

Abstract
Objective
Injury‐associated axon‐intrinsic signals are thought to underlie pathogenesis and progression in many neuroinflammatory and neurodegenerative diseases, including multiple sclerosis (MS). Retrograde interferon gamma (IFN γ) signals are known to induce expression of major histocompatibility class I (MHC I) genes in murine axons, thereby increasing the susceptibility of these axons to attack by antigen‐specific CD8+ T cells. We sought to determine whether the same is true in human neurons.
Methods
A novel microisolation chamber design was used to physically isolate and manipulate axons from human skin fibroblast‐derived induced pluripotent stem cell (iPSC)‐derived neuron‐enriched neural aggregates. Fluorescent retrobeads were used to assess the fraction of neurons with projections to the distal chamber. Axons were treated with IFN γ for 72 h and expression of MHC class I and antigen presentation genes were evaluated by RT‐PCR and immunofluorescence.
Results
Human iPSC‐derived neural stem cells maintained as 3D aggregate cultures in the cell body chamber of polymer microisolation chambers extended dense axonal projections into the fluidically isolated distal chamber. Treatment of these axons with IFN γ resulted in upregulation of MHC class I and antigen processing genes in the neuron cell bodies. IFN γ‐induced MHC class I molecules were also anterogradely transported into the distal axon.
Interpretation
These results provide conclusive evidence that human axons are competent to express MHC class I molecules, suggesting that inflammatory factors enriched in demyelinated lesions may render axons vulnerable to attack by autoreactive CD8+ T cells in patients with MS. Future work will be aimed at identifying pathogenic anti‐axonal T cells in these patients.
Introduction
Axonal injury, metabolic dysfunction, and transport disruption are thought to underlie pathogenesis and progression in an array of neuroinflammatory and neurodegenerative diseases.1, 2, 3, 4 Likewise, anterograde and retrograde signaling events propagating in stressed, damaged, or dysfunctional axons are thought to contribute separately to processes involved in both injury and repair in the central nervous system (CNS)5, 6, 7, 8, 9, 10 (reviewed in 11). While the expression and function of MHC class I on adult human neurons remains disputed,12, 13, 14, 15 we previously demonstrated that retrograde interferon gamma (IFNγ) signaling induced the expression of major histocompatibility class I (MHC I) molecules in murine axons.10 Moreover, we found that increased surface expression of peptide‐loaded MHC I rendered axons more susceptible to attack by antigen‐specific CD8+ T cells. We have proposed that this mechanism underlies the injury of demyelinated axons in multiple sclerosis (MS).1, 2, 4, 10 Evidence also suggests that MHC I is involved with synaptic pruning during cortical development12, 13, 14, 16, 17, 18, 19, 20, 21 and following injury22, 23, 24, 25 and contributes to neuronal plasticity.16, 17, 26, 27 These findings also suggest that retrogradely induced MHC class I may contribute to the diffuse synaptopathy observed in MS and other neurodegenerative diseases.28, 29, 30, 31, 32, 33, 34, 35 However, whether human neurons and axons respond to inflammatory stress in this way is unknown, and the dearth of methods for efficiently culturing human neurons and isolated axons has prevented robust analysis. Indeed, the study of human CNS axon biology in general is hindered by the technical challenges of generating isolated fields of patient‐specific axons.
Current evidence suggests that three‐dimensional (3D) culture techniques provide a distinct advantage for the maintenance and expansion of neurogenic neural stem cells (NSCs). Methods for 3D cell culture include the use of nano‐carriers, microencapsulation, microisolation chambers (MICs), and cellular aggregates or organoids (reviewed in 36). Cellular aggregates and MICs are particularly amenable to the culturing of neural cells in 3D. Previously it was shown that high‐density NSC cultures spontaneously form neural aggregates that recapitulate several NSC niche components.37 In line with this, large neural aggregates or neural organoids exhibiting CNS tissue architecture have recently been described.38, 39 It has also been proposed that MICs could offer further benefits for culturing NSCs and NSC‐derived cells. MICs offer the ability to create growth factor or hormone gradients that mimic the in vivo microenvironment and the ability to compartmentalize extracellular matrix composition to enhance physical and functional differentiation. For neuron cultures in particular, MICs that separate axons from cell bodies have allowed scientists to study axon biology in greater detail,40, 41, 42, 43, 44, 45 permitting the analysis of factors that impact neurite outgrowth46 and the analysis of retrograde and anterograde axonal signaling.10 Building on these concepts, we provide a robust and efficient paradigm for transferring iPSC‐derived, NSC‐derived, neuron‐rich neural aggregates into MICs that routinely achieves dense axonal outgrowth into distal chambers (Fig. 1A). We also present a novel MIC design that is more amenable to culturing neural aggregates without the need for labor‐intensive and error‐prone modification of existing chambers (Fig. 1B). More importantly, we show that human axons isolated in this culture platform retrogradely transport fluorescent microbeads and demonstrate for the first time that human neurons respond to distal IFNγ stimulation by retrograde induction of antigen processing, transport, and presentation genes, and that MHC I is subsequently expressed on the axolemma. We also discuss the implications of retrograde induction of MHC I in MS in terms of the potential role in lesional axon injury, neuron loss in normal appearing gray matter, and diffuse synaptopathy.
Figure 1.
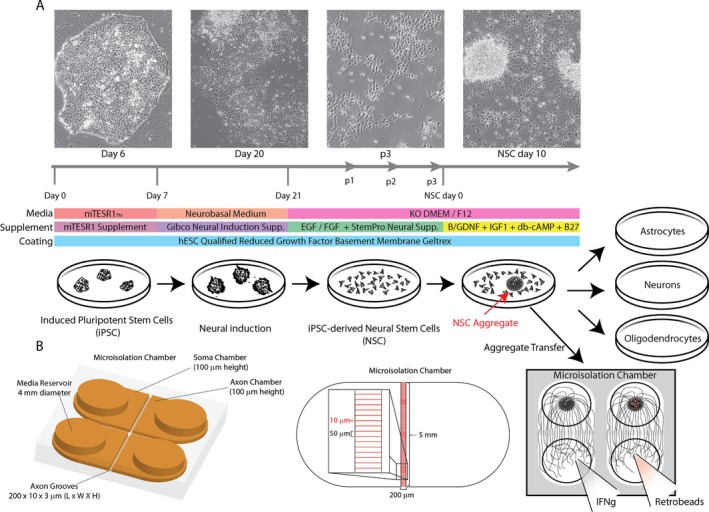
Tissue culture strategy for differentiating human neurons in microisolation chambers. (A) Schematic showing the differentiation strategy for generating neural aggregates from human‐induced pluripotent stem cells. (B) Schematic of microisolation chamber design and dimensions showing 3D representation (left) and top view (center). Open chamber design enables transfer of larger neural aggregates in direct apposition to interchamber grooves for increased axonal outgrowth to distal chamber.
Experimental Procedures
Cell culture
Previously characterized iPSC cell lines (100‐BIOTR‐0012) generated using Sendai virus‐based methodology were obtained from the Mayo Clinic Center for Regenerative Medicine BioTrust. All studies were approved by the Mayo Clinic Institutional Review Board, Mayo Clinic Stem Cell Research Oversight Committee and Mayo Clinic Center for Regenerative Medicine Biotrust Oversight group. All experimental methods were carried out in accordance with the approved guidelines. For iPSC and NSC cultures, tissue‐culture‐treated 6‐well plates were coated with hESC Qualified Geltrex (Gibco, ThermoFisher) diluted to 83 μg/mL in DMEM/F12 by incubating at 37°C and 5% CO2 for a minimum of 60 min. iPSCs were expanded as diffuse colonies in mTESR1 media (Stemcell Technologies, Vancouver, BC, Canada). Media was changed daily and differentiated cells were manually removed as needed. Following neural induction, NSCs were cultured in NSC expansion media consisting of KO DMEM/F‐12, 1% Glutamax, 2% StemPro Neural Supplement (Gibco, ThermoFisher), 1% Anti‐Anti (Gibco, ThermoFisher) and 10 ng/mL FGFb (PreproTech) and EGF (Gibco, Invitrogen). All cells were supplemented with 5 μmol/L Rock Inhibitor (Y27632, Stemcell Technologies) for the first 24 h after thawing. NSCs and neurons were maintained in their respective media with freshly added growth factors and half of their media replaced every other day. Neurons were differentiated in media composed of KO DMEM/F‐12, 2% B27 supplement (Gibco, ThermoFisher), 1% Glutamax, 100 U/mL penicillin, 100 μg/mL streptomycin, 1 mmol/L dibutyryl cyclic‐adenosine monophosphate (cAMP; Sigma), 5 μg/mL plasmocin (Invivogen), 1 mmol/L sodium pyruvate and 10 ng/mL each of BDNF (PreproTech), IGF1 (Corning) and GDNF (PeproTech).
