2 family of RTKs strongly suggest that the decrease of intermediate metabolites in PPP and glycolysis such as lactate, FBP, G6P or R5P and the impairment of glycolysis-related enzymes such as GLUT1 and HK1 are not events resulting from inhibited proliferation but could potentially serve as biomarkers to predict the response to and, more importantly, efficacy of EGFR and HER2 TKI treatment.
BCR-ABL
BCR-ABL harbours a constitutively active form of the ABL TK and is present in more than 90% of chronic myeloid leukemia (CML) patients [43]. CML treatment was revolutionized by the use of the BCR-ABL TKI imatinib (formerly STI571, Gleevec®), a compound that was writing the first success stories in the field of targeted neoplastic treatment [43]. Imatinib provides effective and durable therapy: the treatment resulted in 5-year survival of approximately 90% for CML patients in clinical trials [44].
In 2004, Gottschalk et al. reported that imatinib treatment changed glucose metabolism from anaerobic glycolysis to the aerobic mitochondrial TCA cycle in two human BCR-ABL-positive cell lines CML-T1 and K562 but not in BCR-ABL-negative cell line HC-1 [45]. Interestingly, metabolic responses to imatinib were dependent on the concentration of the molecule. When using a concentration of 0.25 μmol/L, which is below imatinib’s IC50 value (for CML-T1 IC50 is 0.69 ± 0.06 μmol/L and for K562 IC50 is 0.47 ± 0.04 μmol/L), lactate production was reduced in BCR-ABL-positive cell lines and concurrently, the production of glutamate increased, thus suggesting increased employment of the mitochondrial glucose pathway; when using a concentration above its IC50 value (2.5 μmol/L), no activation of TCA cycle was observed [45]. Moreover, imatinib was able to increase extracellular glucose in the lyophilized media of BCR-ABL-positive cell lines in contrary to media coming from the BCR-ABL-negative cell line, where the concentration of extracellular glucose did not change [45]. Consequently, this resulted in an increased ratio of extracellular to intracelullar glucose and decreased glucose uptake in BCR-ABL-positive cells [45]. These data correlate with previous findings of Boros et al. who showed that imatinib regulates glycolysis through downregulation of GLUT1 in human leukemia cells [46]. In fact, BCR-ABL-positive haemopoietic cells TonB210 express high affinity GLUT1 and demonstrate increased glucose uptake [47]. Following the treatment in vitro, imatinib led to the internalization of 90% of GLUT1 and drastically decreased hexose uptake [47]. A study carried out by the group of Serkova et al. aimed at understanding of development of imatinib resistance metabolic phenotype in CML, using imatinib-sensitive K562-s and LAMA84-s and imatinib-resistant K562-r and LAMA84-r cell lines [48]. By using nuclear magnetic resonance spectroscopy and gas chromatography mass spectrometry to assess 13C glucose uptake and metabolism, they showed that in both imatinib-sensitive cell lines, imatinib treatment (1 μmol/L) significantly decreased glucose uptake and lactate export together with reduced [4-13C]glutamate, in contrast to imatinib-resistant cell lines, suggesting a decrease in the activity of glycolysis together with TCA cycle [48]. To confirm their findings, they used 2-deoxy-d-glucose uptake assay and showed that imatinib-sensitive cell lines displayed decreased uptake of glucose, compared to imatinib-resistant cell lines that exhibit even higher glucose uptake, as a possible consequence of imatinib resistance progress [48]. To explain the drop in glucose uptake in imatinib-sensitive cell lines, they reported that imatinib inhibits glycolysis and translocates GLUT1 from the membrane into the cytosol, whereas GLUT1 remains located at the plasma membrane in resistant cell lines [48]. Interestingly, a decrease in 18-fluoro-2-deoxy-D-glucose (FDG) uptake was previously described in a case report of a patient with a jejunal gastrointestinal stromal tumor with multiple hepatic metastases treated with imatinib [49].
Studies employing BCR-ABL targeted therapy provided a rationale for the combined use of an inhibitor of glucose metabolism and kinase inhibitors for treatment of BCR-ABL-positive patients who acquired resistance either to classical chemotherapy or to the targeted treatment.
Met
The MET RTK for hepatocyte growth factor (HGF) is, analogously to other RTKs, actively involved in cell growth, migration and proliferation and additionally functions also as a main regulator of embryogenesis [50]. In a study published in 2011, Lui et al. used two nasopharyngeal cancer (NPC) cell lines, HK1-LMP1 and CNE-2, and described that protein levels of regulator of apoptosis and glycolysis, TP53-induced Glycolysis and Apoptosis Regulator (TIGAR), were reduced after the treatment with two MET TKIs (by AM7, a MET inhibitor binding to the kinase linker region and extending to a hydrophobic binding site and by a tool compound SU11274), indicating that the effect is induced by METi itself and does not depend on the exact nature of inhibitor used [51]. Previously, it was proposed that TIGAR inhibits apoptosis by regulation of cellular NADPH levels and via regulation of the PPP [52]. Indeed, they explored METi reduction of intracellular NADPH, a protector from oxidative stress and a driver of the force of most biosynthetic enzymatic reactions, responsible for biosynthesis of DNA, RNA, cholesterol and fatty acids [53, 54], in both NPC cell lines [51]. Interestingly, using a METi-sensitive SNU5 and METi-resistant SNU1 gastric cancer cell lines, expression of several glycolysis-related mitochondrial enzymes, such as voltage-dependent anion-selective channel protein 1 (VDAC1) and adenine nucleotide translocase 2 (ANT2), was significantly regulated in response to MET inhibitor PHA665752 [55]. Impact of MET inhibition on glucose metabolism was confirmed using H1975 NSCLC cancer cells in a xenograft model (Ncr-nu mice) monitored in vivo by FDG-PET (glucose analogue [18F]fluoro-2-deoxy-D-glucose-positron emission tomography) analysis with MRI [56]. Indeed, MET inhibitor SU11274-treated xenografts displayed a 45% drop in glucose metabolism compared to untreated controls [56].
