

Abstract
Previously we have shown that loss of non-canonical NF-κB signaling impairs self-renewal of hematopoietic stem/progenitor cells (HSPCs). This prompted us to investigate whether persistent activation of the non-canonical NF-κB signaling will have supportive effects on HSPC self-renewal. NF-κB-inducing kinase (NIK) is an important kinase that mainly activates the non-canonical pathway through directly phosphorylating IKKα. In contrast to our expectations, constitutive activation of NIK in the hematopoietic system leads to bone marrow failure and postnatal lethality due to intrinsic impairment of HSPC self-renewal and extrinsic disruption of bone marrow microenvironment through enhancing osteoclastogenesis. The impaired HSPC function is associated with reduced cell proliferation and increased apoptosis and inflammatory cytokine responses. RNAseq analysis of control and NIK-activated HSPCs reveals that these effects are through non-canonical NF-κB signaling without significant changes in the canonical pathway. Gene set expression analysis of RNAseq data reveals globally decreased stem cell signature, increased maturation signature, and increased inflammatory responses. Many genes (Mpl, Tifab, Emcn, Flt3, Bcl2, and others) that regulate HSPC self-renewal, lineage commitment, and apoptosis are significantly downregulated—and those genes that regulate inflammatory responses and cell cycle inhibition (Cdkn2a and Cdkn2b) are significantly upregulated—by activation of NIK. Importantly, our data demonstrate that activation of NIK-non-canonical signaling has distinct phenotypes – smaller spleen size, decreased white blood cell counts, and reduced HSPC proliferation – compared to activation of canonical signaling. Collectively, these data indicate that the balanced non-canonical NF-κB signaling is essential for maintaining normal hematopoiesis and NIK-non-canonical signaling contributes to the development of bone marrow failure.
Keywords: NIK, Non-cononical NF-κB, hematopoietic stem/progenitor cell, self-renewal, bone marrow failure
Introduction
Adult hematopoiesis is maintained by the self-renewing hematopoietic stem/progenitor cells (HSPCs) within the bone marrow (BM). HSPCs include long-term stem cells, which have the unique capacity of self-renewal, short-term stem cells, and multipotent progenitor cells, which differentiate further to produce different lineage cells [1, 2]. Long-term stem cells are predominantly quiescent and undergo slow cell-cycle progression throughout life to maintain and protect the stem cell pool from premature exhaustion in response to variety of pathologic stresses [1, 3, 4]. The mechanisms regulating HSPC self-renewal involve extrinsic and intrinsic signals. Extrinsic signals are derived from the BM microenvironment or niche where HSPCs undertake cell fate decisions: self-renewal or differentiation. These cell fate decisions occur in osteoblastic, vascular, and stromal niches where HSPCs integrate and respond to different cytokines including thrombopoietin, Flt3 ligand, and stem cell factor (SCF) [5, 6].
The NF-κB transcription factor family includes RelA/p65, RelB, c-Rel, p105, and p100. In response to receptor ligation, p105 is processed constitutively to p50, which forms dimers typically with RelA in the canonical pathway. p100 typically is bound to RelB and undergoes proteasomal processing to p52 through receptor-mediated activation of NF-κB-inducing kinase (NIK) in the non-canonical pathway, thus releasing p52/RelB complexes to translocate to nuclei. Canonical and non-canonical signaling play critical roles in many physiological processes, including innate and adaptive immune responses, cell proliferation, apoptosis, and bone homeostasis [7–9]. However, whether and how NF-κB signaling regulates HSPCs has been overlooked for decades. Using genetic Relb and p52 double-deficient mice, we previously made the novel observation that non-canonical NF-κB signaling positively and intrinsically regulates HSPC self-renewal and maintains stromal/osteoblastic niches [10]. The role of the non-canonical pathway in regulating HSPC self-renewal was supported by a later study using NIK-inactive mutant mice [11]. In addition, the indispensable role of canonical NF-κB signaling in HSPCs was demonstrated by another group shortly later using conditional RelA-deficient mice [12]. Taken together, these results indicate that both canonical and non-canonical pathways positively regulate HSPC self-renewal. More recently, two groups showed that activation of the canonical pathway by deletion of Tnfaip3 (A20), an inhibitor of the canonical pathway, also impairs HSPC self-renewal, indicating that there is a fine balance between positive and negative control of canonical NF-κB signaling for HSPC homeostasis [13, 14]. The question remained to be answered is, what are the effects of activation of the non-canonical pathway on HSPC self-renewal?
NIK is a key signaling molecule in the non-canonical NF-κB pathway by directly phosphorylating IKKα. It is constitutively degraded by tumor necrosis factor receptor-associated factor 3 (TRAF3) in unstimulated cells to prevent unnecessary NF-κB activation [15]. In this study we designed series of experiments to investigate whether enhancing NIK-NF-κB non-canonical signaling will have a supportive role on HSPC self-renewal. In contrast to our expectations, our results demonstrated that constitutive activation of NIK in the hematopoietic system impairs HSPC self-renewal, and induces BM failure and postnatal lethality. Further RNAseq analysis revealed that NIK-NF-κB non-canonical signaling affects several pathways involved in HSPC self-renewal both intrinsically and extrinsically. Importantly, our data demonstrate that activation of NIK-non-canonical signaling has distinct phenotypes compared to activation of canonical signaling.
Materials and Methods
Mice
NIKΔT3flSTOP and Vav-Cre mice have been described previously [16, 17]. ROSA-CreERT2 mice were purchased from Jackson laboratory. NIKΔT3flSTOP mice were crossed with Vav-Cre or ROSA-CreERT2 mice to generate mice with hematopoietic-specific or inducible whole body activation of NIK (hereafter caNIK and NIKERT2 mice, respectively). All these mice are in a C57BL6 background. All the mice used throughout our experiments were NIKΔT3flSTOP heterozygous, therefore only one allele of NIKΔT3 was activated. caNIK mice were used at 5–7 days old due to postnatal lethality. Transplant recipients (C57BL/Ka CD45.1) were 8–10 weeks old. Tamoxifen (5mg, Sigma, St. Louis, MO) was administered daily by oral gavage for 4 days at indicated time points to induce NIK activation. The Institutional Animal Care and Use Committee of the University of Iowa approved all animal experiments.