Microisolation chambers
Chamber dimensions are provided in Figure 1. Chambers were fabricated by curing polydimethylsiloxane elastomer (PDMS; Sylgard 184 Elastomer Kit, Dow Corning) on etched silicon wafer molds (SIMTech Microfluidics Foundry, Distributed by APP Systems, Singapore), using one part curing reagent per 10 parts PDMS and incubating at 37–55°C overnight. Cured gel imprints were cut from molds and media reservoirs were created using a 4 mm biopsy punch (Miltex) on a cutting mat (Electron Microscopy Sciences). To sterilize, imprinted gels were autoclaved in glass petri dishes and stored until use. German 22 × 22 mm cover slips (Neuvitro, GG‐2222‐oz) were acid washed with 0.1 N HCl for 3 h, and serially washed with H2O, 100% ethanol, and 95% ethanol before storage in 70% ethanol. When needed, individual glass coverslips were placed in 6 well plates using sterile forceps and allowed to dry in a biosafety cabinet under UV light. Coverslips were coated overnight at 37°C with 0.5 mg/mL poly‐ornithine (Sigma, P4638) in 100 μmol/L borate buffer (Polysciences). The next day coverslips were washed 3 times with sterile water and allowed to dry completely. To assemble the MIC, autoclaved PDMS imprints were tightly adhered to dried coverslips using sterile forceps. Ice‐cold reduced growth factor Geltrex (Gibco, A1569601) diluted to 83 μg/mL in DMEM/F12 was added to each chamber (10 μL), and chambers were incubated at 37°C for at least 60 min to ensure coating.
Purification of neural aggregates
Mature aggregated cultures of differentiating NSCs were gently detached from the tissue culture vessel surface using cell lifters (Sigma) in differentiation media. The resulting suspension was minimally triturated (once or twice) with a 5 mL serological pipette and expelled into a 40 μm micron cell strainer. The strainer was then serially washed by dipping into 3 wells of a 6‐well plate each containing 8–10 mL prewarmed growth media. After washing, cell strainers were flipped and back‐flushed into an empty tissue culture vessel taking care to maintain sterility. Eluted aggregates were transferred to a conical tube and allowed to settle for 1–2 min. Excess media and nonaggregated cells were aspirated, and aggregates were gently resuspended in 50 μL supplemented growth media per cm2 original growth area in the TC vessel from which the aggregates were prepared. For MIC plating, the suspension was gently mixed and quickly aliquoted into a 96‐well V‐bottom plate (100 μL/well), mixing again as needed. For each chamber, 10 μL from the bottom of each well was collected and plated immediately following aspiration of Geltrex solution to prevent drying of the Geltrex matrix.
Real‐time polymerase chain reaction
All lysate samples were stored at −80°C in 1% β‐mercaptoethanol in Buffer RLT Plus (Qiagen, Germantown, MD, USA 1053393) prior to disruption and homogenization with QIAShredder columns (Qiagen, 79656). For experiments involving microisolation chambers (MICs), 7 cell culture lysates of the same treatment were pooled prior to RNA isolation using the RNeasy Plus Micro Kit (Qiagen, 74034). RNA concentration was estimated with a NanoDrop spectrophotometer (ThermoFisher). Transcriptor First Strand cDNA Synthesis kit (Roche, 4897030001) was used to synthesize cDNA from RNA samples using oligo‐dT primers to target mRNA. Equal amounts of sample template RNA (0.5–2.5 μg depending on the experiment) were used for each cDNA reaction. The reactions were placed in a thermal block cycler and incubated at 55°C for 45 min and inactivated by heating at 85°C for 5 min. Samples were diluted with PCR grade water (1:10–1:50) and stored at −20°C. RT‐PCR was performed according to the protocol outlined with SsoAdvanced Universal SYBR Green Supermix (Bio‐Rad, 172‐5271) and each sample was run in triplicate. Briefly, 20 μL reactions were prepared adding 10 μL of SsoAdvanced universal SYBR Green 2 × master, 2 μL of 5 μmol/L Forward and 5 μmol/L Reverse Primers (total of 4 μL), 1 μL Nuclease‐free water, and 5 μl sample template containing 2–10 ng cDNA. Samples were run on a Bio‐Rad CFX Connect system for 50 cycles with the following protocol: 15 sec at 95°C, 45 sec at 55°C, and 5 sec at 65°C. Primers (Table S1) were selected using Primer BLAST (NCBI) to have melt temperatures around 60°C. Melt curve analysis was used to determine specificity of each reaction. Data were exported and analyzed in Excel using the Pfaffl method to determine relative quantitation based on an estimated amplification efficiency of 95%.10 Expression across all samples was normalized to the GAPDH housekeeping gene.
Immunofluorescent staining and confocal microscopy
To preserve neuronal integrity, cells were first fixed by adding 1 volume fresh 4% para‐formaldehyde (PFA) in PBS into the cell culture media and incubating for 25 min at room temperature (2% PFA final concentration). After this initial fixation, media and fixative were aspirated, replaced with fresh ice‐cold 4% PFA in PBS, and incubated for 25 min. Cells were subsequently permeabilized overnight at 4°C with 0.1% Triton X‐100 in PBS. Cells were blocked for at least 30 min in blocking buffer containing 5–8% normal donkey serum, 1% bovine serum albumin, 0.01% sodium azide, and 0.1% Triton X‐100 or 0.1% Tween 20 in PBS with calcium and magnesium. After blocking, cells were incubated overnight at 4°C with primary antibodies (Table S2) diluted to 5 micrograms/μL in blocking buffer. The next day, cells were washed 3 times with 0.1% Tween 20 in PBS and incubated for 2 h at room temperature with secondary antibodies diluted 1:200 in blocking buffer. Cells were washed and counterstained with DAPI prior to imaging. Images were acquired on an LSM 780 confocal fluorescent microscope using Zen software (Zeiss). Images were minimally processed with NIH ImageJ software prior to publication.
Retrobead experiments
Fluorescent 40 nm TransFluoSphere (Thermofisher, T‐8864; retrobeads) suspensions were centrifuged for 10 min at >20,000 g and resuspended in 10 volumes neuronal differentiation media. Prior to loading into axonal chambers, 10 μL of media was removed from the axon chamber and 20–40 μL media was added to the cell body chamber to create a positive pressure gradient across the chamber grooves. Subsequently, 10 μL of the diluted retrobeads were added to the axon chambers. After 20–24 h, chambers were removed from 6 to well tissue culture plates and high magnification images were acquired on LSM 780 confocal microscope by directly imaging through the cover glass comprising the chamber bottom.
Statistical analyses
Here, α = 0.05 and β = 0.2 were established a priori. Post hoc power analysis was performed for all experiments and significance was only considered when power ≥0.8. Normality was determined by the Shapiro–Wilk test or Kolmogorov–Smirnov test. For multiple comparisons, one‐way analysis of variance (ANOVA) or nonparametric (Kruskal–Wallis) tests were performed where appropriate. Reported P values were corrected for multiple comparisons (Holm‐Sidak correction for ANOVA; Dunn's correction for Kruskal–Wallis). Unpaired two‐tailed Student's t‐tests were used for comparisons made between two groups. Curran‐Everett guidelines were followed.
