

Abstract
The continuing rise of multidrug resistant pathogens has made it clear that in the absence of new antibiotics we are moving toward a “postantibiotic” world, in which even routine infections will become increasingly untreatable. There is a clear need for the development of new antibiotics with truly novel mechanisms of action to combat multidrug resistant pathogens. Experimental evolution to resistance can be a useful tactic for the characterization of the biochemical mechanism of action for antibiotics of interest. Herein, we demonstrate that the use of a diverse panel of strains with well-annotated reference genomes improves the success of using experimental evolution to characterize the mechanism of action of a novel pyrrolizidinone antibiotic analog. Importantly, we used experimental evolution under conditions that favor strongly polymorphic populations to adapt a panel of three substantially different Gram-positive species (lab strain Bacillus subtilis and clinical strains methicillin-resistant Staphylococcus aureus MRSA131 and Enterococcus faecalis S613) to produce a sufficiently diverse set of evolutionary outcomes. Comparative whole genome sequencing (WGS) between the susceptible starting strain and the resistant strains was then used to identify the genetic changes within each species in response to the pyrrolizidinone. Taken together, the adaptive response across a range of organisms allowed us to develop a readily testable hypothesis for the mechanism of action of the CJ-16 264 analog. In conjunction with mitochondrial inhibition studies, we were able to elucidate that this novel pyrrolizidinone antibiotic is an electron transport chain (ETC) inhibitor. By studying evolution to resistance in a panel of different species of bacteria, we have developed an enhanced method for the characterization of new lead compounds for the discovery of new mechanisms of action.
Keywords: antibiotic resistance, antibiotics, experimental evolution, mechanism of action, pyrrolizidinone
Graphical abstract
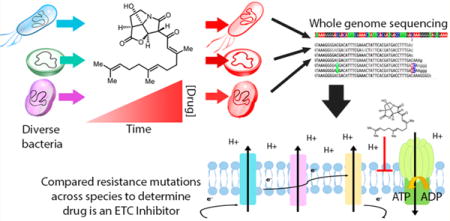
The increasing prevalence of antibiotic resistance has resulted in a global health crisis due to the difficulty in treating infections caused by resistant pathogens.1 In the United States alone, each year two million illnesses and 23 000 deaths are caused by antibiotic-resistant microbes.2 Compounding the problems caused by the rapid evolution of multidrug resistant pathogens, the pipeline of FDA approved antibiotics has been declining for several decades, meaning fewer new therapies are coming to market to treat infections.1 Nearly every aspect of modern medicine from surgery to cancer treatment to treating an infected surface wound from the prick of a rose thorn (sporotrichosis) relies upon the effectiveness of antimicrobials to treat or prevent infection. Additionally, though there are currently 37 new antibiotics in development, only two of these kill pathogens through novel mechanisms.3 Developing antibiotics with novel mechanisms of action is important, as pathogens are unlikely to harbor resistance against mechanisms they have never been exposed to. Thus, there is a clear need for the development of new antibiotics, especially those with novel mechanisms of action, to combat infections.
With an emphasis on new lead compounds and novel mechanisms of action, we wished to develop an enhanced and widely applicable method to elucidate in vivo mechanisms of action that did not require an a priori hypothesis about how a compound might work. For this study, “mechanism of action” refers to the biochemical targets of a compound that can then be used to infer the general basis for activity. For example, tetracyclines inhibit translation, while β-lactams alter cell wall biosynthesis. Established approaches for the identification of mechanisms include screening overexpression libraries for mutants with increased resistance, as increased expression of the molecular target of the antibiotic will sometimes confer cells with resistance.4–11 However, to efficiently and quickly complete such overexpression screens, robotic high-throughput technology is necessary. Additionally, sometimes overexpression of a target may not confer resistance, and thus, this approach will fail to identify the target in these cases.12 Proteomic and transcriptomic approaches have also been employed to evaluate the responses of bacteria to an antibiotic, which subsequently gives insights into the target of the antibiotic.13–15 However, these approaches require specialized instrumentation and complex data analysis and can be costly. The mechanism of action behind potential leads can also be confirmed by identifying resistance alleles using classical genetic techniques, such as resistor generation or experimental evolution and comparative genomics.16,17 However, resistance mutations do not always provide insights into an antibiotic’s mechanisms of action. For example, mutations in efflux pumps or in poorly characterized hypothetical proteins can be difficult to interpret. From our previous evolution studies with established antibiotics, such as daptomycin and tigecycline, we observed that some strains tend to acquire adaptive alleles that are more insightful than others.18–21 This led us to believe that using a diverse panel of strains, instead of a single organism, could greatly enhance the usefulness of experimental evolution as a tool to characterize the mechanism of action of any candidate antibiotic. With the increasing need for antibiotics with novel mechanisms, it would be extremely valuable to have innovative and robust methods that identify novel antibiotic mechanisms of action efficiently.
Here, we describe the successful use of an approach to characterize the mechanism of an antibiotic using experimental evolution across a diverse group of bacteria with relatively well-annotated genomes (Figure 1). To validate this approach, we characterized a synthetic analog of CJ-16,264 (KCN-AAS-35, Figure 2) based on its parent natural product (CJ-16,264, Figure 2) that was originally isolated and characterized from an unknown fungus in Japan.22 CJ-16,264 was previously shown to be highly active against several different Gram-positive bacterial species but not against Gram-negative species, with the exception of Moraxella catarrhalis and an E. coli strain with increased membrane permeability.22 Additionally, CJ-16,264 has a unique pyrrolizidinone structure differing from all other known antibiotics, which allows for a possibly novel mechanism of action.22 The Nicolaou group developed a synthetic pathway for this natural product and several derivatives of CJ-16,264 to assess their biological activity and potential as therapeutic agents.23 By comparing the adaptive mutations identified across Bacillus subtilis 168 and clinical strains methicillin-resistant Staphylococcus aureus 131 and Enterococcus faecalis S613, we were able to develop a hypothesis and then validate the mechanism of action of the CJ-16,264 analog (KCN-AAS-35, Figure 2). By performing oxygen consumption assays on the three bacterial strains in the presence of KCN-AAS-35, we were able to confirm that the compound inhibits bacterial respiration. Furthermore, mitochondrial inhibition studies show that this novel pyrrolizidinone antibiotic (i.e., KCN-AAS-35) is likely an electron transport chain (ETC) inhibitor. We also surveyed previously published experimental evolution studies with different organisms and drugs to show that combining results from across species is a general and effective approach to the identification of in vivo mechanisms-of-action for antimicrobials. In light of the decreasing cost of genome sequencing and the improved quality of genomic annotation, experimental evolution with diverse strains of bacteria should prove to be an effective method for the evaluation of new lead compounds and the identification of novel mechanisms of action.
Figure 1.
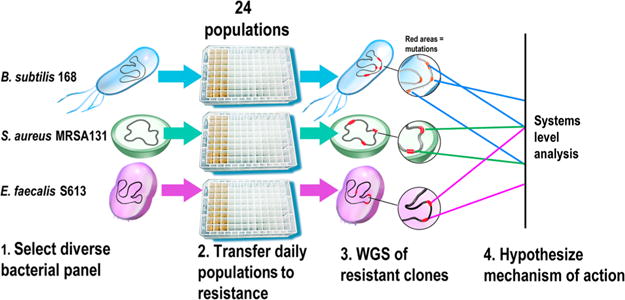
Summary of approach to identify the mechanism of action of a novel antibiotic. Identifying an antibiotic’s mechanism of action can be challenging, as antibiotics can target a diverse range of processes in bacteria, such as cell wall synthesis, protein translation, and nucleic acid metabolism. Therefore, we developed an approach that can be used to efficiently develop a hypothesis for the mechanism of action of any novel antibiotic. (1) A panel of at least three different susceptible species of bacteria is selected on the basis of several different criteria. Ideally, the different strains should have well-annotated genomes that would allow mutated genes and pathways to be readily identified. Model organisms also allow experimental validation of the proposed mechanism. Thus, it is beneficial to include at least one model organism, such as a laboratory strain of Bacillus subtilis or Escherichia coli. Additionally, the panel should consist of phylogenetically diverse species, since distantly related organisms are more likely to adapt through different mechanisms and thus reveal more pathways and targets affected by the antibiotic. Finally, some of the selected strains should be clinically relevant species that the antibiotic would potentially be used against. (2) Replicate populations of each species are adapted to become resistant to the antibiotic under study. Including replicate populations of the same strain is important since it allows for the identification of genes and pathways that are mutated reproducibility across the different populations, which in turn reveals which targets are most important to resistance. During adaptation, conservative increases in antibiotic concentration should be used to favor genetically diverse populations. (3) Comparative WGS between the adapted resistant strains and the ancestral susceptible starting strains is used to determine what adaptive mutations were acquired. (4) The different adaptive mutations are compared across replicate populations and the different species to develop a hypothesis for the mechanism of action of the antibiotic. Loci and pathways mutated across the different replicate populations and species are more likely to be important to resistance and thus targets of the antibiotic. Experimental validation is then performed to confirm the proposed mechanism of action.
Figure 2.
