

Abstract
Objective
Inhibitors of the voltage‐gated K+ channel Kv1.3 are currently in development as immunomodulators for the treatment of autoimmune diseases. As Kv1.3 is also expressed on microglia and has been shown to be specifically up‐regulated on “M1‐like” microglia, we here tested the therapeutic hypothesis that the brain‐penetrant small‐molecule Kv1.3‐inhibitor PAP‐1 reduces secondary inflammatory damage after ischemia/reperfusion.
Methods
We studied microglial Kv1.3 expression using electrophysiology and immunohistochemistry, and evaluated PAP‐1 in hypoxia‐exposed organotypic hippocampal slices and in middle cerebral artery occlusion (MCAO) with 8 days of reperfusion in both adult male C57BL/6J mice (60 min MCAO) and adult male Wistar rats (90 min MCAO). In both models, PAP‐1 administration was started 12 h after reperfusion.
Results
We observed Kv1.3 staining on activated microglia in ischemic infarcts in mice, rats, and humans and found higher Kv1.3 current densities in acutely isolated microglia from the infarcted hemisphere than in microglia isolated from the contralateral hemisphere of MCAO mice. PAP‐1 reduced microglia activation and increased neuronal survival in hypoxia‐exposed hippocampal slices as effectively as minocycline. In mouse MCAO, PAP‐1 dose‐dependently reduced infarct area, improved neurological deficit score, and reduced brain levels of IL‐1β and IFN‐γ without affecting IL‐10 and brain‐derived nerve growth factor (BDNF) levels or inhibiting ongoing phagocytosis. The beneficial effects on infarct area and neurological deficit score were reproduced in rats providing confirmation in a second species.
Interpretation
Our findings suggest that Kv1.3 constitutes a promising therapeutic target for preferentially inhibiting “M1‐like” inflammatory microglia/macrophage functions in ischemic stroke.
Introduction
The voltage‐gated K+ channel Kv1.3 was first discovered in human T‐cells in 19841 and has since then been pursued as a target for immunosuppression. After the channel was cloned, the pharmaceutical industry initiated Kv1.3 discovery programs in the mid‐1990s but largely failed to identify compounds that were suitable for development.2 Interest in Kv1.3 afterward waned but revived following reports from our group that Kv1.3 is overexpressed in activated CCR7− effector memory T‐cells (TEM) and that Kv1.3 blockers therefore constitute immunomodulators rather than general immunosuppressants.3, 4, 5 Subsequent proof‐of‐concept animal studies demonstrated that the Kv1.3 blocking sea anemone Stichodactyla helianthus peptide ShK and its derivatives treat rat models of multiple sclerosis and rheumatoid arthritis,4, 5, 6 while our small molecule Kv1.3 blocker PAP‐1 prevents autoimmune diabetes4 in rats and treats psoriasis in a mouse xenograft model.7 As Kv1.3 blockers preferentially target TEM cells, Kv1.3 blockers do not impair the ability to clear acute infections or develop vaccine responses. For example, Kv1.3 blockers do not affect the ability of rats to clear influenza virus and Chlamydia trachomatis 5 or prevent rhesus macaques from developing protective, flu‐specific central memory T‐cell responses following immunization with a live influenza vaccine.8 Based on these encouraging target validation studies, ShK‐186, now called Dalazatide, was moved into clinical trials, where it exhibited no major safety findings in Phase‐1a and showed efficacy in Phase‐1b studies in plaque psoriasis and is now being developed further.9
In addition to T‐cells, Kv1.3 is also expressed in microglia, where in vitro experiments with rodent microglia have shown the channel to be involved in inflammatory cytokine and nitric oxide production and in microglia‐mediated neuronal killing.10, 11, 12, 13, 14 Electrophysiological experiments on slices15 or acutely isolated microglia16 have further demonstrated Kv1.3 currents on activated microglia following MCAO. In humans, strong Kv1.3 staining has been reported on microglia in the frontal cortex of patients with Alzheimer's disease17 and on CD68+ cells in multiple sclerosis brain lesions.18 Using cultured neonatal mouse microglia, our group recently demonstrated that Kv1.3 expression increases following stimulation with the M1‐polarizing stimuli LPS or LPS plus IFN‐γ, while IL‐4 stimulation induced a down‐regulation of Kv1.3 and an increase in currents for the inward‐rectifier K+ channel Kir2.1,19 suggesting that Kv1.3 blockers might be able to preferentially target detrimental proinflammatory M1 microglia functions. We therefore hypothesized that Kv1.3 blockers could be useful for ameliorating pathology in ischemic stroke, where activated microglia significantly contribute to the secondary expansion of the infarct.20, 21 In order to test this therapeutic hypothesis, we here confirmed Kv1.3 expression on microglia in infarcted rodent and human brain and then evaluated our small molecule blocker PAP‐122 first in organotypic brain slices and then in middle cerebral artery occlusion (MCAO) with reperfusion. PAP‐1 is currently the most potent and selective small molecule Kv1.3 blocker available. It inhibits Kv1.3 with an IC50 of 2 nmol/L,22 has a half‐life of 3 h in rodents, is orally available, brain penetrant and does not exhibit any long‐term toxicity in rodents or primates.4, 8, 23
Materials and Methods
Transient Focal Ischemia
This study was approved by the University of California, Davis, Animal Use and Care Committee and conducted in accordance with the guidelines for survival surgery. Adult 12‐week‐old male C57BL/6J mice (Jackson Laboratory) or adult male Wistar rats (Charles River) were acclimatized to the vivarium for a week and used for surgery when they were 12–14 weeks old (mice) or weighed 200–230 g (rats). Animals were anesthetized with 5% isoflurane and then maintained on 0.5 to 1.5% isoflurane. To assure consistent and continuous reduction in cerebral blood flow (CBF), a laser Doppler (Moor Instruments) was used throughout the surgery. Reversible focal cerebral ischemia was induced by occlusion of the left middle cerebral artery (MCA) according to Zea Longa.24 Briefly, the left common carotid artery was exposed, the external carotid artery ligated distally from the common carotid artery, and a silicone rubber‐coated nylon monofilament with a tip diameter of 0.21 ± 0.02 mm for mice and of 0.43 ± 0.02 mm for rats (Doccol Corp.) inserted into the external carotid artery and advanced into the internal carotid artery to block the origin of the MCA.24, 25 The filament was kept in place for 60 min (mice) or 90 min (rats) and then withdrawn. Animals received subcutaneous Buprenex at 0.02 mg/kg every 12 h to limit postsurgical pain for 24 h after surgery. For sham surgeries, the filament was placed into the external carotid artery but not advanced into the internal carotid artery.
Four MCAO surgeries were performed per day. The survival rate was 85% in mice and 90% in rats. Animals where CBF was not reduced by at least 70% and which did not display any obvious neurological deficit 12 h after reperfusion were excluded. Animals that met these inclusion criteria were assigned to treatment or vehicle groups based on a computer‐generated randomization scheme. Based on our previous work with the KCa3.1 blocker TRAM‐34 in MCAO,16, 25 sample size was powered to detect a reduction of 40% in mean percentage infarct area with 80% power. Starting 12 h after reperfusion animals received the vehicle miglyol 812 neutral oil (Neobee M5®, Spectrum Chemicals) or PAP‐1 at 10 or 40 mg/kg intraperitoneally every 12 h until sacrifice on day‐8. PAP‐1 was synthesized in our laboratory.22 Miglyol 812 is a low viscosity oily vehicle, which is widely used as a pharmaceutical excipient and well tolerated for i.p., s.c. or oral administration. It can be autoclaved and allows for the dissolution of lipophilic compounds like PAP‐1 at high concentrations (5 and 20 mg/mL for this study). Neurological deficits were scored according to a tactile and proprioceptive limb‐placing test26 as previously described.25 The investigator performing the drug administration and neurological scoring was blinded to the treatments.