Results
Differentiation of human fibroblast‐derived iPSCs into NSCs
In order to generate stably expandable human NSCs, well‐characterized fibroblast‐derived iPSCs were differentiated via a rapid (14 day) nonrosette‐based neuroinduction protocol relying on dual SMAD inhibition.47 Following neuroinduction, NSCs were maintained in serum‐free media supplemented with StemPro NeuroSupplement and freshly administered FGFb and EGF (10 ng/mL). Under these conditions, we observed that NSCs exhibited morphological stability for >13 passages (p13). We compared expression of the NSC marker genes nestin, SOX1, and PAX6 to the iPSC marker gene OCT3/4 in p13 NSCs, p3 NSCs, and parental iPSCs by RT‐PCR. p3 and p13 NSCs expressed significantly lower levels of OCT3/4 (p3: P = 0.0229, p13: P = 0.0229 vs. iPSC; Fig. 2A) and significantly higher levels of PAX6 (p3: P = 0.1887, p13: P < 0.0001 vs. iPSC; Fig. 2B), nestin (p3: P = 0.0191, p13: P = 0.0055 vs. iPSC; Fig. 2C), and SOX1 (p3: P = 0.0002, p13: P < 0.0001 vs. iPSC; Fig. 2D). Expression levels of OCT3/4, nestin, and SOX1 did not differ significantly between p3 and p13 NSC, whereas PAX6 expression levels continued to increase from p3 to p13 (p3 vs. p13: P < 0.0001). Expression of the NSC marker genes nestin and Sox2 was verified in p3 NSC by immunofluorescent staining (Fig. 2E).
Figure 2.
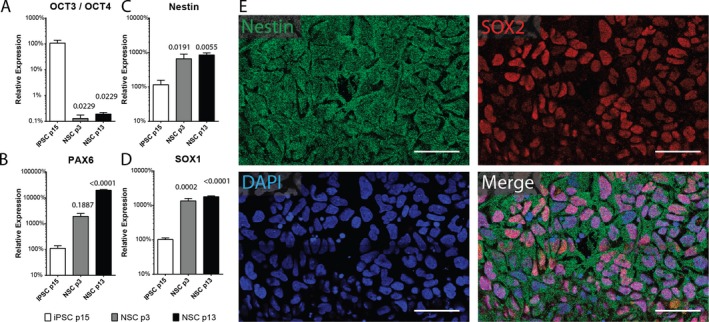
Validation of neural induction of human iPSCs. Relative expression of (A) OCT3/4, (B) PAX6, (C) nestin, and (D) SOX1 in early (p3) or late (p13) passage NSC and parent iPSCs (n = 3–5 per group). Data are representative of 2 independent experiments. Error bars represent SEM. P‐values shown above individual histograms are relative to iPSC expression levels. (E) Representative confocal images of p3 NSCs immunostained for nestin and SOX2 and counterstained with DAPI. Scale bar = 50 μm.
Efficient differentiation of astrocytes, oligodendrocytes, and neurons from iPSC‐derived NSCs
To verify multipotency, NSCs were switched to neural differentiation media containing B27 (2%), cAMP (1 mmol/L), and growth factors (BDNF, IGF1, GDNF; 10 ng/mL). NSCs rapidly differentiated into oligodendrocytes, astrocytes, and neurons as assessed by immunofluorescence (Fig. 3A). We also compared expression levels of genes indicative of astrocytes (GLT1, GLAST), oligodendrocytes (GALC, MBP) and neurons (NeuN, MAP2, VGLUT1, VGAT) in early passage (p3‐5), late passage (p13) and terminally differentiated (14 days) NSCs. Compared to p3‐5 NSC, terminally differentiated cells expressed significantly higher levels of GLT1 (P = 0.0259), GLAST (P = 0.0048), MBP (P = 0.0034), GalC (P = 0.0009), NeuN (P = 0.0001), MAP2 (P = 0.0023), VGAT (P < 0.0001), and VGLUT1 (P = 0.0153) (Fig. 3B). Late passage NSC (p13) exhibited intermediate expression levels of VGAT (P = 0.0061 vs. p3‐5) and NeuN (P = 0.0380 vs. p3‐5), indicating that some differentiation had occurred spontaneously in these later‐passage cultures. We also noted that differentiating cells tended to stochastically form neural aggregates reminiscent of neurospheres but adherent to the culture dish. These neural aggregates showed a higher degree of neurite outgrowth than adjacent areas. When the neural aggregates were purified by back‐flushing through 40 μm cell strainers and reseeded onto geltrex‐coated tissue culture wells they remained viable and rapidly regrew neurites. Immunostaining also revealed that compared to monolayer cultures of differentiated NSCs, the aggregates exhibited a higher density of neurofilament light chain‐positive axonal projections and NeuN‐positive neuronal cell bodies (Fig. 3A).
Figure 3.
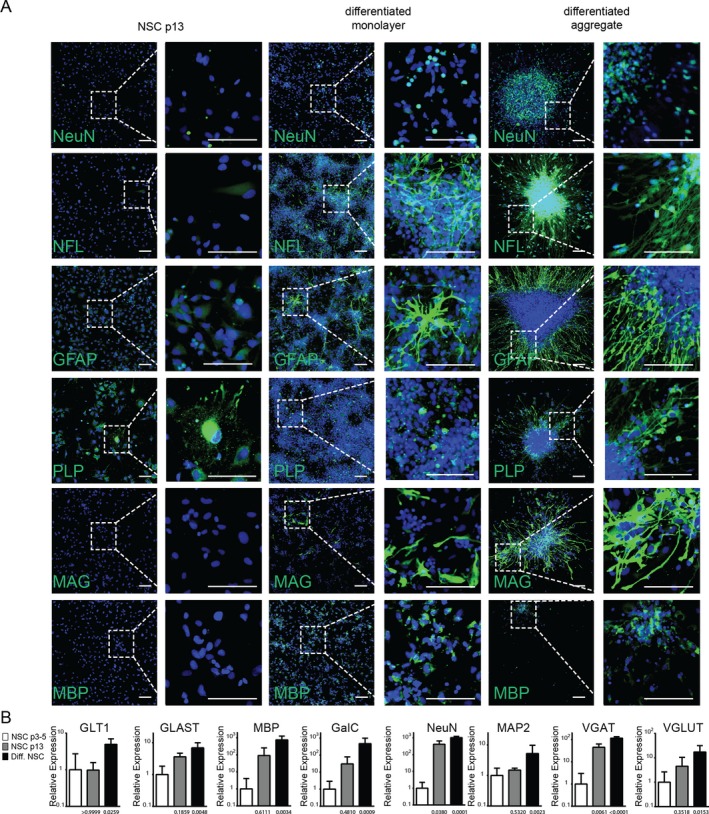
Validation of NSC multipotency. Human iPSC‐derived neural stem cells were differentiated for 7–10 days in KO DMEM/F12‐containing db‐cAMP (1 mmol/L), BDNF, IGF1, GDNF (10 ng/mL each), and 2% B27 until aggregate formation was observed. Aggregates were isolated, using 40 μm cell strainers and seeded on Geltrex coated glass chamber slides. (A) Expression of differentiation markers for neurons (NF‐L, NeuN), Astrocytes (GFAP), and oligodendrocytes (MAG, MBP, PLP) was assessed by immunostaining in undifferentiated late passage NSC as well as monolayer cultures or purified aggregates after 14–21 days differentiation. Representative confocal images are shown. Scale bar 100 μm. (B) mRNA expression levels of neuronal (NeuN, MAP2, VGAT, VGLUT1), oligodendrocyte (GALC, MBP), and astrocyte (GLT1, GLAST) marker genes as assessed by RT‐PCR in early (NSC p3‐5) and late (NSC p13) passage NSC as well as NSC after 14–21 days differentiation (Diff. NSC). n = 3–8 samples per group. Data are presented as mean with 95% confidence interval.
