

Abstract
Objective
To investigate transplantation of rat Schwann cells or human iPSC‐derived neural crest cells and derivatives into models of acquired and inherited peripheral myelin damage.
Methods
Primary cultured rat Schwann cells labeled with a fluorescent protein for monitoring at various times after transplantation. Human‐induced pluripotent stem cells (iPSCs) were differentiated into neural crest stem cells, and subsequently toward a Schwann cell lineage via two different protocols. Cell types were characterized using flow cytometry, immunocytochemistry, and transcriptomics. Rat Schwann cells and human iPSC derivatives were transplanted into (1) nude rats pretreated with lysolecithin to induce demyelination or (2) a transgenic rat model of dysmyelination due to PMP22 overexpression.
Results
Rat Schwann cells transplanted into sciatic nerves with either toxic demyelination or genetic dysmyelination engrafted successfully, and migrated longitudinally for relatively long distances, with more limited axial migration. Transplanted Schwann cells engaged existing axons and displaced dysfunctional Schwann cells to form normal‐appearing myelin. Human iPSC‐derived neural crest stem cells and their derivatives shared similar engraftment and migration characteristics to rat Schwann cells after transplantation, but did not further differentiate into Schwann cells or form myelin.
Interpretation
These results indicate that cultured Schwann cells surgically delivered to peripheral nerve can engraft and form myelin in either acquired or inherited myelin injury, as proof of concept for pursuing cell therapy for diseases of peripheral nerve. However, lack of reliable technology for generating human iPSC‐derived Schwann cells for transplantation therapy remains a barrier in the field.
Introduction
Myelin damage or dysfunction is a key component of a variety of peripheral nerve diseases in humans, including immune‐mediated neuropathies,1 and in a diverse set of genetic lesions of neurons and Schwann cells collectively referred to as Charcot–Marie–Tooth disease (CMT).2 CMT is the most frequent one among all the hereditary neurological disorders with an estimated worldwide prevalence of 1 per 2500 population, and results from mutations in ~80 disease‐associated genes, most of which are involved in Schwann cell development or myelin maintenance.3, 4, 5, 6, 7 The most common cause of CMT is from duplication of a 1.4 Mb segment on chromosome 17p11.2 harboring the PMP22 gene (CMT 1A), found in about 50% of all patients with CMT.8, 9, 10 Although the precise disease mechanism is not clear, it is suspected that overproduction of the PMP22 protein by the extra gene copy leads to abnormal Schwann cell development and myelin sheath maintenance, ultimately resulting in secondary axon loss and loss of sensory and motor function.11, 12 CMT is typically not life‐threatening but the patients’ symptoms impact their quality of life profoundly, and there is no effective treatment.7, 13 Several pharmacological approaches for CMT1A have attempted to reduce PMP22 expression levels with progesterone antagonism14 or ascorbic acid treatment.15 Unfortunately, ascorbic acid failed to show any benefit in clinical trials.16 Other therapeutic strategies described for CMT1A include treatment with neurotrophin‐3,17 neuregulin 1,18, or a combination drug regime containing baclofen, naltrexone, and D‐sorbitol.19
Despite these efforts to mitigate secondary axon loss or enhance the ability of endogenous Schwann cells to form myelin, they will likely fail if Schwann cells have died or senesced, or if endogenous Schwann cells carry a genetic predisposition to form abnormal myelin as in CMT1A. While intraneural Schwann cell transplantation could potentially address this problem, thus far most work investigating Schwann cell transplantation has occurred in the setting of spinal cord injury, rather than peripheral nerve disease.20, 21 While early studies investigated the use of nerve grafts in dysmyelinated animal models,22 or seeded Schwann or other cells into sites of nerve injury to enhance axonal regrowth23, 24, few have investigated whether intraneural injection of Schwann cells into demyelinated or dysmyelinated nerve could lead to successful engraftment, or examined key parameters of this approach including the ability of transplanted cells to survive, migrate and form functional myelin sheaths.
Devising strategies for Schwann cell transplantation into peripheral nerve is of increasing importance, as technology for genetic manipulation of human‐induced pluripotent stem cells (iPSCs) and the ability to differentiate them into neural crest cells and Schwann cell precursors has improved rapidly in recent years. Here, we describe a platform for intraneural delivery of rat Schwann cells or human iPSC derivatives into (1) a model of focal demyelination from lysolecithin (LPC),25 and (2) a transgenic rat model of inherited dysmyelination due to PMP22 overexpression.26 These studies provide evidence that direct intraneural delivery of engineered cells is a feasible strategy to rejuvenate or replace myelin in inherited or acquired demyelinating peripheral nerve disorders, while also identifying limitations that will need to be addressed for moving forward with cell therapy for nerve disorders.
Materials and Methods
Cell lines for transplantation
Rat Schwann cells (RSCs) were harvested from sciatic nerves of P0‐P2 rat pups.27 and were infected with a lentiviral vector expressing red fluorescent protein (RFP) mCherry. The RSCs were grown in Dulbecco's Modified Eagle Medium (DMEM), with 5% fetal bovine serum (FBS), 1% nonessential amino acids (NEAA), and passaged weekly. On the day of surgery, RSCs were trypsinized, and live cells counted following Trypan blue staining and kept on ice for ~4 h.
Human iPSCs were differentiated into neural crest stem cells by incubating for 11–14 day in defined conditions containing cocktail of growth factors and two small‐molecule compounds, to activate Wnt signaling (BIO), and inhibit Activin A/Nodal pathway (SB431542).28 The DC‐HAIF culture media consisted of DMEM/F‐12, 2% FBS, 1xNEAA, 50U/mL penicillin, 50 μg/mL streptomycin, 50 μg/mL ascorbic acid, 10 μg/mL transferrin, 0.1 mmol/L 2‐mercaptoethanol, 1X trace elements A,B,C, 10 ng/mL heregulin β‐1, 10 ng/mL activin A, 200 ng/mL insulin‐like growth factor 1, and 8 ng/mL fibroblast growth factor 2.
Cell samples from various passages were fixed in 4% paraformaldehyde (PFA) and processed for immunocytochemistry (ICC) using antibodies (Table S1) against nestin, activator protein‐2 (AP2), paired box 6 protein (Pax6), neurotrophin receptor (p75), and human natural killer‐1 cell marker (HNK1). Flow cytometry was run on NCSCs with antibodies for p75 and HNK1, using an LSRFortessa cell analyzer (BD Biosciences) and FlowJo data analysis software. Additionally, mRNA was isolated for RNA‐seq analysis on Illumina Hi‐Seq platform for 100‐bp paired‐end reads. The resultant reads were aligned to hg19 with BOWTIE and imported to Partek software for gene annotation and generation of reads per kilobase per million values.
Human iPSC‐derived NCSCs were differentiated toward Schwann cell precursors as previously described29 using MesenPRO medium supplemented with heregulin for 40 days. Differentiation of motor neurons (MNs) from human iPSCs was performed as previously described.30 Briefly, pluripotent stem cells were propagated on matrigel with StemBeads. On the day of differentiation, iPSC colonies were dissociated into single cells using Accutase and plated at 400K per 10 cm plate to form embryoid bodies in DMEM/F12 containing 2% B27 and 1% N2. The embryoid bodies were treated with Dorsomorphin and SB431542 to induce a neural lineage and, additionally, with retinoic acid at day 5 and a smoothened agonist 1.3 at day 7. After 11 days of neural aggregate formation, colonies were plated on poly‐ornithine, fibronectin, and laminin‐coated plates to differentiate neural progenitors to motor neurons. At day 13, DAPT (2.5 μmol/L) was added, and at day 20, Ara‐C (2 μmol/L) was added to eliminate progenitors or dividing cells in culture. After 30 days of differentiation, human MNs were then cocultured with hNCSCs (GFP‐labeled) in neural induction medium supplemented with heregulin, SB431542, forskolin, and FBS for 2 weeks, then dissociated and replated to allow for the isolation of cells cocultured with MNs. List of the reagents used for cell culture is shown in Table S2.