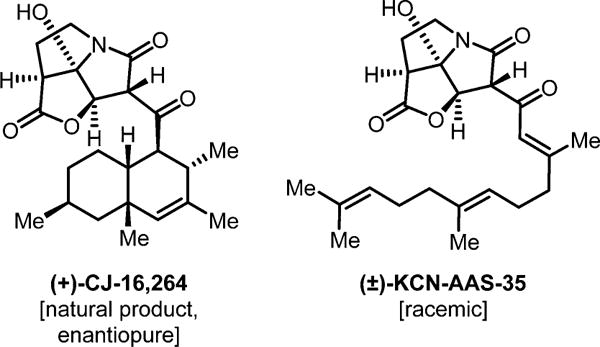
Structures of the natural product CJ-16,264 (left) and the synthetic analog KCN-AAS-35 (right). To demonstrate the effectiveness of our approach, an antibiotic with a unique structure and unknown mechanism of action was characterized. The selected antibiotic is a synthetic derivative of the natural pyrrolizidinone antibiotic CJ-16,264 (left) and is referred to as KCN-AAS-35 (right). The pyrrolizidinone structure of these compounds is not present in any other class of antibiotics and therefore indicates they likely have a unique mechanism of action. KCN-AAS-35 was selected for these studies since it has a simpler structure and was easier to synthesize.22
RESULTS AND DISCUSSION
Analog KCN-AAS-35 Has Similar Activity to the CJ-16,264 Pyrrolizidinone Natural Product
The antibiotic used for these studies was an analog of CJ-16,264 [(+)-CJ-16,264], referred to as KCN-AAS-35, that was developed by the Nicolaou group and was easier to synthesize than the natural product due to its less complex structure [(±)-KCN-AAS-35, Figure 2]. Using minimum inhibitory concentrations (MICs), we determined that KCN-AAS-35 was as effective as CJ-16,264 against four different Gram-positive species and was even more effective against E. faecalis S613 (MIC = 2 μg/mL) than the natural product (MIC = 8 μg/mL) (Table 1). Importantly, the natural product and KCN-AAS-35 had similar efficacy against the strains as the clinical therapy minocycline (Table 1). To further characterize both pyrrolizidinone compounds, we performed time kill assays and determined that KCN-AAS-35 and the natural product have similar killing kinetics against S. aureus MRSA131 (see Figure S1). The MIC testing and time kill assays both indicate that KCN-AAS-35 has similar, or better, potency than the natural product. Combining the unique structure, easy synthesis, and high potency of KCN-AAS-35, it was a promising candidate for further characterization. Therefore, we decided to use experimental evolution to resistance and comparative genomics to gain insights in the mechanism of action of KCN-AAS-35.
Table 1.
Minimum Inhibitory Concentration (MIC) Data of Pyrrolizidinone Natural Product (CJ-16,264) and KCN-AAS-35 against Ancestral and KCN-AAS-35 Adapted Strains
| antibiotic MIC (μg/mL)
|
|||||
|---|---|---|---|---|---|
| species | clone | tetracycline | minocycline | CJ-16,264 | KCN-AAS-35 |
| B. subtilis 168 | ancestor | 4 | 0.125 | 0.5 | 0.5 |
| A10-4 | – | – | – | 2 | |
| A12-3 | – | – | – | 2 | |
| B11-3 | – | – | – | 2 | |
| C10-1 | – | – | – | 1 | |
| D11-1 | – | – | – | 1 | |
| G11-2 | – | – | – | 2 | |
| S. aureus MRSA131 | ancestor | >128 | 0.5 | 2 | 2 |
| E10-1 | – | – | – | 8 | |
| E10-3 | – | – | – | 4 | |
| F10-4 | – | – | – | 8 | |
| B11-3 | – | – | – | 8 | |
| C11-2 | – | – | – | 8 | |
| E11-1 | – | – | – | 4 | |
| E. faecalis S613 | ancestor | 8 | 8 | 8 | 2 |
| A2-1 | – | – | – | 8 | |
| H2-4 | – | – | – | 4 | |
| A3-1 | – | – | – | 8 | |
| C3-4 | – | – | – | 8 | |
| H3-2 | – | – | – | 4 | |
| H3-3 | – | – | – | 16 | |
| E. faecium 105 | ancestor | – | 4 | 2 | 2 |
Selecting the Panel of Strains Used for Identification of the Biochemical Mechanism of Action for KCN-AAS-35
Our approach can be used to characterize the mechanism of action of any candidate antibiotic agent (Figure 1). However, the panel of bacteria used in the experimental evolution studies should consist of phylogenetically distinct organisms. Additionally, it is important to select strains with well-assembled and well-annotated genomes, as this makes it easier to quickly and accurately identify mutated genes and pathways (Figure 1). MIC testing suggested that KCN-AAS-35 was not effective against Gram-negatives, and therefore, our panel of three strains did not include them (Table 1). While B. subtilis 168, E. faecalis S613, and S. aureus MRSA131 are all Gram-positive species, they span different phylogenetic orders and families, therefore possessing substantially different genomes. While B. subtilis 168 is not a pathogen, it was selected for its well-annotated genome and for its ability to serve as a good model organism in the event that we wished to further validate our hypothesis on the biochemical mechanism by knocking out or knocking in specific genes.
Stepwise Adaptation to Increasing Concentrations of KCN-AAS-35
To produce cells with increased tolerance for KCN-AAS-35, the cells are grown at subinhibitory concentrations of the antibiotic and then transferred daily to increasing concentrations until they are sufficiently resistant (typically 2–8 times their original MIC) (Figure 3). To identify the biochemical processes that are affected by a drug, it is very useful to use selection conditions that provide a broad range of successful evolutionary outcomes for resistance. By increasing KCN-AAS-35 concentrations at steps well below the MIC, the maintenance of a highly polymorphic population with many competing evolutionary trajectories for resistance can be favored.19–21
Figure 3.
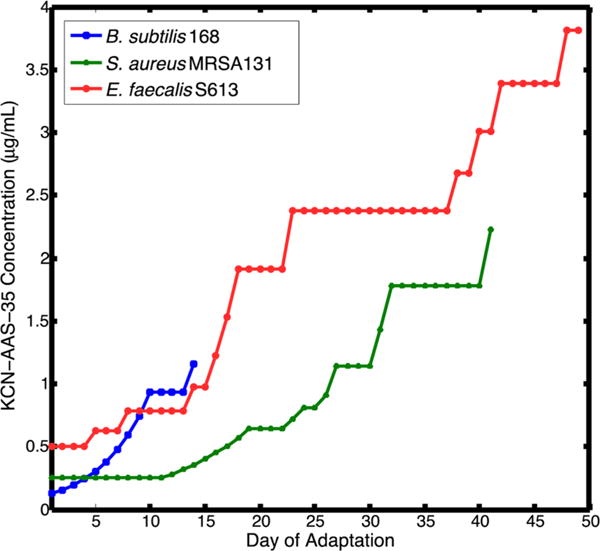
Adaptation timeline for B. subtilis 168, S. aureus MRSA131, and E. faecalis S613 to resist KCN-AAS-35. 24 replicate populations of each strain were initially grown at subinhibitory concentrations of drug. A portion of each population was transferred daily to the same concentration and an elevated concentration. Once all of the populations could grow to a high cell density at the elevated drug concentration, the populations were transferred from that concentration to a higher concentration (see Methods). Transfers were continued until all of the populations could grow well at a concentration above their starting MIC. Therefore, if some of the populations were struggling to grow at the elevated drug concentration, all of the populations were maintained at a lower concentration until the slower adapting populations could also grow well at the higher concentration. The clinical strains, S. aureus MRSA131 and E. faecalis S613, struggled to adapt to KCN-AAS-35 and took 40 or more days to reach growth at their respective final concentrations. Additionally, many of the populations of S. aureus MRSA131 and E. faecalis S613 died during adaption, with only eight and five populations remaining viable by the end of adaptation for S613 and MRSA131, respectively. The lab strain, B. subtilis 168, achieved growth at an elevated concentration after 14 days.
While increased resistance was eventually achieved, both the E. faecalis S613 and S. aureus MRSA131 strains adapted slowly to KCN-AAS-35, taking 40 or more days to reach growth at their respective final concentrations (Figure 3). In previous studies with established antibiotics, such as daptomycin and tigecycline, populations adapted much more rapidly, typically within 15–25 days.18–21 Additionally, during the course of adaptation many of the E. faecalis S613 and S. aureus MRSA131 populations slowly went to extinction. Again, this was unusual and suggested that it was exceptionally difficult for the cells to accumulate mutations that would allow them to sustain viability even at sub-MIC concentrations of KCN-AAS-35. Initially, all 24 S. aureus MRSA131 populations grew well at 1.8 μg/mL on day 32 of adaptation. However, in the following days, many of the populations began growing poorly and failed to reach a high cell density, despite remaining at the same drug concentration (Figure 3). Eventually, the struggling MRSA131 populations failed to achieve any growth, leaving only eight of 24 populations still viable by day 37 of adaptation. Similarly, all 24 E. faecalis S613 populations began growing well at 2.4 μg/mL on day 23 of adaptation. However, like the MRSA131 populations, in the following days, many of the S613 populations began to falter and were unable to reach a high cell density, despite remaining at the same drug concentration (Figure 3). Eventually, the struggling S613 populations went to extinction, leaving only five of 24 populations viable by day 35 of adaptation. In total, two-thirds of the MRSA131 populations and more than three-fourths of the S613 populations were lost before reaching the final drug concentrations, demonstrating that adaptation to KCN-AAS-35 was difficult for these strains to achieve. In contrast, the B. subtilis 168 populations adapted relatively quickly and without the loss of any populations, taking only 14 days to reach growth at a sufficiently elevated drug concentration (Figure 3). At the completion of adaptation, clones isolated from all three species demonstrated a 2- to 8-fold increase in KCN-AAS-35 MIC values compared to their respective ancestral starting strains, indicating that the clones had indeed adapted to the antibiotic (Table 1). The comparatively different ability of specific species to become resistant highlights the extent to which a wide panel of strains can survey substantially different evolutionary pathways to resistance. This observation will be reinforced further in the subsequent sections when the genetic bases for resistance is described.