Robust axonal outgrowth following transfer of neuron‐enriched iPSC‐derived neural aggregates into microisolation chambers
We transferred purified neural aggregates into polymer‐based MICs constructed such that two large, readily accessible fluidic compartments are separated by 3 μm high, 10 μm wide, 200 μm long grooves (Fig. 1B). We have previously shown that these dimensions exclude the migration of cell bodies into the distal chamber.10 The transferred aggregates adhered to the Geltrex‐coated MICs within 4 h and sprouted neurites within 24 h (Fig. 4A, top two panels). Neurites typically reached the proximal face of the microgrooves by 3 days after plating and emerged into the distal chamber as early as 4 days posttransfer (Fig. 4A, bottom two panels).
Figure 4.
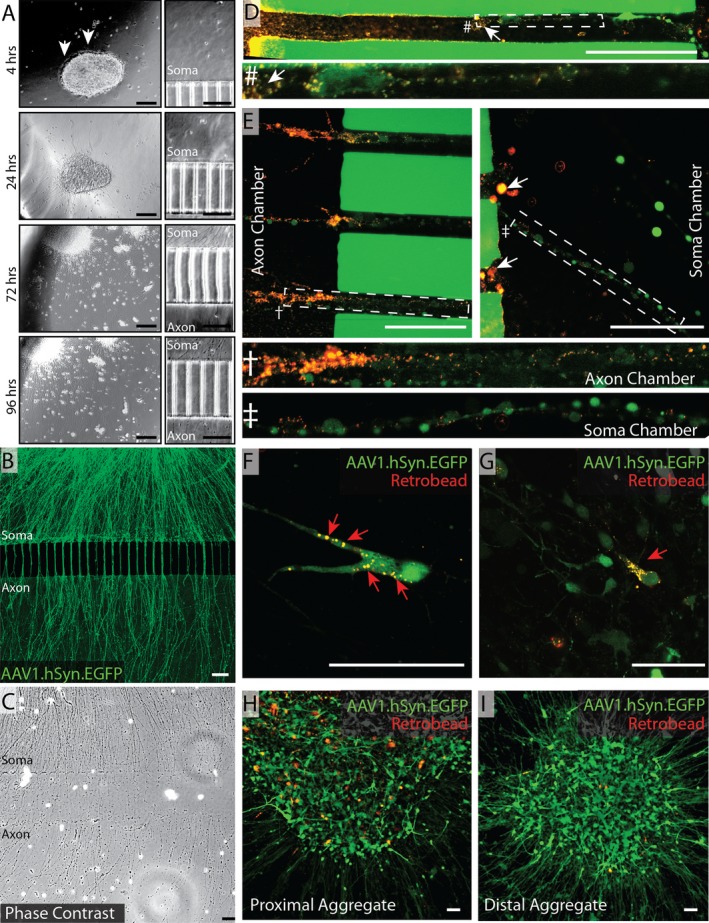
Robust axonal outgrowth from neural aggregates in microisolation chambers. (A) Neural aggregates were isolated from differentiating NSC cultures using 40 μm cell strainers and seeded into microisolation chambers at a density of 5–10 aggregates per chamber. Phase contrast images of aggregates (left) and proximal grooves (right) were captured 4 h, 24 h, 72 h, and 96 h postplating. Representative images are shown. Arrows in top panel indicate earliest neurite outgrowth. (B) Neural aggregates were infected with AAV1.hSyn.EGFP (2000 MOI) and allowed to differentiated for 14 days. Representative live confocal image shows robust axonal outgrowth into distal chamber. Scale bars A–C = 100 μm. Fluorescent retrobeads (0.04 μm) were then diluted and loaded into the axonal chamber. Representative confocal images of intra‐axon and intrasoma retrobead‐laden vesicles were acquired 20–24 h later. (C) 11 days after neural aggregates were transferred, cells were fixed with 2–4% PFA and microisolation chambers were carefully disassembled and imaged by phase contrast microscopy. Photomerged image shown is representative of multiple high magnification tile scanning images. (D) Top panel shows interchamber groove—higher magnification of the indicated area is shown below. (E) Groove‐proximal regions of axon and soma chambers are shown. Higher magnification images of the indicated areas are shown below. White arrows (D–E) indicate axonal swellings containing multiple fluorescent retrobeads. Representative images of retrobeads laden cell bodies in isolated neurons (F,G) as well as in distal (H) and proximal (I) aggregates. Scale bars D–I = 50 μm.
To better visualize axon outgrowth we transduced the aggregates with an adeno‐associated viral construct driving expression of enhanced green fluorescent protein (EGFP) under transcriptional control of the human synapsin 1 promoter (AAV1.hSyn.EGFP). Using this approach, we readily visualized dense axonal outgrowth into the distal chamber (Fig. 4B), which corresponded well with phase contrast imaging of fixed cells in disassembled chambers (Fig. 4C).
In vitro retrograde axonal transport of fluorescent retrobeads by human iPSC‐derived neurons
Small fluorescently labeled microbeads (retrobeads) have been used for decades to label neurons based on the principle of retrograde axonal transport.48, 49, 50 We exploited this material to verify the transport competency of the neural aggregate‐derived axons. Aggregates were plated as above and transduced with AAV1.hSyn.EGFP. After 7–11 days we observed robust axonal outgrowth in a majority of chambers. Chambers in which cell‐free axons were not observed due to occlusion of the grooves or leakage of the chamber were excluded from experiments. Red fluorescent microspheres resuspended in neuronal differentiation media were added to the axon chamber and a volume differential of 20:40 μL (axon chamber:cell body chamber) was established to prevent diffusion‐based transfer of material from the axon chamber, as previously described.10 At 24 h, we observed strong uptake of the fluorescent retrobeads by axons at the distal groove opening (Fig. 4D and E left panel), with modest levels present within healthy axonal segments, as would be expected with continuous retrograde and anterograde vesicle trafficking. We also noted significant accumulation of retrobeads within axonal swellings (white arrows Fig. 4D and E right panel). Finally, we observed numerous GFP‐positive neuronal cell bodies containing retrobeads (Fig. 4G red arrows), indicating distal uptake and retrograde transport. The proportion of neuronal cell bodies containing retrobeads varied from aggregate to aggregate, with aggregates closest to the grooves showing more labeled neurons (Fig. 4H) compared to distal aggregates (Fig. 4I).
Stimulation of axons with IFNγ induces retrograde upregulation of antigen processing and presentation genes in human iPSC‐derived neurons
We previously demonstrated that stimulation of primary murine axons with IFNγ for 72 h induced surface expression of peptide‐loaded MHC class I molecules.10 Extending this paradigm to the human neural aggregates, we found that IFNγ (100 ng/mL) added to the axon chamber in the presence of a differential fluidic gradient (20:40 μL, axon chamber:cell body chamber) stimulated the upregulation of antigen processing genes (immunoproteasome subunits PSMB8, PSMB9), antigen transport genes (TAP1, TAP2), and antigen presentation genes (NLRC5, B2M, HLA‐A, HLA‐B, HLA‐C) (Fig. 5A). For example, the immunoproteasome gene PSMB9 was upregulated nearly 60‐fold over vehicle‐treated neurons (P < 0.0001). PSMB8 and B2M were upregulated over fivefold (P < 0.0001). TAP1 was also upregulated nearly threefold (P < 0.0001). While more modest, HLA‐A (P = 0.048), HLA‐B (P = 0.0004), and HLA‐C (P = 0.0006), along with the master MHC class I transcriptional regulatory factor NLRC5 (P = 0.0135) were upregulated more than twofold. Given the relative number of neurons in the aggregates extending axons into the distal chamber versus the total number of cells from which RNA was prepared, these values likely underestimate the size of the induction. Of note, expression of the housekeeping gene GAPDH did not differ between IFNγ‐ and vehicle‐stimulated cultures (data not shown). Unexpectedly, the genes NRTN (P = 0.6513) and PACRG (P = 0.4204), which have been shown to decrease in neurons following IFNγ exposure, were not impacted in our system (data not shown). Again, this may reflect the relative number of neurons responding to the retrograde signal versus the total cells in the culture.