Animal subjects
Homozygous PMP22 transgenic rats (homo‐PMP22)
Hemizygous PMP22 rat breeders were imported from Max‐Planck Institute, Germany, and crossed to homozygosity. Soon after birth, the homo‐PMP22 rats were readily distinguished from their wild‐type (WT) or hemizygous littermates due to smaller size, muscle weakness, convulsive episodes, ataxia, and sometimes sudden death. The Kaplan–Meier survival curves for homo‐PMP22 rats are shown in Figure S1. The homo‐PMP22 rats became progressively disabled and were unable to walk normally but could right themselves with difficulty. The hind limbs were primarily affected, resulting in severe muscle wasting and weakness; elongated sipper tubes were fitted on water bottles, and moist chow was used. Homo‐PMP22 rats that survived beyond 8 weeks did not show a further increased risk of sudden death, and cell transplantations were performed on 10‐week‐old homo‐PMP22 rats.
Rowett nude rats
One‐month‐old male Rowett nude rats were purchased from Envigo RMS that were athymic and T‐cell deficient. They were housed in pairs with free access to food and water. All animal experiments were performed after prior approval by the institutional review board and institutional animal care and use committee at Cedars‐Sinai Medical Center and conformed to the National Institutes of Health (NIH) guidelines.
Surgical procedures
Under anesthesia, the thigh was shaved and prepped and a 2‐cm skin incision was made. Using microscope, blunt dissection was carried out to expose the sciatic nerve; a self‐retaining retractor was placed to keep muscle masses separate (Fig. S2). An improvised malleable ribbon retractor was placed under the nerve at middle level, and a 10 μL syringe (Hamilton, Cat #80330) fitted with 33G needle (Hamilton, Cat#7803‐05) was mounted on micromanipulator (Model MM‐3) attached to magnetic stand (Model GJ‐8, Narishige Scientific Instrument Lab). The needle tip was introduced for about 1.5 mm, and intraneural injections were performed very slowly under continuous visual observation to confirm the absence of injectate leakage; the needle was left in place for 3 min and slowly withdrawn. The muscle layers were opposed with absorbable sutures and skin stapled. Finally, the fully recovered rats were returned to the animal care facility and examined daily.
In 1‐m‐old nude rats, 2 μL of LPC (1% solution; Sigma‐Aldrich, Cat #L0906) was injected in sciatic nerve for toxin‐induced focal demyelination. The injection site was marked with carbon and after 7 days, the mark was readily visible, aiding two separate injections of cells into the LPC‐lesioned area.
For homo‐PMP22 rats, immunosuppression was started 2 days prior to a injection of cells into the midsciatic nerve. Cyclosporine injections were continued daily until euthanasia.
Tissue harvest and processing
At predetermined time points (1 week, 3 weeks, or 6 weeks following cell transplantation), the sciatic, tibial, and peroneal nerves were harvested. Additionally, nerves were harvested from naive homo‐PMP22 and WT age–sex‐matched litter mates to generate comparative data. The samples were fixed overnight in 4% PFA, and using 20x magnification water immersion objectives of Olympus BX51 upright fluorescent microscope, ~1‐mm‐long segments from within the RFP+ transplant were selected for plastic blocks. The remaining specimens were embedded in O.C.T. compound and frozen sectioned; a set of the cryosections were cover slipped using Prolong Gold reagent with DAPI. Additional sections were processed for ICC for myelin basic protein (MBP), glial fibrillary acidic protein (GFAP), and neurofilament H nonphosphorylated (SMI32).
Microscopy
A series of low magnification images of longitudinal sections encompassing the entire transplant were acquired with Olympus BX51 upright microscope and were then montaged using Adobe Photoshop CS3 software. Additionally, the Nikon Eclipse Ti inverted microscope was used to acquire stitched images encompassing the entire nerve cross section in high resolution from toluidine blue‐stained plastic sections.
Histomorphometric analysis
The length and width of transplants were determined from longitudinal and axial cryosections. Quantification of myelinated axons was carried on toluidine blue‐stained plastic sections with a semiautomated method using ImageJ; briefly, “threshold function” was used to highlight axoplasm, and “analyze particle function” was then utilized to count the myelinated axons and measure the “Feret's diameter.” For quantifying the total axon count, the number of nonmyelinated axons was added manually to myelinated axon count employing the “Wand tool.”
Nerve conduction studies
Sciatic nerve electrophysiology was conducted with a VikingQuest EMG machine; stimulating electrodes were placed at the sciatic notch or just above ankle, and recording electrodes were placed in the foot pad between second and third digits.
Statistical analysis
Statistical analysis was conducted with Prism (Graph Pad software). All data are shown as mean ± standard error of the mean (SEM) and statistical tests indicated in the figure legends.
Results
Models of acquired and inherited peripheral nerve myelin damage for cell transplantation
To examine cell transplantation in the context of an acquired demyelinating injury, we utilized the LPC injection model, which damages myelin sheaths but largely spares myelinated axons (Fig. 1A).25, 31 LPC was injected unilaterally, and 7 days later, nerves from both sides were harvested and analyzed. The LPC‐lesioned area appeared swollen (Fig. 1C) with fragmented MBP staining (Fig. 1D), in contrast to the normal appearance in areas away from lesion (Fig. 1E). Plastic sections of the LPC‐lesioned area revealed lysis of myelin sheaths and the appearance of numerous membranous vesicles in the remaining cells which was absent from the contralateral control nerve (Fig. 1F,G).32 NCV across the demyelinated region was slowed 6d after demyelination, but distal CMAP amplitudes were preserved consistent with minimal large caliber axon loss (Fig. S3). Cell transplants were performed 7 days after LPC injection and harvested 1, 3, and 6 weeks later.
Figure 1.

Cell transplantation paradigms in toxic and inherited disorders of Schwann cell myelin. (A), schematic of toxic demyelination model with lysolecithin (LPC). (B), schematic of cell transplantation in inherited hypomyelination. (C–E), myelin basic protein (MBP) staining of the sciatic nerve day seven after LPC injection. (C), image in longitudinal section of the LPC‐lesioned area showing local swelling in comparison with the distal nerve portion (arrow). (D), inset of C showing area of LPC‐induced myelin damage. (E), normal pattern of MBP immunoreactivity adjacent to LPC‐lesioned area. (F,G), longitudinal semithin plastic sections of the nerve showing normal untreated nerve (F) and the LPC‐lesioned area (G) with loss of myelin sheaths and debris‐laden cells lying in parallel to one another along the long axis of nerve. (H–K), plastic sections of normal distal tibial nerve (H,I) and age–sex‐matched homo‐PMP22 rat (J,K). The nerve was much smaller in caliber (over 2.5‐fold) compared to wild type and showed axons with completely lacking compact myelin (open arrows) with increased endoneurial connective tissue (closed arrows). (L), graph of total axons (myelinated and nonmyelinated axons) from tibial nerves of homo‐PMP22 rats and their wild‐type littermates. Data shown are mean ± SEM; P < 0.001 (unpaired t test with Welch's correction). (M), size frequency histogram of myelinated axons showing only rare small myelinated axons in the homo‐PMP22 rat, indicating severe loss of both sensory and motor axons. Abbreviations used: WT, wild‐type litter mates; Homo‐PMP22, homozygous PMP22 rat model of hereditary dysmyelination; Scale: C = 500 μm; E = 100 μm (same in D); G = 25 μm (same in F); H = 150 μm; J = 50 μm; K = 25 μm (same in I).