Whole Genome Sequencing of Individual Clones Isolated from the End Point of Experimental Evolution Show a Wide Range of Genetic Changes
The mutations associated with adaptation were identified by whole genome sequencing of clones isolated from the end of the adaptation experiments. A total of 18 resistant clones were sequenced, with six clones that were randomly selected from each of the three different species. Each clone had a unique genotype and carried at least two adaptive mutations (Table S1). As shown in Tables 2 and S1, whole genome sequencing of 18 individual isolates exhibited a broad range of genetic changes. Typically, end-point strains had from 2 to 13 mutations per isolate. One of the resistant MRSA131 strains had acquired 50 mutations, including a frame shift at position Y82 in the DNA mismatch repair gene mutS, suggesting that the strain had likely become a hypermutator (Table S1). Hypermutators are not uncommon in both laboratory and clinical settings and allow lineages to adapt very rapidly to selection conditions. While hypermutators adapt quickly, they also accumulate mutations that “hitchhike” with those responsible for increased resistance, which can burden the organisms with additional mutations whose overall effect will be largely deleterious to cellular fitness.20,24 What was striking about the 124 total mutations isolated across the 18 adapted strains, 16 adapted populations, and three species was that there were very different biochemical processes undergoing change during adaptation. Mutations were identified in genes that affected a variety of cellular processes, including membrane efflux, membrane homeostasis, metabolism, cell division, and peptidoglycan layer synthesis (Table S1). The diversity of changes reflects the diversity of the strains themselves and highlights how a broad survey of adaptive strategies to drug selection can be achieved readily by starting with at least three strains. Importantly, the breadth of the changes observed within each strain, however, also makes it difficult to confidently ascribe the biochemical mechanism of action for KCN-AAS-35 based on the results from any one strain.
Table 2.
Resistance Mutations to KCN-AAS-35 that Affect Components of the Electron Transport Chain (ETC)
| species | gene | adapted clone | mutation |
|---|---|---|---|
| B. subtilis 168 | cymR | A10-4 | deletion of RBS and start codon |
| A12-3 | S27L | ||
| B11-3 | E43D | ||
| C10-1 | S38F | ||
| cysK | D11-1 | +C (frame shift at H219) | |
| G11-2 | R47C | ||
| S. aureus MRSA131 | cymR | E10-1 | complete deletion of gene |
| E10-3 | complete deletion of gene | ||
| F10-4 | G64R | ||
| B11-3 | +G (frame shift at Q23) | ||
| C11-2 | +A (frame shift at E21) | ||
| E11-1 | 9-bp upstream of gene (A → G) | ||
| E. faecalis S613 | ATP Synthase | A2-1 | β-chain: V305D |
| H2-4 | F0 subunit c: G15C | ||
| A3-1 | β-chain: V305D | ||
| C3-4 | F0 subunit a: G188* | ||
| H3-2 | α-chain: F395C | ||
| H3-3 | α-chain: F395C |
Genomic Analysis of KCN-AAS-35 Adapted S. aureus MRSA131 Strains
We identified a total of 79 different mutations across the six adapted S. aureus MRSA131 clones (Table S1). Although many of these mutations occurred in distinct and diverse locations within the genome, we noted that each clone had acquired a mutation within cymR, a master regulator and repressor of cysteine synthesis and sulfur assimilation (Table 2). Importantly, cymR was the only gene that was mutated in all six of the adapted S. aureus MRSA131 clones (Tables 2 and S1). This suggests that mutation of cymR was particularly important to acquiring KCN-AAS-35 resistance, since adaptive alleles at this loci emerged independently and reproducibly across the five replicate populations. Some of the adaptive cymR alleles suggest loss-of-function, such as a complete or partial deletion of cymR in S. aureus E10-1 and S. aureus E10-3 and frame-shift mutations in S. aureus B11-3 and S. aureus C11-2 (Table 2). Therefore, we reasoned that the mutations in cymR were reducing the activity of CymR and subsequently leading to the derepression of cysteine metabolism.
Genomic Analysis of KCN-AAS-35 Adapted B. subtilis 168 Strains
Among the six adapted B. subtilis 168 strains, we identified a total of 14 alleles (Table S1). Looking for conserved adaptive alleles across the resistant B. subtilis strains, we noted that four had acquired mutations within cymR (Table 2). B. subtilis A10-4 had a 32-bp deletion that eliminated the ribosome binding site and start codon of cymR (Table 2). This deletion suggests loss-of-function, similar to the cymR mutations observed in the adapted S. aureus MRSA131. Even et al. showed that complete deletion of cymR in B. subtilis leads to an increase in the expression of genes involved in cysteine intake, cysteine synthesis, sulfur incorporation, and a shift toward anaerobic growth.25 Therefore, it is likely that the cymR mutations in our resistant strains are leading to an increase in cellular cysteine metabolism in response to KCN-AAS-35. Interestingly, two of the resistant B. subtilis strains (D11-1 and G11-2) maintained the intact ancestral cymR sequence but instead had mutations in cysK (Table 2). The cysK gene encodes cysteine synthetase, which catalyzes the last step of cysteine production by condensing sulfide with O-acetyl-L-serine. In B. subtilis, CysK also acts as a global regulator in conjunction with CymR to repress cysteine metabolism when cysteine is in excess.26,27 We speculate that the cysK mutations observed in B. subtilis D11-1 and G11-2 impact the regulatory functions and not the catalytic functions of cysK. Since derepression of cysteine metabolism was observed in the other resistant B. subtilis 168 and MRSA131 strains, it is likely that the same evolutionary path was utilized by B. subtilis D11-1 and G11-2. Consistent with this hypothesis, B. subtilis D11-1 and G11-2 were able to grow in Spizizen salts minimal media lacking cysteine, demonstrating that the cells were still capable of synthesizing cysteine despite harboring mutations in cysteine synthetase (see Figure S2).
Genomic Analysis of KCN-AAS-35 Adapted E. faecalis S613 Strains
We identified a total of 30 different mutations across the six adapted E. faecalis S613 clones (Table S1). In contrast to the results obtained for B. subtilis 168 and S. aureus MRSA131, the resistant E. faecalis S613 strains did not have mutations affecting cysteine synthesis. However, all of the E. faecalis strains did have mutations in various subunits of ATP synthase (Table 2). It is striking that each isolate had at least one mutation within the ATP synthase holoenzyme. Specifically, two of the resistant E. faecalis strains acquired mutations in the β-chain (A2-1 and A3-1), two acquired mutations in the α-chain (H3-2 and H3-3), and two acquired mutations in different subunits of the F0 sector (C3-4 and H2-4) (Table 2). While the role of ATP synthase in E. faecalis is not entirely clear, the species has a functional electron transport chain in the presence of heme, indicating it is capable of generating a proton gradient.28,29 Since cellular fitness under aerobic conditions is tightly linked to the function of ATP synthase in most species, it seemed unlikely that an improvement in ATP synthase function could be achieved across so many subunits by single nucleotide changes when earlier studies of adaptation in rich aerobic conditions have not observed changes in ATP synthase.21 Therefore, we reasoned that it was more likely that these mutations reduced the synthesis of ATP and transport of protons into the resistant E. faecalis cells. Consistent with this hypothesis, E. faecalis C3-4 acquired a stop codon at position 188 of the F0 a subunit, eliminating the last 52 amino acids of this subunit (Table 2). Vik et al. showed that E. coli with similar truncations at the carboxy-terminal of the a subunit had greatly reduced proton permeability, indicating that the proton translocation activity of ATP synthase was reduced in these cells.30
Successful Adaptive Strategies Identified from All Three Strains Suggests KCN-AAS-35 Acts by Inhibiting Cellular Respiration
Having evaluated the adaptive alleles across the different populations and species, next we wanted to use this information to determine the likely mechanism of action of KCN-AAS-35. Cysteine plays many diverse and essential roles in the cell as an important amino acid for protein synthesis and as a substrate for the formation of iron–sulfur clusters and essential cofactors and coenzymes. Therefore, it would have been challenging to determine why KCN-AAS-35 led to the selection of mutations in cysteine regulation in the adapted B. subtilis 168 and S. aureus MRSA131 strains without having knowledge of the adaptive mutations acquired by E. faecalis S613. However, ATP synthase was mutated in all of the adapted E. faecalis S613 strains, which strongly suggested that KCN-AAS-35 was impacting cellular respiration. Cysteine metabolism is also important to cellular respiration, as the biogenesis of the iron–sulfur clusters for the electron transfer chain (ETC) requires cysteine.31,32 By derepressing cysteine metabolism, the B. subtilis 168 and S. aureus MRSA131 cells were likely increasing the formation of the ETC complexes. Therefore, we hypothesized that KCN-AAS-35 could be acting as an inhibitor of cellular respiration. It is important to note that if mutations from only one species were evaluated we may have reached the wrong conclusion. For example, if only the S. aureus MRSA131 mutations were considered, we may have incorrectly concluded that KCN-AAS-35 specifically targets CymR, since the cymR gene was mutated in each of the adapted S. aureus strains. Therefore, it would have been difficult to generate our hypothesis using the results from any one species, as the context of the mutations between the different species was necessary.