Figure 5.
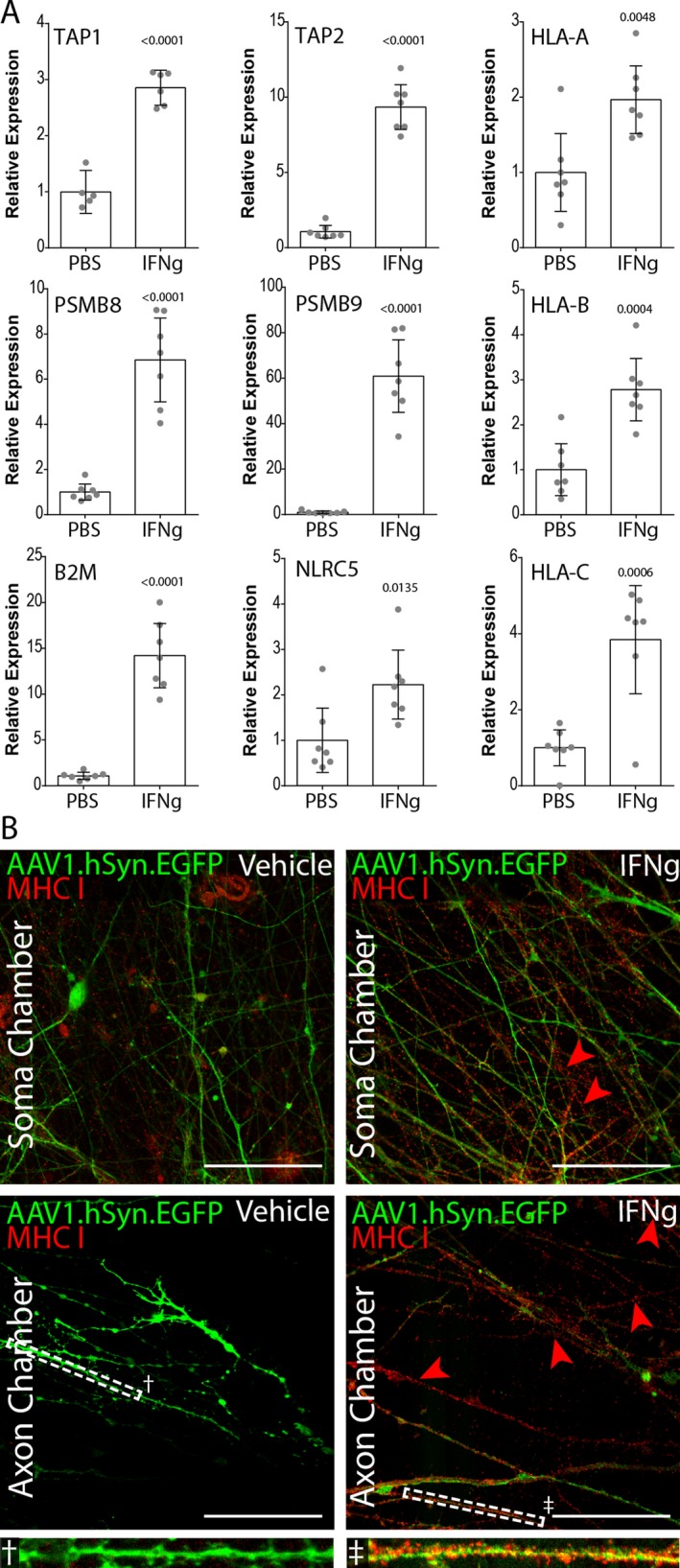
Retrograde IFN γ induction of MHC class I genes in human neural aggregates using microisolation chambers. (A) Nontransduced neural aggregates cultured as in Figure 4 were treated with 100 ng/mL IFN γ on the axon side chamber, isolated by fluidic drive. After 72 h, RNA was isolated from the soma‐side chamber and converted into cDNA. RT‐PCR was performed using primers for the indicated genes normalizing to GAPDH expression levels. (n = 7 per group). Data are representative of 3 independent experiments. P‐values shown above individual histograms are relative to iPSC expression levels. Data are presented as mean with 95% confidence interval. (B) AAV1.Syn.EGFP transduced neural aggregates were treated with IFN γ in the axon chamber as in (A). 72 h later cells and axons were fixed and labeled with anti‐HLA‐A,B,C (W6/32). Representative confocal images are shown for soma and axon chambers. Scale bars 50 μm.
Stimulation of axons with IFNγ induces expression of MHC class I molecules on human axons
Given the upregulation of the entire transcriptional program involved in antigen presentation, we asked whether the retrograde IFNγ response resulted in anterograde trafficking of MHC I proteins to the distal axon. Using the same conditions employed above, we found that GFP‐positive axons in both chambers exhibited high levels of MHC I as revealed by immunostaining with a pan‐HLA antibody (i.e. anti‐HLA‐A,B,C) (Fig. 5B). Essentially no axonal MHC I was detected in vehicle‐treated cultures, either in the cell body or axon chamber. The MHC I staining pattern in the IFNγ‐stimulated MICs was punctate and densely distributed on GFP‐positive axons in the distal chamber (Fig. 5B, inset). This pattern was also observed on GFP‐negative axons in the IFNγ‐stimulated cultures, reflecting the presence of projections from neurons that were not transduced with the AAV1.hSyn.EGFP vector.
Discussion
Reynold and Weiss were the first to show that adherent cells derived from fetal rodent brain proliferated and eventually formed floating colonies termed neurospheres capable of giving rise to neurons, astrocytes, and oligodendrocytes.51 It has been speculated that part of the success of neurosphere cultures is due to the fact that the 3D architecture more closely recapitulates the in vivo microenvironment of the NSC niche.52, 53 Recently, techniques have been established for reliably generating monolayers of induced pluripotent stem cell (iPSC)‐derived NSCs using supplementation with basic fibroblast growth factor (FGFb) and epidermal growth factor (EGF).54, 55, 56 However, such cultured monolayer NSCs are more transcriptionally homogeneous than in vivo NSCs, and it has been speculated that this lack of heterogeneity is responsible, at least in part, for the challenges of promoting the differentiation of cultured NSCs into diverse neuronal subtypes.56, 57, 58, 59
Using a novel strategy to isolate axons from human iPSC‐derived neuron‐enriched neural aggregates we have shown that such axons respond to local inflammatory stress by retrogradely signaling to upregulate expression of a transcriptional program involved in presentation of antigenic peptides on MHC class I molecules. It is important to note that retrograde axonal IFNγ signaling may trigger activation of adjacent nonneuronal cells via the release of secondary factors by neurons. In this regard, it is possible that some of the observed changes in MHC I mRNA expression in our experiments may be associated with upregulation in other neural cell types responding to such secondary signals. Future studies will be necessary to clarify this issue. Additionally, whether retrograde induction of MHC class I contributes to neuronal dysfunction or neuroprotection is not clear. For example, Sabha et al. demonstrated that deficient upregulation of MHC I in neurons following axotomy was associated with reduced axonal outgrowth, suggesting that increased expression may facilitate repair.23 On the other hand, a pathogenic role for retrograde induction of MHC I by IFNγ in MS is likely to be associated with gray matter pathology that occurs remotely from active demyelinating lesions. Diffuse synapse loss with altered cortical connectivity, as well as diffuse neuron and axon loss, are all commonly found in MS tissue 28, 29, 30, 31, 32, 33, 34, 60, 61, 62, 63 and could be driven by somatic or axonal MHC I expression. For example, neuronal MHC I has an established role in neuronal plasticity16 via mechanisms involving postsynaptic interactions with glutamate receptors and presynaptic interactions with the immunoreceptor tyrosine‐based inhibitory motif‐containing leukocyte immunoglobulin‐like receptor PirB.64 Neuronal MHC I has also been shown to inhibit glutamatergic receptor function in hippocampal circuits65 and studies in the retinogeniculate system indicate a role for somatic MHC class I in synapse elimination.66, 67 Likewise, in vitro overexpression and knock down experiments have inversely associated MHC I expression with synapse density in cortical neurons.68 Somatic upregulation of MHC I on neurons may also mark these cells for elimination by anti‐neuronal T cells, leading to loss of neurons in otherwise normal appearing gray matter. Indeed several studies have shown that targeting neoantigens to CNS neuronal subsets results in rapid clearance by antigen‐specific CD8+ T cells.15, 69, 70, 71 Finally, anterograde transport of MHC class I molecules into the distal axon makes these molecules available near the site of inflammation, where they may increase vulnerability to attack by autoreactive anti‐axonal CD8+ T cells.10 As discussed below, these findings have significance for the study of neurological conditions in which axonal injury processes may contribute to clinical progression.