To examine cell transplantation in a model of inherited dysmyelination (Fig. 1B), we utilized a transgenic rat model that overexpresses PMP22.26 While rats heterozygous for the PMP22 transgene had a mild demyelinating neuropathy with a minimal clinical phenotype, homo‐PMP22 rats had severe ataxia and muscle wasting apparent at weaning, with severe hypomyelinating neuropathy on pathology (Figs. 1J and K) with absent M responses in the foot muscles. Toluidine blue‐stained plastic sections of homo‐PMP22 nerves showed they were ~2.5‐fold smaller in caliber than WT nerves, and almost all the axons were completely devoid of myelin (Fig. 1H–K). Quantification of total axon numbers visible with light microscopy revealed that tibial nerves in homo‐PMP22 had decreased numbers of axons compared to WT littermates (Fig. 1L and M). We utilized this severe hypomyelinating neuropathy model to assess the ability of transplanted cells to engraft and produce myelin in the context of a nerve with no functional endogenous Schwann cells.
Rat Schwann cell graft survival and migration in toxic and inherited myelin injury
To assess the optimal ability of transplanted cells to engraft and myelinate in both acquired and inherited myelin injury models, we first utilized rat Schwann cells cultured from Sprague–Dawley rats and transduced with cytoplasmic RFP as a marker to assess graft fate. LPC was injected in midsciatic nerve of 1‐m‐old nude rats to induce focal demyelination, 4x105 RSCs were transplanted in the lesioned site 7 days later, and sciatic, tibial, and peroneal nerves were harvested after 1 week, 3 weeks, or 6 weeks (see Fig. 1A). Robust numbers of spindle‐shaped elongated RFP‐positive cells with a low cytoplasmic‐to‐nuclear volume ratio were observed in the transplant site at all time points evaluated (Fig. 2A–F).
Figure 2.
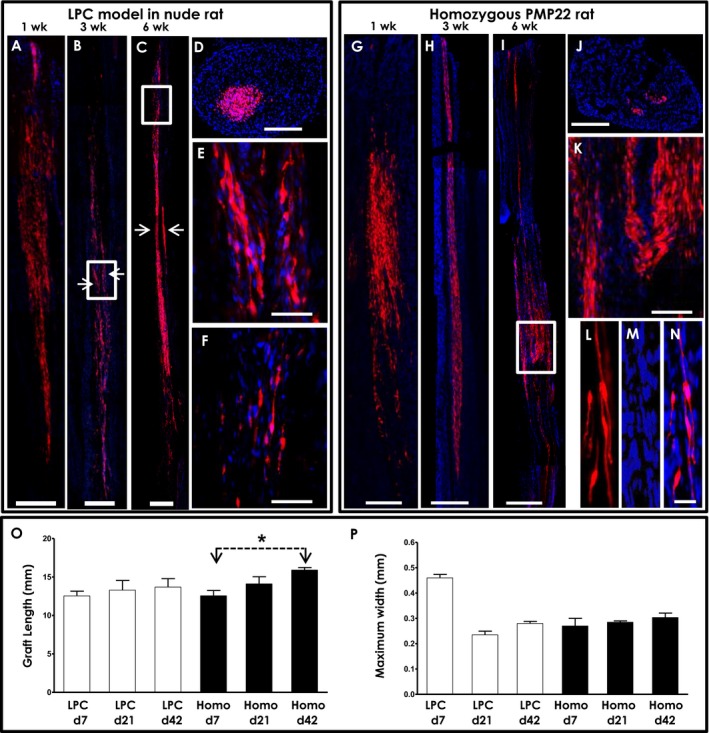
Survival and migration of heterologous rat Schwann cell (RSC) transplants into toxic and hereditary demyelination models. A,B,C, representative longitudinal montages of sciatic nerve from the nude rat LPC model at 1, 3, and 6 weeks after transplant demonstrating survival and longitudinal migration of RFP‐labeled heterologous RSCs. In B,C, the two separate injection sites performed to deliver RSC are readily discernible (arrows). (D), axial image of sciatic nerve from the same rat as A showing more limited axial migration. (E,F), inset of B and C shown at higher magnification. (G,H,I), representative longitudinal montage images of sciatic nerve in homo‐PMP22 rats at 1, 3, and 6 weeks after transplantation. (J), cross section of sciatic nerve from I. (K–N), higher magnification images displaying elongated and spindle‐shaped morphology with low cytoplasmic‐to‐nuclear volume ratios of RFP‐labeled RSCs. (O,P), quantitation of graft lengths and width at different time points after transplantation. Graft length did not increase significantly in the LPC model over time, whereas in the homo‐PMP22 rats, the graft lengths increased over time suggesting continued longitudinal migration out to at least 6 weeks after transplant. Axial width did not change other than resolution of the initial swelling seen in the LPC model at the earliest time point. *P < 0.05, two‐tailed unpaired t test; data shown is mean ± standard error of mean; n = 2–4/group. Abbreviations used: LPC, lysolecithin‐induced focal demyelination model in nude rat; Homo, homozygous PMP22 rat. Scale: A,B,D,G,H,J = 250 μm; C,I = 500 μm; E = 50 μm; F, K = 100 μm; L,M,N = 25 μm.
Transplanted RSCs migrated longitudinally over long distances (~1.0–1.5 cm), but axial migration was limited (~0.25 mm), possibly due to connective tissue barriers in the perineurium given the fascicular appearance of the grafts on cross section (Fig. 2D). In the LPC model, transplant length did not increase significantly over time, likely because engraftment was limited to the location of the demyelination induced by the toxin (Fig. 2O). The width of the graft was maximum early after transplant, likely due to persistent swelling in the nerve after LPC injection, which resolved by 21 days and did not change further (Fig. 2P).
We similarly assessed engraftment and migration of transplanted RSCs in the homo‐PMP22 rats with severe hypomyelination; 10‐wk‐old homo‐PMP22 rats were treated with cyclosporine 2 days prior to surgery, and 2 × 105 RSCs were transplanted in midsciatic nerve. Sciatic, tibial, and peroneal nerves were harvested after 1 week, 3 weeks, or 6 weeks. Graft survival was observed in the homo‐PMP22 rats out to 6‐wk post‐transplant (Fig. 2G–N). Transplants again showed extensive longitudinal migration (~1.0–1.5 cm), which increased over time (42 days vs. 7 days; P < 0.05, two‐tailed unpaired t test), possibly because no endogenous Schwann cells could impede migration of transplanted cells, unlike the LPC model (Fig. 2O). Again, axial migration was limited and did not change in time after transplant (Fig. 2P). While transplanted Schwann cells could lose fluorescent protein expression over time leading to an underestimation of graft survival, we did not observe a decrease in fluorescence per cell or contraction of graft size to indicate loss of marker expression occurred during the 6‐week follow‐up period.