In conclusion, analogously to findings pertaining to inhibition of ErbB2 family of receptors, MET inhibition also seems to modulate glucose metabolism and this observation could potentially serve as a mean to predict cancer cells responses to MET targeting-based treatments.
Other protein TKs
Anaplastic lymphoma kinase (ALK) is engaged in the induction and progression of various cancer types, including non-small cell lung cancer (NSCLC), neuroblastomas and lymphomas. ALK is usually targeted in clinical practice by crizotinib, approved for use in ALK-positive NSCLC [57]. Some preliminary work on impacts of ALK inhibition on cellular metabolism was carried out by McDonnell et al., focusing on anaplastic large cell lymphoma (ALCL) cell lines SU-DHL-1, DEL, Karpas299, SUPM2 and using ALK inhibitor CEP-26939 (CEP, unknown mechanism of action, Cephalon) [58]. Metabolomic analysis by both gas chromatography–mass spectrometry and liquid chromatography-mass spectrometry displayed a significant decrease in lactate following 3 h of treatment by 300 nM of CEP, which was accompanied by a decrease in phosphorylated LDH detected by phosphoproteomics via metal oxide affinity chromatography (MOAC) [58]. Using 13C–glucose, they could demonstrate that lactate in these cell lines was derived directly from glucose, suggesting reduction of glycolytic flux following ALK inhibition. Moreover, reduced glycolytic flux occurred to be due to a decreased glucose uptake and reduced metabolites such as FBP, G6P and F6P [58]. In addition, ribose-5-phosphate and xylulose-5-phosphate, main metabolites in PPP, were significantly downregulated following inhibition of ALK [58]. On contrary, no similar metabolic changes were detected in ALK-negative Jurkat cells treated by CEP, used as a negative control [58]. Of clinical importance is the fact that comparable results were observed also using crizotinib [58]. Altogether, the data in this study provided a rationale that PKM2 is functioning as a mediator of ALK-regulated metabolic switch as inhibition of ALK resulted in reduction of pY105 PKM2, without a change in total PKM2 levels [58].
Differently from what was reported previously using other TKIs, Hudson and colleagues treated mouse pancreatic ductal adenocarcinoma (PDAC) cell lines from pancreatic cancer mouse model (KrasG12DPdx1-cre) with axitinib (Inlyta®, mechanism of action through VEGFR, c-KIT and PDGFR) and did not observe the expected effect on glycolysis and [C-14] deoxyglucose uptake was increased in axitinib-treated cells after 24 and 48 h [59]. It has to be considered, however, that these experiments were performed with axitinib-resistant PDAC clones, surviving after longer incubation times or higher concentrations of axitinib [59]. These results suggest that the increased glucose uptake following axitinib treatment is involved in the resistance mechanism towards the inhibitor-induced anti-cancer effect. Moreover, the treatment with increasing concentrations of axitinib upregulated GLUT1 together with ECAR, proposing a way through which axitinib induces glucose uptake [59].
Sorafenib (Nexavar®), a multikinase inhibitor targeting BRAF, PDGFR and VEGFR, enhanced in the hepatocholangiocarcinoma cell line LCSC-2 the expression of GLUT3, Enolase 2 (ENO2), and the platelet phosphofructokinase (PFKP), three genes directly associated with glycolysis, hence suggesting a metabolic shift towards glucose metabolism [60]. Indeed, the response to sorafenib also induced uptake of the fluorescent glucose analogue 6NDBG, glucose consumption and lactate production [60]. The gene signature that emerges following treatment with sorafenib indicates an induction of glycolytic readjustment as a response to mitochondrial collapse [60].
In another study, FGFR1 inhibition by TKI258/dovitinib, a multikinase inhibitor (VEGFR, FGFR, PDGF, c-KIT, CSF-1R), significantly increased enzymatic activity of PKM2 in human myeloid leukemia cell line KG1, breast cancer cell line MDA-MB-134 and a lung cancer cell line NCI-H1299, all three of them overexpressing FGFR1 [61]. Additional data that suggest a role for FGFR1 in modulating glucose energy metabolism were provided recently by Fumarola et al. [62]. Using squamous cell lung cancer (SQCLC) cell lines H1703 and H520 after FGF2 induction, they could show that the protein expression of both HIF-1α and GLUT1 correlated with elevated glucose uptake, glycolysis, lactate production and elevated PKM2 activity. Treatment with a selective FGFR inhibitor NVP-BGJ398 or with a multikinase inhibitor dovitinib hindered all these processes, pointing towards AKT/mTOR pathway as a key player in this regard. Importantly, the involvement of FGFR1 signaling affecting glucose metabolism was equally confirmed in vivo with LENTI-4 cells with FGFR1 amplification generated from SQCLC SKMES-1 cells by lentiviral expression [62].
TCA cycle and energy metabolism
TCA cycle is commonly presented in a simple viewpoint of a cyclic mitochondrial pathway continually oxidizing acetyl-CoA to CO2, spawning NADH and FADH2, whose electrons are used in electron transport chain (ETC) to generate ATP for chemical and physical work within the cell [16]. Mitochondrial metabolism plays a role in tumorigenesis [63] and furthermore, major mitochondrial enzymes and pathways reinforce tumor progression induced by key oncogenic drivers [64, 65]. Dominant defects associated with oncogenesis were reported for succinate dehydrogenase (SDH), fumarate hydratase (FH) and isocitrate dehydrogenase (IDH) [66]. These mutations in enzymes underlie the mechanistic rationale on how alterations in the mitochondrial pathway can potentially change bioenergetics of the cell itself. In this chapter we discuss potent TKIs that were shown to perturb pathways and metabolites included in mitochondria metabolism such as TCA components, ETC complexes and metabolites related to oxidative phosphorylation (OXPHOS).