Infarct area
Animals were euthanized with an overdose of isoflurane, brains quickly removed, and sectioned into four (mice) or eight (rats) 2‐mm thick coronal slices starting from the frontal pole. Slices were fixed in 10% buffered formalin for 24 h in order to make it possible to prepare undamaged sections from the otherwise often brittle aged infarcts. Slices were stored in 70% ethanol, embedded in paraffin, and sectioned at 5 μm by the Histology Laboratory of the UC Davis School of Veterinary Medicine. Infarct area was quantified in a blinded fashion by analyzing the NeuN‐negative area using a pixel‐based method as previously described.16
Isolation of CD11b+ cells and electrophysiology
CD11b+ cells from the infarct area were isolated as previously described.16, 27 Briefly, mice were anesthetized with isoflurane 2, 5, and 8 days after reperfusion MCAO and killed by cervical dislocation. Brains were removed quickly and the infarcted hemispheres dissected and kept in ice‐cold Hank's balanced salt solution (HBSS) after removing cerebellum and brain stem. The brain tissue was chopped into small pieces and centrifuged at 300 g for 2 min to remove the supernatant. Following digestion with enzyme mix 1 for 15 min and enzyme mix 2 for 10 min at 37°C, brain tissue was mechanically dissociated by pipetting up and down. The tissue suspension was centrifuged at 300 g at 4°C for 10 min to spin down the cells. Magnetic microbeads conjugated to CD11b‐antibody were added to the cell suspension, and CD11b+ cells were isolated with the Miltenyi magnet according to the manufacturer's protocol (Miltenyi Biotec Inc.). Acutely isolated microglia were over 94% pure (CD11b+) based on flow cytometry assessments.
Acutely isolated CD11b+ cells were plated on poly‐L‐lysine‐coated glass coverslips immediately and then incubated at 37°C for 10 min before electrophysiological recordings. Overall, the isolation procedure lasted 90 min from the removal of the brains until the microglia were on the microscope stage ready to be patch‐clamped. Currents were recorded at room temperature using the whole‐cell configuration of the patch‐clamp technique with an EPC‐10 HEKA amplifier. Normal Ringer solution (160 mmol/L NaCl, 4.5 mmol/L KCl, 2 mmol/L CaCl2, 1 mmol/L MgCl2, 10 mmol/L HEPES, pH 7.4, 300 mOsm) was used as an external. Patch pipettes were filled with an internal solution containing 145 mmol/L KF, 2 MgCl2, 10 mmol/L HEPES, 10 EGTA (pH 7.2, 290 mOsm). K+ currents were elicited with voltage ramps from −120 to 40 mV of 200‐ms duration or by voltage steps from −80 to +40 mV applied every 30 sec.3 Access resistance was monitored continuously and no series resistance compensation was used as access resistances typically stayed below 10 MΩ. Kv1.3 current densities were determined by dividing the current amplitude at +40 mV by the cell capacitance.
Hippocampus slice culture and hypoxia treatment
Hippocampal slice cultures (400 μm thick) were prepared from 7‐day‐old C57BL/6J mice as described.28 After 3 days, culture medium was changed to a hypoxic/hypoglycemic medium (75% Neurobasal, 25% HBSS, 1% L‐glutamine, bubbled with 99.4% nitrogen). Slices were placed in a hypoxia incubation chamber (Stemcell Technologies, Vancouver), and nitrogen gas was flowed into the chamber, followed by incubation at 37o for 1 h. The culture medium was replaced by normal culture medium containing glucose, and slices were placed in a tissue culture incubator. After 2 h of culture, 1 μmol/L of PAP‐1 or 10 μmol/L of minocycline were added and slices cultured for 3 days. Slices were fixed in 4% formaldehyde and stained with anti‐NeuN (1:400; Chemicon) and anti‐Iba1 (1:400; Wako Chemical) followed by secondary Alexa‐Fluor®488‐conjugated anti‐mouse or Alexa‐Fluor®568‐conjugated anti‐rabbit antibodies (1:700; Life Technologies). For Western blotting, slices were lysed and protein electrophoresed and visualized as previously described.14 The following primary antibodies were used as follows: anti‐PSD95 (1:1000) and anti‐GAPDH (1: 3000; both Cell Signaling Technology). The secondary antibody was an HRP‐conjugated anti‐rabbit antibody (1:3000; Amersham Biosciences).
Immunohistochemical (IHC) and immunofluorescence (IF) staining
All IHC or IF staining in this study was performed after antigen retrieval (heating in 10 mmol/L Na citrate for 15 min). NeuN (1:1000, A60, Millipore) staining to assess infarct area was performed as described.16 For the mouse brain IF images in Figure 1A, Kv1.3 was stained with a polyclonal anti‐Kv1.3 antibody (1:300, APC‐101, Alomone) and Iba1 with a monoclonal anti‐Iba1 antibody (1:1000, NCNP24, Wako Chemical). For Figure 1B, CD68 was stained with a monoclonal anti‐mouse CD68 antibody (1:200, FA‐11, Bio‐Rad) and Kv1.3 with a polyclonal anti‐Kv1.3 antibody (1:300, APC‐002, Alomone). For Figure 1C, Kv1.3 was stained with a mouse monoclonal anti‐Kv1.3 antibody (1:300, 1D8, Bio‐Rad) and iNOS with a polyclonal rabbit antibody (1:100, ab3523, Abcam). Bound primary antibodies were detected with Alexa‐Fluor®546‐ or Alexa‐Fluor®647‐conjugated secondary antibodies (Life Technologies). The IHC staining for rat CD68 in Figure 7A was performed as described.25
Figure 1.
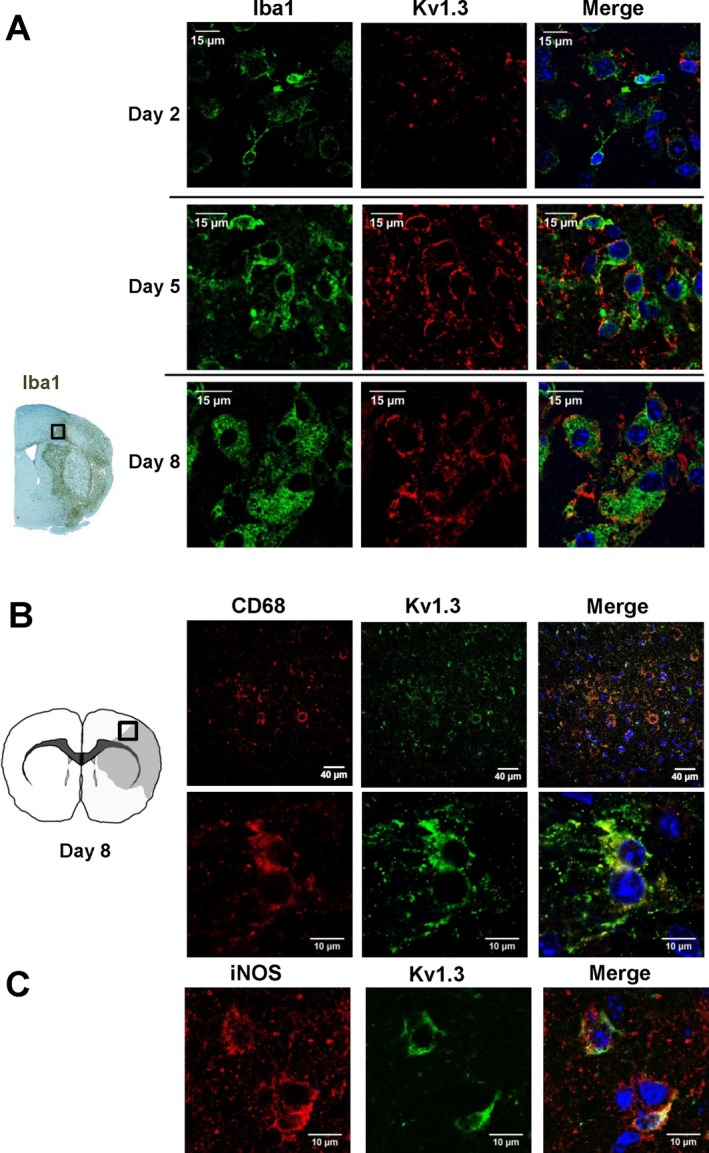
Kv1.3 expression on microglia in the border zone of ischemic infarcts in mice. (A) Representative immunofluorescent staining showing Iba1 and Kv1.3 expression 2, 5 and 8 days after MCAO with reperfusion. (B) Kv1.3 and CD68 or Kv1.3 and iNOS double staining 8 days after MCAO with reperfusion.