Myelin formation by transplanted Schwann cells on chronically hypomyelinated axons
Cell therapy for inherited demyelinating neuropathies will require that exogenously delivered Schwann cells displace dysfunctional Schwann cells and engage axons that have been chronically hypomyelinated or dysmyelinated. To examine the capacity of transplanted Schwann cells to form myelin in this context, we examined myelin formation by engrafted RSCs in the homo‐PMP22 model, in which no endogenous myelin is present. RFP‐labeled RSCs were transplanted into midsciatic nerves of homo‐PMP22 rats, harvested 6 weeks later, and cryo‐sectioned and processed for immunofluorescence of myelin basic protein (MBP). While MBP staining was nearly completely absent in untreated homo‐PMP22 nerves and regions distant from the graft, we observed strong MBP immunoreactivity selectively in the region of the RFP‐labeled RSC grafts indicative of myelin formation by transplanted cells (Fig. 3A–E).
Figure 3.

Schwann cell transplants in homo‐PMP22 rats showed myelin formation in the engrafted regions. (A), longitudinal image of sciatic nerve of homo‐PMP22 rat showing the presence of myelin basic protein (MBP) staining (green) in the region of the transplanted RSCs (red), but almost complete lack of MBP staining (green) in locations away from the graft. (B), inset in “A” is shown in higher magnification. (C–E), unmerged images showing RSC (C), MBP staining (D), and DAPI staining of nuclei (E). (F–J), regions of sciatic nerve for plastic embedding were selected from the transplanted area after direct visualization of the RFP‐tagged RSCs. (F), image of contralateral uninjected sciatic nerve revealed near total absence of myelinated axons. (G), inset in F at higher magnification showing amyelination with enlarged Schwann cell nuclei in the homo‐PMP22 rat. (H), image of sciatic nerve 6 weeks after RSC transplantation revealed numerous myelinated axons at the cell therapy delivery site, which was never seen in uninjected control nerves. (I), higher magnification of inset showing myelinated axons of various sizes (surrounded by dark blue‐stained myelin sheaths) intersperse with amyelinated fibers with enlarged Schwann cell nuclei. (J), higher magnification of region distant from the graft site. (K), quantitation of myelinated axons in untreated (“naïve”) and treated sciatic nerves. (L), size frequency histogram of myelinated axon size showing increased number of smaller diameter axons (<3 μm) and the exclusive presence of larger diameter (3–7 μm) myelinated axons in the sciatic nerve on the transplanted side compared to the untreated contralateral side. Data shown are mean ± SEM; P < 0.05 in K (unpaired t test) and L (two‐way ANOVA with Bonferroni post‐tests). Scale: A = 250 μm; B = 100 μm; E = 100 μm; F,H = 50 μm; J = 25 μm (same in G,I). *P<0.05; **P<0.01; ***P<0.001
To further confirm that the engrafted Schwann cells had engaged axons and formed compact myelin, we analyzed toluidine blue‐stained plastic sections of nerve 6 weeks after transplant into homo‐PMP22 rat nerves. While contralateral untransplanted sciatic nerves from homo‐PMP22 rats showed essentially no compact myelin formation (Fig. 3F and G), we observed clusters of myelinated axons on the transplanted side (Fig. 3H–J), with similar size and appearance to axial migration of transplanted cells seen on RFP fluorescence (Fig. 2J). Occasional myelinated axons were also seen further away from the graft site cluster, suggesting that a small number of engrafted cells did migrate axially toward the periphery of the nerve, or possibly the graft‐induced endogenous Schwann cells to myelinate (Fig. 3J). Quantification across the entire nerve cross section showed that myelinated fibers were almost exclusively present on the transplanted side (Fig. 3K). This was true across all axon diameters, with larger diameter (3–7 μm) axons present only on the transplanted side (Fig. 3L).
These data indicate that exogenously transplanted Schwann cells can engraft into a nerve with an inherited defect in myelin formation, displace endogenous dysfunctional Schwann cells, and form myelin around chronically hypomyelinated axons.
In vitro and in vivo characterization of iPSC‐derived neural crest precursors for peripheral nerve cell therapy
Schwann cells are derived from neural crest stem cells (NCSCs), which migrate along with axons during development to form Schwann cell precursors, and subsequently differentiate into mature myelinating and nonmyelinating Schwann cells.33 As protocols for differentiating neural crest precursor cells from iPSCs are already established,28, 34, 35 we first examined iPSC‐derived human NCSCs as a candidate cell type for Schwann cell replacement therapy in demyelinated nerves. iPSCs from two normal subjects (lines 00i and 14i) were directly differentiated into human NCSCs via a protocol utilizing Wnt signaling activation and Activin/Nodal signaling inhibition (Fig. 4A). The iPSC‐derived NCSCs were immuno‐positive for nestin and AP2 (Fig. 4B), and 99% were double positive for p75 and HNK1 on flow cytometry by passage 9 (Fig. 4C), indicating they express several key neural crest markers as previously reported.28 Furthermore, RNA‐seq analysis on NCSCs differentiated independently from four different iPSC lines (and flow sorted for p75/HNK1 double positivity) revealed the expression of a variety of neural crest markers including SOX9, AP2, SNAI2, ITBG1, and NGFR, but not markers of iPSCs or of Schwann cells (Fig. 4D). These results indicate that NCSC differentiation is robust and reproducible across a variety of iPSC lines.
Figure 4.

Human neural crest stem cells (hNCSC) engraft and migrate in LPC‐lesioned sciatic nerve but do not express Schwann cell markers, align with axons or form myelin. (A), schematic representation of iPSC differentiation to human neural crest stem cells (hNCSCs) adapted from.28 (B), immunostaining for NCSC markers AP2 and nestin in two different control lines (00i and 14i) and phase contrast image at passage 9. (C), flow cytometry of iPSC‐derived NCSC for markers p75 (NGFR) and HNK1 showed that over 99% were double positive by passage 9. (D), RNA‐seq analysis of iPSC‐derived NCSCs (n = 3) showed expression of a variety of canonical neural crest markers including SOX9, SOX10, AP2, SNAI2, ITBG1, and NGFR, but not markers of iPSCs or Schwann cell precursors. (E‐H), representative montage images of sciatic nerve transplants of nuclear GFP‐labeled hNCSCs (4 x 105) harvested at 1, 4, and 9 weeks after surgery showing longitudinal migration, but no change in nuclear morphology or alignment along axons. (H), inset of G. (I–M), transplanted hNCSCs did not express GFAP (marker of nonmyelinating Schwann cells) and formed clusters of cells that did not interact with axons (SMI32). The GFAP and MBP staining surrounding NCSC clusters presumably arose from endogenous nonmyelinating and myelinating Schwann cells surrounding the grafted cell clusters. Abbreviations used: iPS, induced pluripotent stem; DC‐HAIF, cell culture media in defined conditions HAIF (heregulin‐1β, activin A, insulin‐like growth factor 1, fibroblast growth factor 2); Pax6, paired box 6 protein; NGFR, nerve growth factor receptor; HNK1, human natural killer‐1 cell marker; AP2, activator protein‐2; p9, passage 9; APC, allophycocyanin; GFAP, glial fibrillary acidic protein; MBP, myelin basic protein; SMI32, neurofilament H nonphosphorylated. Scale: E,G = 400 μm; F = 300 μm; H = 100 μm; I = 50 μm; K3 = 500 μm (same in J‐K2); M = 100 μm (same in L).