HSPC Isolation and Analysis
We used the following antibodies from e-Bioscience and BD Biosciences: CD3, CD4, CD8, CD11b, CD11c, CD34, CD45.1, CD45.2, CD48, CD117, CD127, CD135, CD150, B220, FcγII/III, Gr-1, Sca1, and Ter119. For isolation of c-kit+lineage−Sca-1+ (KLS) cells, WBM cells were incubated with a cocktail of lineage antibodies from BD Biosciences (biotinylated anti-mouse antibodies directed against CD3e, CD11b, CD45R/B220, Gr-1, Ter119) followed by lineage depletion using BD IMag streptavidin particles Plus-DM, then stained with Sca-1 Percp-Cy5.5, c-kit BV421, and streptavidin PE-CF594. For HSPC and apoptosis analyses, lineage depletion was omitted and the following antibodies were used when appropriate: PE-conjugated lineage markers (CD3, CD4, CD5, CD8, B220, Gr-1, Mac-1, and Ter119) or biotinylated lineage markers plus streptavidin PE-CF594, Sca-1 PE-Cy5.5, c-kit-BV421, CD34-A700, Flk2-APC, or CD150-PECy7, CD48-APC, or CD48-PE and Annexin V-APC. LSRII was used for all the analyses, and FACSAria was used for cell sorting. Data was analyzed using FlowJo (TreeStar).
BM Transplant Experiments
For non-competitive BM transplantation (BMT), 1×106 WBM cells (approximately equivalent to BM cells harvested from an individual caNIK mouse) from control or caNIK mice were injected into retro-orbital venous sinuses of lethally irradiated CD45.1 recipients. For competitive BMT, either WBM cells (2×105, 5×105, or 10×105/recipient, 5–6 recipients per dose per genotype) or sorted lin−kit+Sca1+ cells (2500 cells/recipient, 6 recipients per genotype) from caNIK mice along with 2×105 competing BM cells were injected into retro-orbital venous sinuses of lethally irradiated CD45.1 recipients. Beginning 4 weeks after transplantation and continuing for 16 weeks, blood was obtained from recipients, subjected to ammonium-chloride potassium red cell lysis, and stained with CD45.2 A700 and CD45.1 APC along with B220 PECy5 and CD3 PE or Mac-1 PE and Gr1 PECy5 for monitoring donor engraftment and donor-derived lymphoid or myeloid cells, respectively. Successful engraftment was defined as the presence of a distinct CD45.2+CD45.1− population above a background set by parallel analyses of animals transplanted with only competitor cells. For secondary transplantation, 3×106 WBM cells from the primary transplants were injected into lethally irradiated congenic CD45.1 recipients. To investigate the activation of NIK in adult hematopoiesis, 3×106 WBM cells from NIKERT2 mice were transplanted into congenic recipients and NIK activation was induced by tamoxifen treatment 16 weeks post BMT.
Colony-Forming Assay
Hematopoietic clonogenic progenitor frequencies were determined by plating sorted KLS cells (1000/6-well) or WBM cells (1×105/6-well) in methylcellulose media (Methocult M3434, StemCell Technologies). Colonies were counted after 7–10 days.
Measurement of Serum Cytokine Concentration
Concentrations of pro-inflammatory cytokines were measured using a ProcartaPlex Mouse Cytokine & Chemokine Panel 1A (36 plex) from eBioscience according to the manufacturer’s instructions.
BrdU Labeling and Cell Proliferation Analysis
For in vivo BrdU labeling, 5-day-old matched control or caNIK mice were injected intraperitoneally with BrdU (10 mg/ml) to a final concentration of 50 ug/g body weight. One hour later, mice were sacrificed and BM cells were stained first with surface marker, followed by BrdU staining using a BrdU-APC staining kit (BD Biosciences). For Ki67 staining, BM cells were first stained with surface markers to define the progenitor population, followed by fixation and permeabilization with Cytofix/Cytoperm (BD Biosciences) and intracellular staining with Ki67-PE.
Histology
Femurs or tibias from age-matched control or caNIK mice were fixed in 10% neutral buffered formalin. Following decalcification and fixation (approximately 5 days), tissues were routinely processed, paraffin-embedded, sectioned at 4 μm, and stained with hematoxylin and eosin (H&E) and tartrate-resistant acid phosphatase (TRAP) staining.
RNA-Seq and Transcriptome Analysis
Total RNA was extracted from sorted Lin−cKit+Sca1+ (KLS) cells from caNIK or control mice, and two biological replicates (each was pooled from 2–3 wild-type [WT] and 6–8 caNIK mice) were obtained for each genotype. cDNA synthesis and amplification were performed using SMARTer Ultra Low Input RNA Kit (Clontech) starting with 20 ng of total RNA per sample following the manufacturer’s instructions. cDNA was fragmented with Q800R sonicator (Qsonica) and used as input for NEBNext Ultra DNA Library Preparation Kit (NEB). Libraries were sequenced on Illumina’s HiSeq2000 in single-read mode with the read length of 50 nucleotides producing 60–70 million reads per sample. Sequence data in fastq format were generated using CASAVA 1.8.2 processing pipeline from Illumina. We considered genes differentially expressed between two groups of samples when the DESeq2 analysis resulted in an adjusted p-value of <0.01 and the fold-change in gene expression was 2-fold. Heat map was generated with normalized data of RNA-seq analyses. Gene set enrichment analysis (GSEA) is according to the previous report using the software downloaded from GSEA website (http://software.broadinstitute.org) [18].
Statistical Analysis
Student’s t test was used for all but survive curve statistical analyses and significance was set at p<0.05. Values are mean ± standard deviation (SD). For the survival curve (Fig. 1D and Fig. 3B), the log-rank test was used.
Figure 1. NIK Activation induces postnatal lethality and bone marrow (BM) failure.