In the already mentioned study focusing on imatinib-treated BCR-ABL-positive cells, the increase of mitochondrial glucose metabolism following treatment by high imatinib concentration (above the IC50 value of 2.5 μmol/L) was accompanied by a higher energy state (e.g., with an increase of all phosphate nucleoside triphosphates (NTPs)), being possibly a result of an activation of the TCA cycle together with dysregulation of glucose metabolism [45]. The energy metabolism in BCR-ABL-negative HC-1 cell line was not affected by imatinib [45]. The TCA cycle metabolite α-ketoglutaric acid was significantly reduced upon treatment with the selective MET inhibitor capmatinib in two TNBC mesenchymal-like cell lines MDA-MB-231 and Hs578. Similarly, TCA cycle and central carbon metabolites such as aspartate, fumarate and malate were decreased following erlotinib treatment [30]. Impact on TCA cycle was described in another study using LAD adenocarcinoma cell lines treated with either erlotinib or gefitinib [27]. Despite the unchanged levels of acetyl-CoA following the distribution of these TKIs, other metabolites such as fumarate, malate and citrate were downregulated in EGFRi-responsive HCC827 and PC-9 cells [27]. This suggests that glutaminolysis is decreased after inhibition of EGFR signaling, consistent with the lower expression levels of glutaminase [27]. Moreover, although the inhibition of EGFR signaling downregulated de novo pyrimidine biosynthesis (reported downregulation of phosphorylation of ribosomal protein S6 kinase 1 (S6K), CAD trifunctional multi-domain protein (carbamoyl-phosphate synthetase 2, aspartate transcarbamoylase and dihydroorotase)), adenosine triphosphate levels (ATP) were not affected [27]. It was proposed, that after the treatment with WZ4002, an EGFR inhibitor, ATP levels increased in H1975 cell line. The results were constant with the results for H1993 cell line, exposed to another MET inhibitor, PHA665752, suggesting a reactivation effort of mitochondrial respiration following the treatment with the inhibitors [32]. To support this hypothesis, it has been further shown that ALK inhibition induces upregulation in total ATP levels while downregulating ADP in favour of biomass production (amino acids, lipids) [58]. The evidence from these data points towards the possibility that the reduction in glycolytic flux following ALK inhibition is not a characteristic feature of a viable cell since ATP levels are normally used as a representation of viability [67].
However, similarly to a previous study [27], an enhanced expression of ETC complexes II, III, IV and V was observed using erlotinib for the treatment of EGFR-sensitive HCC827 cells along with increased citrate levels, while no alterations of malate values were detected [32]. Comparable results indicating dysregulation of mitochondria by a TKI were obtained by Guo et al., who reported a deregulation of eight mitochondrial proteins (SLC25A13, NDUFS3, SDHB, UQCRC1, UQCRC2, COX2, COX5A, CYC1) representative of all four components of ETC and a decrease of mitochondrial permeability transition pore (mPTP) in response to the MET inhibitor PHA665752 in gastric carcinoma cell line SNU5 [55]. In a more recent study, Tesori and colleagues described a dose-dependent increase of reactive oxygen species (ROS), 12 h after exposure of the rat hepatocholangiocarcinoma cell line LCSC-2 to sorafenib [60]. Since mitochondria are a major source of ROS, they indicated that the observed increase of ROS is reflecting an impact of sorafenib on these energy sources [60]. Indeed, sorafenib was shown to depolarize mitochondria, interfering with the mitochondrial function and deregulating one of the mitochondrial enzymes, pyruvate dehydrogenase alpha 1 (PDHA1), which catalyses the production acetyl-CoA [60]. Furthermore, ATP levels were reduced, proposing that LCSC-2 cells strongly depend on mitochondrial functionality and that this drug interacts directly with mitochondria [60]. In addition, a 2017 study by Fumarola et al. using FGFR-amplified cell line H1703 reported, that FGFR1 inhibition by dovitinib or NVP-BGJ398 prevented ATP production and that decreased ATP levels caused activation of AMPK, a master energy sensor activated by elevated AMP:ADP ratio within the cell [62]. Aforementioned evidence uncovered novel mechanisms through which inhibitors act on mitochondrial biomarkers such as TCA cycle, NTPs and acetyl-CoA. Although the reported results are not always consistent across distinct TK systems, most of these studies agree, that upon TKI treatment cancer cells develop efforts to reactivate mitochondria and functionality of mitochondrial respiration as a potential saving mechanism against rapidly lethal effects of targeted therapies.
Metabolism of amino acids and their products
High demand for protein synthesis in tumors boosts the enormous need for amino acids. The mTOR pathway, a signaling cascade mobilized by many different oncogenes, is a one of the major pathways strongly associated with amino acid metabolism [68]. Tumor cells have a particular interest in amino acids such as serine and glycine, which fuel synthesis of nucleotides, proteins and lipids needed for proliferation [69, 70] and asparagine, which regulates the uptake of amino acids, hence the increased asparagine synthetase having role in a drug resistance [71]. Interestingly, amino acid deregulation plays an important function in immune tolerance in cancer [17]. Since T cells need for their proliferation tryptophan, amino acid depleted in many types of cancers, their response to fight this neoplastic phenotype is limited [72]. Furthermore, some cancers are auxotrophic for arginine, an amino acid playing role in urea, ornithine and citrulline production [17, 73]. Considering the influence that amino acid metabolism has on the reprogramming of neoplastic metabolism, we discuss in this section known effects of TKIs on amino acids and their related metabolites and appropriate enzymes.