Figure 7.
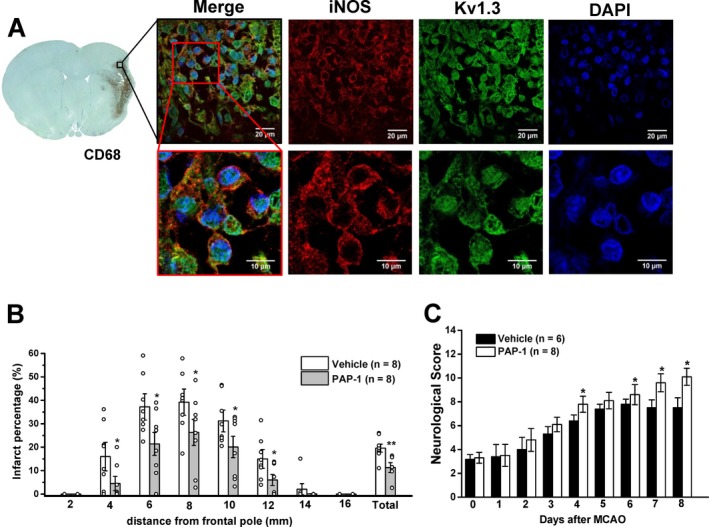
Kv1.3 inhibition with PAP‐1 reduces infarction and improves neurological deficit in rats. (A) Fluorescent staining for Kv1.3 and iNOS in the border zone of an ischemic infarct from a rat, which contains many ED1 (= CD68) positive microglia/macrophages. (B) Infarct area in brain slices 2, 4, 6, 8, 10, 12 and 16 mm from the frontal pole in vehicle‐treated rats or rats treated with 40 mg/kg PAP‐1 twice daily started 12 h after reperfusion. (C) Neurological deficit in the 14‐score system (normal rat = 14). Values are mean ± S.E.M.
The human brain autopsies and the biopsy (Fig 3) were obtained from the University of California Biorepository, which provides sections from archived paraffin blocks for research. The needle biopsy is a 7‐day‐old ischemic infarct from a 37‐year‐old patient. The surgery was conducted due to clinical uncertainty, and ischemic infarct was confirmed by a board‐certified neuropathologist (Dr. Lee‐Way Jin). Human macrophages/microglia were stained with mouse anti‐human macrophages antibody (1:100, MAC387, Bio‐Rad), Kv1.3 with a mouse monoclonal anti‐Kv1.3 antibody (1:100, 1D8, Bio‐Rad) and iNOS with a polyclonal rabbit antibody (1:100, ab3523, Abcam).
Figure 3.
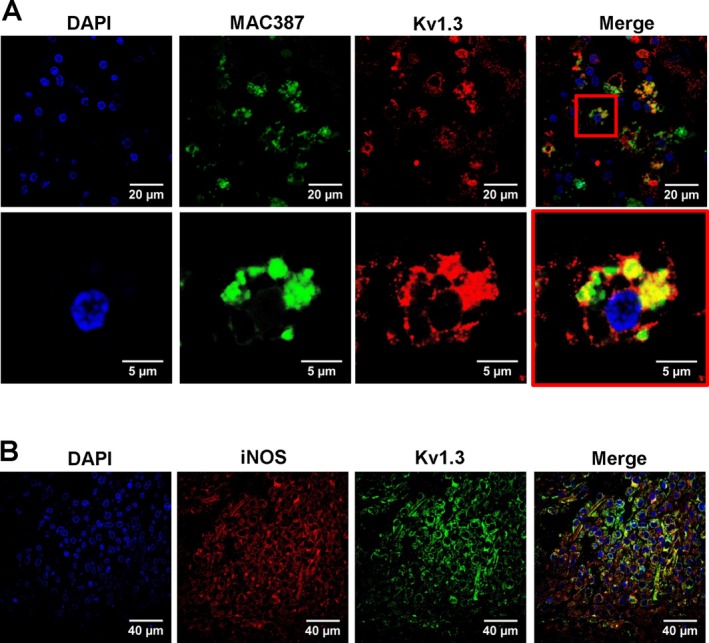
Kv1.3 expression in human infarcts. (A) Double immunofluorescent staining for activated microglia/macrophages (MAC387) and Kv1.3 in a roughly 2‐week‐old microinfarct. (B) Staining for Kv1.3 and iNOS in a needle biopsy from a 37‐year‐old man 7 days after an ischemic stroke.
Phagocytosis was evaluated by the association of CD68‐positive cells with TUNEL+ cells or the internalization of TUNEL+ material. Paraffin sections of the infarct zone were stained with rat anti‐mouse CD68 antibody conjugated with Alexa Fluor®488 (1:200, FA‐11, Bio‐Rad) and In situ BrdU‐Red DNA Fragmentation (TUNEL) Assay Kit (ab66110, Abcam). The percentage of CD68‐positive cells adjacent to TUNEL+ cells or containing TUNEL+ material was calculated by analyzing confocal images with Image J.29
Brain cytokines
Cytokines (IFN‐γ, IL‐1β, IL‐6, IL‐10, TNF‐α), BDNF, and TGF‐β1 in brain extract were analyzed with Millipore Milliplex mouse magnetic bead kits and a Luminex 200TM reader and the results normalized to pg/mg of total protein content in each sample (assayed by the bicinchoninic (BCA) procedure) as described.16
Statistical analyses
Statistical analysis of infarct area, neurological deficit scoring, and IHC were performed with one‐way analysis of variance (ANOVA; Origin software) followed by post hoc pair‐wise comparison of the different groups using Tukey's method, also referred to as honestly significant difference test, as recommended by Schlattmann and Dirnagl for MCAO studies.30 Shown are mean ± S.E.M. P < 0.05 was used as the level of significance. *=P < 0.05, **=P < 0.01, ***=P < 0.001.
Results
Kv1.3 Expression in ischemic stroke
In order to investigate Kv1.3 expression in ischemic stroke, we subjected adult male mice to reversible focal cerebral ischemia induced by 60‐min occlusion of the left middle cerebral artery (MCA) and performed immunohistochemistry for Kv1.3 on brains removed 2, 5, and 8 days after reperfusion. In normal rodent brain, Kv1.3 staining is most prominent in mitral cells of the olfactory bulb31 and presynaptic terminals of brain stem auditory neurons,32 while lower levels of Kv1.3 expression are observed in cortical interneurons.33 However, compared to activated T‐cells and microglia in inflammatory lesions,18, 34 neuronal Kv1.3 staining is typically faint and often only observable with higher antibody concentrations and long incubation times.33 In agreement with these findings, we did not note any significant Kv1.3 staining on neurons, astrocytes, or surveillant microglia in normal mouse brain (data not shown) when using anti‐Kv1.3 antibody concentrations that stain activated lymphocytes in our hands.7, 35
Following MCAO, staining with a polyclonal anti‐Kv1.3 antibody directed against an extracellular epitope revealed strong Kv1.3 immunoreactivity on hypertrophic Iba1+ microglia/macrophages in the infarct border of paraffin sections prepared five and eight days after reperfusion (Fig. 1A). Two days after MCAO, Iba1 and Kv1.3 staining was fainter (Fig. 1A top) but started to be observable suggesting that Kv1.3 expression in vivo increases following microglia activation with a similar time course as described in vitro following stimulation of microglia with LPS or astrocyte conditioning medium.36 At the peak of microglia activation on day‐8, Kv1.3 expression was found to be localized to cells positive for inducible nitric oxide synthase (iNOS) and CD68 using both a polyclonal and a monoclonal anti‐Kv1.3 antibody directed against the intracellular C‐terminus of the channel (Fig. 1B,C).