(A) Representative images of 5-day-old wild-type (WT) and caNIK mice showing body size. (B) Total body weight of 5-day-old WT and caNIK mice (n=6, p<0.01). (C) Representative images of spleen (left) and the spleen weight (right) of 5-day-old WT and caNIK mice (n=5, p<0.05). (D) Kaplan-Meier survival curve analysis of WT and caNIK mice (n=20, p<0.0001). (E) Complete blood count (CBC) analysis of 5-day-old WT (n=5–7) and caNIK (n=5–8) mice (**p<0.01). WBC, white blood cells; HGB, hemoglobin; MCV, mean corpuscular volume; PLT, platelet. (F) Cellularity of bone marrow (BM, left) and spleen (right) of 5-day-old WT (n=9) and caNIK (n=12) mice (**p<0.01).
Figure 3. NIK-activated HSPCs are functional compromised.

(A) Number of colonies formed in methycellulose (M3434) from KLS cells of WT or caNIK mice (n=4, p<0.01). (B) Kaplan-Meier survival curve analysis of lethally irradiated (9.5 Gy) congenic recipients (CD45.1+) that received WBM cells of either WT or caNIK mice (both CD45.2+) (n=10, p<0.0001). (C, D) Equal numbers (10×105) of WT or caNIK WBM cells along with competitor cells (2×105) were transplanted into lethally irradiated congenic recipients. FACS plots show representative donor-derived chimerisms from mice receiving WT or caNIK cells at 16 weeks (C). Donor-derived chimerism was monitored for 16 weeks (n=5, **p<0.01) (D). (E) Kinetic lineage (including CD3+ T cells, B220+ B cells, Mac1+Gr1− monocytes, and Mac1+Gr1+ granulocytes) contribution of WT or caNIK cells after transplantation (n=5, **p<0.01). In D and E, each dot represents one individual recipient. (F) Frequencies of donor (CD45.2+)-derived cells in the peripheral blood 16 weeks post-secondary BMT. Each recipient received WBM cells (3×106) from WT or caNIK cells transplanted primary recipients (n=5, **p<0.01).
Results
Activation of NIK induces BM failure and postnatal lethality
To more definitively investigate whether enhancing non-canonical NF-κB signaling will have a supportive role in HSPC self-renewal, mice carrying NIKΔT3flSTOP allele were crossed with Vav-Cre mice (caNIK mice). Mouse carrying NIKΔT3flSTOP allele is generated to allow cell type–specific expression of a mutated form of NIK lacking the TRAF3-binding domain under the control of the ROSA26 promoter after Cre-mediated deletion of the LoxP-flanked STOP cassette [16]. The caNIK mice were born at a normal Mendelian ratio but quickly developed growth retardation with reduced body and organ sizes (Fig. 1A–C). Almost all caNIK mice died before postnatal day 7 compared to control littermates (either Cre-positive alone or NIK mice without deletion of STOP cassette) (Fig. 1D). Peripheral complete blood count showed that caNIK mice had pancytopenia with increased mean corpuscular volume (MCV) (Fig. 1E). Consistent with pancytopenia, the BM cellularity was markedly decreased in these mutant mice (Fig. 1F). These features are reminiscent of the findings in human BM failure syndrome and indicate that constitutive activation of NIK is detrimental to normal hematopoiesis.
NIK controls lineage commitment and the HSPC pool
Consistent with the markedly decreased BM cellularity, the absolute numbers of CD3+ T cells, B220+ B cells, Ter119+ erythrocytes, and Mac1+Gr1+ granulocytes in caNIK mice were also significantly decreased compared to controls (Fig. 2A, upper panel). However, the ability of differentiation to CD3+ T cells, Ter119+ erythrocytes, and Mac1+Gr1+ granulocytes was maintained. Interestingly, the ability of commitment to B220+ B cells was diminished and of differentiation to Mac1+Gr1− monocytes/macrophages was relatively increased with an only modestly reduced absolute number of Mac1+Gr1−monocytes/macrophages (Fig. 2A). Further analysis of BM B cells using the well-established methods showed that the differentiation from pre-pro-/early pro-B cells to late pro-/early pre-B cells was partially blocked and the ability of differentiation to late pre-/immature B cells was accelerated, indicating NIK regulates B cells differentially at different stages (Fig. 2B and 2C) [19–21]. It is interesting that both gain- (current study) and loss-of-function of NIK impair B cell development [22, 23].
Figure 2. NIK controls lineage commitment and HSPC pool size.

(A) Absolute counts (upper panel) and frequencies (lower panel) of BM CD3+ T cells, B220+ B cells, Ter119+ erythrocytes, Mac1+Gr1− monocytes/macrophages and Mac1+Gr1+ granulocytes in WT or caNIK mice (n=6, **P<0.01). (B, C) Representative fluorescence-activated cell sorting (FACS) plots (B) and frequencies (C) of BM B cells at different developmental stages in WT and caNIK mice: I) prepro-/early pro-B; II) late pro-/early pre-B; III) late pre-/immature B and IV) mature recirculating B (n=3, **p<0.01). (D–G) Representative FACS plots (D, F), frequencies (upper) and absolute numbers (lower) (E, G) of KLS (Lin−cKit+Sca1+) cells, long-term stem cells (CD150+CD48−KLS), and short-term stem cells (CD150−CD48−KLS) from BM (D, E) or spleen (F, G) of 5-day-old WT and caNIK mice (n=10 for BM and n=7 for spleen analysis, *p<0.05, **p<0.01).
To determine whether the HSPC pool is altered by NIK activation, we performed immunophenotyping experiments of HSPCs using flow cytometry. Analysis of HSPC-enriched KLS (Lin−cKit+Sca1−) compartment revealed that the absolute numbers of HSPCs were significantly reduced without significant difference in HSPC frequency (calculated as Lin−%xKLS%) in caNIK mice compared to controls (Fig. 2D and E). Further analysis revealed decreased frequency of long-term stem cells (defined as CD150+CD48−KLS, calculated as Lin−%xKLS%xCD150+CD48−%) but not of short-term stem cells (defined as CD150−CD48−KLS, and calculated as Lin−%xKLS%xCD150−CD48−) with decreased absolute number of both long-term- and short-term-stem cells (Fig. 2D and E) [24]. The decreased BM HSPC pool was not due to peripheral mobilization of HSPCs, as the frequency and absolute number of long-term stem cells from spleen were also decreased (Fig. 2F and 2G) in caNIK mice.