To follow NCSCs after in vivo transplantation, we differentiated an iPSC line genetically engineered to express nuclear GFP (nuc‐GFP) under a ubiquitous human β‐actin promoter36 into NCSCs. Seven days after LPC injection in midsciatic nerve of nude rats, nuc‐GFP‐labeled NCSCs (4 × 105) were transplanted in the lesioned site. Similar to rat Schwann cells, human NCSCs largely migrated longitudinally, but not axially, and survived through 9 weeks after transplantation (Fig. 4E–H). However, iPSC‐derived NCSCs maintained an enlarged nuclear morphology distinct from that seen in RSCs (Fig. 4H). Co‐staining of NCSC grafts harvested after 4 weeks revealed they did not express markers of nonmyelinating (GFAP) or myelinating Schwann cells (MBP), although rare NCSCs aligned themselves along the orientation of GFAP and MBP immunoreactivity (Fig. 4I–M). Cross sections of the grafts primarily showed clusters of nuclei surrounded by MBP staining, likely arising from endogenous Schwann cells that were devoid of SMI32‐positive axons (Fig. 4J–M).
These data indicated that while human iPSC‐derived NCSCs successfully engrafted after transplantation into demyelinated peripheral nerves in rats, they either did not have the appropriate environmental cues or intrinsic cellular capacity to further differentiate into Schwann cells. Therefore, we pursued two additional approaches to further differentiate NCSCs toward Schwann cell precursors prior to transplantation.
Characterization of putative iPSC‐derived Schwann cell precursors in vitro and after in vivo transplantation
Several methods have been published to differentiate human iPSCs into cells which possess markers of Schwann cells, typically as a minor component of a neuronal differentiation, or directed from NCSCs to generate cultures enriched in Schwann precursors.37 An earlier report demonstrated that prolonged culture of NCSCs in mesenchymal stem cell medium supplemented with heregulin‐β1 (a key instructive signal for Schwann cell development) generated cells expressing markers of Schwann cell precursors and found that these cells could form myelin after coculture with sensory neurons.29 Utilizing the “MESH” protocol (MesenPRO with heregulin), we attempted to differentiate Schwann cell precursors from human NCSCs generated above (Fig. 5A). Phase contrast images of hNCSCs before (left panel) and after differentiation (right panel) showed these cells developed an elongated spindle‐like morphology as previously reported (Fig. 5B). However, transplantation of nuc‐GFP iPSCs differentiated via the MESH protocol into the PMP22 transgenic rat model showed poor engraftment, with cells remaining in isolated clusters with enlarged nuclear morphology (Fig. 5C and D). Given their poor engraftment and lack of Schwann‐like morphology, we further characterized these cells using whole transcriptome analysis with RNA‐seq. While NCSC markers were lost after differentiation as expected, cells did not express any markers of Schwann cell precursors or immature Schwann cells (Fig. 5E). Given the appearance of the cells and the fact that this culture utilizes mesenchymal stem cell (MSC) medium, we examined markers of MSCs and found that MESH‐derived cells expressed all key markers of these cells (CD105/CD90/CD73/CD166/CD54/CD44/CD13 positive, CD34/CD45/CD31/CD11 negative) (Fig. 5F). These data indicate that while occasional cells with Schwann cell may appear in MESH‐derived cultures, the primary cell type resulting from this protocol are mesenchymal stem cells, making them not appropriate attempts at cell therapy for peripheral nerve demyelination.
Figure 5.
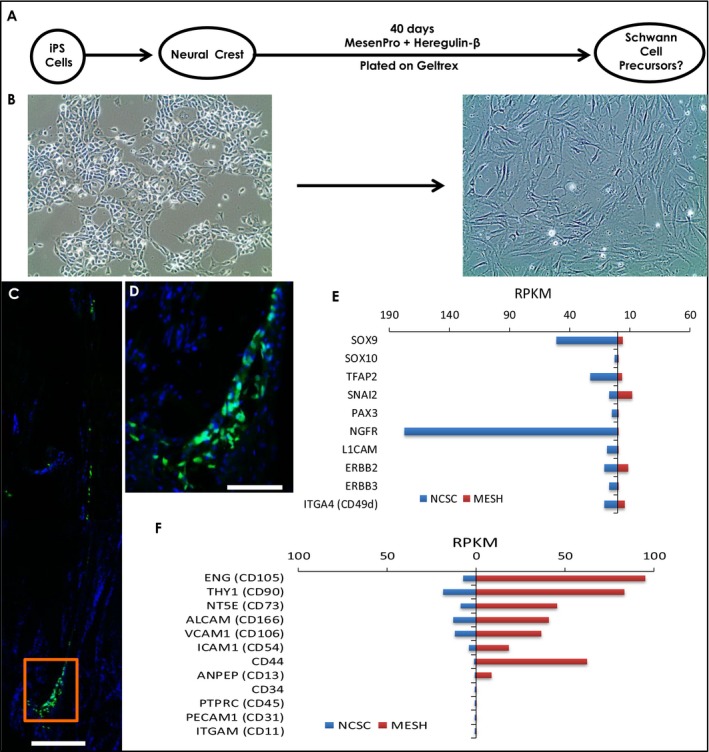
The MESH protocol promotes mesenchymal stem cell rather than Schwann cell precursor fate from neural crest stem cells. (A), schematic representation of protocol for NCSC differentiation to Schwann cells using MesenPRO + heregulin (MESH) from.29 (B), phase contrast images of NCSCs prior to MESH protocol (left) and afterward (right) showing change in morphology to an elongated spindle shape. (C,D), MESH cells (2 × 105) grafted into 6‐month‐old heterozygous PMP22 transgenic rat harvested 1 week after transplantation showed poor engraftment and enlarged nuclear morphology. (E), Comparison of RPKM values on RNA sequencing of neural crest markers in NCSCs vs. MESH cells differentiated from control iPSC lines (n = 3). Markers of NCSCs were decreased after culture in MesenPRO and heregulin. (F), MESH‐derived cells expressed all key markers of mesenchymal stem cells (CD105/CD90/CD73/CD166/CD54/CD44/CD13 positive, CD34/CD45/CD31/CD11 negative) rather than Schwann cells. Abbreviations used: iPS, induced pluripotent stem; NCSC, neural crest stem cell; MESH, protocol using MesenPRO + heregulin; RPKM, reads per kilobase per million values. Scale: C = 200 μm; D = 100 μm.
We next attempted a novel strategy to further differentiate NCSCs toward a Schwann lineage by coculture with human iPSC‐derived MNs, given that axonal signals provide an instructive signal for Schwann cell development (Fig. 6A).33 NCSCs cocultured with iPSC‐derived MNs developed a thick, elongated shape which was maintained after dissociation and replating (Fig. 6B,C). To investigate their potential to form myelin in vivo, GFP‐labeled NCSCs were cocultured with MNs were transplanted (4 × 105 cells) into the LPC‐lesioned nude rat model and the graft was harvested after 7 weeks. The transplanted cells showed good survival and longitudinal more than axial migration and appeared to align along axons (Fig. 6D and E), but did not co‐stain for myelin markers. To further characterize these cells, we performed RNA‐seq and again found that they lost markers of NCSCs as expected (Fig. 6F). However, analysis of the top 250 most highly expressed genes indicated they were heavily enriched for processes related to skeletal muscle function, and indeed a survey of key transcription factors and genes encoding contractile apparatus unique to skeletal muscle strongly indicated that MN coculture had directed the NCSCs to a muscle rather than Schwann cell fate (Fig. 6F).