In a study published in 2015, where the objective was to comparatively profile the metabolite composition of hepatocellular carcinoma HepG2 cells treated solely with sorafenib or everolimus (formerly RAD001, a mTOR inhibitor), and the combination of these two drugs using a NMR-based metabolomic approach, the group of Ji-Xiang Zhang reported that key metabolites are significantly altered in everolimus-treated cells [74]. Aspartate and glutathione disulfide were not changed in sorafenib-treated cells, however, alanine, arginine and glycine were significantly decreased in everolimus-treated cells. When comparing changes occurring between sorafenib and combination treatment, the combination therapy significantly downregulated molecules such as leucine, alanine, arginine and glycine. Combination-treated cells encountered decrease in arginine and increase in valine, lysine, tyrosine and aspartate as compared to the changes induced by the everolimus therapy, thus proposing that sorafenib and everolimus may, in addition to their individually induced effects on the cells, act on the metabolism of HepG2 cells also synergistically [74]. Further, it has been reported that amino acids proline and aspartate were increased following erlotinib treatment in EGFR-sensitive LAD cells [27]. Supporting these findings, a study looking for potential RTK inhibition biomarkers for TNBC models reported that in the basal-like cell line MDA-MB-231, perturbation of amino acid metabolism (e.g., glycine, alanine, cystine, glycolic acid, valine, leucine, proline and tryptophan) occurs upon erlotinib or capmatinib treatment [30]. Moreover, the authors of this study could further demonstrate that suppression of tryptophan metabolism enhances capmatinib treatment [30]. Other recent work highlights significant changes in glycine, serine and threonine metabolism in response to ALK inhibition as a consequence of deregulation of PKM2 [58], which may regulate de novo serine synthesis via 3-phosphoglycerate [75].
Comparable to the aforementioned, metabolic profiling of gefitinib-sensitive myeloma cancer cells LP-1 revealed upregulation of threonine, histidine, proline, asparagine and tyrosine following EGFR inhibition by gefitinib [34]. Related to gefitinib treatment, it has been reported that the concentration of arginine in breast cancer patients is significantly reduced [76]. The results of this study suggest that depletion of arginine in malignancies, for which arginine is auxotrophic, can be exploited as a potential targeted therapy [77]. At this point it is important to clarify that arginine is a nonessential amino acid in a healthy environment, however it is essential for highly proliferating cells [77]. In the aforementioned report by Gent et al., tryptophan, a major determinant marker of metastasis competence, did not change upon EGFR inhibition with the small molecule inhibitor gefitinib, widening the gap between the in vitro findings and their in vivo translation [78].
To fulfil biosynthetic demands associated with proliferation, tumors increase the import of nutrients including amino acids for their survival. Studies discussed in this section suggest that many amino acids are consistently decreased following the treatment with TKs inhibitors. Since most of these reports have been primarily focused on changes in glucose and mitochondrial metabolism, we are only starting to unravel the extent to which amino acids contribute to tumors’ pathology and if fluctuations in their levels that occur upon administration of TKIs could be plausibly considered as markers of therapy efficacy, or are rather merely passengers of events that take place upon inhibition of the respective oncogenic kinases.
Lipid metabolism
Although phospholipids, fatty acids and cholesterol represent extensive energetic storage and important building blocks for plasma membrane, impact on lipid metabolism in cancer cells received less attention than changes in glucose or amino acid metabolism. At the same time it has been well established that cancerous tissues are defined also by an increased rate of lipid synthesis [79]. Transcription factor sterol regulatory element-binding protein 1c (SREBP-1c) regulated by mTORC1 promotes tumor progression by increasing de novo lipid synthesis [80], which potentially implicates mTORC2 in the control of lipogenesis. Although lipids are extensively used as cancer biomarkers (e.g., phospholipid levels for breast cancer [81] or apolipoprotein A-I for colorectal cancer [82]), our current knowledge concerning the impact of TKIs on lipid metabolites and pathways is rather limited. The aforementioned study by Gottschalk et al. reported a significant decrease of phosphocholine, a precursor for membrane synthesis, as a consequence of the inhibition of cell proliferation in imatinib-treated BCR-ABL-positive cells [45]. At the same time, no changes were detected is BCR-ABL-negative HC-1 cell line following imatinib treatment [45]. It has been proposed that phosphocholine accumulates in different types of tumors (for example in breast, ovary or colon) as a result of an enhanced choline transport into the cells [83–85] and the high increase of phosphocholine is used as a marker for various cancers with higher proliferation rate. Imatinib-induced drop in phosphocholine reported by Gottschalk was accompanied by an upregulation glycerophosphocholine [45], related to apoptotic processes and membrane degradation [86]. In this respect, a 2015 study by Zheng et al. revealed that low dosage of sorafenib treatment affects glycerophospholipid metabolism in hepatocellular carcinoma cells HepG2 [74]. Interestingly, the treatment with non-tyrosine kinase inhibitors, including inhibitors of PI3K and RAS, mostly lead to downregulation of choline-containing metabolite levels, composed of total choline, phosphocholine and glycerophosphocholine [87–89]. In addition, a study conducted by Lanning et al. reported perturbed lipid metabolism which was present in more than 15% of total hits in a metabolomics study assessing responses of TNBC cancer cell lines to EGFR and MET inhibition. Interestingly, MDA-MB-231 and Hs578T cell lines were sensitive to the knockdown of fatty acid genes upon erlotinib treatment whereas capmatinib (INC280) sensitized MDA-MB-468 cells to knockdown of arachidonic and linoleic acid metabolism rate limiting enzymes, providing an additional motivation for co-targeting the metabolic and kinase pathways in TNBC patients [30].
Taken together, although our current expertise regarding alterations in lipid metabolism upon distribution of distinct TKIs is rather limited, the aforementioned results strongly suggest that TK inhibition often leads to a decrease in levels of fatty acid metabolites such as phosphocholine. Given the central role that lipids are playing in tumor development and tumor progression, further investigations regarding potential clinical relevance of TKI-related modulations in lipid metabolism are needed.
Conclusions
The introduction of TKIs to the armamentarium for the modulation of growth factor signaling has revolutionized treatment outcome of many cancer patients. Nevertheless, acquisition of drug resistance and reported side effects strongly limit their clinical use. Importantly, molecular mechanisms responsible for these complex processes induced by TKIs are not sufficiently understood yet. Metabolomics, either as a unique approach or in use in combination with other omics technologies, is a highly effective approach not only for biomarker discovery but has also the potential to unravel molecular processes that underlie mechanisms of action of various compounds including TKIs.