NIK-activated HSPCs are functionally compromised
To assess the function of NIK-activated HSPCs, we first compared the in vitro colony-forming ability of NIK-activated HSPCs to controls. Using sorted KLS cells revealed a significantly decreased colony-forming ability (Fig. 3A). To investigate the function of NIK-activated HSPCs in vivo, we performed BM transplantation experiments in the presence or absence of competitor cells. Pooled WBM cells from caNIK mice when transplanted into lethally irradiated congenic recipients in the absence of rescue cells failed to provide radioprotection compared to controls, and all recipients died within 2 weeks (Fig. 3B). In the presence of rescue cells, we observed markedly impaired BM reconstitution ability of NIK-activated WBM cells (Fig. 3C). In fact, we transplanted three different doses of donor cells (2×105, 5×105, and 10×105) along with 2×105 competitor cells into each recipient, but even the highest dose was unable to efficiently reconstitute lethally irradiated recipients (Fig. 3C, 3D and data not shown). Kinetic analysis of lineage commitment after BM transplantation demonstrated impaired B cell, T cell, and monocyte/granulocyte commitment of NIK-activated BM cells (Fig. 3E). We also tested flow-sorted HSPC-enriched KLS cells (2500/recipient) isolated from caNIK mice, but the BM reconstitution efficiency was still low (Supporting information Fig. 1). We also transplanted the WBM cells from primary recipients into secondary recipients (3 million WBM cells/mouse) and found donor cells from the primary recipients receiving caNIK cells were almost completely exhausted in the secondary recipients (Fig. 3F). Overall, these results suggest that NIK intrinsically controls HSPC self-renewal and lineage commitment.
We next investigated if activation of NIK in adult hematopoiesis would also impair HSPC function. To this end, we crossed NIKΔT3flSTOP mice with ROSA-CreERT2 mice (NIKERT2 mice). We first induced NIK activation by directly delivery tamoxifen into 2 months old NIKERT2 mice. Due to the systemic activation of NIK in NIKERT2 mice, these mice begun to die 5 days after last tamoxifen treatment due to the inflammatory “storm” induced by NIK activation (Supporting information Fig. 2). Analyzing NIKERT2 mice at day 4 after the last tamoxifen delivery showed that the BM cell numbers and the frequencies of KLS, long-term and short-term stem cells were decreased in tamoxifen-treated NIKERT2 mice (Fig. 4A–C). To further investigate the effect of NIK activation on adult HSPC function, we transplanted NIKERT2 WBM cells into lethally irradiated recipients and started tamoxifen treatment 16 weeks post BM transplantation when recipient mice were stably engrafted. Similar to the mice transplanted with caNIK cells, the chimerisms were decreased in tamoxifen treated recipients compared to control mice with decreased frequencies of CD3+ T cells and B220+ B cells, but increased frequencies of Mac+Gr1− macrophages and Mac+Gr1+ neutrophils (Fig. 4D and E). This may be due to the effects of elevated inflammatory cytokines or the difference between activation of NIK in early and adult hematopoiesis. Importantly, the frequency of HSPCs was reduced by tamoxifen-induced NIK activation, indicating NIK activation also impairs adult HSPC function (Fig. 4F).
Figure 4. Activation of NIK in adult hematopoiesis has similar effects as in early hematopoiesis.

(A, B) BM numbers (A) and HSPC (Lin−cKit+Sca1+) frequencies (B) of control or NIKERT2 mice 4 days post tamoxifen treatment (n=4, *p<0.05, **p<0.01). (C) Representative FACS plots of long-term stem cells (CD150+CD48−KLS) and short-term stem cells (CD150−CD48−KLS) from BM of control or NIKERT2 mice 4 days post tamoxifen treatment. (D) 3×106 of NIKERT2 WBM cells were transplanted into lethally irradiated congenic recipients. 16 weeks post BM transplantation mice were treated with tamoxifen or corn oil (Con) by oral gavage. Donor-derived chimerisms were monitored 12 weeks post tamoxifen treatment. (E) Kinetic lineage (including CD3+ T cells, B220+ B cells, Mac1+Gr1− monocytes, and Mac1+Gr1+ granulocytes) contribution of control or NIK-activated cells 12 weeks after tamoxifen treatment. Each dot represents one individual recipient. In D and E, control n=8, NIKERT2 n=10, *p<0.05, **p<0.01. (F) Representative FACS plots of HSPC (Lin−cKit+Sca1+) population from BM of control (0.3 ± 0.1) or NIKERT2 (0.03±0.01) mice 12 weeks post tamoxifen treatment. Numbers are mean ± sem., p<0.01.
Activation of NIK up-regulates inflammatory cytokines, increases cell apoptosis, and impairs proliferation
It has been shown that elevated inflammatory cytokines impair HSPC self-renewal, accelerate apoptosis, and contribute to both inherited and acquired BM failure syndrome; and NF-κB plays critical roles in regulating the production of inflammatory cytokines [8, 25–27]. To determine whether the altered inflammatory cytokine secretion contributes to the phenotypes observed in caNIK mice, we simultaneously measured the expression of 36 cytokines using peripheral blood serum prepared from control or caNIK mice. We found G-CSF, interferon-γ, TNFα and IL6 along with others are significantly increased in caNIK mice compared to controls (Fig. 5A and Supporting information Fig. 3). Importantly, culturing sorted control Lin−cKit+ BM cells with serum prepared from caNIK mice induced decreased KLS population, increased HSPC apoptosis, but increased overall cell number (Fig. 5B–D). These results indicate that the decreased BM cellularity and pancytopenia observed in caNIK mice cannot be explained by solely increased cytokines, which would be expected to exhibit decreased long-term HSCs but increased short-term and multipotent progenitors with increased myeloid proliferation [13, 14, 25, 26]. To further understand the underlying cellular mechanisms of activation of NIK-induced BM failure and HSPC self-renewal defects, we compared cell apoptosis and proliferative status of progenitor cells from caNIK mice to controls. For these assays we compared Lin−cKit+ progenitor cells between control and caNIK mice, as there are few KLS cells from a single mutant mouse (Fig. 2D). As expected, we found that NIK-activated progenitor cells were more predisposed to apoptosis (Fig. 5E). In general, HSPCs once exposed to an inflammatory environment proliferate faster [26]. Therefore we would expect to see that the progenitor cells from mutant mice are more proliferative. However, both Ki67 staining and BrdU labeling showed that NIK-activated progenitor cells had a lower proliferative index (Fig. 5F and G). Collectively, these results indicate that other unknown factors along with inflammatory cytokines are responsible for the impaired HSPC self-renewal observed in caNIK mice.