Oxygen Consumption Assay Demonstrates Inhibition of Bacterial Respiration
To test our hypothesis that KCN-AAS-35 could act as an inhibitor of cellular respiration, we tested the effect of the drug on the rate of oxygen consumption in B. subtilis, S. aureus, and E. faecalis. Since KCN-AAS-35 is a bacteriostatic agent (Figure S1), a decrease in oxygen consumption would indicate inhibition of respiration and not cell death. Respiring cells were exposed to 62.5 μg/mL KCN-AAS-35, and the change in rate of oxygen consumption was measured. Although the MIC of the drug is 0.5 or 2 μg/mL for the three strains studied, we chose to use a higher concentration (62.5 μg/mL) for this assay. This is because the density of cells used for oxygen consumption measurements is approximately 75 times higher than the cell density used in MIC tests. It has been demonstrated previously that the efficacy of some antibiotics decreases with an increase in bacterial cell density,33 referred to as the inoculum effect.34 The higher drug concentration adjusts for any decrease in efficacy of this drug that may be associated with the high density of the bacterial culture used for measuring oxygen consumption.
Figure 4 shows the oxygen consumption curves for the different bacterial strains. Whole cells were allowed to respire either by themselves or in the presence of one of the compounds added when 60% oxygen remained in the oxygen electrode chamber. Oxygen consumption was measured for KCN-AAS-35; potassium cyanide, a known respiratory inhibitor (positive control); tetracycline, a bacteriostatic drug that is not known to target cellular respiration; DMSO, the vehicle used to solubilize KCN-AAS-35. The immediate decline in the rate of oxygen consumption in B. subtilis 168 and S. aureus MRSA131 by potassium cyanide showed that bacterial respiration was indeed being measured in this assay. The uninhibited respiratory curve of E. faecalis in the presence of cyanide was expected as E. faecalis S613 has a cytochrome bd oxidoreductase that is known to be resistant to inhibition by cyanide.29,35 Lack of respiratory inhibition by a ribosomal inhibitor, tetracycline, suggested that agents affecting cellular mechanisms other than respiration did not affect the ability of the cells to consume oxygen. This, in turn, implied that the decrease in respiration rate seen in the presence of KCN-AAS-35 was not caused by a nonspecific effect of the drug acting on another cellular component. Upon addition of 62.5 μg/mL KCN-AAS-35, the rates of oxygen consumption in E. faecalis S613 and S. aureus MRSA131 decreased 2.2 ± 0.11-fold and 2.3 ± 0.05-fold, respectively, while a 10.9 ± 0.85-fold decrease in rate of oxygen consumption was observed in B. subtilis (normalized to change in respiration rate upon addition of DMSO). The reason the effect of the drug on respiration in B. subtilis is more pronounced than its effect on S. aureus or E. faecalis is not fully understood, but it does corelate with the lower MIC of KCN-AAS-35 for B. subtilis (0.5 μg/mL) compared to the other strains (2 μg/mL) (Table 1). We speculate that the peptidoglycan layer surrounding the cell membrane of B. subtilis is more permeable to KCN-AAS-35 allowing the drug better access to the respiratory machinery of the cell than that of S. aureus or E. faecalis.
Figure 4.
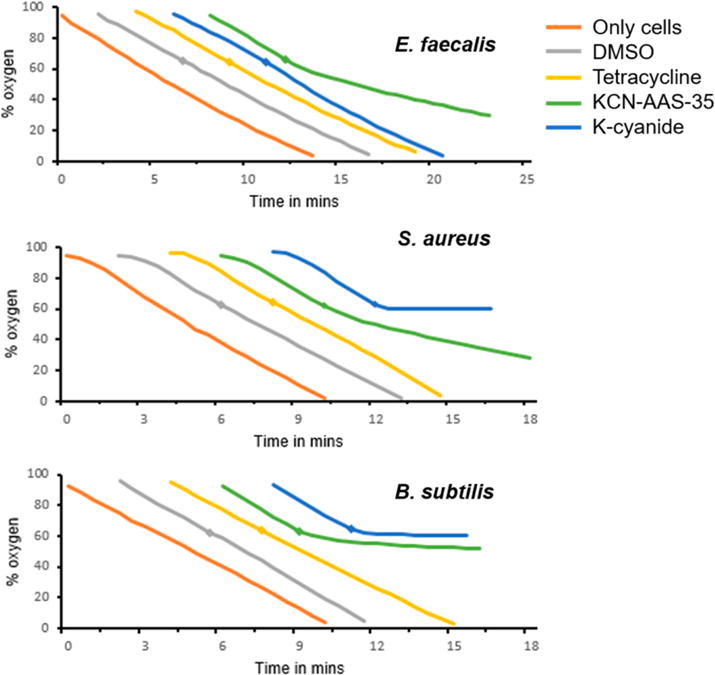
Oxygen consumption assays in the three bacterial strains show that KCN-AAS-35 inhibits cellular respiration. Aerobic respiration in E. faecalis S613, S. aureus MRSA 131, and B. subtilis 168 was measured in an oxygen electrode chamber by recording the percentage of extracellular oxygen remaining in the chamber as a function of time after addition of cells. The intrinsic respiration rate of the cells was recorded in the absence of any compounds (orange trace). Oxygen consumption was then measured by the addition of 12.5 μL of DMSO (gray trace), 105 μg/mL tetracycline (yellow trace), 10 mM potassium cyanide (blue trace), or 62.5 μg/mL KCN-AAS-35 (green trace), with the addition being made when 60% oxygen remained in the chamber (represented by diamonds on the respective traces). All assays were done in triplicate. The addition of DMSO (vehicle) or tetracycline did not affect bacterial respiration. The addition of cyanide, a known respiratory inhibitor, aborted respiration in S. aureus and B. subtilis but not in E. faecalis, which uses a cyanide insensitive cytochrome bd terminal oxidoreductase. The addition of KCN-AAS-35 caused a decrease in the rate of respiration in all the bacterial strains, with the effect being more pronounced in B. subtilis.
Although KCN-AAS-35 is known to be a Gram-positive specific antibacterial, we observed that it could inhibit NADH dependent respiration of inverted membrane vesicles obtained from a Gram-negative bacterium, Escherichia coli (data not shown). We did not observe this inhibition in whole cells of E. coli respiring in the presence of the drug (data not shown). The parent compound of KCN-AAS-35 is known to be effective against E. coli with reduced membrane permeability,22 which suggests that the outer membrane of Gram-negative bacteria protects them from this parent compound and its derivative. This data further supports our hypothesis that KCN-AAS-35 targets bacterial respiration.
Functional Mitochondrial Toxicity Testing Demonstrates Toxicity at Concentrations Similar to Effective MIC
To confirm our hypothesis that KCN-AAS-35 could affect cellular respiration, the Cyprotex Company tested how a range of concentrations (0.4–400 μg/mL) of KCN-AAS-35 could produce changes in the respiration efficiency of liver mitochondria. The Cyprotex team assayed for changes in the oxygen consumption rate, reserve capacity, and extracellular acidification rate of the eukaryotic mitochondria (Figure 5). The reserve capacity, or ability of mitochondria to respond to changes in energy, can be determined by detecting fluctuations in the oxygen consumption rate when mitochondria are exposed to the uncoupler FCCP. Upon addition of FCCP, cells treated with relatively low concentrations of KCN-AAS-35 (0.4 and 1.27 μg/mL) produced over a 100% increase in oxygen consumption rate. However, the oxygen consumption rate did not increase in the presence of FCCP when the cells were treated with higher concentrations of KCN-AAS-35 (4.0, 12.7, and 40 μg/mL) (Figure 5). This indicates mitochondrial dysfunction in the presence of 4.0–40 μg/mL KCN-AAS-35, as the reserve capacity decreases at those concentrations. Additionally, a dose dependent increase in the extracellular acidification rate was detected, indicating an increase in cellular glycolysis at higher concentrations of KCN-AAS-35 (Figure 5). Combined, these data show that, at 4.0–40 μg/mL KCN-AAS-35, the compound inhibits oxidative phosphorylation. Specifically, the bioenergetic profile of KCN-AAS-35 suggests that the compound is an ETC inhibitor, since KCN-AAS-35 decreased the oxygen consumption rate, decreased the reserve capacity, and increased the extracellular acidification rate in a dose dependent manner.36 These results confirmed that KCN-AAS-35 is capable of inhibiting cellular respiration in eukaryotes. While there are notable differences between Gram-positive and mitochondrial ETCs, many inhibitors, such as antimycin A, rotenone, and sodium cyanide, are capable of inhibiting both types of ETCs.37–40 Thus, this data suggests that KCN-AAS-35 kills bacteria and mammalian cells through the same mechanism. While an interesting observation, it does suggest that KCN-AAS-35 may have limited therapeutic use.
Figure 5.