We further subjected microglia/macrophages acutely isolated with CD11b‐mangetic beads to whole‐cell patch‐clamp in order to directly study microglia Kv currents and determine the contribution of Kv1.3 to the total Kv current based on its pharmacological and biophysical properties. In order to avoid contributions from calcium‐activated K+ channels or chloride currents, we used a KF‐based pipette solution. While Kv current amplitudes were very low or often barely detectable in microglia isolated from normal, nonischemic brains (Fig. 2A), Kv current amplitudes recorded from microglia isolated from the infarcted area were significantly increased on day‐2, day‐5 and peaked on day‐8 after MCAO (Fig. 2D). The current was predominantly carried by Kv1.3 based on its threshold voltage of activation around −40 mV, sensitivity to PAP‐1 (Fig. 2B and C) and its use‐dependence (data not shown). When current amplitudes were converted into current densities in order to account for the increase in cell size during microglia activation, Kv1.3 current density was significantly higher on day‐8 after MCAO compared to microglia isolated from the contralateral side of the same animals or to microglia from normal control brains (Fig 2E).
Figure 2.

Functional Kv1.3 channel expression on acutely isolated MCAO microglia. Representative whole‐cell current traces showing Kv1.3 currents recorded from microglia acutely isolated from either (A) normal control mouse brain, (B) day‐2 ipsilateral MCAO mouse brain or (C) day‐8 ipsilateral MCAO mouse brain. (D) Scatterplot showing Kv1.3 current amplitude measured from individual microglial cells for each condition. A significant increase in Kv1.3 current expression is observed starting on day‐2 (51.41 ± 29.24 pA, n = 12) and day‐5 (41.78 ± 26.49 pA, n = 19) on ipsilateral microglia compared to normal control (14.46 ± 8.60 pA, n = 15) and day‐8 contralateral microglia (15.16 ± 10.38 pA, n = 18). ***P < 0.001. Day‐8 ipsilateral microglia (154.53 ± 94.46 pA, n = 32) express the largest Kv1.3 increase compared to all other conditions. ***P < 0.001. (E) Scatterplot showing Kv1.3 current density calculated for individual microglial cells for each condition. A significant increase in Kv1.3 current density is observed only in day‐8 ipsilateral microglia (36.40 ± 22.42 pA, n = 32) compared to normal control (5.06 ± 3.77 pA, n = 15), day‐2 ipsilateral (6.70 ± 3.98 pA, n = 12), day‐5 ipsilateral (5.27 ± 4.08 pA, n = 19) and day‐8 contralateral microglia (5.73 ± 4.44 pA, n = 18). **P < 0.01. All values are presented as mean ± SD. The significance of the difference between two conditions was tested using two‐tailed paired Student's t‐test.
Kv1.3 expression Human Infarcts
In order to confirm that Kv1.3 is also expressed on human microglia, we stained postmortem brain sections from patients with vascular dementia, which contained microinfarcts estimated to be roughly 2 weeks old, with the monoclonal anti‐Kv1.3 antibody. Strong, positive Kv1.3 staining was localized to MAC387‐positive microglia/macrophages (Fig. 3A). We were further able to obtain a needle biopsy sample from a 37‐year‐old patient, which was conducted due to clinical uncertainty. A 7‐day‐old ischemic infarct was confirmed by a board‐certified neuropathologist. The needle biopsy contained many clustered MAC387‐positive macrophages/microglia (not shown), which stained positive for both Kv1.3 and iNOS (Fig. 3B).
Kv1.3 Inhibition reduces microglia activation and increases neuronal survival in organotypic hippocampal slices
As a first test of our hypothesis that Kv1.3 inhibition could be beneficial in ischemic stroke, we investigated the effect of PAP‐1 on organotypic hippocampal slice cultures. Hippocampal slices reflect conditions in the brain in terms of microglia interactions with neurons and astrocytes but allow no additional monocytes or T‐cells to infiltrate from the blood. Exposure of hippocampal slices to hypoxia/aglycemia for 60 min resulted in strong microglia activation and a reduction in surviving NeuN+ neurons 3 days later as determined by Iba‐1 and NeuN staining (Fig. 4A). Treatment of slices with PAP‐1 or the microglia inhibitor minocycline resulted in a significant increase in neuronal survival and a reduction in microglia activation (Fig. 4A,B). PAP‐1 further reduced the hypoxia‐induced loss of PSD95 expression suggesting increased preservation of excitatory synapses (Fig. 4C). PAP‐1 is unlikely to have exerted any direct neuroprotective effects in this assay because experiments performed with PAP‐1 by the NIH Anticonvulsant Screening program did not report any neuroprotection with 10 μmol/L of PAP‐1 in organotypic hippocampal slices (n = 8) exposed to 20 μmol/L of the excitotoxic agent kainic acid for 4 h and evaluated for propidium iodide uptake in the CA1 and CA3 pyramidal cell layers and the dentate gyrus at 24 h.37 Taken together, these findings and their time course confirm previous observations made with Kv1.3‐blocking peptides and LPS‐stimulated microglia in Transwell cell culture systems11 and suggest that Kv1.3 inhibition increases neuronal survival by reducing microglia‐mediated neuronal killing.
Figure 4.
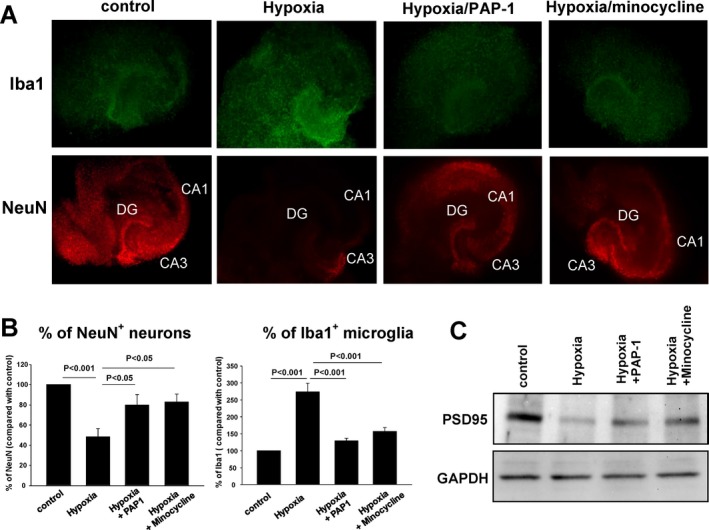
PAP‐1 reduces microglia activation and neuronal death in organotypic hippocampal mouse slices. (A) Hippocampal slices stained for Iba‐1 (green) and NeuN (red) 3 days after a 60 min exposure to hypoxia/aglycemia. PAP‐1 (1 μmol/L) and minocycline (10 μmol/L) were added 2 h after the end of the hypoxia. (B) Quantification of data shown in (A) from five slices. Shown are means ± S.E.M. (C) Western blot analysis for PSD95 of lysates from control slices or slices exposed to hypoxia in the presence of absence of PAP‐1 or minocycline.
