

Abstract
A new series of blood-brain barrier permeable molecules designed to mimic the activity of Pleiotrophin in the CNS has been designed and synthesized. These compounds exert their action by interacting with the intracellular domain PD1 of the Protein Tyrosine-Phosphatase Receptor Z1 (PTPRZ1), and inhibiting its tyrosine phosphatase activity. The most potent compounds 10a and 12b (IC50 = 0,1 μM) significantly increase the phosphorylation of key tyrosine residues of PTPRZ1 substrates involved in neuronal survival and differentiation, and display protective effects against amphetamine-induced toxicity. Docking and molecular dynamics experiments have been used to analyze the binding mode and to explain the observed selectivity against PTP1B. An In vivo experiment has demonstrated that 10a can cross the BBB, thus promoting the possibility of moving forward these candidates for the development of drugs for the treatment of CNS disorders, such as drug addiction and neurodegenerative diseases.
Keywords: PTPRZ1, CNS disorders, Drug addiction, Molecular Dynamics, Synthesis
INTRODUCTION
Pleiotrophin (PTN) and Midkine (MK) are neurotrophic factors that share over 50% identity in amino acid sequence [1]. Both PTN and MK play important roles in development and repair of the central nervous system (CNS) [2]. The PTN and MK genes are widely expressed at different times in different cell types during development [3]. However, their expression levels are highly restricted to a few cell types in adults [4]. Both PTN and MK are upregulated at sites of injury, inflammation and repair in different cells of the CNS [5]. It is important to note that both PTN and MK regulate neuroinflammation depending on the inflammatory stimulus and the brain area considered [6]. In addition, MK and PTN are expressed in senile plaques of patients with Alzheimer’s disease [7] and PTN upregulation has been also observed in the substantia nigra of Parkinson’s Disease (PD) patient [8]. Both cytokines are also upregulated in different brain areas after administration of different drugs of abuse [9], suggesting PTN and MK signaling may be critical in different steps of wound repair in neurotoxic and neurodegenerative processes. Accordingly, in studies carried out in PTN knockout (PTN-/-) mice, it was shown that PTN prevents amphetamine-induced dopaminergic injury in the nigrostriatal pathway [10]. In concordance with these data, striatal overexpression of PTN in different mouse models of dopaminergic injury exerts neuroprotective effects [11].
These cytokines also modulate addictive behaviors. MK and PTN contribute to the extinction of cocaine- and amphetamine-seeking behaviors, respectively [10c, 12] PTN limits morphine withdrawal syndrome [13] and both, PTN and MK, are potent regulators of behavioral effects induced by ethanol [14]. Particularly, it has been demonstrated that PTN transgenic overexpression in the brain blocks the rewarding effects of alcohol [14a, 14b]. Overall, the data suggest that PTN and MK could be used for the treatment of neurodegenerative diseases, for prevention of drugs of abuse-induced neurotoxicity and for the treatment of a wide variety of drug addiction disorders. Like other proteins, PTN and MK exert suboptimal drug-like properties/inability to cross the blood-brain barrier (BBB). Although strategic drug delivery to the brain has been successfully used with other proteins [15], the risks in many occasions outweigh the benefits. If possible, pharmacological modulation of the signaling pathways triggered by PTN and MK may be preferred [9].
Both PTN and MK bind to the Protein Tyrosine-Phosphatase Receptor Z1 (PTPRZ1; a.k.a. (R)PTPβ or RPTPβ/ζ), induce its oligomerization and inactivate its phosphatase activity [16]. This leads to an increase in tyrosine phosphorylation of substrates critical for the effects of these cytokines such as β-catenin16b, Fyn kinase [17] and Anaplastic Lymphoma Kinase (ALK) [18]. We hypothesize that PTN and MK actions on neurodegenerative diseases and drug addiction disorders described above can be reproduced with rationally designed small molecule inhibitors of PTPRZ1 [5a, 9].
PTPRZ1 inhibitors have been recently tested in tumor models [19]. However, these molecules were unable to cross the cell membrane and required intracellular delivery by liposome carriers precluding use for the treatment of CNS diseases. In the present work, we propose to modulate the phosphatase activity of PTPRZ1 through the design and synthesis of small molecules that can cross the BBB and mimic the actions of PTN and MK.
