

Abstract
We describe a family with Charcot Marie Tooth disease type 4J presenting with features of Charcot Marie Tooth disease plus parkinsonism and aphemia. Genetic testing found two variants in the FIG4 gene: c.122T>C (p.I41T) – the most common Charcot Marie Tooth disease type 4J variant – and c.1949‐10T>G (intronic). Proband fibroblasts showed absent FIG4 protein on western blot, and skipping of exon 18 by RT‐PCR. As most patients with Charcot Marie Tooth disease type 4J do not have central nervous system deficits, we postulate the intronic variant and I41T mutation together are causing loss of FIG4 protein and subsequently the central nervous system findings in our family.
Introduction
Charcot Marie Tooth disease type 4J (CMT4J) is a rare inherited peripheral neuropathy, affecting 0.24% of people with CMT,1 caused by biallelic disease causing variants in the FIG4 gene. CMT4J may present either as an early and severe, or a late onset and more slowly progressive disease. Patients typically present with a length dependent progressive demyelinating motor and sensory polyneuropathy.2 There have been isolated case reports of people with CMT4J also having central nervous system (CNS) features, such as parkinsonism.3 In addition, people with disease‐causing variants in FIG4 have also been identified with Yunis‐Varon syndrome and familial epilepsy with polymicrogyria syndrome, both early onset diseases with severely abnormal CNS development and pathology. Here, we describe a family with both CMT4J and unique CNS features.
Patients and Methods
All aspects of this study, including Video S1, were performed with approval by the Stanford University Institutional Review Board and with written patient consent.
The proband is a 52‐year‐old right‐handed woman who was diagnosed with Charcot Marie Tooth disease (CMT) at 12 years of age. She experienced slowly progressive weakness in distal legs until the severity of her symptoms accelerated in her late forties, at which time she noticed more rapidly progressive weakness and also developed various CNS abnormalities. On examination she had slow, hesitant speech consistent with aphemia without significant dysarthria, and word finding difficulties and long pauses when either speaking spontaneously or reading. Additional pertinent findings on physical examination included: slow resting hand tremor (right more than left); slow and dysrhythmic finger tapping bilaterally; and bilateral upper extremity rigidity with cogwheeling. Strength testing (MRC – L/R) showed normal strength proximally with distal weakness: FDI: 4‐/1, ADM: 4‐/4‐; APB: 4‐/4‐; ankle dorsiflexion 3/3; plantarflexion 5/5. Tendon reflexes were absent. Sensory exam normal pinprick sensation in hands and feet and reduced perception of vibration to knees. (Video S1)
Nerve conduction studies (NCS) at 51 years of age showed slow motor conduction velocities (ulnar and median: 19 m/sec) with relatively preserved compound muscle action potential (CMAP) amplitudes (ulnar: 6.7 mV, median: 3.7 mV), suggesting de‐/dysmyelination. The radial sensory response was absent. Her CMT Neuropathy Score (version 2) was 17 (moderate impairment). Her brain MRI showed diffuse cortical volume loss with pronounced focal left temporal lobe atrophy (Fig. 1A).
Figure 1.
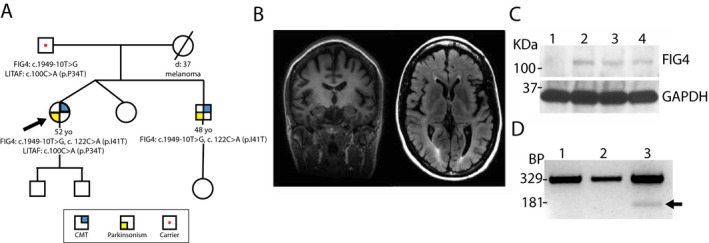
Family pedigree, proband MRI brain, RT‐PCR, and western blot. (A) Family pedigree. (B) MRI brain T1 weighted coronal section and T2 FLARE axial section demonstrate diffuse cortical atrophy with pronounced focal left temporal lobe atrophy. (C) RT‐PCR of FIG4 from fibroblasts collected from a healthy age‐matched control (Lane 1), an individual with CMT4J heterozygous for the I41T variant (Lane 2), and our patient with the I41T and intronic c.1949‐10T>G variants in trans. The upper band (329 bp) is a PCR product containing exon 18, while the lower band (181 bp) only seen in Lane 3 is the PCR product with the smaller size consistent with exon 18 skipping. (D) Western blot staining for Fig4 protein expression along with GAPDH as a loading control from patient fibroblast samples. Lane 1 is our patient and demonstrates Fig4 immunostaining. Lanes 2–4 are age and gender‐matched controls and demonstrate normal endogenous Fig4 immunostaining. Lanes 2 and 4 are healthy individuals, and lane 3 is an individual with CMT2 with unknown genetic cause.