Figure 6.
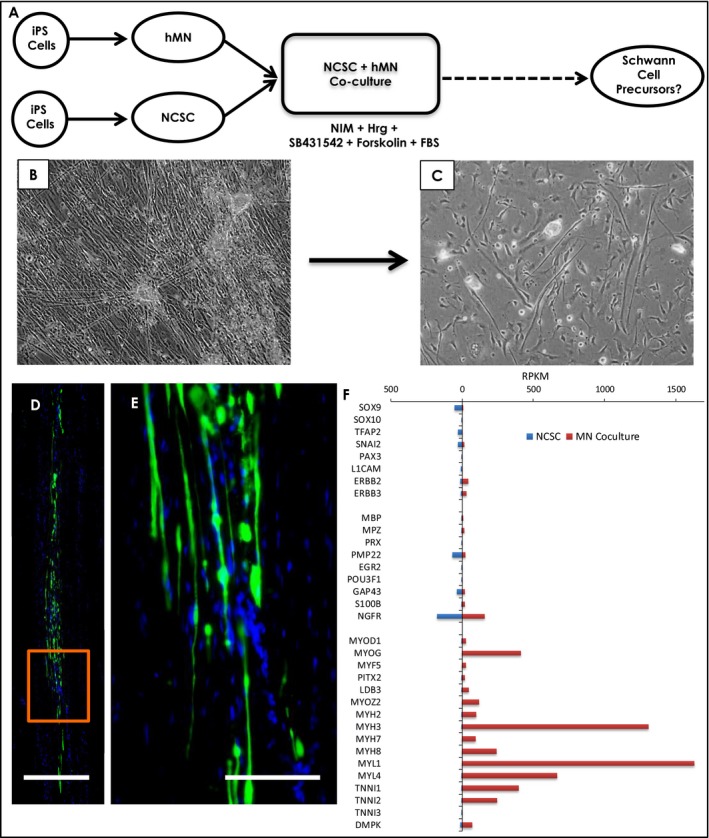
Coculture of neural crest stem cells with human motor neurons in Schwann cell induction medium. (A), schematic of coculture strategy. Control iPSC lines (N = 3) were separately differentiated into motor neurons (unlabeled) and NCSCs (nuclear GFP‐labeled), then cocultured in neural induction medium with heregulin, SB431542, forskolin, and growth factors for 2 weeks to drive Schwann cell differentiation. Upon dissociation and replating, only GFP‐positive cells survived indicating they were derived from NCSCs. (B), phase contrast images of NCSCs cocultured with motor neurons, showing clusters of MNs with axonal projections, and NCSCs. (C), phase contrast images of cells after dissociation, showing a change in morphology to long spindle shapes. (D), montage image of GFP‐labeled NCSCs cocultured with MNs and transplanted into LPC demyelinated sciatic nerve, harvested 7 weeks after injection. (E), inset in D. (F), comparison of RPKM values on RNA‐seq of neural crest markers (SOX9 through ERBB3), Schwann cell markers (MBP through NGFR), and muscle cell markers (MYOD1 through DMPK) in NCSCs and MN‐coculture‐derived cells. Marked enrichment was observed in genes expressed selectively in skeletal muscle, indicating that MN coculture had directed NCSCs to a muscle rather than a Schwann cell fate. Abbreviations used: iPS, induced pluripotent stem; NCSC, neural crest stem cell; hMN, human iPSC‐derived motor neurons; NIM, neural induction medium; Hrg, heregulin; FBS, fetal bovine serum. Scale: D = 250 μm; E = 100 μm.
Discussion
Cell replacement therapy for myelin disorders of the CNS is being investigated for conditions including multiple sclerosis and inherited hypomyelinating leukodystrophies like Pelizaeus–Merzbacher disease (PMD).38 Indeed, in a recent phase I trial conducted in four patients with PMD and treated with human neural stem cell transplantation in their brains, the procedure was reported to be safe after 1 year of transplant and showed evidence of donor‐derived myelination on MRI.39 Given the rapidly advancing technology for differentiating iPSCs into a variety of cell types, and the ability to genetically modify these cells, we investigated strategies for cell transplantation into both a toxin‐induced and genetic model of peripheral nerve myelin damage in rats. We observed that transplanted rat Schwann cells (as a “gold standard” cell type) were capable of successful engraftment in either model, migrated for relatively long distances longitudinally in nerves, and formed normal‐appearing myelin. Several earlier studies elegantly demonstrated the ability of human or rodent cell or nerve transplants to ensheath regenerating axons resulting from nerve transection.22, 23 Likewise, use of proliferating Schwann cells has been shown to be useful to bridge acellular nerve allografts, as senescent Schwann cells are less capable of promoting axonal regrowth in the setting of nerve injury.40 However, to our knowledge, no prior studies investigated the ability of transplanted Schwann cells to myelinate axons in the context of inherited dysmyelination, and only one study demonstrated engraftment of human skin‐derived Schwann cells after LPC‐induced peripheral nerve injury.41 These data together support that Schwann cells transplanted into nerve can effectively engage existing axons, displace dysfunctional or damaged Schwann cells, and form structurally normal myelin, indicating that cell transplantation for inherited disorders of peripheral myelin is feasible, if the right cell type can be introduced.
A variety of methods have been investigated to develop a source of Schwann cells for transplantation therapy or in vitro study.42 Schwann cells can be cultured from a sural nerve biopsy as an expandable autologous cell source, and this strategy has been extensively investigated for use in spinal cord injury.43 However, autologous Schwann cells would not be appropriate for patients with inherited myelin disorders, and obtaining them necessitates damage of a functioning nerve. Other cells from easier to harvest sources have been differentiated into Schwann‐like cells, including skin‐derived precursors, mesenchymal stem cells, and adipose tissue‐derived stem cells,41, 44, 45 although these too would have limited utility in inherited myelin disorders, and difficulty with scale of production. iPSCs are a highly attractive source for cell therapy because they are easy to generate, are infinitely expandable, and can be readily manipulated using genome‐editing techniques to correct disease gene defects.46 Several groups have reported differentiating Schwann‐like cells from iPSCs.29, 47, 48 Characterization of iPSC‐derived Schwann cells from these protocols varied widely, in most cases being limited to expression of a few markers such as S100b or GFAP (reviewed in37). We attempted to reproduce a protocol previously reported to generate primarily Schwann cell precursors with the capacity to myelinate axons in vitro;29 however, we found placing NCSCs into MesenPRO with heregulin‐β1‐generated cells most closely resembling mesenchymal precursor cells that did not have the capacity to form myelin after in vivo transplantation. Likewise, when we attempted to instruct iPSC‐derived NCSCs toward a Schwann cell fate by coculture with MNs to recapitulate axonal signals present during development, we found that instead these cells differentiated into skeletal muscle cells. These results strongly support the use of whole transcriptome analysis to characterize iPSC derivatives, as examination of only a small panel of Schwann cell markers can be misleading, and many are expressed in other cell types, including neural crest stem cells. Additionally, it is important to point out that while transplanted rat Schwann cells could form myelin in a host environment of either toxic or inherited demyelination, this environment may be less hospitable to human Schwann cell precursors and contribute to the lack of further differentiation or myelin formation we observed.