Many known phosphatase inhibitors contain multi-charged phosphate-mimicking components, and show poor cellular uptake. Huang et al. [20] described, for the first time, one compound carrying an uncharged phosphate mimic (compound 1, Table 1) with a moderate activity against PTPRZ1 (IC50 = 3.5 μM), and a certain degree of selectivity against other related phosphatases. Considering 1 as a hit compound, we have followed a classical medicinal hit to lead optimization for the discovery of new PTPRZ1 inhibitors with a potential increased activity and selectivity and, more importantly, capable of crossing the BBB.
Table 1.
Values of calculated TPSA, logP/logD and results of the Phospho-Tyr and PTPRZ1 inhibition test at 1.0 μM
| Comp. | TPSA | LogP /LogD | Phospho-Tyr | PTPRZ1 inhibition at 1.0 μM |
|---|---|---|---|---|
| 1 | 94.27 | 5.35 | X | - |
| 4a | 79.70 | 7.26 | √ | X |
| 4b | 79.70 | 7.37 | √ | X |
| 4c | 79.70 | 6.99 | √ | √ |
| 4d | 79.70 | 6.88 | √ | √ |
| 5a | 114.14 | 4.88 | X | - |
| 5b | 114.14 | 5.00 | √ | √ |
| 5c | 114.14 | 4.62 | √ | X |
| 10a | 59.83 | 7.73 | √ | √ |
| 10b | 51.75 | 6.54 | X | - |
| 10c | 47.42 | 4.34 | √ | X |
| 12a | 62.63 | 7.55 | √ | X |
| 12b | 62.63 | 7.50/6.64 | √ | √ |
| 12c | 41.91 | 5.43/4.41 | X | |
| 12d | 37.33 | 5.48 | √ | √ |
| 12e | 37.33 | 5.99 | X | |
| 13 | 79.14 | 11.72/11.56 | √ | X |
RESULTS AND DISCUSSION
Design Rationale
The structure of compound 1 and all the analogs synthesized in this work is depicted in Schemes 1–3.
Scheme 1. Synthetic route to 4 and 5a.
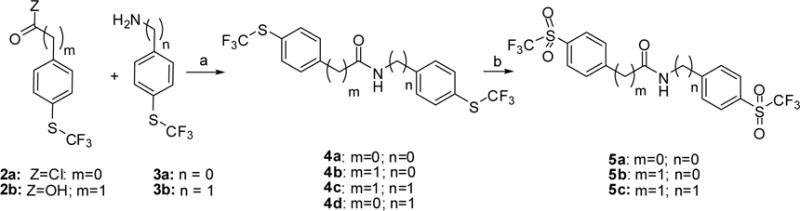
a Reagents and conditions: (a) for 4a and 4d: K2CO3, THF; for 4c and 4b: HOBt, EDCI, DMF; (b) MCPBA, DCM, 80 °C, sealed tube.
Scheme 3. Synthetic route to 12a-ea.
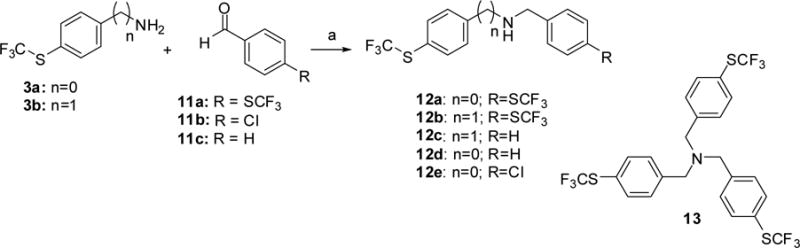
a Reagents and conditions: (a) NaBH(OAc)3, CH3CO2H or CF3CO2H, DCM.