Neuropsychiatric testing at 52 years of age found an IQ of 78, less than her estimated average baseline functioning, with impaired performance in auditory attention, expressive language, and executive functioning. She met the criteria for mild cognitive impairment.
Family history
The patient's unaffected fraternal twin sister was available for physical evaluation, but not her affected younger brother, aged 48 years with CMT, speech abnormalities, Parkinsonism (status post deep brain stimulation implantation), and behavioral difficulties (he was available for genetic evaluation). Parents were unaffected, and reportedly had normal NCS. The patient's mother died at 37 years of melanoma. The maternal and paternal ethnicities were Irish/English and Belgian/Italian, respectively, with no known consanguinity (Fig. 1B).
Genetic evaluation
Genetic testing was performed by Invitae (San Francisco, CA) for a 24 gene CMT demyelinating panel.
Biochemical evaluation of the intronic variant
RNA and protein were acquired from fibroblasts of the proband and control subjects. RT‐PCR was utilized to amplify the exon downstream of our patient's intronic mutation [c.1949‐10T>G], which is exon 18. The forward primer was designed at the boundary of exons 16 and 17 [CCAACAAGAAGAAGTTATACTTACTGGTG], and reverse primer designed at the boundary of exons 19 and 20 [TATTGCTTTTGTTTCCCAATACTGA]. Western blot was performed to evaluate for presence or absence of FIG4 protein.
Results
Genetic testing
There were three genetic variants found in the proband: FIG4 c.122T>C (p.I41T) and c.1949‐10T>G (intronic); and LITAF c.100C>A (p.P34T). The affected brother carried both FIG4 variants but not the LITAF variant. The unaffected father was found to have both the FIG4 intronic and the LITAF variants – excluding the LITAF mutation as pathogenic. The FIG4 c.122T>C variant (I41T) is one of the most common variants in patients with CMT4J, and leads to rapid degradation of the FIG4 proteins.4 The intronic FIG4 variant was recently reported in a person with CMT4J, also with the I41T variant5 and was absent in >60,000 exomes [exac.broadinstitute.org].
Biochemical evaluation of the intronic variant
Pathogenic mutations in FIG4 are usually loss‐of‐function,6 raising the suspicion that the intronic mutation would eliminate FIG4 expression via altered splicing. The amplified exon primers generated a band at 329 bp that would be equivalent to the sum of exons 17, 18, and 19. This allele size was detected in fibroblasts from a healthy control and a CMT4J patient homozygous for the I41T missense variant (Fig. 1C: lane 1 and 2, respectively). However, a second band at 181 bp, equivalent to the sum of exons 17 and 19, was detected only in our patient (Fig. 1C: lane 3). This size is consistent with the skipping of exon 18. As also demonstrated in the previous report of these variants,5 western blot showed minimal expression of FIG4 protein for our patient, suggesting a natural decay of FIG4 mRNA with the skipped exon 18 (Fig. 1D).
Discussion
Recessive mutations (usually compound heterozygous mutations) in FIG4 have caused three distinct clinical phenotypes, CMT4J,7 Yunis‐Varon syndrome,8 and familial epilepsy with polymicrogyria.9 Both Yunis‐Varon syndrome and familial epilepsy with polymicrogyria syndrome are early onset diseases with severely abnormal CNS development and pathology. Late onset CNS abnormalities in multiple functional domains with concomitant de‐/dysmyelinating polyneuropathy have not been reported to date. This includes the recent report of another individual with the same geneotype as our patient, who has a severe form of CMT4J without CNS abnormalities.5 The parkinsonism and cognitive language impairment with a moderate CMT phenotype are unique to our family. Thus, our case study expands the phenotypic spectrum of patients with FIG4 deficiency. The CNS abnormalities in our patient did not exclusively fit into the spectrum of parkinsonism, thus a genetic test for inherited Parkinson diseases would not help.