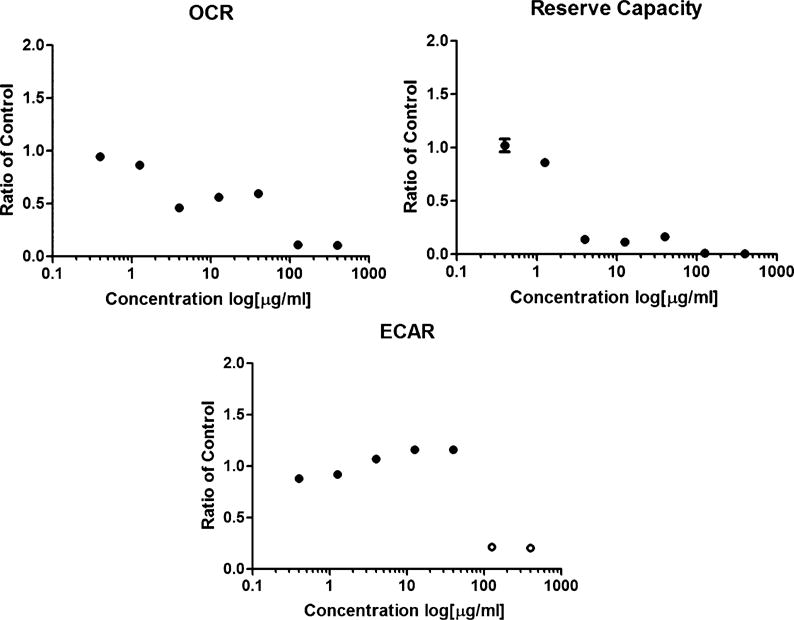
Functional mitochondrial toxicity assay (XFe96 Seahorse) reveals that KCN-AAS-35 likely acts as an ETC inhibitor. The Cyprotex Company assayed for changes in the oxygen consumption rate (OCR), reserve capacity, and extracellular acidification rate (ECAR) of HepG2 liver mitochondria treated with vehicle and 0.4–400 μg/mL KCN-AAS-35 using solid-state sensors to simultaneously measure effects on oxidative phosphorylation (OXPHOS) and glycolysis as described in Eakins et al.36 The data is represented as the average ratio to the vehicle (DMSO) with the error bars showing the standard deviation between three replicates. Open circles were excluded due to data plateau. Dose dependent decreases in OCR and reserve capacity were observed, indicating that KCN-AAS-35 inhibits mitochondrial respiration and causes mitochondrial dysfunction. Additionally, a dose dependent increase in ECAR was observed, indicating the release of more protons into the media due to an increase in glycolysis. Combined, these results are consistent with known inhibitors of the ETC, such as rotenone, suggesting that KCN-AAS-35 is likely an inhibitor of the ETC.
Results from Previous Experimental Evolution Studies Suggest That Predictions for Biochemical Mechanisms of Action Are More Robust When Genomic Data from Multiple Genera Are Included
Previous studies have used experimental evolution to identify the adaptive trajectories leading to resistance to antibiotics with characterized mechanisms of action. Interestingly, in many of these cases, if the antibiotic mechanism of action had not been known, it could have been inferred from the resistance data. For example, studies evaluating tigecycline resistance in different bacterial species, including several studies from our lab, have identified mutations on a loop of the ribosomal S10 protein and mutations leading to increased expression of the ribosomal protection gene tetM (Table 3).18–20,41–43 Tigecycline is a tetracycline derivative that inhibits translation by binding to the bacterial 16S rRNA and blocking the entry of tRNA into the A-site.44–46 The identified mutations in the ribosomal S10 protein were in close proximity to the 16S rRNA that composes the tigecycline binding pocket. Therefore, if the mechanism of action of tigecycline had been unknown, these S10 mutations would have strongly suggested that tigecycline was acting on the ribosome. Likewise, TetM is a protein with homology to elongation factor-G and binds to the ribosome.47 Thus, the identified resistance mutations that increased tetM expression would have also suggested that tigecycline was acting on the ribosome.19 Importantly, studying resistance in multiple different species would have made the identification of the mechanism of action of tigecycline more likely. This is because some species, such as Acinetobacter baumannii and E. coli, frequently become resistant to tigecycline by overexpressing efflux pumps, which indicates that the antibiotic must enter the cell but does not indicate the specific pathway or target the antibiotic is acting on (Table 3).18,20,48 Conversely, some species, such as E. faecalis and E. faecium, more frequently become resistant to tigecycline by acquiring the mutations in the ribosomal S10 protein or mutations that lead to increased tetM expression, which in turn would have provided more useful insights into the mechanism of action of tigecycline (Table 3).18,19,49 Therefore, using many replicate populations and a panel of different bacterial species increases the likelihood of gaining useful insights into the mechanism of an antibiotic.
Table 3.
Examples of Tigecycline Resistance Mutations Identified in Different Species from Experimental Evolution
| species | tigecycline resistance mutation | references |
|---|---|---|
| Enterococcus faecalis | S10: R53QΔ54-57ATHK TetM: increased tetM copy number; regulatory mutations leading to increased tetM expression |
Beabout et al.19 |
| Enterococcus faecium | S10: R53QΔ54-57ATHK; A54E-T55A; H56R-K57N; K57E; K57R; Y58S; Y58D; D60Y TetM: increased tetM copy number efflux: increased tetL copy number |
Beabout et al.18 Cattoir et al.41 Fiedler et al.43 |
| Staphylococcus aureus | S10: K57M, K57E, K57M-Y58F, K57Q-Y58F, Y58M, Y58S, D60Y TetM: regulatory mutations leading to increased tetM expression efflux: increased expression of MepA pump |
Beabout et al.18 McAleese et al.49 |
| Acinetobacter baumannii | S10: V57L, V57I efflux: increased expression of AdeABC pump |
Hammerstrom et al.20 Beabout et al.18 |
| Escherichia coli | S10: V57D, V57I efflux: increased expression of AcrAB-TolC pump |
Beabout et al.18 Keeney et al.48 |
Another example in the literature where the mechanism of action of an antibiotic could have been inferred from experimental evolution studies across multiple species of bacteria includes the lipopeptide daptomycin, which disrupts cell membrane integrity in a calcium and phosphatidylglycerol dependent manner.50 E. faecalis adapted to daptomycin resistance reproducibly acquires mutations in LiaFSR, a three-component signaling system and membrane-stress response pathway (Table 4).21,51 Similarly, B. subtilis strains frequently become daptomycin-resistant through mutations in the VraRS system, which is a signaling pathway orthologous to the E. faecalis LiaFSR pathway (Table 4).52 Additionally, daptomycin-resistant E. faecalis often acquire mutations in cardiolipin synthase (cls), while daptomycin-resistant B. subtilis often acquire mutations in pgsA, both of which result in reduced levels of phosphatidylglycerol on the cell membrane (Table 4).53,54 Conversely, in experimental evolution studies of S. aureus to daptomycin, mutations have been observed in the gene mprF that increase the levels of lysyl-phosphatidylglycerol on the cell membrane (Table 4).55 Therefore, while mutations identified in the different species frequently affected different genes, they all suggested that daptomycin was acting upon the membrane. Additionally, the different alleles reveal different aspects of the mechanism through which daptomycin acts, such as the cls and pgsA alleles in E. faecalis and B. subtilis, which suggest that phosphatidylglycerol levels are important to the activity of daptomycin.
Table 4.
Examples of Daptomycin Resistance Mutations Identified in Different Species from Experimental Evolution
| species | daptomycin resistance mutation | references |
|---|---|---|
| Enterococcus faecalis | signaling: mutations upregulating LiaFSR signaling | Miller et al.21 Arias et al.51 |
| membrane lipid remodeling: mutations in cls | Davlieva et al.54 | |
| Staphylococcus aureus | membrane lipid remodeling: mutations in mprF | Friedman et al.55 |
| Bacillus subtilis | signaling: mutations upregulating VraRS signaling | Hachmann et al.52 |
| membrane lipid remodeling: mutations in pgsA | Hachmann et al.53 |
SUMMARY
In this study, we have demonstrated that experimental evolution of a panel of Gram-positive bacterial species when complemented by comparative whole genome sequencing (WGS) can be used to efficiently identify the biochemical mechanism of action for a novel antibiotic (Figure 1). We adapted three strains of Gram-positive bacteria, including two clinical pathogens, to resistance against a novel and biologically uncharacterized pyrrolizidinone antibiotic (KCN-AAS-35) (Figures 2 and 3). Comparative WGS of the resistant clones to the susceptible ancestral strains revealed adaptive mutations within genes leading to the derepression of cysteine synthesis in both B. subtilis 168 and S. aureus MRSA131 and a reduction in the activity of ATP synthase in E. faecalis S613 (Table 2). Combining the results across the different species, we hypothesized that KCN-AAS-35 affected cellular respiration, as increased cysteine metabolism could subsequently lead to an increase in iron–sulfur cluster and ETC complex formation and reduced activity in ATP synthase could mitigate the effects of inhibiting respiration by helping to preserve the electrochemical gradient (Table 2). Furthermore, the inhibition led to the down-regulation of ATP synthesis within the ETC of E. faecalis S613 so that energy could be produced by other means (Table 2). Therefore, we were able to hypothesize that KCN-AAS-35 likely inhibits the ETC of these organisms. Oxygen consumption assays confirmed our hypothesis that the drug does, indeed, inhibit bacterial respiration (Figure 4). Subsequent Cyprotex toxicity testing also showed that KCN-AAS-35 is capable of inhibiting the mitochondrial ETC (Figure 5).