Despite these challenges for generating iPSC‐derived Schwann cells, we found that generation of iPSC‐derived NCSCs using a single‐step method involving Wnt signaling and Smad pathway blockade was highly reliable and reproducible across four different iPSC lines.28 However, we were unsuccessful in further differentiating these cells into Schwann cells. It is notable that SOX10 levels were relatively low in these cells, despite the fact they expressed a variety of other transcription factors and cell surface markers associated with NCSCs including SOX9, TFAP2, SNAI2, ITGB1, and NGFR. Neural crest cells are highly plastic in vivo,49 but may become fate restricted even before migration has occurred in the embryo.33 It is possible that NCSCs generated from iPSCs via Wnt activation and Smad inhibition are not equivalently multipotent for generating all neural crest derivatives and are biased toward other cell fates. Notably, both methods we used to differentiate NCSCs into Schwann cells instead generated mesenchymal derivatives, and the original report of this one step protocol also primarily demonstrated mesenchymal derivatives including muscle, chondrocytes, osteocytes, and adipocytes.28 The higher expression of SOX9 than SOX10 in these NCSCs, a critical regulator of mesenchymal cell development,50 may be a key determinant of this fate restriction. Further studies will be required to investigate alternative methods for generating iPSC‐derived Schwann cells, including directed differentiation using transcription factors, or bypassing the need for hNCSCs as was recently demonstrated.48
Taken together, our data support the feasibility of surgical delivery of Schwann cells for inherited disorders of peripheral myelin, demonstrating good survival, migration, and myelination of chronically amyelinated axons. Further studies are needed to address whether cell therapy for peripheral nerves can mitigate secondary axon loss which leads to disability in Charcot–Marie–Tooth disease and to identify appropriate cell sources for transplantation therapy.
Author Contributions
A.K.M.G.M., K.K, I.E., J.S., and R.H.B conceived and designed the experiments. A.K.M.G.M, K.K., I.E., A.A., S.B., J.L., M.E.P., S.C., and R.H.B. performed the experiments and participated in data analysis. A.K.M.G.M and R.H.B. wrote the manuscript. All authors read and approved the final manuscript.
Conflict of Interest
Nothing to report.
Supporting information
Table S1: List of primary antibodies used in the study.
Table S2: List of reagents used for cell culture in the study.
Figure S1: Kaplan–Meier survival curves for homozygous PMP22 rats of both sexes (females, n = 14, solid circles; male, n = 13, open circles) showing that about 60% of them survived for over 3 months.
Figure S2: View of the surgical table and operative field during cell therapy in nude and homozygous PMP22 rats.
Figure S3: Representative tracings of sciatic nerve conduction studies (NCS) performed on the different rat subject groups.
Acknowledgments
We thank the iPSC Core at Cedars‐Sinai Medical Center for their assistance and Klaus Nave for providing PMP22 transgenic rats. R.H.B. is supported by grants RN3‐06530 (California Institute for Regenerative Medicine) and NS097545 (National Institutes of Health).
Funding information
R.H.B. is supported by grants RN3‐06530 (California Institute for Regenerative Medicine) and NS097545 (National Institutes of Health).
Funding Statement
This work was funded by California Institute for Regenerative Medicine grant RN3‐06530; National Institutes of Health grant NS097545.
References
- 1. Dalakas MC. Pathogenesis of immune‐mediated neuropathies. Biochem Biophys Acta 2015;1852:658–666. [DOI] [PubMed] [Google Scholar]
- 2. Baets J, De Jonghe P, Timmerman V. Recent advances in Charcot‐Marie‐Tooth disease. Curr Opin Neurol 2014;27:532–540. [DOI] [PubMed] [Google Scholar]
- 3. Jerath NU, Shy ME. Hereditary motor and sensory neuropathies: understanding molecular pathogenesis could lead to future treatment strategies. Biochem Biophys Acta 2015;1852:667–678. [DOI] [PubMed] [Google Scholar]
- 4. Martyn CN, Hughes RA. Epidemiology of peripheral neuropathy. J Neurol Neurosurg Psychiatry 1997;62:310–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Timmerman V, Strickland AV, Zuchner S. Genetics of Charcot‐Marie‐Tooth (CMT) disease within the frame of the human genome project success. Genes. 2014;5:13–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. DiVincenzo C, Elzinga CD, Medeiros AC, et al. The allelic spectrum of Charcot‐Marie‐Tooth disease in over 17,000 individuals with neuropathy. Mol Genet Genomic Med 2014;2:522–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Saporta MA, Shy ME. Inherited peripheral neuropathies. Neurol Clin 2013;31:597–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Harel T, Lupski JR. Charcot‐Marie‐Tooth disease and pathways to molecular based therapies. Clin Genet 2014;86:422–431. [DOI] [PubMed] [Google Scholar]
- 9. Tousignant R, Trepanier A, Shy ME, Siskind CE. Genetic testing practices for Charcot‐Marie‐Tooth type 1A disease. Muscle Nerve 2014;49:478–482. [DOI] [PubMed] [Google Scholar]
- 10. van Paassen BW, van der Kooi AJ, van Spaendonck‐Zwarts KY, et al. PMP22 related neuropathies: charcot‐Marie‐Tooth disease type 1A and hereditary neuropathy with liability to pressure palsies. Orphanet J Rare Dis 2014;19:38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Robaglia‐Schlupp A, Pizant J, Norreel JC, et al. PMP22 overexpression causes dysmyelination in mice. Brain 2002;125(Pt 10):2213–2221. [DOI] [PubMed] [Google Scholar]
- 12. Watila MM, Balarabe SA. Molecular and clinical features of inherited neuropathies due to PMP22 duplication. J Neurol Sci 2015;355:18–24. [DOI] [PubMed] [Google Scholar]
- 13. Gutmann L, Shy M. Update on Charcot‐Marie‐Tooth disease. Curr Opin Neurol 2015;28:462–467. [DOI] [PubMed] [Google Scholar]
- 14. Sereda MW, Meyer zu Horste G, Suter U, et al. Therapeutic administration of progesterone antagonist in a model of Charcot‐Marie‐Tooth disease (CMT‐1A). Nat Med 2003;9:1533–1537. [DOI] [PubMed] [Google Scholar]
- 15. Passage E, Norreel JC, Noack‐Fraissignes P, et al. Ascorbic acid treatment corrects the phenotype of a mouse model of Charcot‐Marie‐Tooth disease. Nat Med 2004;10:396–401. [DOI] [PubMed] [Google Scholar]
- 16. Gess B, Baets J, De Jonghe P, et al. Ascorbic acid for the treatment of Charcot‐Marie‐Tooth disease. Cochrane Database Syst Rev. 2015. Dec 11; (12):CD011952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sahenk Z, Nagaraja HN, McCracken BS, et al. NT‐3 promotes nerve regeneration and sensory improvement in CMT1A mouse models and in patients. Neurology 2005;65:681–689. [DOI] [PubMed] [Google Scholar]
- 18. Fledrich R, Stassart RM, Klink A, et al. Soluble neuregulin‐1 modulates disease pathogenesis in rodent models of Charcot‐Marie‐Tooth disease 1A. Nat Med 2014;20:1055–1061. [DOI] [PubMed] [Google Scholar]