A majority of patients with CMT4J typically presents with no or negligible CNS symptoms.3, 6 It has been suggested that residual FIG4 mutant proteins, such as I41T, may protect CMT patients from CNS deficits.6 However, our patient and her brother have prominent CNS abnormalities with no other reported family history. Nearly undetectable FIG4 protein in the proband's fibroblasts (Fig. 1D) would be consistent with the CNS pathology in our case. While the I41T allele is rapidly degraded, we postulated exon skipping by the intronic mutation (c.1949‐10T>G) in the other allele of our patient might explain the near absence of FIG4 protein on western blot. Because the case reported by Gentil et al. showed no CNS symptom, additional intrinsic and/or extrinsic factors could contribute to the CNS abnormalities in our case.
FIG4 has been shown in mice to be expressed widely in both CNS and PNS.10 Thus, variable clinical phenotypes with different degrees of CNS involvement are not unexpected, and new clinical phenotypes may continue to emerge in the future studies.
Author Contributions
James P Orengo, MD, PhD contributed to the drafting and revising of the manuscript for content, acquisition of data, and study concept and design. Pravin Khemani, MD contributed to the drafting and revising of the manuscript for content and acquisition of data. John W Day, MD, PhD contributed to the drafting and revising of the manuscript for content, acquisition of data, and obtaining funding for the study. Jun Li, MD, PhD contributed to the drafting and revising of the manuscript for content, study concept and design, acquisition, analysis and interpretation of data, contribution of vital reagents, and, obtaining funding for this study. Carly E Siskind, MS contributed to the the drafting and revising of the manuscript for content, acquisition of data, and study supervision and coordination.
Conflict of Interest
None declared.
Supporting information
Video S1. Patient's neurological dysfunction.
Acknowledgments
We gratefully acknowledge the following grants for supporting this study: NIH R01NS066927 and NINDS/ORDR, NCATS U54NS065712.
Funding Statement
This work was funded by NIH grant R01NS066927; NINDS/ORDR grant ; NCATS grant U54NS065712.
References
- 1. Fridman V, Bundy B, Reilly MM, et al. CMT subtypes and disease burden in patients enrolled in the Inherited Neuropathies Consortium natural history study: a cross‐sectional analysis. J Neurol Neurosurg Psychiatry 2015;86:873–878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Vaccari I, Carbone A, Previtali SC, et al. Loss of Fig 4 in both Schwann cells and motor neurons contributes to CMT4J neuropathy. Hum Mol Genet 2015;24:383–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Nicholson G, Lenk GM, Reddel SW, et al. Distinctive genetic and clinical features of CMT4J: a severe neuropathy caused by mutations in the PI(3,5)P(2) phosphatase FIG 4. Brain 2011;134(Pt 7):1959–1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ikonomov OC, Sbrissa D, Fligger J, et al. ArPIKfyve regulates Sac3 protein abundance and turnover: disruption of the mechanism by Sac3I41T mutation causing Charcot‐Marie‐Tooth 4J disorder. J Biol Chem 2010;285:26760–26764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Gentil BJ, O'Ferrall E, Chalk C, et al. A new mutation in FIG 4 causes a severe form of CMT4J involving TRPV4 in the pathogenic cascade. J Neuropathol Exp Neurol 2017;76:789–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zhang X, Chow CY, Sahenk Z, et al. Mutation of FIG 4 causes a rapidly progressive, asymmetric neuronal degeneration. Brain 2008;131(Pt 8):1990–2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chow CY, Zhang Y, Dowling JJ, et al. Mutation of FIG 4 causes neurodegeneration in the pale tremor mouse and patients with CMT4J. Nature 2007;448:68–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Campeau PM, Lenk GM, Lu JT, et al. Yunis‐Varon syndrome is caused by mutations in FIG 4, encoding a phosphoinositide phosphatase. Am J Hum Genet 2013;92:781–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Baulac S, Lenk GM, Dufresnois B, et al. Role of the phosphoinositide phosphatase FIG 4 gene in familial epilepsy with polymicrogyria. Neurology 2014;82:1068–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Guo J, Ma YH, Yan Q, et al. Fig 4 expression in the rodent nervous system and its potential role in preventing abnormal lysosomal accumulation. J Neuropathol Exp Neurol 2012;71:28–39. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Video S1. Patient's neurological dysfunction.