Our approach has not only identified the likely mechanism of action of the novel antibiotic KCN-AAS-35 but also has provided a clear understanding of how resistance will likely emerge to a previously uncharacterized antibiotic. These methods provide an advantage to antibiotic discovery because they are translatable to a broad range of laboratory settings. In principle, any culturable species of bacteria can be potentially included in the microbe panel. Having a diverse panel of bacteria reveals a range of adaptive strategies, and by performing experimental evolution on replicate populations under selection conditions that favor a polymorphic population (well below the current MIC of the evolving population), more evolutionary trajectories are identified among and within populations further strengthening hypothesis development. For example, while the resistant E. faecalis S613 strains all had mutations in ATP synthase, the specific mutations identified across the different E. faecalis populations were distinct (Table 2).
There are limitations to using experimental evolution to characterize the mechanisms of action of antibiotics. Depending on the types of mutations that emerge, it could be difficult to determine the mechanism. For example, if a strain evolves into a hypermutator and acquires hundreds of mutations, it would be difficult to identify which mutations are critical for resistance and which are hitchhikers. Alternatively, mutations may not always occur in target genes and could therefore be difficult to interpret. While not completely removing these potential pitfalls, using a panel of diverse strains increases the likelihood of identifying informative alleles by exploring more evolutionary outcomes. As the quality of genomic annotation increases, the power of experimental evolution to inform hypotheses on the action of lead compounds will also improve. In addition, as the cost of WGS continues to decrease and the accessibility of sequencing technology increases with each year, experimental evolution should be among the earliest studies performed on a novel drug candidate. Currently, the number of antibiotics in development is decreasing, while the number of resistant bacteria is increasing.56 Therefore, it is necessary to develop innovative, low-cost methods to characterize novel small-molecule antibiotics to help address this discrepancy. The lack of new antimicrobials currently in development suggests that new and scalable methods are needed to identify compounds of interest that challenge the resistance capabilities of pathogens and that the methods currently being used should be expanded.57 Importantly, these methods will allow for more participation in antibacterial discovery during a time when the development of novel antimicrobial therapeutics is essential.
METHODS
Strains, Growth Conditions, and Minimal Inhibitory Concentration (MIC) Testing
Freezer stocks of B. subtilis 168 and S. aureus MRSA131 were streaked onto LB agar plates and incubated at 37 °C overnight. The next day, 10 mL of LB media was inoculated with a single colony from each plate and incubated at 37 °C while shaking. Using a previously described protocol from Nicolaou et al.,58 B. subtilis 168 and MRSA131 were exposed to increasing concentrations of the tested antibiotic (0.125–64.0 μg/mL; prepared in the Nicolaou laboratories) in LB broth and incubated overnight at 37 °C. The lowest concentration at which the antibiotic appeared to be inhibiting growth was determined to be the MIC for the drug. This same procedure was used for E. faecalis S613 except a mix of 80% Lysogeny and 20% Brain Heart Infusion (LBHI) agar and broth media was used and MICs were determined by reading the OD600 on the Synergy 2 (BioTek) Gen5 plate reader. To test the cysteine dependency of the adapted B. subtilis 168 strains, they were cultured overnight in Spizizen minimal media (17 mM K2HPO4, 8 mM KH2PO4, 3 mM (NH4)2SO4, 1.2 mM sodium citrate, 0.16 mM MgSO4, 1 μg/mL tryptophan, and 0.4% glucose at pH 7.0) at 37 °C on a Synergy 2 (BioTek) Gen5 plate reader.
Experimental Evolution by Stepwise Increases in Drug Concentrations Well below the MIC
In this procedure, stepwise increases in subinhibitory concentrations, or concentrations below the MIC of KCN-AAS-35, were used to gradually adapt each strain to resistance. B. subtilis 168 and MRSA131 were plated on LB agar and incubated overnight at 37 °C. Twenty-four individual colonies were inoculated in 1 mL of LB media in 96-well plates and grown overnight at 37 °C on a shaker to initiate 24 populations. A portion of the 24 populations (10 μL) was then transferred to 990 μL of LB media containing one-fourth of the initial MIC to KCN-AAS-35. MRSA131 was transferred to one-eighth of MIC to KCN-AAS-35 as the strain was unable to grow robustly even at one-fourth of the MIC for KCN-AAS-35. The concentration of KCN-AAS-35 was then increased 1.25×, and the strains were grown overnight in a 37 °C shaker. These transfers (10 μL of overnight culture to 990 μL of media with antibiotic) were repeated daily until the cells grew to saturating density overnight at the new drug concentration. On the day when all of the populations of cells grew to full density overnight at the current drug concentration, they were then transferred to a higher concentration of KCN-AAS-35 (1.25 times the prior concentration of KCN-AAS-35). If only some of the populations for a particular strain were growing well at an elevated drug concentration, those populations were held at the lower drug concentration until the slower adapting populations could also achieve good growth at the elevated concentration. If the cells in any of the populations continued to struggle to reach a high cell density at 1.25 times the current drug concentration for more than 3 days, a lower increase, such as 1.125 times, would be used. This same procedure was repeated for E. faecalis S613 except LBHI plates and broth media were used and the cells were grown overnight in a 37 °C incubator instead of a shaker.
Whole Genome Sequencing
At the end of each adaptation experiment, individual colonies were isolated on nonselective LB agar (for B. subtilis 168 and MRSA131) and LBHI (for E. faecalis S613) agar plates. The individual colonies were then grown overnight in broth media. Genomic DNA was isolated from 10 mL of the overnight culture using the UltraClean Microbial DNA Isolation Kit (MoBio) with an extra lysis step to improve yields for Gram-positive cells. The additional lysis step consisted of incubating the cells at 37 °C for 1 h in 300 μL of MicroBead Solution with 78 U/mL mutanolysin (Sigma) and 7.8 μg/mL lysozyme (Sigma). Whole genome sequencing was then performed (GeneWiz) using a Nextera XT DNA Library Preparation Kit (Illumina, San Diego, CA) and an Illumina MiSeq Sequencer. Genomic data analysis was completed using Breseq-0.27.0, a computational pipeline developed by the Barrick lab (University of Texas, Austin, TX) to compare the genomes of the ancestral starting strains to that of the adapted resistant strains.59
Oxygen Consumption Assay
B. subtilis 168 and S. aureus MRSA131 were grown in LB, and E. faecalis S613 was grown in LBHI broth at 37 °C with shaking at 225 rpm until they reached exponential phase. Cells were then harvested and washed in the appropriate buffers: cold LB for B. subtilis, 0.1 M K-phosphate buffer (pH 7.0) for S. aureus, and 50 mM HEPES (pH 7.4) + 100 mM NaCl, 5 mM KCl, 1 mM MgCl2, 1 mM NaH2PO4, and 1 mM CaCl2 for E. faecalis. Cells were resuspended in their respective buffers at equal cell densities. Oxygen consumption was measured using a Clark type oxygen electrode (YSI model 5300 Biological Oxygen Monitor) at room temperature. 20, 3, and 1 mM glucose was added to B. subtilis, S. aureus, and E. faecalis cells, respectively, in the electrode chamber, and rate of respiration was measured as the percentage of oxygen consumed by the cells per unit time. Respiring cells were exposed to 62.5 μg/mL KCN-AAS-35, 12.5 μL of DMSO (solvent for KCN-AAS-35), 10 mM potassium cyanide, or 105 μg/mL tetracycline when 60% oxygen remained in the chamber.
Functional Mitochondrial Toxicity Assay (XFe96 Seahorse)
To assess the effect of KCN-AAS-35 on cellular respiration, functional mitochondrial toxicity testing was performed by the Cyprotex Company as previously described by Eakins et al.36 Briefly, the Cyprotex team assayed for changes in the oxygen consumption rate (OCR), reserve capacity, and extracellular acidification rate (ECAR) of HepG2 liver mitochondria treated with vehicle (0.5% DMSO) and 0.4–400 μg/mL KCN-AAS-35 using solid-state sensors to simultaneously measure effects on oxidative phosphorylation (OXPHOS) and glycolysis. The oxygen consumption rate (OCR) was determined by measuring the oxygen content in the extracellular media of HepG2 cells treated with KCN-AAS-35 compared to vehicle. By taking a ratio of the maximal achievable OCR, as determined by treating the cells with the uncoupler FCCP, and the basal OCR, the reserve capacity was determined. The ECAR was determined by measuring the pH value of the extracellular media of mitochondria treated with KCN-AAS-35 compared to vehicle.
Time Kill Assays
Using a protocol adapted from Diaz et al. and Pace et al.,60,61 70% glycerol stocks of MRSA131 were streaked onto LB plates and incubated at 37 °C overnight. The next day, 10 mL of LB media was inoculated with a single colony from each plate and incubated at 37 °C while shaking. On the following day, the overnight culture was diluted 100-fold in LB and incubated at 37 °C until the culture reached OD600 of around 0.2. Once this OD600 was reached, the culture was diluted 1:100 into media containing antibiotic at 5 times its MIC. Several dilutions of this mixture were plated onto LB plates and spread by sterile glass beads. The mixture was incubated at 37 °C. Dilutions were plated every 2 to 8 h. Colony forming units (CFUs) per milliliter were derived from the number of CFUs on each dilution plate at each time point.
Supplementary Material
Acknowledgments
This work was funded by grants from the National Institutes of Health (R01AI080714) and the Defense Threat Reduction Agency (HDTRA-1-10-1-0069) to Y.S. and by The Welch Foundation (Grant C-1819) to K.C.N. K.B. was funded under the National Institutes of Health fellowship (F31GM108402NIAID). We would like to thank an anonymous reviewer for their insightful and thoughtful comments, which helped us improve the manuscript. We acknowledge the Cyprotex mitochondrial toxicity testing team for completing the XFe96 Seahorse assays.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsinfecdis.7b00135.