The use of neuron‐rich neural aggregates grown in microfluidic microisolation devices is clearly useful for the study of human axon biology. While we demonstrate this approach, using iPSC‐derived NSCs, it is readily adaptable to NSCs derived from a variety of pluripotent stem cell sources. Currently available MICs are not amenable to neural aggregate culturing due to physical restrictions such as the chamber height proximal to the interchamber grooves and the need to distribute single cell suspensions into the device under differential fluidic pressure10, 43, 72, 73 (e.g. Xona Microfluidics). In contrast, our chamber design and other recent designs provide an open access port that facilitates placement of the large neural aggregates near the microgrooves.41, 46 This design also facilitates media exchange without flow‐induced disruption of the cells or shear force‐induced damage to the axons.
While the aggregate cultures yield high numbers of neurons and hence large numbers of projecting axons that penetrate the distal chamber, variations in size of the aggregates may contribute to mechanical and metabolic stress that is hard to control. This challenge may be circumvented by adapting the current protocol for controlling the size of cultured rodent prenatal neural aggregates.74 It will also be important to determine the clonal heterogeneity of neural aggregates and to determine how this heterogeneity differs between early and late passage NSC‐derived aggregates. For example, early passage neurospheres exhibit high levels of transcriptional heterogeneity, similar to NSCs in vivo,75 but appear to lose neurogenic heterogeneity after a few passages in vitro,76 with the remaining neuronal progenitors more or less restricted to a GABA‐ergic fate.57 Of note, we have anecdotally observed that late passage aggregates exhibit reduced axonal outgrowth compared to early passage NSC aggregates (data not shown). Likewise, compared to the glutamatergic marker VGLUT1, we measured a larger increase in expression of the GABA‐ergic neuron marker VGAT in late passage NSCs relative to early passage cells (Fig. 3B).
There are still many unanswered questions about the initiation factors, mechanism(s), and timing of axon injury in numerous diseases.77, 78, 79, 80, 81, 82, 83, 84 Axonopathy is ultimately driven by a confluence of factors and occurs via several intermediate and potentially reversible stages that eventually lead to irrevocable axonal degeneration.85 In multiple sclerosis, we and others 3, 78, 86 have proposed that disease progression is driven by immune‐mediated injury to demyelinated axons. Specifically, we have hypothesized that inflammatory and/or metabolic stress associated with focal demyelination results in the upregulation of MHC class I molecules presenting neuronal antigens to cytotoxic anti‐axonal CD8+ T cells. We have tested this hypothesis using several different in vivo and in vitro paradigms,1, 2, 4, 10 but all have depended upon the response of murine neurons. To our knowledge, this is the first report of inflammation‐induced MHC class I upregulation in human iPSC‐derived neurons and axons and the first report to show that such an effect is mediated by retrograde signaling from the distal axon to the neuron cell body. Ultimately, it is our goal to use this iPSC‐derived neural aggregate method to prepare patient‐specific axons that can be exposed to the patient's own immune cell repertoire to identify true anti‐axonal CD8+ T cells in patients with multiple sclerosis. However, the neural aggregate platform that we describe is broadly applicable to the analysis of axon injury in many neurologic disease conditions. For example, we anticipate that this system will shed additional light on transport defects and metabolic vulnerability in axons from patients with amyotrophic lateral sclerosis, Alzheimer disease, Parkinson disease, etc. This platform will also be amenable to medium‐throughput screening of drugs that improve axonal transport and may eventually permit tailored therapeutic interventions for many diseases involving axon injury.
Author Contributions
C.L.H and B.D.S.C. contributed to study conception and design. B.D.S.C., M.S.P., and R.L.C. performed experiments. B.D.S.C., M.S.P., R.L.C., and C.L.H analyzed and interpreted the data. B.D.S.C., M.S.P., and C.L.H drafted the manuscript. R.L.C. provided critical revision of the manuscript. C.L.H and B.D.S.C. wrote and edited the final manuscript.
Conflicts of Interests
The authors declare that they have no competing financial interests.
Supporting information
Table S1. Supplementary Materials
Table S2. Supplementary Materials
Acknowledgments
This work was supported by a generous gift from Don and Fran Herdrich to C.L.H. B.D.S.C. was supported by fellowships from the Mayo Clinic Center for Regenerative Medicine and the Center for MS and Autoimmune Neurology. Support was also provided to C.L.H. by a Pilot Award and by a Collaborative MS Research Center Award from the National Multiple Sclerosis Society. We gratefully acknowledge the Mayo Clinic Regenerative Medicine Biotrust for providing characterized iPSCs and for ongoing technical assistance.
Funding information
This work was supported by a generous gift from Don and Fran Herdrich to C.L.H. B.D.S.C. was supported by fellowships from the Mayo Clinic Center for Regenerative Medicine and the Center for MS and Autoimmune Neurology. Support was also provided to C.L.H. by a Pilot Award and by a Collaborative MS Research Center Award from the National Multiple Sclerosis Society.
Funding Statement
This work was funded by Mayo Clinic Center for Regenerative Medicine and the Center for MS and Autoimmune Neurology grant ; National Multiple Sclerosis Society grant .