Nowadays it is relatively well established that TKIs such as imatinib, erlotinib or gefitinib impose metabolic changes on glycolysis profile of cancer cells expressing their respective targets. Indeed, recent studies show that these compounds decrease glucose uptake, potentially affecting major players of glucose metabolism such as transporters and rate limiting enyzmes, and by still unknown mechanisms contribute to side effects such as reactivation effort of mitochondrial respiration. On the contrary, metabolic effects of TKIs on amino acid and lipid metabolism are much less clear and cannot be generalized yet.
In summary, although the current knowledge on TKIs impact on cellular metabolism is continuously expanding, the detailed molecular mechanisms underlying many of the observations described within this review remain largely unknown and further biological investigations are warranted to understand the metabolic on- and off-target effects related to TKIs treatment.
Acknowledgements
None.
Funding
This study was supported by Bernische Krebsliga, Stiftung zur Krebsbekämpfung, The Werner und Hedy Berger-Janser Stiftung (grants to M.M.) and by Swiss National Scientific Foundation (grant no. 31003A_156816 to Y.Z.).
Availability of data and materials
Not applicable.
Abbreviations
- ALK
Anaplastic lymphoma kinase
- ATP
Adenosine triphosphate
- CML
Chronic myeloid leukemia
- ECAR
Extracellular acidification rate
- EGF(R)
Epidermal growth factor (receptor)
- ERBB2
Receptor tyrosine-protein kinase erbB-2 precursor
- ETC
Electron transport chain
- FGF(R)
Fibroblast growth factor (receptor)
- GLUT
Glucose transporter
- GTP
Guanosine triphosphate
- HCC
Hepatocellular carcinoma
- HIF
Hypoxia-inducible factor
- HK2
Hexokinase 2
- HNSCC
Head and neck squamous cell carcinoma
- LAD
Lung adenocarcinoma
- LDHA
Lactate dehydrogenase A
- mTOR
Mammalian target of rapamycin
- NADPH
Nicotinamide adenine dinucleotide phosphate
- NPC
Nasopharyngeal cancer
- NSCLC
Non-small cell lung cancer
- PFKFB2
6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase 2
- PI3K
Phosphatidylinositol 3-kinase
- PKM2
Pyruvate kinase muscle isozyme 2
- PPP
Pentose phosphate pathway
- ROS
Reactive oxygen species
- RTK
Receptors tyrosine kinase
- TCA
Tricarboxylic acid
- TIGAR
TP53-inducible glycolysis and apoptosis regulator
- TKI
Tyrosine kinase inhibitor
- TNBC
Triple-negative breast cancer
- VEGF(R)
Vascular endothelial growth factor (receptor)
Authors’ contributions
MP, MM, and YZ designed and wrote the manuscript. DMA, YZ, and MM provided resources. All authors read and approved the final manuscript.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Michaela Poliaková, Phone: 0041 31 6323565, Email: michaela.medova@dbmr.unibe.ch.
Michaela Medová, Phone: 0041/31/6323565, Email: michaela.medova@dbmr.unibe.ch.
References
- 1.Kroemer G, Pouyssegur J. Tumor cell metabolism: cancer's Achilles' heel. Cancer Cell. 2008;13(6):472–482. doi: 10.1016/j.ccr.2008.05.005. [DOI] [PubMed] [Google Scholar]
- 2.Warburg O. On the origin of cancer cells. Science. 1956;123(3191):309–314. doi: 10.1126/science.123.3191.309. [DOI] [PubMed] [Google Scholar]
- 3.Pavlova NN, Thompson CB. The emerging hallmarks of cancer metabolism. Cell Metab. 2016;23(1):27–47. doi: 10.1016/j.cmet.2015.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.DeBerardinis RJ, Chandel NS. Fundamentals of cancer metabolism. Sci Adv. 2016;2(5):e1600200. doi: 10.1126/sciadv.1600200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Holdsworth CH, et al. CT and PET: early prognostic indicators of response to imatinib mesylate in patients with gastrointestinal stromal tumor. AJR Am J Roentgenol. 2007;189(6):W324–W330. doi: 10.2214/AJR.07.2496. [DOI] [PubMed] [Google Scholar]
- 6.Scott TA, et al. Host-microbe co-metabolism dictates cancer drug efficacy in C. Elegans. Cell. 2017;169(3):442–456. doi: 10.1016/j.cell.2017.03.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wagner W, Ciszewski WM, Kania KD. L- and D-lactate enhance DNA repair and modulate the resistance of cervical carcinoma cells to anticancer drugs via histone deacetylase inhibition and hydroxycarboxylic acid receptor 1 activation. Cell Commun Signal. 2015;13:36. doi: 10.1186/s12964-015-0114-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang J, et al. EGFR modulates monounsaturated fatty acid synthesis through phosphorylation of SCD1 in lung cancer. Mol Cancer. 2017;16(1):127. doi: 10.1186/s12943-017-0704-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sun Y, et al. Metabolic and transcriptional profiling reveals pyruvate dehydrogenase kinase 4 as a mediator of epithelial-mesenchymal transition and drug resistance in tumor cells. Cancer Metab. 2014;2(1):20. doi: 10.1186/2049-3002-2-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ghosh JC, et al. Adaptive mitochondrial reprogramming and resistance to PI3K therapy. J Natl Cancer Inst. 2015;107(3):dju502. [DOI] [PMC free article] [PubMed]
- 11.Bilanges B, et al. Vps34 PI 3-kinase inactivation enhances insulin sensitivity through reprogramming of mitochondrial metabolism. Nat Commun. 2017;8(1):1804. doi: 10.1038/s41467-017-01969-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Alvarez-Calderon F, et al. Tyrosine kinase inhibition in leukemia induces an altered metabolic state sensitive to mitochondrial perturbations. Clin Cancer Res. 2015;21(6):1360–1372. doi: 10.1158/1078-0432.CCR-14-2146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gharwan H, Groninger H. Kinase inhibitors and monoclonal antibodies in oncology: clinical implications. Nat Rev Clin Oncol. 2016;13(4):209–227. doi: 10.1038/nrclinonc.2015.213. [DOI] [PubMed] [Google Scholar]