Figure S1, killing curves for pyrrolizidinone natural product (CJ-16,264) and analog (KCN-AAS-35) against S. aureus MRSA131; Figure S2, growth of B. subtilis D11-1 and G11-2; Table S1, mutations identified in KCN-AAS-35 adapted clones (PDF)
ORCID
Kathryn Beabout: 0000-0003-2353-9685
Akshay A. Shah: 0000-0001-9451-0869
Stephan Rigol: 0000-0003-2470-3512
K. C. Nicolaou: 0000-0001-5332-2511
Yousif Shamoo: 0000-0001-9241-8962
Notes
The authors declare no competing financial interest.
References
- 1.Blair JMA, Webber MA, Baylay AJ, Ogbolu DO, Piddock LJV. Molecular mechanisms of antibiotic resistance. Nat Rev Microbiol. 2015;13(1):42–51. doi: 10.1038/nrmicro3380. [DOI] [PubMed] [Google Scholar]
- 2.CDC. Antibiotic resistance threats in the United States, 2013. CDC; Atlanta, GA: 2013. [Google Scholar]
- 3.The PEW Charitable Trusts. Antibiotics Currently in Clinical Development. 2016 http://www.pewtrusts.org/~/media/assets/2016/05/antibiotics-currently-in-clinical-development.pdf.
- 4.Rine J, Hansen W, Hardeman E, Davis RW. Targeted selection of recombinant clones through gene dosage effects. Proc Natl Acad Sci U S A. 1983;80(22):6750–6754. doi: 10.1073/pnas.80.22.6750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Banerjee A, Dubnau E, Quemard A, Balasubramanian V, Um KS, Wilson T, Collins D, de Lisle G, Jacobs WR. inhA, a gene encoding a target for isoniazid and ethionamide in Mycobacterium tuberculosis. Science. 1994;263(5144):227–230. doi: 10.1126/science.8284673. [DOI] [PubMed] [Google Scholar]
- 6.Belanger AE, Besra GS, Ford ME, Mikusová K, Belisle JT, Brennan PJ, Inamine JM. The embAB genes of Mycobacterium avium encode an arabinosyl transferase involved in cell wall arabinan biosynthesis that is the target for the antimycobacterial drug ethambutol. Proc Natl Acad Sci U S A. 1996;93(21):11919–11924. doi: 10.1073/pnas.93.21.11919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sugden CJ, Roper JR, Williams JG. Engineered gene over-expression as a method of drug target identification. Biochem Biophys Res Commun. 2005;334(2):555–560. doi: 10.1016/j.bbrc.2005.06.117. [DOI] [PubMed] [Google Scholar]
- 8.Shi W, Zhang X, Jiang X, Yuan H, Lee JS, Barry CE, Wang H, Zhang W, Zhang Y. Pyrazinamide Inhibits Trans-Translation in Mycobacterium tuberculosis. Science. 2011;333(6049):1630–1632. doi: 10.1126/science.1208813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Payne DJ, Gwynn MN, Holmes DJ, Rosenberg M. Genomic Approaches to Antibacterial Discovery. In: Woodford N, Johnson AP, editors. Genomics, Proteomics, and Clinical Bacteriology: Methods and Reviews. Humana Press; Totowa, NJ: 2004. pp. 231–259. [DOI] [PubMed] [Google Scholar]
- 10.Luesch H, Wu TYH, Ren P, Gray NS, Schultz PG, Supek F. A Genome-Wide Overexpression Screen in Yeast for Small-Molecule Target Identification. Chem Biol. 2005;12(1):55–63. doi: 10.1016/j.chembiol.2004.10.015. [DOI] [PubMed] [Google Scholar]
- 11.Butcher RA, Bhullar BS, Perlstein EO, Marsischky G, LaBaer J, Schreiber SL. Microarray-based method for monitoring yeast overexpression strains reveals small-molecule targets in TOR pathway. Nat Chem Biol. 2006;2(2):103–109. doi: 10.1038/nchembio762. [DOI] [PubMed] [Google Scholar]
- 12.Palmer AC, Kishony R. Opposing effects of target overexpression reveal drug mechanisms. Nat Commun. 2014;5:4296. doi: 10.1038/ncomms5296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bandow JE, Brötz H, Leichert LIO, Labischinski H, Hecker M. Proteomic Approach to Understanding Antibiotic Action. Antimicrob Agents Chemother. 2003;47(3):948–955. doi: 10.1128/AAC.47.3.948-955.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brazas MD, Hancock REW. Using microarray gene signatures to elucidate mechanisms of antibiotic action and resistance. Drug Discovery Today. 2005;10(18):1245–1252. doi: 10.1016/S1359-6446(05)03566-X. [DOI] [PubMed] [Google Scholar]
- 15.Le CF, Gudimella R, Razali R, Manikam R, Sekaran SD. Transcriptome analysis of Streptococcus pneumoniae treated with the designed antimicrobial peptides, DM3. Sci Rep. 2016;6:26828. doi: 10.1038/srep26828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Miesel L, Greene J, Black TA. Genetic strategies for antibacterial drug discovery. Nat Rev Genet. 2003;4(6):442–456. doi: 10.1038/nrg1086. [DOI] [PubMed] [Google Scholar]
- 17.Belanger AE, Lai A, Brackman MA, LeBlanc DJ. PCR-Based Ordered Genomic Libraries: a New Approach to Drug Target Identification for Streptococcus pneumoniae. Antimicrob Agents Chemother. 2002;46(8):2507–2512. doi: 10.1128/AAC.46.8.2507-2512.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Beabout K, Hammerstrom TG, Perez AM, Magalhães B, de F, Prater AG, Clements TP, Arias CA, Saxer G, Shamoo Y. The ribosomal S10 protein is a general target for decreased tigecycline susceptibility. Antimicrob Agents Chemother. 2015;59:5561. doi: 10.1128/AAC.00547-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Beabout K, Hammerstrom TG, Wang TT, Bhatty M, Christie PJ, Saxer G, Shamoo Y. Rampant Parasexuality Evolves in a Hospital Pathogen during Antibiotic Selection. Mol Biol Evol. 2015;32(10):2585–2597. doi: 10.1093/molbev/msv133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hammerstrom TG, Beabout K, Clements TP, Saxer G, Shamoo Y. Acinetobacter baumannii Repeatedly Evolves a Hypermutator Phenotype in Response to Tigecycline That Effectively Surveys Evolutionary Trajectories to Resistance. PLoS One. 2015;10(10):e0140489. doi: 10.1371/journal.pone.0140489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Miller C, Kong J, Tran TT, Arias CA, Saxer G, Shamoo Y. Adaptation of Enterococcus faecalis to Daptomycin Reveals an Ordered Progression to Resistance. Antimicrob Agents Chemother. 2013;57:5373. doi: 10.1128/AAC.01473-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sugie Y, Hirai H, Kachi-Tonai H, Kim YJ, Kojima Y, Shiomi Y, Sugiura A, Sugiura A, Suzuki Y, Yoshikawa N, et al. New pyrrolizidinone antibiotics CJ-16,264 and CJ-16,367. J Antibiot. 2001;54(11):917–925. doi: 10.7164/antibiotics.54.917. [DOI] [PubMed] [Google Scholar]
- 23.Nicolaou KC, Shah AA, Korman H, Khan T, Shi L, Worawalai W, Theodorakis EA. Total Synthesis and Structural Revision of Antibiotic CJ-16,264. Angew Chem, Int Ed. 2015;54(32):9203–9208. doi: 10.1002/anie.201504337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tanaka MM, Bergstrom CT, Levin BR. The evolution of mutator genes in bacterial populations: the roles of environmental change and timing. Genetics. 2003;164(3):843–854. doi: 10.1093/genetics/164.3.843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Even S, Burguière P, Auger S, Soutourina O, Danchin A, Martin-Verstraete I. Global Control of Cysteine Metabolism by CymR in Bacillus subtilis. J Bacteriol. 2006;188(6):2184–2197. doi: 10.1128/JB.188.6.2184-2197.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Albanesi D, Mansilla MC, Schujman GE, de Mendoza D. Bacillus subtilis Cysteine Synthetase Is a Global Regulator of the Expression of Genes Involved in Sulfur Assimilation. J Bacteriol. 2005;187(22):7631–7638. doi: 10.1128/JB.187.22.7631-7638.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tanous C, Soutourina O, Raynal B, Hullo MF, Mervelet P, Gilles AM, Noirot P, Danchin A, England P, Martin-Verstraete I. The CymR Regulator in Complex with the Enzyme CysK Controls Cysteine Metabolism in Bacillus subtilis. J Biol Chem. 2008;283(51):35551–35560. doi: 10.1074/jbc.M805951200. [DOI] [PubMed] [Google Scholar]
- 28.Brooijmans R, Smit B, Santos F, van Riel J, de Vos WM, Hugenholtz J. Heme and menaquinone induced electron transport in lactic acid bacteria. Microb Cell Fact. 2009;8(1):28. doi: 10.1186/1475-2859-8-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Winstedt L, Frankenberg L, Hederstedt L, von Wachenfeldt C. Enterococcus faecalis V583 Contains a Cytochrome bd-Type Respiratory Oxidase. J Bacteriol. 2000;182(13):3863–3866. doi: 10.1128/jb.182.13.3863-3866.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vik SB, Lee D, Marshall PA. Temperature-sensitive mutations at the carboxy terminus of the alpha subunit of the Escherichia coli F1F0 ATP synthase. J Bacteriol. 1991;173(14):4544–4548. doi: 10.1128/jb.173.14.4544-4548.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Beinert H, Holm RH, Münck E. Iron-Sulfur Clusters: Nature’s Modular, Multipurpose Structures. Science. 1997;277(5326):653–659. doi: 10.1126/science.277.5326.653. [DOI] [PubMed] [Google Scholar]