References
- 1. Howe CL, Ure D, Adelson JD, et al. CD8 + T cells directed against a viral peptide contribute to loss of motor function by disrupting axonal transport in a viral model of fulminant demyelination. J Neuroimmunol 2007;188(1–2):13–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Howe CL, Adelson JD, Rodriguez M. Absence of perforin expression confers axonal protection despite demyelination. Neurobiol Dis 2007;25:354–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Howe CL. Immunological aspects of axon injury in multiple sclerosis. Curr Top Microbiol Immunol 2008;318:93–131. [DOI] [PubMed] [Google Scholar]
- 4. Deb C, LaFrance‐Corey RG, Zoecklein L, et al. Demyelinated axons and motor function are protected by genetic deletion of perforin in a mouse model of multiple sclerosis. J Neuropathol Exp Neurol 2009;68:1037–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Carlos AJ, Cotman CW, Prieto GA, Tong L. IL‐1beta impairs retrograde flow of BDNF signaling by attenuating endosome trafficking. J Neuroinflammation 2017;14:29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zahavi EE, Ionescu A, Gluska S, et al. A compartmentalized microfluidic neuromuscular co‐culture system reveals spatial aspects of GDNF functions. J Cell Sci 2015;128:1241–1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Poon WW, Blurton‐Jones M, Tu CH, et al. Beta‐Amyloid impairs axonal BDNF retrograde trafficking. Neurobiol Aging 2011;32:821–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Perlson E, Maday S, Fu MM, et al. Retrograde axonal transport: pathways to cell death? Trends Neurosci 2010;33:335–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. von Bartheld CS. Axonal transport and neuronal transcytosis of trophic factors, tracers, and pathogens. J Neurobiol 2004;58:295–314. [DOI] [PubMed] [Google Scholar]
- 10. Sauer BM, Schmalstieg WF, Howe CL. Axons are injured by antigen‐specific CD8(+) T cells through a MHC class I‐ and granzyme B‐dependent mechanism. Neurobiol Dis 2013;59:194–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ito K, Enomoto H. Retrograde transport of neurotrophic factor signaling: implications in neuronal development and pathogenesis. J Biochem 2016;160:77–85. [DOI] [PubMed] [Google Scholar]
- 12. Zhang A, Yu H, Shen Y, et al. The expression patterns of MHC class I molecules in the developmental human visual system. Neurochem Res 2013;38:273–281. [DOI] [PubMed] [Google Scholar]
- 13. Zhang A, Yu H, He Y, et al. Developmental expression and localization of MHC class I molecules in the human central nervous system. Exp Brain Res 2015;233:2733–2743. [DOI] [PubMed] [Google Scholar]
- 14. Zhang A, Yu H, He Y, et al. The spatio‐temporal expression of MHC class I molecules during human hippocampal formation development. Brain Res 2013;1529:26–38. [DOI] [PubMed] [Google Scholar]
- 15. Cebrián C, Zucca FA, Mauri P, et al. MHC‐I expression renders catecholaminergic neurons susceptible to T‐cell‐mediated degeneration. Nat Commun 2014;5:3633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Shatz CJ. MHC class I: an unexpected role in neuronal plasticity. Neuron 2009;64:40–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Huh GS, Boulanger LM, Du H, et al. Functional requirement for class I MHC in CNS development and plasticity. Science 2000;290:2155–2159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Goddard CA, Butts DA, Shatz CJ. Regulation of CNS synapses by neuronal MHC class I. Proc Natl Acad Sci USA 2007;104:6828–6833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Liu J, Shen Y, Li M, et al. The expression pattern of classical MHC class I molecules in the development of mouse central nervous system. Neurochem Res 2013;38:290–299. [DOI] [PubMed] [Google Scholar]
- 20. McAllister AK. Major histocompatibility complex I in brain development and schizophrenia. Biol Psychiat 2014;75:262–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Elmer BM, McAllister AK. Major histocompatibility complex class I proteins in brain development and plasticity. Trends Neurosci 2012;35:660–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Spejo AB, Oliveira AL. Synaptic rearrangement following axonal injury: Old and new players. Neuropharmacology 2015; 96(Pt A): 113–123. [DOI] [PubMed] [Google Scholar]
- 23. Sabha M Jr., Emirandetti A, Cullheim S, De Oliveira AL. MHC I expression and synaptic plasticity in different mice strains after axotomy. Synapse 2008;62:137–148. [DOI] [PubMed] [Google Scholar]
- 24. Thams S, Oliveira A, Cullheim S. MHC class I expression and synaptic plasticity after nerve lesion. Brain Res Rev 2008;57:265–269. [DOI] [PubMed] [Google Scholar]
- 25. Inacio RF, Zanon RG, de Castro MV, et al. Astroglioma conditioned medium increases synaptic elimination and correlates with major histocompatibility complex of class I (MHC I) upregulation in PC12Cells. Neurosci Lett 2016;634:160–167. [DOI] [PubMed] [Google Scholar]
- 26. Ribic A, Zhang M, Schlumbohm C, et al. Neuronal MHC class I molecules are involved in excitatory synaptic transmission at the hippocampal mossy fiber synapses of marmoset monkeys. Cell Mol Neurobiol 2010;30:827–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lv D, Shi Q, Liu J, et al. The similar expression pattern of MHC class I molecules in human and mouse cerebellar cortex. Neurochem Res 2014;39:180–186. [DOI] [PubMed] [Google Scholar]
- 28. Santo Araújo SE, Mendonça HR, Wheeler NA, et al. Inflammatory demyelination alters subcortical visual circuits. J Neuroinflammation 2017;14:162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Musella A, Mandolesi G, Mori F, et al. Linking synaptopathy and gray matter damage in multiple sclerosis. Mult Scler 2016;22:146–149. [DOI] [PubMed] [Google Scholar]
- 30. Mandolesi G, Grasselli G, Musumeci G, Centonze D. Cognitive deficits in experimental autoimmune encephalomyelitis: neuroinflammation and synaptic degeneration. Neurol Sci 2010;31(Suppl 2):S255–S259. [DOI] [PubMed] [Google Scholar]
- 31. Mandolesi G, Gentile A, Musella A, et al. Synaptopathy connects inflammation and neurodegeneration in multiple sclerosis. Nat Rev Neurol 2015;11:711–724. [DOI] [PubMed] [Google Scholar]
- 32. Jürgens T, Jafari M, Kreutzfeldt M, et al. Reconstruction of single cortical projection neurons reveals primary spine loss in multiple sclerosis. Brain 2016; 139(Pt 1): 39–46. [DOI] [PubMed] [Google Scholar]
- 33. Centonze D, Muzio L, Rossi S, et al. Inflammation triggers synaptic alteration and degeneration in experimental autoimmune encephalomyelitis. J Neurosci 2009;29:3442–3452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Barnett MH, Mathey E, Kiernan MC, Pollard JD. Axonal damage in central and peripheral nervous system inflammatory demyelinating diseases: common and divergent pathways of tissue damage. Curr Opin Neurol 2016;29:213–221. [DOI] [PubMed] [Google Scholar]
- 35. Cebrian C, Loike JD, Sulzer D. Neuronal MHC‐I expression and its implications in synaptic function, axonal regeneration and Parkinson's and other brain diseases. Front Neuroanat 2014;8:114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Karimi M, Bahrami S, Mirshekari H, et al. Microfluidic systems for stem cell‐based neural tissue engineering. Lab Chip 2016;16:2551–2571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lutolf MP, Blau HM. Artificial stem cell niches. Adv Mater 2009;21(32–33):3255–3268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Camp JG, Badsha F, Florio M, et al. Human cerebral organoids recapitulate gene expression programs of fetal neocortex development. Proc Natl Acad Sci USA 2015;112:15672–15677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lancaster MA, Renner M, Martin CA, et al. Cerebral organoids model human brain development and microcephaly. Nature 2013;501:373–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Park JW, Kim HJ, Byun JH, et al. Novel microfluidic platform for culturing neurons: culturing and biochemical analysis of neuronal components. Biotechnol J 2009;4:1573–1577. [DOI] [PubMed] [Google Scholar]
- 41. Park J, Koito H, Li J, Han A. Microfluidic compartmentalized co‐culture platform for CNS axon myelination research. Biomed Microdevice 2009;11:1145–1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Park HS, Liu S, McDonald J, et al. Neuromuscular junction in a microfluidic device. Conference proceedings: Annual International Conference of the IEEE Engineering in Medicine and Biology Society. IEEE Engineering in Medicine and Biology Society. Annual Conference 2013; 2013: 2833–2835. [DOI] [PubMed] [Google Scholar]