Figure 5. Activation of NIK up-regulates inflammatory cytokines, increases cell apoptosis, and impairs proliferation.

(A) Serum concentrations of pro-inflammatory cytokines. Mouse serum was prepared by centrifugation of clotted mouse peripheral blood from WT (n=4) or caNIK (n=7) mice and the cytokines were measured using ProcartaPlex Mouse Cytokine & Chemokine Panel 1A (36 plex) (*p<0.05, **p<0.01). (B–D) Sorted Lin−cKit+ WT cells were cultivated in vitro in the presence or absence of caNIK serum for 2 days and the frequencies of KLS cells (B), apoptosis (Annexin+) of progenitor cells (C), and the relative cell expansion (D) were examined (n=4, *p<0.05, **p<0.01). Representative FACS plots of KLS frequency and annexin V staining are shown in (B) and (C), respectively. (E–G) Analysis of apoptosis (E), ki67 proliferation index (F), and BrdU incorporation (G) of Lin−cKit+ progenitor cells in WT or caNIK mice (n=4 for apoptosis and Ki67; n=5 for BrdU labeling, *p<0.05, **p<0.01). Representative FACS plots and the frequencies of apoptosis, Ki67, and BrdU staining are shown in the left and right panels of E–G, respectively.
NIK indirectly regulates BM microenvironment
It is well known that constitutive activation of NIK in osteoclast precursors drives enhanced osteoclastogenesis and bone resorption, both in basal conditions and in response to inflammatory stimuli [28]. Here we found that activation of NIK in HSPCs from early hematopoiesis not only intrinsically impaired HSPC self-renewal, but also disrupted bone development in caNIK mice, which have pale shorter femurs (Fig. 6A). Histologic examination showed that activation of NIK enhanced osteoclastogenesis in caNIK mice, in accordance with the relatively preserved Mac1+Gr1− macrophage differentiation – precursors of osteoclasts and the role of NIK in osteoclastogenesis (Fig. 6B, 6C and Fig. 2A) [28]. Importantly, the number of osteoclasts with more than two nuclei, which correlates with increased osteoclast activity, was significantly increased in caNIK mice (Fig. 6D). Consistent with the enhanced osteoclastogenesis, the trabecular bone number was decreased, suggesting NIK activation destroys osteoblastic niche (Fig. 6B). The likely decreased osteoblast number is different from the previous report, which showed that activation of NIK specifically in osteoclast lineage induced a high turnover osteoporosis with increased activity of both osteoclasts and osteoblasts [28]. This may be because NIK is activated in the early hematopoietic developmental stage and because of the effects of the elevated inflammatory cytokines. In addition, when NIK was systemically activated in NIKERT2 mice by tamoxifen treatment, the trabecular bone volume was significantly decreased, and the volume of BM adipose tissue was increased, with increased osteoclast activity (Supporting information Fig. 4). Collectively, these data indicate that one way that NIK activation influences BM microenvironment may be through enhancing osteoclast activity. However, further functional confirmation—and whether activation of NIK also affects other niches—need to be answered in future studies.
Figure 6. NIK indirectly regulates BM microenvironment.
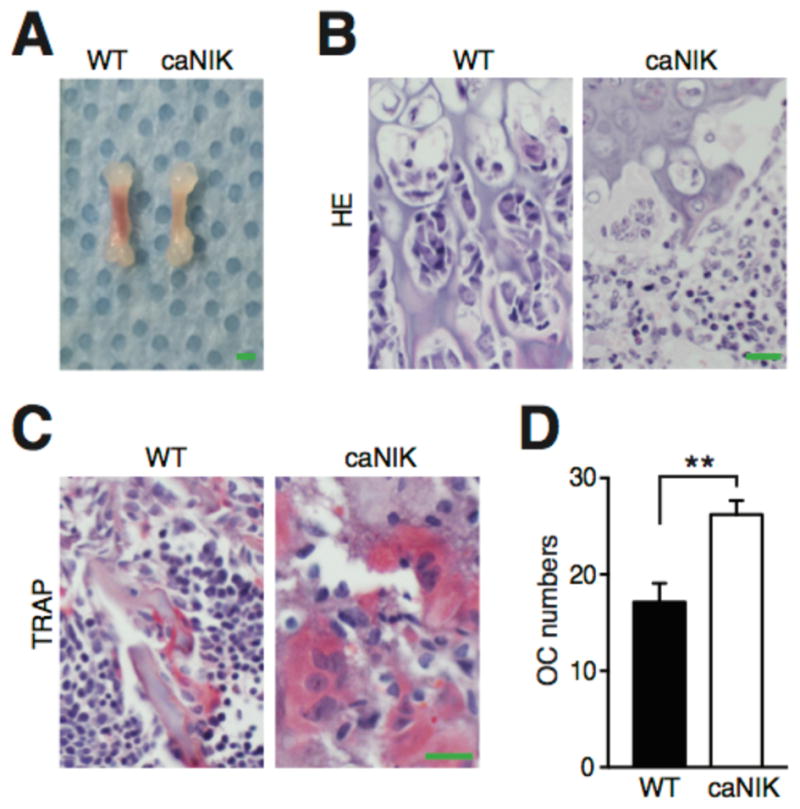
Representative gross pictures of femurs (A, scale bar 1mm), H&E (B) and TRAP (tartrate resistant acid phosphatase) (C) stains of 5-day-old femurs from WT or caNIK mice (n=5). Scale bar 20 μm in (B) and (C). (D) Average number of osteoclasts with two or more nuclei per cross-section of femur metaphysis of WT or caNIK mice (n=5, two levels of each, *p=0.0024).