- 14.Cairns RA, Harris IS, Mak TW. Regulation of cancer cell metabolism. Nat Rev Cancer. 2011;11(2):85–95. doi: 10.1038/nrc2981. [DOI] [PubMed] [Google Scholar]
- 15.Hutton JE, et al. Oncogenic KRAS and BRAF drive metabolic reprogramming in colorectal cancer. Mol Cell Proteomics. 2016;15(9):2924–2938. doi: 10.1074/mcp.M116.058925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cardaci S, Ciriolo MR. TCA cycle defects and cancer: when metabolism tunes Redox state. Int J Cell Biol. 2012;2012:161837. doi: 10.1155/2012/161837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nagarajan A, Malvi P, Wajapeyee N. Oncogene-directed alterations in cancer cell metabolism. Trends Cancer. 2016;2(7):365–377. doi: 10.1016/j.trecan.2016.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang GL, et al. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci U S A. 1995;92(12):5510–5514. doi: 10.1073/pnas.92.12.5510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Semenza GL. Hypoxia-inducible factors: coupling glucose metabolism and redox regulation with induction of the breast cancer stem cell phenotype. EMBO J. 2017;36(3):252–259. doi: 10.15252/embj.201695204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yamamoto T, et al. Over-expression of facilitative glucose transporter genes in human cancer. Biochem Biophys Res Commun. 1990;170(1):223–230. doi: 10.1016/0006-291X(90)91263-R. [DOI] [PubMed] [Google Scholar]
- 21.Kocdor MA, et al. Progressive increase of glucose transporter-3 (GLUT-3) expression in estrogen-induced breast carcinogenesis. Clin Transl Oncol. 2013;15(1):55–64. doi: 10.1007/s12094-012-0882-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hay N. Reprogramming glucose metabolism in cancer: can it be exploited for cancer therapy? Nat Rev Cancer. 2016;16(10):635–649. doi: 10.1038/nrc.2016.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Galluzzi L, et al. Metabolic targets for cancer therapy. Nat Rev Drug Discov. 2013;12(11):829–846. doi: 10.1038/nrd4145. [DOI] [PubMed] [Google Scholar]
- 24.Rahman M, Hasan MR. Cancer metabolism and drug resistance. Meta. 2015;5(4):571–600. doi: 10.3390/metabo5040571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nicholson RI, Gee JM, Harper ME. EGFR and cancer prognosis. Eur J Cancer. 2001;37(Suppl 4):S9–15. doi: 10.1016/S0959-8049(01)00231-3. [DOI] [PubMed] [Google Scholar]
- 26.Mendelsohn J, Baselga J. The EGF receptor family as targets for cancer therapy. Oncogene. 2000;19(56):6550–6565. doi: 10.1038/sj.onc.1204082. [DOI] [PubMed] [Google Scholar]
- 27.Makinoshima H, et al. Epidermal growth factor receptor (EGFR) signaling regulates global metabolic pathways in EGFR-mutated lung adenocarcinoma. J Biol Chem. 2014;289(30):20813–20823. doi: 10.1074/jbc.M114.575464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhou W, et al. Novel mutant-selective EGFR kinase inhibitors against EGFR T790M. Nature. 2009;462(7276):1070–1074. doi: 10.1038/nature08622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lim SO, et al. EGFR signaling enhances aerobic Glycolysis in triple-negative breast cancer cells to promote tumor growth and immune escape. Cancer Res. 2016;76(5):1284–1296. doi: 10.1158/0008-5472.CAN-15-2478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lanning NJ, et al. Metabolic profiling of triple-negative breast cancer cells reveals metabolic vulnerabilities. Cancer Metab. 2017;5:6. doi: 10.1186/s40170-017-0168-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xue M, et al. Chemical methods for the simultaneous quantitation of metabolites and proteins from single cells. J Am Chem Soc. 2015;137(12):4066–4069. doi: 10.1021/jacs.5b00944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.De Rosa V, et al. Reversal of Warburg effect and reactivation of oxidative Phosphorylation by differential inhibition of EGFR signaling pathways in non-small cell lung cancer. Clin Cancer Res. 2015;21(22):5110–5120. doi: 10.1158/1078-0432.CCR-15-0375. [DOI] [PubMed] [Google Scholar]
- 33.Momcilovic M, et al. Targeted inhibition of EGFR and Glutaminase induces metabolic crisis in EGFR mutant lung cancer. Cell Rep. 2017;18(3):601–610. doi: 10.1016/j.celrep.2016.12.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen Y, et al. Multiple myeloma acquires resistance to EGFR inhibitor via induction of pentose phosphate pathway. Sci Rep. 2015;5:9925. doi: 10.1038/srep09925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Necela BM, et al. The antineoplastic drug, trastuzumab, dysregulates metabolism in iPSC-derived cardiomyocytes. Clin Transl Med. 2017;6(1):5. doi: 10.1186/s40169-016-0133-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li H, et al. Efficacy and safety of trastuzumab combined with chemotherapy for first-line treatment and beyond progression of HER2-overexpressing advanced breast cancer. Chin J Cancer Res. 2016;28(3):330–338. doi: 10.21147/j.issn.1000-9604.2016.03.07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Maher M. Current and emerging treatment regimens for HER2-positive breast cancer. P T. 2014;39(3):206–212. [PMC free article] [PubMed] [Google Scholar]
- 38.Zhao Y, et al. Overcoming trastuzumab resistance in breast cancer by targeting dysregulated glucose metabolism. Cancer Res. 2011;71(13):4585–4597. doi: 10.1158/0008-5472.CAN-11-0127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhao YH, et al. Upregulation of lactate dehydrogenase a by ErbB2 through heat shock factor 1 promotes breast cancer cell glycolysis and growth. Oncogene. 2009;28(42):3689–3701. doi: 10.1038/onc.2009.229. [DOI] [PubMed] [Google Scholar]