Kv1.3 blockade reduces infarction and improves neurological deficit in middle cerebral artery occlusion with reperfusion in mice
As our Kv1.3 blocker PAP‐1 penetrates well into the brain (Cbrain/Cplasma = 1.1), we next used it as a pharmacological tool in a mouse model of ischemic stroke. Adult male C57BL/6J mice were subjected to reversible focal cerebral ischemia induced by 60‐min occlusion of the left MCA and scored daily for neurological deficit until sacrifice on day‐8. Starting 12 h after reperfusion, mice were treated intraperitoneally twice daily with either vehicle or the Kv1.3 blocker PAP‐1 at 10 or 40 mg/kg. PAP‐1 treated mice exhibited a significant reduction in infarct area as defined by NeuN‐negativity (Fig. 5A). The mean infarct area was reduced from 25.0 ± 3.0% in vehicle‐treated mice (n = 12) to 8.9 ± 1.2% in mice administered 10 mg/kg PAP‐1 (n = 7, P = 0.0001) and to 7.3 ± 1.7% in mice administered 40 mg/kg PAP‐1 (n = 9, P = 0.0006).
Figure 5.
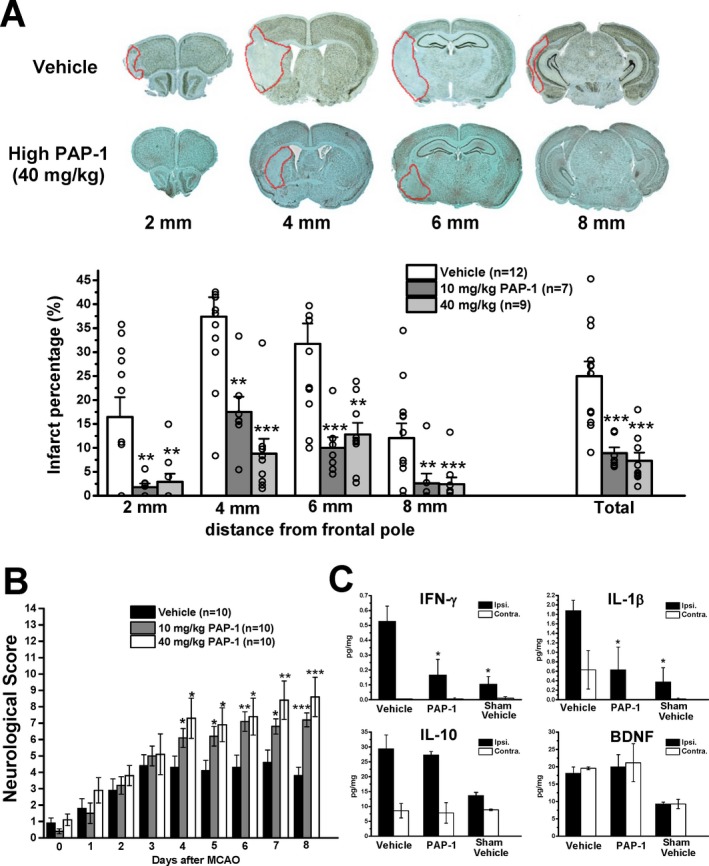
The Kv1.3 inhibitor PAP‐1 reduces infarct area, improves neurological deficit, and reduces inflammatory cytokine production in mice. (A) NeuN‐defined infarct area in brain slices 2, 4, 6, and 8 mm from the frontal pole in vehicle‐treated mice or mice treated intraperitoneally with 10 mg/kg or 40 mg/kg PAP‐1 twice daily started 12 h after reperfusion. (B) Neurological deficit in the 14‐score system (normal mouse = 14). (C) Brain cytokine concentrations in the ipsi‐ and contralateral side 8 days after MCAO from vehicle or PAP‐1 (40 mg/kg)‐treated mice or shams (n = 3). All values are mean ± S.E.M.
As filament MCAO induces infarction not only in the cortex, but also in the striatum, we used a 14 score tactile and proprioceptive limb‐placing test26 to evaluate mice in a blinded fashion every 24 h. All mice exhibited scores of ~1 when tested 12 h after the surgery before drug administration was commenced (Fig. 5B). While vehicle‐treated animals only improved to an average score of 4 by day‐4 and then leveled off (Fig. 5B), PAP‐1 treatment significantly improved neurological scores starting from day‐4 onwards. Mice treated with 10 mg/kg PAP‐1 exhibited a score of 7.2 ± 0.43 (P = 0.0005 vs vehicle). Mice treated with 40 mg/kg scored 8.6 ± 1.20 (P = 0.0001 vs vehicle) by day‐8 of a maximal possible score of 14 for a normal mouse.
Kv1.3 blockade preferentially reduces inflammatory cytokine production but does not affect phagocytosis of TUNEL‐positive Cells
We further determined brain cytokine levels on day‐8 in the ipsi‐ and contralateral hemisphere from vehicle‐treated sham‐operated animals as well as vehicle‐ and PAP‐1‐treated mice (40 mg/kg) subjected to MCAO (Fig. 5C). MCAO significantly increased levels of the inflammatory cytokines IL‐1β and IFN‐γ and of the anti‐inflammatory cytokine IL‐10 in the infarcted hemisphere. PAP‐1 reduced IL‐1β and IFN‐γ concentrations without affecting IL‐10 (Fig. 5C). PAP‐1 also did not reduce levels of brain‐derived nerve growth factor (BDNF) in both the ipsi‐ and contralateral side. We also tried to measure TNF‐α and IL‐6, but found that levels were below detection of the multiplex assay on day‐8 after MCAO. Taken together, these findings suggest that Kv1.3 inhibition preferentially reduces proinflammatory cytokine levels.
As phagocytosis is a beneficial function of microglia, which ideally should not be inhibited by a drug used for stroke, we further investigated whether PAP‐1 treatment reduced the frequency at which CD68+ phagocytes in the infarct area closely contacted TUNEL+ dying cells or had incorporated TUNEL+ material (Fig. 6). In keeping with our macroscopic observation that infarct areas in PAP‐1‐treated mice are shrunken on day‐8 after reperfusion MCAO and not swollen like in mice where phagocytosis is impaired (e.g., TREM2−/− mice29), PAP‐1 treatment at both the low and the high dose did not reduce the percentage of CD68+ phagocytes closely contacting TUNEL+ cells or containing TUNEL+ material on day‐8. However, in animals treated with the higher dose of PAP‐1, overall fewer CD68+ phagocytes and less TUNEL+ staining was observed (not shown), but the percentage of association/internalization was not significantly changed (Fig. 6A). The CD68+ cells containing TUNEL+ apoptotic cell material mostly exhibited intact, regularly shaped nuclei (Fig. 6B) when scanning through the cells in the confocal images suggesting that the phagocytes themselves are not undergoing apoptosis. These findings suggest that Kv1.3 inhibition does not affect phagocytosis in vivo confirming a recent study reporting that Kv1.3 blockers to not impair the ability of isolated brain mononuclear cells to phagocytose polystyrene microspheres.38
Figure 6.