Activation of NIK alters the expression of genes regulating HSPC homeostasis, lineage commitment, and inflammatory responses
To investigate the underlying molecular mechanisms, we performed RNAseq experiments using KLS cells from control or caNIK mice to globally analyze the genes regulated by NIK. First, we confirmed that Map3k14 (NIK) was overexpressed in NIK-activated HSPCs (Fig. 7A). RNAseq data revealed that the expression of major non-canonical NF-κB pathway genes Nfκb2, Relb, and Traf3 was upregulated, but the expression of the major canonical pathway genes (Nfkb1, RelA, Rel) was not significantly upregulated in NIK-activated HSPCs (Fig. 7A). This was attributable at least partially to the NIK-induced overexpression of some canonical pathway inhibitors like Nfkbia, Nfkbie, and Tnfaip3 (A20) (Fig. 7A). Increased nuclear localization of RelB, but not RelA, by immunofluorescence microscopic examination and accelerated p100 processing and nuclear translocation by Western blotting further supported the RNAseq results (Supporting information Fig. 5), indicating that NIK exerts its role in HSPCs through the non-canonical pathway.
Figure 7. Activation of NIK alters the expression of genes regulating HSPC homeostasis, lineage commitment, and inflammatory responses.

(A, C) RNA-seq analyses of sorted KLS cells from 5-day-old WT or caNIK mice. Heat maps were generated from selected genes of interest. Genes with * are significantly up- or down-regulated. (B, E) Gene set enrichment analysis (GSEA) of statistically significant gene sets enriched in the KLS cells of caNIK mice (up in caNIK) or WT mice (down in caNIK). (D) Selected relative expression of genes that are important for HSPC homeostasis from RNAseq analysis (**p<0.01).
Furthermore, our RNAseq data revealed many genes that are important for HSPC self-renewal were significantly down-regulated, for example, Emcn, Crispld1, Myct1, Mpl, Hlf, CD34, Flt3, and Tifab (Fig. 7C and D). Knockdown of Emcn, Crispld1, or Myct1 has been recently shown to impair HSPC self-renewal and BM reconstitution ability [29]. Tifab (TRAF-interacting protein with forkhead-associated domain, family member B), a gene involved in myelodysplastic syndrome (MDS) with 5q deletion, has recently been shown to regulate hematopoiesis through Toll-like receptor (TLR)-TRAF6 signaling, and loss of Tifab induces BM failure with dysplastic changes in some transplanted mice [30]. In addition, the increased expression of Gzmb may also contribute to HSPC damage, as Gzmb is induced by lipopolysaccharide and associated with increased cell death of HSPCs (Fig. 7C) [31].
In support of the increased HSPC apoptosis and impaired HSPC proliferation, the anti-apoptotic gene Bcl2 was downregulated, and proapoptotic genes Fas along with the cell cycle inhibitors (Cdkn2a and Cdkn2b) were upregulated (Fig 7C and D). The increased expression of TLRs (Tlr1, Tlr9 and Tlr13) and TNF family members along with activated non-canonical NF-κB pathway suggested that inflammatory responses were also involved in the observed HSPC defects (Fig. 7A) [25, 26, 32]. Consistent with the enhanced inflammatory response, Ly6a (Sca1), a direct target of interferon-γ, was upregulated (Fig. 2D protein level; Fig. 7C) [26]. Compatible with these gene expression changes, gene set expression analysis (GSEA) of RNAseq data revealed significant enrichment for inflammatory response (TNFα and INFγ)–related genes, hematopoietic maturation–related genes, and decreased stemness–related genes in NIK-activated HSPCs compared to controls (Fig. 7B, 7E and Supporting information Fig. 6).
Our RNAseq analyses also revealed the underlying mechanisms of the reduced B cell differentiation of NIK-activated HSPCs. The genes regulating B cell commitment and differentiation (Flt3, Ebf1, Pax5, Rag1, and Vpreb1) were significantly decreased (Fig. 7C). The increased expression of Tnfrsf11a (RANK) correlated with relatively preserved Mac1+Gr1− monocytes and contributed to enhanced osteoclast formation (Fig. 6). It should be noted that the dysregulated expression of multiple HSPC markers (CD34, Ly6a, Flt3 in Fig. 7C) prevented us from accurately analyzing the common lymphoid-, common myeloid-, granulocyte-macrophage-, and megakaryocyte-erythrocyte progenitors.
Discussion
The critical role of canonical NF-κB signaling in regulating HSPC self-renewal has been well characterized through loss- and gain-of-function studies, and the role of non-canonical signaling in this process has been demonstrated through loss-of function studies [10–14]. Our studies investigating the role of activation of the NIK-NF-κB non-canonical signaling in hematopoiesis have timely filled the knowledge gap on the role of NF-κB signaling in hematopoiesis. Specifically, we demonstrated that activation of NIK is sufficient to induce BM failure through dysregulating multifaceted pathways regulating HSPC self-renewal, apoptosis, cell proliferation, inflammatory responses, and the BM microenvironment through enhancing osteoclast formation, and these effects are mediated by NF-κB non-canonical signaling. Additionally, we also showed that activation of NIK impairs HSPC self-renewal in adult hematopoiesis (Fig. 4).
Deletion of A20 in the hematopoietic system, which is supposed to mainly activate canonical NF-κB signaling, has been demonstrated to induce significant HSPC defects, with dramatic enlarged spleens due to extramedullary hematopoiesis and development of myeloproliferative disorder [13, 14]. The phenotypes of our caNIK mice display some similarities with A20-deficient mice, like postnatal lethality and increased inflammatory cytokines [13, 14]. However, the phenotype of caNIK mice, which all died before day 7 after birth, was more severe than A20-deficient mice, which all died before day 21 after birth. Importantly, caNIK mice exhibited distinct phenotypes—smaller spleen size, decreased white blood cell counts, and reduced HSPC proliferation—compared to A20-deficient mice displaying enlarged spleen, increased white blood cell counts and increased HSPC proliferation [13, 14]. Overall, these results indicate that activation of NIK-NF-κB non-canonical pathway has a distinct role in hematopoietic system compared to A20 deficiency–induced activation of canonical NF-κB pathway. It is well known that the canonical NF-κB signaling is often transiently activated and due to feed-forward mechanisms; it can prime the activity of the non-canonical pathway by inducing the expression of p52 and RelB genes. Therefore, it is possible that some effects of canonical signaling have been observed—in fact result from—the non-canonical pathway and non-canonical signaling has unique roles in hematopoiesis. Interestingly, the expression of p52 and RelB was upregulated in A20-deficient mice, and this may partially explain the similarities between these two mutant mice [13].