- 40.Medina PJ, Goodin S. Lapatinib: a dual inhibitor of human epidermal growth factor receptor tyrosine kinases. Clin Ther. 2008;30(8):1426–1447. doi: 10.1016/j.clinthera.2008.08.008. [DOI] [PubMed] [Google Scholar]
- 41.Komurov K, et al. The glucose-deprivation network counteracts lapatinib-induced toxicity in resistant ErbB2-positive breast cancer cells. Mol Syst Biol. 2012;8:596. doi: 10.1038/msb.2012.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ruprecht B, et al. Lapatinib resistance in breast cancer cells is accompanied by Phosphorylation-mediated reprogramming of Glycolysis. Cancer Res. 2017;77(8):1842–1853. doi: 10.1158/0008-5472.CAN-16-2976. [DOI] [PubMed] [Google Scholar]
- 43.An X, et al. BCR-ABL tyrosine kinase inhibitors in the treatment of Philadelphia chromosome positive chronic myeloid leukemia: a review. Leuk Res. 2010;34(10):1255–1268. doi: 10.1016/j.leukres.2010.04.016. [DOI] [PubMed] [Google Scholar]
- 44.Hochhaus A, et al. Six-year follow-up of patients receiving imatinib for the first-line treatment of chronic myeloid leukemia. Leukemia. 2009;23(6):1054–1061. doi: 10.1038/leu.2009.38. [DOI] [PubMed] [Google Scholar]
- 45.Gottschalk S, et al. Imatinib (STI571)-mediated changes in glucose metabolism in human leukemia BCR-ABL-positive cells. Clin Cancer Res. 2004;10(19):6661–6668. doi: 10.1158/1078-0432.CCR-04-0039. [DOI] [PubMed] [Google Scholar]
- 46.Boros LG, Lee WN, Go VL. A metabolic hypothesis of cell growth and death in pancreatic cancer. Pancreas. 2002;24(1):26–33. doi: 10.1097/00006676-200201000-00004. [DOI] [PubMed] [Google Scholar]
- 47.Barnes K, et al. Chronic myeloid leukaemia: an investigation into the role of Bcr-Abl-induced abnormalities in glucose transport regulation. Oncogene. 2005;24(20):3257–3267. doi: 10.1038/sj.onc.1208461. [DOI] [PubMed] [Google Scholar]
- 48.Kominsky DJ, et al. Abnormalities in glucose uptake and metabolism in imatinib-resistant human BCR-ABL-positive cells. Clin Cancer Res. 2009;15(10):3442–3450. doi: 10.1158/1078-0432.CCR-08-3291. [DOI] [PubMed] [Google Scholar]
- 49.Hughes B, et al. Cerebral relapse of metastatic gastrointestinal stromal tumor during treatment with imatinib mesylate: case report. BMC Cancer. 2004;4:74. doi: 10.1186/1471-2407-4-74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Organ SL, Tsao MS. An overview of the c-MET signaling pathway. Ther Adv Med Oncol. 2011;3(1 Suppl):S7–S19. doi: 10.1177/1758834011422556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lui VW, et al. Inhibition of c-met downregulates TIGAR expression and reduces NADPH production leading to cell death. Oncogene. 2011;30(9):1127–1134. doi: 10.1038/onc.2010.490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bensaad K, et al. TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell. 2006;126(1):107–120. doi: 10.1016/j.cell.2006.05.036. [DOI] [PubMed] [Google Scholar]
- 53.Panday A, et al. NADPH oxidases: an overview from structure to innate immunity-associated pathologies. Cell Mol Immunol. 2015;12(1):5–23. doi: 10.1038/cmi.2014.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Spaans SK, et al. NADPH-generating systems in bacteria and archaea. Front Microbiol. 2015;6:742. doi: 10.3389/fmicb.2015.00742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Guo T, et al. Quantitative proteomics discloses MET expression in mitochondria as a direct target of MET kinase inhibitor in cancer cells. Mol Cell Proteomics. 2010;9(12):2629–2641. doi: 10.1074/mcp.M110.001776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tang Z, et al. Dual MET-EGFR combinatorial inhibition against T790M-EGFR-mediated erlotinib-resistant lung cancer. Br J Cancer. 2008;99(6):911–922. doi: 10.1038/sj.bjc.6604559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hallberg B, Palmer RH. Mechanistic insight into ALK receptor tyrosine kinase in human cancer biology. Nat Rev Cancer. 2013;13(10):685–700. doi: 10.1038/nrc3580. [DOI] [PubMed] [Google Scholar]
- 58.McDonnell SR, et al. Integrated phosphoproteomic and metabolomic profiling reveals NPM-ALK-mediated phosphorylation of PKM2 and metabolic reprogramming in anaplastic large cell lymphoma. Blood. 2013;122(6):958–968. doi: 10.1182/blood-2013-01-482026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hudson CD, et al. Resistance to the tyrosine kinase inhibitor axitinib is associated with increased glucose metabolism in pancreatic adenocarcinoma. Cell Death Dis. 2014;5:e1160. doi: 10.1038/cddis.2014.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tesori V, et al. The multikinase inhibitor Sorafenib enhances glycolysis and synergizes with glycolysis blockade for cancer cell killing. Sci Rep. 2015;5:9149. doi: 10.1038/srep09149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hitosugi T, et al. Tyrosine phosphorylation inhibits PKM2 to promote the Warburg effect and tumor growth. Sci Signal. 2009;2(97):ra73. doi: 10.1126/scisignal.2000431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Fumarola C, et al. Enhancement of the anti-tumor activity of FGFR1 inhibition in squamous cell lung cancer by targeting downstream signaling involved in glucose metabolism. Oncotarget. 2017;8(54):91841–91859. doi: 10.18632/oncotarget.19279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tan AS, et al. Mitochondrial genome acquisition restores respiratory function and tumorigenic potential of cancer cells without mitochondrial DNA. Cell Metab. 2015;21(1):81–94. doi: 10.1016/j.cmet.2014.12.003. [DOI] [PubMed] [Google Scholar]