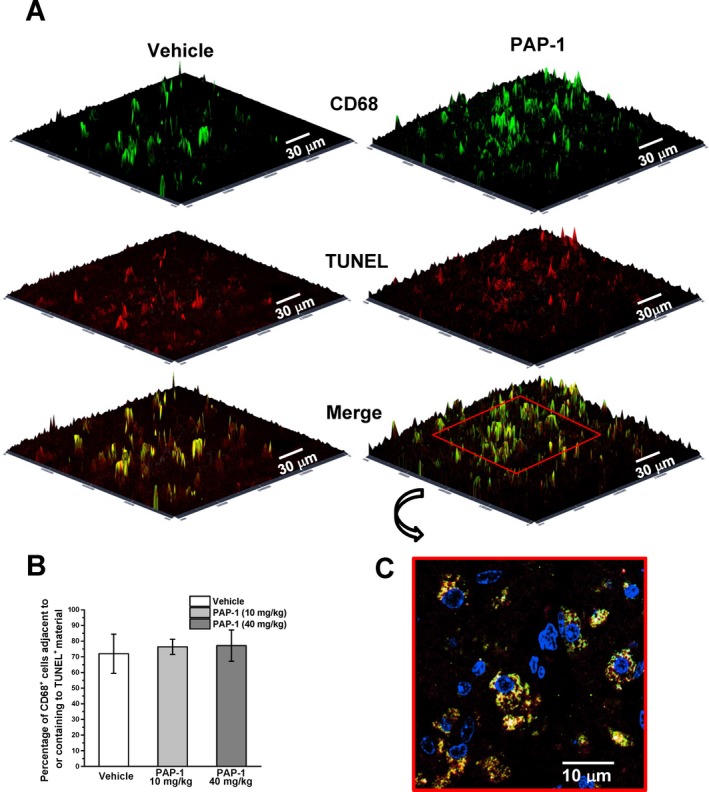
Kv1.3 inhibition does not affect phagocytosis in vivo. (A) Sections obtained 4 mm from the frontal pole on day‐8 after MCAO were stained for activated microglia/macrophages with CD68 (green) and apoptosis associated TUNEL fragments (red). (B) Percentage of CD68+ cells adjacent to or containing TUNEL + material. Three fields along the edge of the infarct area were analyzed in each section (n = 3 animals per treatment group). Values are mean ± SD, P > 0.05. (C) Two‐dimensional image showing DAPI, CD68 and TUNEL staining in a PAP‐1 (10 mg/kg)‐treated mouse at higher magnification.
Confirmation in a second species: PAP‐1 also reduces infarction and improves neurological deficit in rats
Based on the difficulty of translating observations from animal models of ischemic stroke into humans, any therapeutic finding should be reproduced in a second species. We therefore also tested whether Kv1.3 inhibition with PAP‐1 could reduce infarction and improve neurological deficit in rats. Male Wistar rats were subjected to 90 min of MCAO with 8 days of reperfusion. Similar to mice and humans, activated microglia in the infarct border zone, identified by CD68 (= ED1) positivity, exhibited strong Kv1.3 staining on iNOS+ microglia (Fig. 7A). PAP‐1 treatment at 40 mg/kg started 12‐hours after reperfusion significantly reduced infarct area on day‐8 (Fig. 7B) with the mean infarct area being reduced from 23.0 ± 5.2% in vehicle‐treated controls (n = 8) to 11.3 ± 2.1% in PAP‐1‐treated rats (n = 8, P = 0.03). The hemisphere shrinkage was reduced from 9.2 ± 2.3% in vehicle‐treated controls (n = 8) to 4.7 ± 1.3% in PAP‐1‐treated rats (n = 8, P = 0.07).
Rats were also evaluated for neurological deficit using the same 14‐score test26 described above for mice. In contrast to mice, rats were less severely affected by MCAO and exhibited average scores of 3.2 ± 0.4 when tested 12 h after the surgery before starting drug or vehicle administration (Fig. 7C). Vehicle‐treated rats improved to an average score of 7.4 ± 0.4 by day‐5 and did not show any further improvement until day‐8 (Fig. 7C). PAP‐1 treatment improved neurological scores starting from day‐4 onwards and rats in the PAP‐1 group exhibited a score of 10.1 ± 0.7 (P = 0.023) by day‐8.
Discussion
In addition to directly causing neuronal damage, acute ischemic stroke elicits a strong neuroinflammatory response. In both rodent models of cerebral ischemia and in human postmortem brain, activated microglia/macrophages are abundant in the infarcted area between 18 and 96 h after the insult39 and are still present months and sometimes even years after a stroke.40 PET imaging in ischemic stroke patients demonstrated microglia activation in the peri‐infarct zone on a similar time scale41 and along affected pyramidal tracts for up to 6 months.42 Similar time courses for microglia activation with long‐term persistence have been reported for hemorrhagic stroke and traumatic brain injury.43 Although microglia can perform beneficial functions such as phagocytosing debris and releasing neurotrophic factors, activated microglia are also the main sources of inflammatory cytokines, reactive oxygen species, and nitric oxide. In rodent models, “M1‐like” microglia have been described to begin increasing around day‐3 after MCAO and dominate at 7 and 14 days after the insult, especially in the infarct border, where they presumably expand neuronal injury.44 Based on this delayed time course, modulation of this secondary inflammatory damage has been proposed as a more realistic target for stroke therapy21 than acute neuroprotection, which has largely failed in the clinic but is currently being reconsidered in the context of reperfusion. So far a limited number of anti‐inflammatory therapies have advanced from animal studies to clinical trials in acute ischemic stroke and intracerebral hemorrhage. While enlimomab presumably failed due to the use of a murine antibody, the IL‐1 receptor antagonist anakinra, the microglia inhibitor minocycline, and the sphingosine‐1‐phosphate receptor agonist fingolimod have shown signs of efficacy in open‐label Phase‐II studies.45 Additionally, a large body of mechanistic work suggests that more selective immunomodulatory therapies, especially approaches that have shown some efficacy in multiple sclerosis or its animal models, should be evaluated in stroke models.21, 45 We here explored such an alternative pharmacological target for immunomodulation in stroke by demonstrating that the Kv1.3 inhibitor PAP‐1 started 12 h after an ischemic insult reduces proinflammatory cytokine levels in the infarcted hemisphere, and significantly reduces infarct volume and improves neurological deficit in two species. Although, this treatment window should be further explored in future as Kv1.3 blockade might still be effective when started at 24 or 48 h, it clearly exceeds the 4.5 h for intravenous tissue plasminogen activator and we consider these findings encouraging as few experimental stroke treatments still show efficacy when administered that late.
The exact mechanisms of how Kv1.3 inhibition reduces infarction and neuroinflammation of course need to be further investigated. Specifically, it will need to be sorted out using tissue‐specific knockouts whether effects on T‐cells and infiltrating monocytes contribute to the beneficial actions of Kv1.3 inhibitors. Based on the observed Kv1.3 expression on iNOS+ microglia/macrophages in both rodents and humans and the finding that PAP‐1 reduced IL‐1β and IFN‐γ but not IL‐10 and BDNF in the postischemic brain, we propose that Kv1.3 inhibitors preferentially suppress proinflammatory “M1‐like” microglia responses and spare beneficial microglia functions. This hypothesis is supported by our recent mechanistic in vitro studies showing that stimulation of cultured mouse microglia with M1‐polarizing stimuli greatly increased Kv1.3 expression, while IL‐4 treatment down‐regulated Kv1.3 and increased functional expression of the inward‐rectifier K+ channel Kir2.1.19 In the same study, PAP‐1 reduced IL‐1β, TNF‐α and NO production and reduced iNOS and COX‐2 expression by LPS‐stimulated microglia, which is in keeping with the observed reduction in hypoxia‐stimulated microglia activation by PAP‐1 in organotypic hippocampal slices in our current study, where no infiltration of blood‐derived monocytes or T‐cells is possible. However, we can of course not completely exclude effects of PAP‐1 on T‐cells and infiltrating monocytes in vivo, but would like to point out that they are also likely beneficial given the fact that Kv1.3 inhibitors, similar to fingolimod, effectively treat rodent models of multiple sclerosis and other memory T‐cell‐mediated autoimmune diseases.4
As pointed out by a reviewer, we can further not completely rule out PAP‐1 effects on other Kv1‐family channels such as Kv1.2 and Kv1.5. However, we would like to point out that PAP‐1 inhibits Kv1.3 with an IC50 of 2 nmol/L, while the IC50 for Kv1.2 is 250 nmol/L.22 Pharmacokinetic studies using UPLC/MS have shown that total PAP‐1 brain concentrations peak between 2 and 10 μmol/L at 2 h after administration of 10 or 40 mg/kg but that brain concentrations for most of the 12‐h time period between drug administrations are in the range of 200 nmol/L to 1 μmol/L. Considering PAP‐1's plasma protein binding of 98%, these total concentrations translate to effective free concentrations of 40–200 nmol/L at the Cmax and of 4–20 nmol/L for most of the time in this study.4, 23 We therefore believe that Kv1.2 inhibition is not likely to significantly contribute to the effects of PAP‐1. Kv1.5, which PAP‐1 inhibits with an IC50 of 45 nmol/L22 is more likely to contribute. However, despite previous reports of Kv1.5 expression in microglia,46 we have not been able to detect Kv1.5 message in cultured microglia from C57BL6 mice19 and can only speculate that there is a strain difference between C57BL6 and the 129S6 background of the Kv1.5−/− mice.46 We further previously observed16 that the Kv current in acutely isolated microglia from MCAO mice is not only blocked by PAP‐1 but also by 100 pM ShK‐L5, a peptidic inhibitor of Kv1.3, that has no effect on Kv1.5.47 However, we are sure that inhibition other microglia ionic currents such as KCa3.1 currents or Hv1‐mediated proton currents does not contribute to the PAP‐1 effects. PAP‐1 exhibits 5000‐fold selectivity for Kv1.3 over KCa3.122 and, as we recently tested based on a reviewer suggestion, does not affect proton currents in primary mouse microglia at concentrations of 1 and 10 μmol/L (data not shown).