- 19. Attarian S, Tran LC, Moore A, et al. The neurodevelopmental impact of neonatal morphine administration. Brain Sci 2014;4:321–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bastidas J, Athauda G, De La Cruz G, et al. Human Schwann cells exhibit long‐term cell survival, are not tumorigenic and promote repair when transplanted into the contused spinal cord. Glia 2017;65:1278–1301. [DOI] [PubMed] [Google Scholar]
- 21. Bunge MB, Monje PV, Khan A, Wood PM. From transplanting Schwann cells in experimental rat spinal cord injury to their transplantation into human injured spinal cord in clinical trials. Prog Brain Res 2017;231:107–133. [DOI] [PubMed] [Google Scholar]
- 22. Aguayo AJ, Kasarjian J, Skamene E, et al. Myelination of mouse axons by Schwann cells transplanted from normal and abnormal human nerves. Nature 1977;268:753–755. [DOI] [PubMed] [Google Scholar]
- 23. Levi AD, Guenard V, Aebischer P, Bunge RP. The functional characteristics of Schwann cells cultured from human peripheral nerve after transplantation into a gap within the rat sciatic nerve. J Neurosci 1994;14(3 Pt 1):1309–1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gersey ZC, Burks SS, Anderson KD, et al. First human experience with autologous Schwann cells to supplement sciatic nerve repair: report of 2 cases with long‐term follow‐up. Neurosurg Focus 2017;42:E2. [DOI] [PubMed] [Google Scholar]
- 25. Hall SM, Gregson NA. The in vivo and ultrastructural effects of injection of lysophosphatidyl choline into myelinated peripheral nerve fibres of the adult mouse. J Cell Sci 1971;9:769–789. [DOI] [PubMed] [Google Scholar]
- 26. Sereda M, Griffiths I, Puhlhofer A, et al. A transgenic rat model of Charcot‐Marie‐Tooth disease. Neuron 1996;16:1049–1060. [DOI] [PubMed] [Google Scholar]
- 27. Brockes JP, Fields KL, Raff MC. Studies on cultured rat Schwann cells. I. Establishment of purified populations from cultures of peripheral nerve. Brain Res 1979;165:105–118. [DOI] [PubMed] [Google Scholar]
- 28. Menendez L, Yatskievych TA, Antin PB, Dalton S. Wnt signaling and a Smad pathway blockade direct the differentiation of human pluripotent stem cells to multipotent neural crest cells. Proc Natl Acad Sci USA 2011;108:19240–19245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Liu Q, Spusta SC, Mi R, et al. Human neural crest stem cells derived from human ESCs and induced pluripotent stem cells: induction, maintenance, and differentiation into functional schwann cells. Stem Cells Transl Med 2012;1:266–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ding Q, Lee YK, Schaefer EA, et al. A TALEN genome‐editing system for generating human stem cell‐based disease models. Cell Stem Cell 2013;12:238–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Arroyo EJ, Sirkowski EE, Chitale R, Scherer SS. Acute demyelination disrupts the molecular organization of peripheral nervous system nodes. J Comp Neurol 2004;479:424–434. [DOI] [PubMed] [Google Scholar]
- 32. Allt G, Ghabriel MN, Sikri K. Lysophosphatidyl choline‐induced demyelination. A Freeze‐Fracture Study. Acta Neuropathol 1988;75:456–464. [DOI] [PubMed] [Google Scholar]
- 33. Monk KR, Feltri ML, Taveggia C. New insights on Schwann cell development. Glia 2015;63:1376–1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lee G, Chambers SM, Tomishima MJ, Studer L. Derivation of neural crest cells from human pluripotent stem cells. Nat Protoc 2010;5:688–701. [DOI] [PubMed] [Google Scholar]
- 35. Menendez L, Kulik MJ, Page AT, et al. Directed differentiation of human pluripotent cells to neural crest stem cells. Nat Protoc 2013;8:203–212. [DOI] [PubMed] [Google Scholar]
- 36. Hatada S, Subramanian A, Mandefro B, et al. Low‐dose irradiation enhances gene targeting in human pluripotent stem cells. Stem Cells Transl Med 2015;4:998–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ma MS, Boddeke E, Copray S. Pluripotent stem cells for schwann cell engineering. Stem Cell Rev 2015;11:205–218. [DOI] [PubMed] [Google Scholar]
- 38. Pouwels PJ, Vanderver A, Bernard G, et al. Hypomyelinating leukodystrophies: translational research progress and prospects. Ann Neurol 2014;76:5–19. [DOI] [PubMed] [Google Scholar]
- 39. Gupta N, Henry RG, Strober J, et al. Neural stem cell engraftment and myelination in the human brain. Sci Transl Med 2012;4:155ra37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Saheb‐Al‐Zamani M, Yan Y, Farber SJ, et al. Limited regeneration in long acellular nerve allografts is associated with increased Schwann cell senescence. Exp Neurol 2013;247:165–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Krause MP, Dworski S, Feinberg K, et al. Direct genesis of functional rodent and human schwann cells from skin mesenchymal precursors. Stem Cell Reports 2014;3:85–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Walsh SK, Kumar R, Grochmal JK, et al. Fate of stem cell transplants in peripheral nerves. Stem Cell Research. 2012;8:226–238. [DOI] [PubMed] [Google Scholar]
- 43. Kanno H, Pearse DD, Ozawa H, et al. Schwann cell transplantation for spinal cord injury repair: its significant therapeutic potential and prospectus. Rev Neurosci 2015;26:121–128. [DOI] [PubMed] [Google Scholar]
- 44. Widgerow AD, Salibian AA, Lalezari S, Evans GR. Neuromodulatory nerve regeneration: adipose tissue‐derived stem cells and neurotrophic mediation in peripheral nerve regeneration. J Neurosci Res 2013;91:1517–1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wakao S, Matsuse D, Dezawa M. Mesenchymal stem cells as a source of Schwann cells: their anticipated use in peripheral nerve regeneration. Cells, Tissues, Organs. 2014;200:31–41. [DOI] [PubMed] [Google Scholar]
- 46. Hockemeyer D, Jaenisch R. Induced pluripotent stem cells meet genome editing. Cell Stem Cell 2016;18:573–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ziegler L, Grigoryan S, Yang IH, et al. Efficient generation of schwann cells from human embryonic stem cell‐derived neurospheres. Stem Cell Rev 2011;7:394–403. [DOI] [PubMed] [Google Scholar]
- 48. Kim HS, Lee J, Lee DY, et al. Schwann cell precursors from human pluripotent stem cells as a potential therapeutic target for myelin repair. Stem Cell Reports 2017;8:1714–1726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Baggiolini A, Varum S, Mateos JM, et al. Premigratory and migratory neural crest cells are multipotent in vivo. Cell Stem Cell 2015;16:314–322. [DOI] [PubMed] [Google Scholar]
- 50. Jo A, Denduluri S, Zhang B, et al. The versatile functions of Sox9 in development, stem cells, and human diseases. Genes & Dis 2014;1:149–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1: List of primary antibodies used in the study.
Table S2: List of reagents used for cell culture in the study.
Figure S1: Kaplan–Meier survival curves for homozygous PMP22 rats of both sexes (females, n = 14, solid circles; male, n = 13, open circles) showing that about 60% of them survived for over 3 months.
Figure S2: View of the surgical table and operative field during cell therapy in nude and homozygous PMP22 rats.
Figure S3: Representative tracings of sciatic nerve conduction studies (NCS) performed on the different rat subject groups.