The inflammatory cytokines are increased in both caNIK and tamoxifen-treated NIKERT2 mice. We think that the increased cytokines definitely contribute to the observed HSPC defects, mainly through increased apoptosis, but not because increased cell proliferation induced HSPC exhaustion (Fig. 5). The reduced cell proliferation in NIK-activated HSPCs is interesting, as we would expect to see the increased cell proliferation given the NIK-activated HSPCs are exposed to an elevated inflammatory cytokine background. One explanation is that as the inflammatory exposure started from early hematopoiesis, the majority of the HSPCs has been exhausted and enters senescence after cytokine-induced accelerated proliferation. However, this is unlikely given the inflammatory cytokine effects—enhancing HSPC proliferation—observed in A20-deficient mice, in which the A20 deletion was also mediated by Vav-Cre and the HSPCs were exposed to inflammatory microenvironment from the same developmental stages as ours [13]. Our gene transcriptome analysis reveals that the reduced HSPC proliferation is due to increased expression of the cell cycle inhibitors Cdkn2a and Cdkn2b.
Our RNAseq sheds many new insights on the underlying molecular mechanisms whereby NIK activation impairs HSPC self-renewal and alters lineage commitment, uncovering many new roles of NIK-NF-κB non-canonical signaling in regulating HSPCs. Identification of the genes that are directly regulated by NIK-NF-κB non-canonical signaling using chromatin immunoprecipitation (ChIP)-RNAseq in the future will help to pinpoint the key molecule(s) involved in maintaining HSPC homeostasis. Tifab is a new hematopoietic regulator and its loss of function induces BM failure and MDS [30]. Our data indicate that Tifab is a downstream target of NIK-NF-κB non-canonical signaling and suggest that chronic inflammation-induced BM failure and MDS may happen through activation of the NF-κB non-canonical pathway to downregulate Tifab.
In addition to the discussed distinct cell-intrinsic effects of NIK-NF-κB non-canonical signaling on HSPC homeostasis, we identified another new role of NIK in regulating HSPC homeostasis—indirectly disrupting the hematopoietic microenvironment. BM microenvironment provides various indispensable niches, for example, vascular niches and osteoblastic niches, for normal hematopoiesis. Although the vascular niches play the main role in maintaining HSPC homeostasis, we think that the enhanced osteoclastogenesis in the early hematopoietic developmental stage will definitely contribute to the profoundly impaired hematopoiesis in caNIK mice, likely through modifying the osteoblast niche [5, 6]. Whether any other type of niche is also involved and how the elevated inflammatory cytokines modify these change is currently unclear, further detailed studies are required in the future. In addition to contributing to HSPC homeostasis, the reduced trabecular bone and osteoblasts also contribute to B cell defects because osteoblasts form B cell niches [33].
Summary
The current data along with our previous study suggest that the balanced non-canonical NF-κB signaling is essential for maintaining normal hematopoiesis [10]. Importantly, our results indicate that activation of the NIK-NF-κB non-canonical signaling has a distinct role compared to activation of canonical signaling in hematopoietic system. Our results have significant relevance for understanding how NIK-NF-κB non-canonical signaling maintains normal hematopoiesis and contributes to the development of BM failure and MDS.
Supplementary Material
Acknowledgments
This work was supported by departmental startup funds to C.Z. and also in part by funds from the Holden Comprehensive Cancer Center and its National Cancer Institute Cancer Center Support Grant (P30-CA86862).
We thank Heath Vignes and Justin Fishbaugh for cell sorting, Tom Bair for RNAseq data analysis and Paul Casella for critical reading of the manuscript. This work was supported by departmental startup funds to C.Z. and also in part by funds from the Holden Comprehensive Cancer Center and its National Cancer Institute Cancer Center Support Grant (P30-CA86862).
Footnotes
Author contribution summary:
Yan Xiu: Conception and design, data analysis and interpretation
Wingel y. Xue: Provision of study material, data analysis and interpretation
Allyn Lambertz and Mariah Leidinger: Provision of histological studies
Katherine Gibson-Corley: Provision of study material
Chen Zhao: Conception and design, data analysis and interpretation, manuscript writing and final approval of manuscript
Disclosure of Potential Conflicts of Interest: The authors declare no competing financial interests.