When proposing that Kv1.3 inhibitors preferentially inhibit “M1‐like” inflammatory microglia/macrophage functions and preserve beneficial “M2‐like” functions, we are fully cognizant of the fact that microglia are highly plastic and, unlike T‐cells, do not achieve stable differentiation states.48 Yet, our results seem to support this simple therapeutic hypothesis by showing that the observed reduction in infarct area and improvement in neurological deficit following PAP‐1 treatment is accompanied by a reduction in brain levels of IL‐1β and IFN‐γ, but not IL‐10 and BDNF or an impairment of phagocytosis. Another attractive feature of using Kv1.3 inhibitors to reduce neuroinflammation in the wake of ischemic stroke is that Kv1.3 inhibitors are known to be immunomodulators rather than immunosuppressants and do not impair the ability of rodents to clear acute infections or of primates to develop vaccine responses.5, 8 Kv1.3 inhibitors are therefore unlikely to further increase the risk of respiratory and urinary tract infections, which are responsible for considerable morbidity and mortality after stroke.45, 49 This phenomenon, which manifests as lymphopenia in stroke patients and rodent models, is mediated by an increased release of cortisol and catecholamines,49 which has recently been shown to induce a higher bone marrow output of inflammatory monocytes and neutrophils and a drastic reduction in lymphoid progenitors.50 In addition, KV1.3 blockade seems to be relatively safe and well tolerated. Toxicity studies with PAP‐1 and the peptidic KV1.3 blocker ShK‐186 have so far not revealed any toxicity despite 6 months or 28 days of continuous administration in rats or rhesus macaques.4, 8 PAP‐1 has further completed IND‐enabling toxicity studies, while ShK‐186 has passed both IND toxicity studies and Phase‐1 safety studies without any adverse findings.6, 9
In conclusion, we are proposing Kv1.3 inhibitors as novel potential therapeutics for reducing secondary inflammatory damage in ischemic stroke by preferentially targeting “M1‐like” microglia functions. Following this first pharmacological proof‐of‐concept study, PAP‐1 and other small molecule Kv1.3 blockers should be further explored in alternative stroke models, that for example establish reperfusion with alteplase to increase translatability to human stroke, and also be studied in females and animals with comorbidities.
Author Contributions
Y.J.C., H.M.H. and H.W. developed the concept and designed the study. Y.J.C., H.M.H., and I.M. acquired and analyzed the data. Y.J.C., I.M., H.M.H, L.W.J and H.W. wrote the manuscript and prepared the figures.
Conflict of Interests
H.W. is an inventor on a University of California patent claiming PAP‐1 for immunosuppression. This patent has just been abandoned by the University of California because of its short remaining patent life. H.W. holds founder stock in Airmid Inc., a company trying to develop the venom peptide‐based Kv1.3 inhibitor ShK‐186 for autoimmune diseases.
Funding information
This work was supported by the National Institute of Neurological Disease and Stroke (R56NS098328 and NS100294 to H.W.), a National Institute on Aging Award (AG043788 to I.M.), and the Alzheimer's Disease Center at the University of California Davis funded by NIH/NIA (P30 AG10129).
Funding Statement
This work was funded by National Institute of Neurological Disease and Stroke grants R56NS098328 and NS100294; National Institute on Aging grants AG043788 and AG10129; Alzheimer's Disease Center grant ; University of California Davis grant ; NIH/NIA grant P30 AG10129.
References
- 1. DeCoursey TE, Chandy KG, Gupta S, Cahalan MD. Voltage‐gated K+ channels in human T lymphocytes: a role in mitogenesis? Nature 1984;307:465–468. [DOI] [PubMed] [Google Scholar]
- 2. Wulff H, Beeton C, Chandy KG. Potassium channels as therapeutic targets for autoimmune disorders. Curr Opin Drug Discovery & Development 2003;6:640–647. [PubMed] [Google Scholar]
- 3. Wulff H, Calabresi PA, Allie R, et al. The voltage‐gated Kv1.3 K+ channel in effector memory T cells as new target for MS. J Clin Invest 2003;111:1703–1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Beeton C, Wulff H, Standifer NE, et al. Kv1.3 channels are a therapeutic target for T cell‐mediated autoimmune diseases. Proc Natl Acad Sci USA 2006;103:17414–17419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Matheu MP, Beeton C, Garcia A, et al. Imaging of effector memory T cells during a delayed‐type hypersensitivity reaction and suppression by Kv1.3 channel block. Immunity 2008;29:602–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Tarcha EJ, Chi V, Munoz‐Elias EJ, et al. Durable pharmacological responses from the peptide drug ShK‐186, a specific Kv1.3 channel inhibitor that suppresses T cell mediators of autoimmune disease. J Pharmacol Exp Ther 2012;342:642–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kundu‐Raychaudhuri S, Chen YJ, Wulff H, Raychaudhuri SP. Kv1.3 in psoriatic disease: PAP‐1, a small molecule inhibitor of Kv1.3 is effective in the SCID mouse psoriasis–xenograft model. J Autoimmun 2014;55:63–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Pereira LE, Villinger F, Wulff H, et al. Pharmacokinetics, toxicity, and functional studies of the selective Kv1.3 channel blocker 5‐(4‐phenoxybutoxy)psoralen in rhesus macaques. Exp Biol Med (Maywood) 2007;232:1338–1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tarcha EJ, Olsen CM, Probst P, et al. Safety and pharmacodynamics of dalazatide, a Kv1.3 channel inhibitor, in the treatment of plaque psoriasis: a randomized phase 1b trial. PLoS ONE 2017;12:e0180762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Khanna R, Roy L, Zhu X, Schlichter LC. K+ channels and the microglial respiratory burst. Am J Physiol Cell Physiol 2001;280:C796–C806. [DOI] [PubMed] [Google Scholar]
- 11. Fordyce CB, Jagasia R, Zhu X, Schlichter LC. Microglia Kv1.3 channels contribute to their ability to kill neurons. J Neurosci 2005;25:7139–7149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kaushal V, Koeberle PD, Wang Y, Schlichter LC. The Ca2+‐activated K+ channel KCNN4/KCa3.1 contributes to microglia activation and nitric oxide‐dependent neurodegeneration. J Neurosci 2007;27:234–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kettenmann H, Hanisch UK, Noda M, Verkhratsky A. Physiology of microglia. Physiol Rev 2011;91:461–553. [DOI] [PubMed] [Google Scholar]
- 14. Maezawa I, Zimin P, Wulff H, Jin LW. A‐beta oligomer at low nanomolar concentrations activates microglia and induces microglial neurotoxicity. J Biol Chem 2011;286:3693–3706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lyons SA, Pastor A, Ohlemeyer C, et al. Distinct physiologic properties of microglia and blood‐borne cells in rat brain slices after permanent middle cerebral artery occlusion. J Cereb Blood Flow Metab 2000;20:1537–1549. [DOI] [PubMed] [Google Scholar]