- 64.Camarda R, et al. Inhibition of fatty acid oxidation as a therapy for MYC-overexpressing triple-negative breast cancer. Nat Med. 2016;22(4):427–432. doi: 10.1038/nm.4055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Corbet C, Feron O. Cancer cell metabolism and mitochondria: nutrient plasticity for TCA cycle fueling. Biochim Biophys Acta. 2017;1868(1):7–15. doi: 10.1016/j.bbcan.2017.01.002. [DOI] [PubMed] [Google Scholar]
- 66.Laurenti G, Tennant DA. Isocitrate dehydrogenase (IDH), succinate dehydrogenase (SDH), fumarate hydratase (FH): three players for one phenotype in cancer? Biochem Soc Trans. 2016;44(4):1111–1116. doi: 10.1042/BST20160099. [DOI] [PubMed] [Google Scholar]
- 67.Petty RD, et al. Comparison of MTT and ATP-based assays for the measurement of viable cell number. J Biolumin Chemilumin. 1995;10(1):29–34. doi: 10.1002/bio.1170100105. [DOI] [PubMed] [Google Scholar]
- 68.Jewell JL, Russell RC, Guan KL. Amino acid signalling upstream of mTOR. Nat Rev Mol Cell Biol. 2013;14(3):133–139. doi: 10.1038/nrm3522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Amelio I, et al. Serine and glycine metabolism in cancer. Trends Biochem Sci. 2014;39(4):191–198. doi: 10.1016/j.tibs.2014.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kalhan SC, Hanson RW. Resurgence of serine: an often neglected but indispensable amino acid. J Biol Chem. 2012;287(24):19786–19791. doi: 10.1074/jbc.R112.357194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zhang B, et al. Asparagine synthetase is an independent predictor of surgical survival and a potential therapeutic target in hepatocellular carcinoma. Br J Cancer. 2013;109(1):14–23. doi: 10.1038/bjc.2013.293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Uyttenhove C, et al. Evidence for a tumoral immune resistance mechanism based on tryptophan degradation by indoleamine 2,3-dioxygenase. Nat Med. 2003;9(10):1269–1274. doi: 10.1038/nm934. [DOI] [PubMed] [Google Scholar]
- 73.Dillon BJ, et al. Incidence and distribution of argininosuccinate synthetase deficiency in human cancers: a method for identifying cancers sensitive to arginine deprivation. Cancer. 2004;100(4):826–833. doi: 10.1002/cncr.20057. [DOI] [PubMed] [Google Scholar]
- 74.Zheng JF, et al. Comparative Metabolomic profiling of Hepatocellular carcinoma cells treated with Sorafenib Monotherapy vs. Sorafenib-Everolimus combination therapy. Med Sci Monit. 2015;21:1781–1791. doi: 10.12659/MSM.894669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ye J, et al. Pyruvate kinase M2 promotes de novo serine synthesis to sustain mTORC1 activity and cell proliferation. Proc Natl Acad Sci U S A. 2012;109(18):6904–6909. doi: 10.1073/pnas.1204176109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Geng D, et al. The therapy of gefitinib towards breast cancer partially through reversing breast cancer biomarker arginine. Afr Health Sci. 2015;15(2):594–597. doi: 10.4314/ahs.v15i2.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Feun L, et al. Arginine deprivation as a targeted therapy for cancer. Curr Pharm Des. 2008;14(11):1049–1057. doi: 10.2174/138161208784246199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zhang L, et al. Tryptophan as the fingerprint for distinguishing aggressiveness among breast cancer cell lines using native fluorescence spectroscopy. J Biomed Opt. 2014;19(3):37005. doi: 10.1117/1.JBO.19.3.037005. [DOI] [PubMed] [Google Scholar]
- 79.Menendez JA, Lupu R. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat Rev Cancer. 2007;7(10):763–777. doi: 10.1038/nrc2222. [DOI] [PubMed] [Google Scholar]
- 80.Porstmann T, et al. SREBP activity is regulated by mTORC1 and contributes to Akt-dependent cell growth. Cell Metab. 2008;8(3):224–236. doi: 10.1016/j.cmet.2008.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Mistry DA, French PW. Circulating phospholipids as biomarkers of breast cancer: a review. Breast Cancer (Auckl) 2016;10:191–196. doi: 10.4137/BCBCR.S40693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Murakoshi Y, et al. Plasma biomarker discovery and validation for colorectal cancer by quantitative shotgun mass spectrometry and protein microarray. Cancer Sci. 2011;102(3):630–638. doi: 10.1111/j.1349-7006.2010.01818.x. [DOI] [PubMed] [Google Scholar]
- 83.Eliyahu G, Kreizman T, Degani H. Phosphocholine as a biomarker of breast cancer: molecular and biochemical studies. Int J Cancer. 2007;120(8):1721–1730. doi: 10.1002/ijc.22293. [DOI] [PubMed] [Google Scholar]
- 84.Bagnoli M, et al. Choline metabolism alteration: a focus on ovarian cancer. Front Oncol. 2016;6:153. doi: 10.3389/fonc.2016.00153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Mori N, et al. Loss of p53 function in colon cancer cells results in increased phosphocholine and total choline. Mol Imaging. 2004;3(4):319–323. doi: 10.1162/1535350042973517. [DOI] [PubMed] [Google Scholar]
- 86.Evelhoch JL, et al. Applications of magnetic resonance in model systems: cancer therapeutics. Neoplasia. 2000;2(1–2):152–165. doi: 10.1038/sj.neo.7900078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Koul D, et al. Cellular and in vivo activity of a novel PI3K inhibitor, PX-866, against human glioblastoma. Neuro-Oncology. 2010;12(6):559–569. doi: 10.1093/neuonc/nop058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Al-Saffar NM, et al. The phosphoinositide 3-kinase inhibitor PI-103 downregulates choline kinase alpha leading to phosphocholine and total choline decrease detected by magnetic resonance spectroscopy. Cancer Res. 2010;70(13):5507–5517. doi: 10.1158/0008-5472.CAN-09-4476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ronen SM, et al. Magnetic resonance detects changes in phosphocholine associated with Ras activation and inhibition in NIH 3T3 cells. Br J Cancer. 2001;84(5):691–696. doi: 10.1054/bjoc.2000.1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.