- 32.Bandyopadhyay S, Chandramouli K, Johnson MK. Iron-Sulphur Cluster Biosynthesis. Biochem Soc Trans. 2008;36(6):1112–1119. doi: 10.1042/BST0361112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Udekwu KI, Parrish N, Ankomah P, Baquero F, Levin BR. Functional relationship between bacterial cell density and the efficacy of antibiotics. J Antimicrob Chemother. 2009;63(4):745–757. doi: 10.1093/jac/dkn554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Brook I. Inoculum Effect. Clin Infect Dis. 1989;11(3):361–368. doi: 10.1093/clinids/11.3.361. [DOI] [PubMed] [Google Scholar]
- 35.Borisov VB, Gennis RB, Hemp J, Verkhovsky MI. The cytochrome bd respiratory oxygen reductases. Biochim Biophys Acta, Bioenerg. 2011;1807(11):1398–1413. doi: 10.1016/j.bbabio.2011.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Eakins J, Bauch C, Woodhouse H, Park B, Bevan S, Dilworth C, Walker P. A combined in vitro approach to improve the prediction of mitochondrial toxicants. Toxicol In Vitro. 2016;34:161–170. doi: 10.1016/j.tiv.2016.03.016. [DOI] [PubMed] [Google Scholar]
- 37.Erickson SK. The respiratory system of the aerobic, nitrogen-fixing, Gram-positive bacterium, Mycobacterium flavum 301. Biochim Biophys Acta, Bioenerg. 1971;245(1):63–69. doi: 10.1016/0005-2728(71)90008-9. [DOI] [PubMed] [Google Scholar]
- 38.Netrusov A, Pestova E. Oxidative phosphorylation in membrane vesicles of a gram-positive methylotroph. Arch Microbiol. 1991;156(2):115–118. [Google Scholar]
- 39.Garbisu C, Alkorta I, Llama MJ, Serra JL. Aerobic chromate reduction by Bacillus subtilis. Biodegradation. 1998;9(2):133–141. doi: 10.1023/a:1008358816529. [DOI] [PubMed] [Google Scholar]
- 40.Frerman FE, White DC. Membrane Lipid Changes During Formation of a Functional Electron Transport System in Staphylococcus aureus. J Bacteriol. 1967;94(6):1868–1874. doi: 10.1128/jb.94.6.1868-1874.1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cattoir V, Isnard C, Cosquer T, Odhiambo A, Bucquet F, Guérin F, Giard JC. Genomic Analysis of Reduced Susceptibility to Tigecycline in Enterococcus faecium. Antimicrob Agents Chemother. 2015;59(1):239–244. doi: 10.1128/AAC.04174-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Villa L, Feudi C, Fortini D, García-Fernández A, Carattoli A. Genomics of KPC-Producing Klebsiella pneumoniae Sequence Type 512 Clone Highlights the Role of RamR and Ribosomal S10 Protein Mutations in Conferring Tigecycline Resistance. Antimicrob Agents Chemother. 2014;58(3):1707–1712. doi: 10.1128/AAC.01803-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fiedler S, Bender JK, Klare I, Halbedel S, Grohmann E, Szewzyk U, Werner G. Tigecycline resistance in clinical isolates of Enterococcus faecium is mediated by an upregulation of plasmid-encoded tetracycline determinants tet(L) and tet(M) J Antimicrob Chemother. 2016;71(4):871–881. doi: 10.1093/jac/dkv420. [DOI] [PubMed] [Google Scholar]
- 44.Bauer G, Berens C, Projan SJ, Hillen W. Comparison of tetracycline and tigecycline binding to ribosomes mapped by dimethylsulphate and drug-directed Fe2+ cleavage of 16S rRNA. J Antimicrob Chemother. 2004;53(4):592–599. doi: 10.1093/jac/dkh125. [DOI] [PubMed] [Google Scholar]
- 45.Jenner L, Starosta AL, Terry DS, Mikolajka A, Filonava L, Yusupov M, Blanchard SC, Wilson DN, Yusupova G. Structural basis for potent inhibitory activity of the antibiotic tigecycline during protein synthesis. Proc Natl Acad Sci U S A. 2013;110(10):3812–3816. doi: 10.1073/pnas.1216691110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Olson MW, Ruzin A, Feyfant E, Rush TS, O’Connell J, Bradford PA. Functional, Biophysical, and Structural Bases for Antibacterial Activity of Tigecycline. Antimicrob Agents Chemother. 2006;50(6):2156–2166. doi: 10.1128/AAC.01499-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Connell SR, Tracz DM, Nierhaus KH, Taylor DE. Ribosomal Protection Proteins and Their Mechanism of Tetracycline Resistance. Antimicrob Agents Chemother. 2003;47(12):3675–3681. doi: 10.1128/AAC.47.12.3675-3681.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Keeney D, Ruzin A, McAleese F, Murphy E, Bradford PA. MarA-mediated overexpression of the AcrAB efflux pump results in decreased susceptibility to tigecycline in Escherichia coli. J Antimicrob Chemother. 2007;61(1):46–53. doi: 10.1093/jac/dkm397. [DOI] [PubMed] [Google Scholar]
- 49.McAleese F, Petersen P, Ruzin A, Dunman PM, Murphy E, Projan SJ, Bradford PA. A Novel MATE Family Efflux Pump Contributes to the Reduced Susceptibility of Laboratory-Derived Staphylococcus aureus Mutants to Tigecycline. Antimicrob Agents Chemother. 2005;49(5):1865–1871. doi: 10.1128/AAC.49.5.1865-1871.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Miller WR, Bayer AS, Arias CA. Mechanism of Action and Resistance to Daptomycin in Staphylococcus aureus and Enterococci. Cold Spring Harbor Perspect Med. 2016;6:a026997. doi: 10.1101/cshperspect.a026997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Arias CA, Panesso D, McGrath DM, Qin X, Mojica MF, Miller C, Diaz L, Tran TT, Rincon S, Barbu EM, et al. Genetic Basis for In Vivo Daptomycin Resistance in Enterococci. N Engl J Med. 2011;365(10):892–900. doi: 10.1056/NEJMoa1011138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hachmann AB, Angert ER, Helmann JD. Genetic Analysis of Factors Affecting Susceptibility of Bacillus subtilis to Daptomycin. Antimicrob Agents Chemother. 2009;53(4):1598–1609. doi: 10.1128/AAC.01329-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hachmann AB, Sevim E, Gaballa A, Popham DL, Antelmann H, Helmann JD. Reduction in Membrane Phosphatidylglycerol Content Leads to Daptomycin Resistance in Bacillus subtilis. Antimicrob Agents Chemother. 2011;55(9):4326–4337. doi: 10.1128/AAC.01819-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Davlieva M, Zhang W, Arias CA, Shamoo Y. Biochemical Characterization of Cardiolipin Synthase Mutations Associated with Daptomycin Resistance in Enterococci. Antimicrob Agents Chemother. 2013;57(1):289–296. doi: 10.1128/AAC.01743-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Friedman L, Alder JD, Silverman JA. Genetic Changes That Correlate with Reduced Susceptibility to Daptomycin in Staphylococcus aureus. Antimicrob Agents Chemother. 2006;50(6):2137–2145. doi: 10.1128/AAC.00039-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schäberle TF, Hack IM. Overcoming the current deadlock in antibiotic research. Trends Microbiol. 2014;22(4):165–167. doi: 10.1016/j.tim.2013.12.007. [DOI] [PubMed] [Google Scholar]
- 57.Silver LL. Challenges of Antibacterial Discovery. Clin Microbiol Rev. 2011;24(1):71–109. doi: 10.1128/CMR.00030-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nicolaou KC, Hale CRH, Nilewski C, Ioannidou HA, ElMarrouni A, Nilewski LG, Beabout K, Wang TT, Shamoo Y. Total Synthesis of Viridicatumtoxin B and Analogues Thereof: Strategy Evolution, Structural Revision, and Biological Evaluation. J Am Chem Soc. 2014;136(34):12137–12160. doi: 10.1021/ja506472u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Deatherage DE, Barrick JE. Identification of mutations in laboratory evolved microbes from next-generation sequencing data using breseq. Methods Mol Biol. 2014;1151:165–188. doi: 10.1007/978-1-4939-0554-6_12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Diaz L, Tran TT, Munita JM, Miller WR, Rincon S, Carvajal LP, Wollam A, Reyes J, Panesso D, Rojas NL, et al. Whole-genome analyses of Enterococcus faecium isolates with diverse daptomycin MICs. Antimicrob Agents Chemother. 2014;58(8):4527–4534. doi: 10.1128/AAC.02686-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Pace JL, Krause K, Johnston D, Debabov D, Wu T, Farrington L, Lane C, Higgins DL, Christensen B, Judice JK, et al. In vitro activity of TD-6424 against Staphylococcus aureus. Antimicrob Agents Chemother. 2003;47(11):3602–3604. doi: 10.1128/AAC.47.11.3602-3604.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.