- 16. Chen YJ, Nguyen HM, Maezawa I, et al. The potassium channel KCa3.1 constitutes a pharmacological target for neuroinflammation associated with ischemia/reperfusion stroke. J Cereb Blood Flow Metab 2016;36:2146–2161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Rangaraju S, Gearing M, Jin LW, Levey A. Potassium channel Kv1.3 is highly expressed by microglia in human Alzheimer's disease. J Alzheimers Dis 2015;44:797–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Rus H, Pardo CA, Hu L, et al. The voltage‐gated potassium channel Kv1.3 is highly expressed on inflammatory infiltrates in multiple sclerosis brain. Proc Natl Acad Sci USA 2005;102:11094–11099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Nguyen HM, Grossinger EM, Horiuchi M, et al. Differential Kv1.3, KCa3.1, and Kir2.1 expression in “classically” and “alternatively” activated microglia. Glia 2017;65:106–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Macrez R, Ali C, Toutirais O, et al. Stroke and the immune system: from pathophysiology to new therapeutic strategies. Lancet Neurol 2011;10:471–480. [DOI] [PubMed] [Google Scholar]
- 21. Iadecola C, Anrather J. The immunology of stroke: from mechanisms to translation. Nat Med 2011;17:796–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Schmitz A, Sankaranarayanan A, Azam P, et al. Design of PAP‐1, a selective small molecule Kv1.3 blocker, for the suppression of effector memory T cells in autoimmune diseases. Mol Pharmacol 2005;68:1254–1270. [DOI] [PubMed] [Google Scholar]
- 23. Azam P, Sankaranarayanan A, Homerick D, et al. Targeting effector memory T cells with the small molecule Kv1.3 blocker PAP‐1 suppresses allergic contact dermatitis. J Invest Dermatol 2007;127:1419–1429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Longa EZ, Weinstein PR, Carlson S, Cummins R. Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke 1989;20:84–91. [DOI] [PubMed] [Google Scholar]
- 25. Chen YJ, Raman G, Bodendiek S, et al. The KCa3.1 blocker TRAM‐34 reduces infarction and neurological deficit in a rat model of ischemia/reperfusion stroke. J Cereb Blood Flow Metab 2011;31:2363–2374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. De Ryck M, Van Reempts J, Borgers M, et al. Photochemical stroke model: flunarizine prevents sensorimotor deficits after neocortical infarcts in rats. Stroke 1989;20:1383–1390. [DOI] [PubMed] [Google Scholar]
- 27. Jin LW, Horiuchi M, Wulff H, et al. Dysregulation of glutamine transporter SNAT1 in Rett syndrome microglia: a mechanism for mitochondrial dysfunction and neurotoxicity. J Neurosci 2015;35:2516–2529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Maezawa I, Nivison M, Montine KS, et al. Neurotoxicity from innate immune response is greatest with targeted replacement of E4 allele of apolipoprotein E gene and is mediated by microglial p38MAPK. FASEB J 2006;20:797–799. [DOI] [PubMed] [Google Scholar]
- 29. Kawabori M, Kacimi R, Kauppinen T, et al. Triggering receptor expressed on myeloid cells 2 (TREM2) deficiency attenuates phagocytic activities of microglia and exacerbates ischemic damage in experimental stroke. J Neurosci 2015;35:3384–3396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Schlattmann P, Dirnagl U. Statistics in experimental cerebrovascular research: comparison of more than two groups with a continuous outcome variable. J Cereb Blood Flow Metab 2010;30:1558–1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Fadool DA, Tucker K, Perkins R, et al. Kv1.3 channel gene‐targeted deletion produces “Super‐Smeller Mice” with altered glomeruli, interacting scaffolding proteins, and biophysics. Neuron 2004;41:389–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Gazula VR, Strumbos JG, Mei X, et al. Localization of Kv1.3 channels in presynaptic terminals of brainstem auditory neurons. J Comp Neuorol 2010;518:3205–3220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Duque A, Gazula VR, Kaczmarek LK. Expression of Kv1.3 potassium channels regulates density of cortical interneurons. Dev Neurobiol 2013;73:841–855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Peng Y, Lu K, Li Z, et al. Blockade of Kv1.3 channels ameliorates radiation‐induced brain injury. Neuro Oncol 2014;16:528–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Chen YJ, Lam J, Gregory CR, et al. The Ca2+‐activated K+ channel KCa3.1 as a potential new target for the prevention of allograft vasculopathy. PLoS ONE 2013;8:e81006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Schlichter LC, Sakellaropoulos G, Ballyk B, et al. Properties of K+ and Cl− channels and their involvement in proliferation of rat microglial cells. Glia 1996;17:225–236. [DOI] [PubMed] [Google Scholar]
- 37. Noraberg J. Organotypic brain slice cultures: an efficient and reliable method for neurotoxicological screening and mechanistic studies. Altern Lab Anim 2004;32:329–337. [DOI] [PubMed] [Google Scholar]
- 38. Rangaraju S, Raza SA, Pennati A, et al. A systems pharmacology‐based approach to identify novel Kv1.3 channel‐dependent mechanisms in microglial activation. J Neuroinflammation 2017;14:128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Campanella M, Sciorati C, Tarozzo G, Beltramo M. Flow cytometric analysis of inflammatory cells in ischemic rat brain. Stroke 2002;33:586–592. [DOI] [PubMed] [Google Scholar]
- 40. Beschorner R, Schluesener HJ, Gozalan F, et al. Infiltrating CD14+ monocytes and expression of CD14 by activated parenchymal microglia/macrophages contribute to the pool of CD14+ cells in ischemic brain lesions. J Neuroimmunol 2002;126:107–115. [DOI] [PubMed] [Google Scholar]
- 41. Price CJ, Wang D, Menon DK, et al. Intrinsic activated microglia map to the peri‐infarct zone in the subacute phase of ischemic stroke. Stroke 2006;37:1749–1753. [DOI] [PubMed] [Google Scholar]
- 42. Jacobs AH. Tavitian B, consortium IN. Noninvasive molecular imaging of neuroinflammation. J Cereb Blood Flow Metab 2012;32:1393–1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wang J. Preclinical and clinical research on inflammation after intracerebral hemorrhage. Prog Neurobiol 2010;92:463–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hu X, Li P, Guo Y, et al. Microglia/macrophage polarization dynamics reveal novel mechanism of injury expansion after focal cerebral ischemia. Stroke 2012;43:3063–3070. [DOI] [PubMed] [Google Scholar]
- 45. Fu Y, Liu Q, Anrather J, Shi FD. Immune interventions in stroke. Nat Rev Neurol 2015;11:524–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Pannasch U, Farber K, Nolte C, et al. The potassium channels Kv1.5 and Kv1.3 modulate distinct functions of microglia. Mol Cell Neurosci 2006;33:401–411. [DOI] [PubMed] [Google Scholar]
- 47. Pennington MW, Beeton C, Galea CA, et al. Engineering a stable and selective peptide blocker of the Kv1.3 channel in T lymphocytes. Mol Pharmacol 2009;75:762–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Murray PJ, Allen JE, Biswas SK, et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity 2014;41:14–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Meisel C, Schwab JM, Prass K, et al. Central nervous system injury‐induced immune deficiency syndrome. Nat Rev Neurosci 2005;6:775–786. [DOI] [PubMed] [Google Scholar]
- 50. Courties G, Herisson F, Sager HB, et al. Ischemic stroke activates hematopoietic bone marrow stem cells. Cir Res 2015;116:407–417. [DOI] [PMC free article] [PubMed] [Google Scholar]