We have analyzed the effect of the substitution in the aromatic rings present in 1, and the nature and length of the connecting linker. In compounds 5a-c, the ether linkage was substituted by amides of different length (Scheme 1). Topological Polar Surface Area (TPSA) for these sulfoxides and the possibility of establishing ten hydrogen bond acceptors (see table 1) predict a low bioavailability for a successful CNS drug [21]. To analyze whether the sulfoxide group is necessary for activity, and with the aim of improving pharmacokinetic properties, sulfides 4a-d, and 10a-b (Scheme 2) were also biologically tested. Calculated logP values for these sulfides are higher than the optimal 5 value. For this reason, sulfide 10c, where an aromatic ring was substituted by a pyridine, and sulfides 12a-e (Scheme 3) carrying an amino linker, were also selected for synthesis and biological evaluation.
Scheme 2. Synthetic route to 1 and 10.
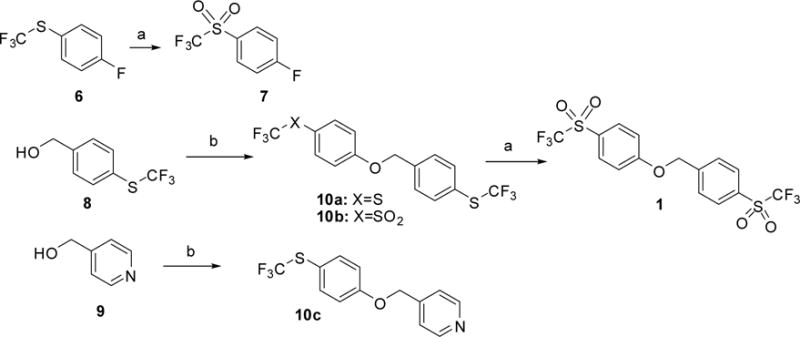
a Reagents and conditions: (a) MCPBA, DCM, 80 °C, sealed tube; (b) NaH, DMF and compound 6 for 10a and 10c respectively, or compound 7 for 10b.
Chemistry
The synthesis of compounds 4 and 5 is depicted in Scheme 1. Sulfides 4a and 4d were synthesized by reaction of 4-((trifluoromethyl)thio) benzoyl chloride (2a) and the corresponding amine 3. For the synthesis of amides 4b and 4c, a (EDCI) catalyzed coupling between 2-(4-((trifluoromethyl)thio)phenyl)acetic acid (2b) and the corresponding amine 3 was followed. Oxidation of 4a-c with m-chloroperbenzoic acid provided sulfones 5a-c.
Compounds 10 were synthesized by arylation of benzylic alcohols 8 and 9 with (4-fluorophenyl)(trifluoromethyl)sulfane (6) or 1-fluoro-4-(trifluoromethylsulfonyl)benzene (7), using sodium hydride as base (Scheme 2). Sulfones 7 and 1 were obtained by oxidation of 6 and 10a with m-chloroperbenzoic acid.
Amines 12a-e were obtained by reductive amination of aldehydes 11 with amines 3 using sodium triacetoxyborohydride as reductive agent (Scheme 3).
Reductive amination of 11a with 3b gave, together with the expected compound 12b, a tertiary amine 13, which was also included for biological evaluation.
Biological Evaluation
As a preliminary screening, we tested the total levels of phosphorylation of tyrosine residues in HeLa cells treated with different concentrations (0.1, 1.0 and 10.0 μM) of all 17 compounds (Phospho-Tyr in Table 1). The compounds that induced at least a 10% increase of Phospho-Tyr levels at any of the used concentrations were selected for further evaluation of inhibition of the phosphatase activity of PTPRZ1 (Figure 1S, see supplementary material). The selected compounds were 4a-d, 5b-c, 10a, 12a-b, 12d, and 13. A limitation of this approach is that HeLa cells constitutively express other tyrosine phosphatases that may compensate for the inhibition of PTPRZ1 induced by these compounds. Thus, it is possible that some of the inhibitors of PTPRZ1 that we designed and synthesized were discarded as false negatives. This may be the case of compound 1, the PTP1B inhibitor synthesized by Huang et al.20 that also exerted some inhibition on PTPRZ1 (IC50 = 3.5 μM) and other phosphatases. However, we chose this strict selection criterion to select the most potent inhibitors for the next phase.
Figure 1.