- 43. Taylor AM, Blurton‐Jones M, Rhee SW, et al. A microfluidic culture platform for CNS axonal injury, regeneration and transport. Nat Methods 2005;2:599–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Taylor AM, Dieterich DC, Ito HT, et al. Microfluidic local perfusion chambers for the visualization and manipulation of synapses. Neuron 2010;66:57–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Millet LJ, Gillette MU. New perspectives on neuronal development via microfluidic environments. Trends Neurosci 2012;35:752–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lee N, Park JW, Kim HJ, et al. Monitoring the differentiation and migration patterns of neural cells derived from human embryonic stem cells using a microfluidic culture system. Mol Cells 2014;37:497–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Chambers SM, Fasano CA, Papapetrou EP, et al. Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nat Biotechnol 2009;27:275–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Liu SL, Zhang ZL, Sun EZ, et al. Visualizing the endocytic and exocytic processes of wheat germ agglutinin by quantum dot‐based single‐particle tracking. Biomaterials 2011;32:7616–7624. [DOI] [PubMed] [Google Scholar]
- 49. Chowdary PD, Che DL, Zhang K, Cui B. Retrograde NGF axonal transport–motor coordination in the unidirectional motility regime. Biophys J 2015;108:2691–2703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Siddique R, Vyas A, Thakor N, Brushart TM. A two‐compartment organotypic model of mammalian peripheral nerve repair. J Neurosci Methods 2014;232:84–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Reynolds BA, Weiss S. Generation of neurons and astrocytes from isolated cells of the adult mammalian central nervous system. Science 1992;255:1707–1710. [DOI] [PubMed] [Google Scholar]
- 52. Campos LS, Leone DP, Relvas JB, et al. Beta1 integrins activate a MAPK signalling pathway in neural stem cells that contributes to their maintenance. Development 2004;131:3433–3444. [DOI] [PubMed] [Google Scholar]
- 53. Bez A, Corsini E, Curti D, et al. Neurosphere and neurosphere‐forming cells: morphological and ultrastructural characterization. Brain Res 2003;993(1–2):18–29. [DOI] [PubMed] [Google Scholar]
- 54. Elkabetz Y, Studer L. Human ESC‐derived neural rosettes and neural stem cell progression. Cold Spring Harb Symp Quant Biol 2008;73:377–387. [DOI] [PubMed] [Google Scholar]
- 55. Elkabetz Y, Panagiotakos G, Al Shamy G, et al. Human ES cell‐derived neural rosettes reveal a functionally distinct early neural stem cell stage. Genes Dev 2008;22:152–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Conti L, Pollard SM, Gorba T, et al. Niche‐independent symmetrical self‐renewal of a mammalian tissue stem cell. PLoS Biol 2005;3:e283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Machon O, Backman M, Krauss S, Kozmik Z. The cellular fate of cortical progenitors is not maintained in neurosphere cultures. Mol Cell Neurosci 2005;30:388–397. [DOI] [PubMed] [Google Scholar]
- 58. Parish CL, Castelo‐Branco G, Rawal N, et al. Wnt5a‐treated midbrain neural stem cells improve dopamine cell replacement therapy in parkinsonian mice. J Clin Investig 2008;118:149–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Bithell A, Finch SE, Hornby MF, Williams BP. Fibroblast growth factor 2 maintains the neurogenic capacity of embryonic neural progenitor cells in vitro but changes their neuronal subtype specification. Stem Cells 2008;26:1565–1574. [DOI] [PubMed] [Google Scholar]
- 60. Calabrese M, Atzori M, Bernardi V, et al. Cortical atrophy is relevant in multiple sclerosis at clinical onset. J Neurol 2007;254:1212–1220. [DOI] [PubMed] [Google Scholar]
- 61. Klaver R, Popescu V, Voorn P, et al. Neuronal and axonal loss in normal‐appearing gray matter and subpial lesions in multiple sclerosis. J Neuropathol Exp Neurol 2015;74:453–458. [DOI] [PubMed] [Google Scholar]
- 62. Inglese M, Ge Y, Filippi M, et al. Indirect evidence for early widespread gray matter involvement in relapsing‐remitting multiple sclerosis. NeuroImage 2004;21:1825–1829. [DOI] [PubMed] [Google Scholar]
- 63. Filippi M, Bozzali M, Rovaris M, et al. Evidence for widespread axonal damage at the earliest clinical stage of multiple sclerosis. Brain 2003;126(Pt 2):433–437. [DOI] [PubMed] [Google Scholar]
- 64. Syken J, GrandPre T, Kanold PO, Shatz CJ. PirB restricts ocular‐dominance plasticity in visual cortex. Science 2006;313:1795–1800. [DOI] [PubMed] [Google Scholar]
- 65. Fourgeaud L, Davenport CM, Tyler CM, et al. MHC class I modulates NMDA receptor function and AMPA receptor trafficking. Proc Natl Acad Sci USA 2010;107:22278–22283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Lee H, Brott BK, Kirkby LA, et al. Synapse elimination and learning rules co‐regulated by MHC class I H2‐Db. Nature 2014;509:195–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Xu HP, Chen H, Ding Q, et al. The immune protein CD3zeta is required for normal development of neural circuits in the retina. Neuron 2010;65:503–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Glynn MW, Elmer BM, Garay PA, et al. MHCI negatively regulates synapse density during the establishment of cortical connections. Nat Neurosci 2011;14:442–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Scheikl T, Pignolet B, Dalard C, et al. Cutting edge: neuronal recognition by CD8 T cells elicits central diabetes insipidus. J Immunol 2012;188:4731–4735. [DOI] [PubMed] [Google Scholar]
- 70. Medana IM, Gallimore A, Oxenius A, et al. MHC class I‐restricted killing of neurons by virus‐specific CD8 + T lymphocytes is effected through the Fas/FasL, but not the perforin pathway. Eur J Immunol 2000;30:3623–3633. [DOI] [PubMed] [Google Scholar]
- 71. Bernard‐Valnet R, Yshii L, Quériault C, et al. CD8 T cell‐mediated killing of orexinergic neurons induces a narcolepsy‐like phenotype in mice. Proc Natl Acad Sci USA 2016;113:10956–10961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Harris J, Lee H, Vahidi B, et al. Non‐plasma bonding of PDMS for inexpensive fabrication of microfluidic devices. J Vis Exp 2007;9:410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Harris J, Lee H, Vahidi B, et al. Fabrication of a microfluidic device for the compartmentalization of neuron soma and axons. J Vis Exp 2007;7:261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Choi YJ, Park J, Lee SH. Size‐controllable networked neurospheres as a 3D neuronal tissue model for Alzheimer's disease studies. Biomaterials 2013;34:2938–2946. [DOI] [PubMed] [Google Scholar]
- 75. Suslov ON, Kukekov VG, Ignatova TN, et al. Neural stem cell heterogeneity demonstrated by molecular phenotyping of clonal neurospheres. Proc Natl Acad Sci USA 2002;99:14506–14511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Ciccolini F. Identification of two distinct types of multipotent neural precursors that appear sequentially during CNS development. Mol Cell Neurosci 2001;17:895–907. [DOI] [PubMed] [Google Scholar]
- 77. Calkins DJ. Critical pathogenic events underlying progression of neurodegeneration in glaucoma. Prog Retin Eye Res 2012;31:702–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Wilkins A, Scolding N. Protecting axons in multiple sclerosis. Mult Scler 2008;14:1013–1025. [DOI] [PubMed] [Google Scholar]
- 79. Gumy LF, Tan CL, Fawcett JW. The role of local protein synthesis and degradation in axon regeneration. Exp Neurol 2010;223:28–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Ma M. Role of calpains in the injury‐induced dysfunction and degeneration of the mammalian axon. Neurobiol Dis 2013;60:61–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Maxwell WL. Development of concepts in the pathology of traumatic axonal and traumatic brain injury In Kobeissy F. H. ed. Brain neurotrauma: molecular, neuropsychological, and rehabilitation aspects. Boca Raton (FL): CRC Press/Taylor & Francis, 2015. [Google Scholar]
- 82. Rishal I, Fainzilber M. Axon‐soma communication in neuronal injury. Nat Rev Neurosci 2014;15:32–42. [DOI] [PubMed] [Google Scholar]
- 83. Stirling DP, Stys PK. Mechanisms of axonal injury: internodal nanocomplexes and calcium deregulation. Trends Mol Med 2010;16:160–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Tsutsui S, Stys PK. Metabolic injury to axons and myelin. Exp Neurol 2013;246:26–34. [DOI] [PubMed] [Google Scholar]
- 85. Stys PK. General mechanisms of axonal damage and its prevention. J Neurol Sci 2005;233(1–2):3–13. [DOI] [PubMed] [Google Scholar]
- 86. Trapp BD, Stys PK. Virtual hypoxia and chronic necrosis of demyelinated axons in multiple sclerosis. Lancet Neurol 2009;8:280–291. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Supplementary Materials
Table S2. Supplementary Materials