References
- 1.Orkin SH, Zon LI. Hematopoiesis: an evolving paradigm for stem cell biology. Cell. 2008;132:631–644. doi: 10.1016/j.cell.2008.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Morrison SJ, Spradling AC. Stem cells and niches: mechanisms that promote stem cell maintenance throughout life. Cell. 2008;132:598–611. doi: 10.1016/j.cell.2008.01.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Passegue E, Wagers AJ, Giuriato S, et al. Global analysis of proliferation and cell cycle gene expression in the regulation of hematopoietic stem and progenitor cell fates. J Exp Med. 2005;202:1599–1611. doi: 10.1084/jem.20050967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kiel MJ, Morrison SJ. Uncertainty in the niches that maintain haematopoietic stem cells. Nat Rev Immunol. 2008;8:290–301. doi: 10.1038/nri2279. [DOI] [PubMed] [Google Scholar]
- 5.Morrison SJ, Scadden DT. The bone marrow niche for haematopoietic stem cells. Nature. 2014;505:327–334. doi: 10.1038/nature12984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Birbrair A, Frenette PS. Niche heterogeneity in the bone marrow. Ann N Y Acad Sci. 2016 doi: 10.1111/nyas.13016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ghosh S, Hayden MS. New regulators of NF-kappaB in inflammation. Nat Rev Immunol. 2008;8:837–848. doi: 10.1038/nri2423. [DOI] [PubMed] [Google Scholar]
- 8.Vallabhapurapu S, Karin M. Regulation and function of NF-kappaB transcription factors in the immune system. Annu Rev Immunol. 2009;27:693–733. doi: 10.1146/annurev.immunol.021908.132641. [DOI] [PubMed] [Google Scholar]
- 9.Boyce BF, Yao Z, Xing L. Functions of nuclear factor kappaB in bone. Ann N Y Acad Sci. 2010;1192:367–375. doi: 10.1111/j.1749-6632.2009.05315.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhao C, Xiu Y, Ashton J, et al. Noncanonical NF-kappaB signaling regulates hematopoietic stem cell self-renewal and microenvironment interactions. Stem cells. 2012;30:709–718. doi: 10.1002/stem.1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gonzalez-Murillo A, Fernandez L, Baena S, et al. The NFKB Inducing Kinase Modulates Hematopoiesis During Stress. Stem cells. 2015;33:2825–2837. doi: 10.1002/stem.2066. [DOI] [PubMed] [Google Scholar]
- 12.Stein SJ, Baldwin AS. Deletion of the NF-kappaB subunit p65/RelA in the hematopoietic compartment leads to defects in hematopoietic stem cell function. Blood. 2013;121:5015–5024. doi: 10.1182/blood-2013-02-486142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nakagawa MM, Thummar K, Mandelbaum J, et al. Lack of the ubiquitin-editing enzyme A20 results in loss of hematopoietic stem cell quiescence. J Exp Med. 2015;212:203–216. doi: 10.1084/jem.20132544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nagamachi A, Nakata Y, Ueda T, et al. Acquired deficiency of A20 results in rapid apoptosis, systemic inflammation, and abnormal hematopoietic stem cell function. PloS one. 2014;9:e87425. doi: 10.1371/journal.pone.0087425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liao G, Zhang M, Harhaj EW, et al. Regulation of the NF-kappaB-inducing kinase by tumor necrosis factor receptor-associated factor 3-induced degradation. J Biol Chem. 2004;279:26243–26250. doi: 10.1074/jbc.M403286200. [DOI] [PubMed] [Google Scholar]
- 16.Sasaki Y, Calado DP, Derudder E, et al. NIK overexpression amplifies, whereas ablation of its TRAF3-binding domain replaces BAFF:BAFF-R-mediated survival signals in B cells. Proc Natl Acad Sci U S A. 2008;105:10883–10888. doi: 10.1073/pnas.0805186105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhao C, Blum J, Chen A, et al. Loss of beta-catenin impairs the renewal of normal and CML stem cells in vivo. Cancer Cell. 2007;12:528–541. doi: 10.1016/j.ccr.2007.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rumfelt LL, Zhou Y, Rowley BM, et al. Lineage specification and plasticity in CD19- early B cell precursors. J Exp Med. 2006;203:675–687. doi: 10.1084/jem.20052444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Clark MR, Mandal M, Ochiai K, et al. Orchestrating B cell lymphopoiesis through interplay of IL-7 receptor and pre-B cell receptor signalling. Nat Rev Immunol. 2014;14:69–80. doi: 10.1038/nri3570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.von Wnuck Lipinski K, Sattler K, Peters S, et al. Hepatocyte Nuclear Factor 1A Is a Cell-Intrinsic Transcription Factor Required for B Cell Differentiation and Development in Mice. J Immunol. 2016;196:1655–1665. doi: 10.4049/jimmunol.1500897. [DOI] [PubMed] [Google Scholar]
- 22.Brightbill HD, Jackman JK, Suto E, et al. Conditional Deletion of NF-kappaB-Inducing Kinase (NIK) in Adult Mice Disrupts Mature B Cell Survival and Activation. J Immunol. 2015;195:953–964. doi: 10.4049/jimmunol.1401514. [DOI] [PubMed] [Google Scholar]
- 23.Hahn M, Macht A, Waisman A, et al. NF-kappaB-inducing kinase is essential for B-cell maintenance in mice. Eur J Immunol. 2016;46:732–741. doi: 10.1002/eji.201546081. [DOI] [PubMed] [Google Scholar]
- 24.Oguro H, Ding L, Morrison SJ. SLAM family markers resolve functionally distinct subpopulations of hematopoietic stem cells and multipotent progenitors. Cell Stem Cell. 2013;13:102–116. doi: 10.1016/j.stem.2013.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Takizawa H, Boettcher S, Manz MG. Demand-adapted regulation of early hematopoiesis in infection and inflammation. Blood. 2012;119:2991–3002. doi: 10.1182/blood-2011-12-380113. [DOI] [PubMed] [Google Scholar]
- 26.King KY, Goodell MA. Inflammatory modulation of HSCs: viewing the HSC as a foundation for the immune response. Nat Rev Immunol. 2011;11:685–692. doi: 10.1038/nri3062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yang L, Qian Y, Eksioglu E, et al. The inflammatory microenvironment in MDS. Cellular and molecular life sciences: CMLS. 2015;72:1959–1966. doi: 10.1007/s00018-015-1846-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yang C, McCoy K, Davis JL, et al. NIK stabilization in osteoclasts results in osteoporosis and enhanced inflammatory osteolysis. PloS one. 2010;5:e15383. doi: 10.1371/journal.pone.0015383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Holmfeldt P, Ganuza M, Marathe H, et al. Functional screen identifies regulators of murine hematopoietic stem cell repopulation. J Exp Med. 2016;213:433–449. doi: 10.1084/jem.20150806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Varney ME, Niederkorn M, Konno H, et al. Loss of Tifab, a del(5q) MDS gene, alters hematopoiesis through derepression of Toll-like receptor-TRAF6 signaling. J Exp Med. 2015;212:1967–1985. doi: 10.1084/jem.20141898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Carnevalli LS, Scognamiglio R, Cabezas-Wallscheid N, et al. Improved HSC reconstitution and protection from inflammatory stress and chemotherapy in mice lacking granzyme B. J Exp Med. 2014;211:769–779. doi: 10.1084/jem.20131072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cannova J, Breslin SJP, Zhang J. Toll-like receptor signaling in hematopoietic homeostasis and the pathogenesis of hematologic diseases. Front Med. 2015;9:288–303. doi: 10.1007/s11684-015-0412-0. [DOI] [PubMed] [Google Scholar]
- 33.Zhu J, Garrett R, Jung Y, et al. Osteoblasts support B-lymphocyte commitment and differentiation from hematopoietic stem cells. Blood. 2007;109:3706–3712. doi: 10.1182/blood-2006-08-041384. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.