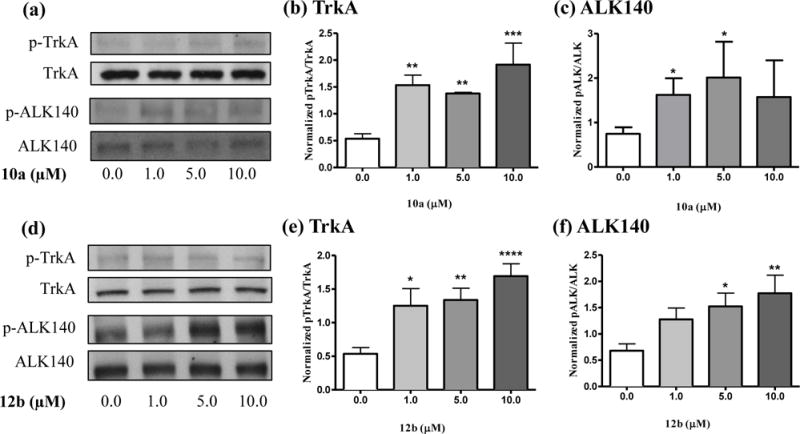
(a) Representative western blots showing 10a-induced increases in phosphorylated TrkA (pTrkA) and the 140 kDa ALK isoform (pALK). Total TrkA and ALK western blots are shown below each phosphorylated protein blot for comparison. (b–c) Quantification of western blots using ImageJ. (d) Representative western blots showing 12b-induced increases in pTrkA and pALK. Total. (e-f) Quantification of western blots using ImageJ. Data are presented as mean ± S.E.M. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 compared to vehicle controls (0.0 μM).
Next, we performed an enzymatic assay of PTPRZ1 using the selected 11 compounds mentioned above. After optimizing the assay conditions, we performed a screening test to assess the capacity of a single concentration (1.0 μM) of each of these compounds to induce at least a 10% inhibition of the phosphatase activity of PTPRZ1. Six compounds, 4c-d, 5b, 10a, 12b and 12d, met this criterion and underwent concentration-response studies to calculate the half-maximal inhibitory concentration (IC50). Among the remaining compounds, we found that 10a, 12b and 4c exhibited the most potent inhibition, with a range of IC50 values between 0.1 – 0.8 μM (Table 2). In contrast with the results of Huang et al [20], it should be noted that compound 1 failed to inhibit PTPRZ1 phosphatase activity in our assays. These apparent discrepancies may reflect significant methodological differences. First, while we have used a commercial PTPRZ1 protein, plasmids containing the PTPRZ1 active domain fragment were used in the previous report to subclone the fragment for expression in insect cells [20]. Second, we have used a different method of detection of inorganic phosphate in the PTPRZ1 enzymatic assay, the phosphate sensor reagent that binds inorganic phosphate in a rapid, tight (Kd ~ 0.1 μM) and more sensitive manner.
Table 2.
IC50 Values (in μM) of PTPRZ1 and PTP1B inhibition.
ND = Non Detected
We also assessed the inhibitor selectivity of all six compounds for PTP1B. We selected PTP1B as the most prominent PTP currently investigated as a pharmacological target [22]. As shown in table 2, 10a and 12b strongly and selectively inhibit PTPRZ1. Compound 4c, although less potent, displays a remarkable selectivity against PTP1B. To further study the selectivity of the most potent compounds, we decided to test their effects on the phosphorylation levels of specific substrates of PTPRZ1 in an in vitro biological assay. As relevant substrates of PTPRZ1, we chose TrkA [23] and anaplastic lymphoma kinase (ALK) [18] because they are known to be involved in the neuroprotective effects of PTN, the endogenous inhibitor of PTPRZ1 [9]. We used neuroblastoma SH-SY5Y cells, which are known to express PTPRZ1 [24]. We stimulated SH-SY5Y cells with different concentrations of 10a and 12b (1.0, 5.0 and 10.0 μM) for 20 minutes, and evaluated using western blots the phosphorylation of those specific tyrosine residues in TrkA (Tyr490) and ALK (Tyr1278), which are involved in the activation of both proteins. It has to be noted that previous evidence suggests that PTPRZ1 preferentially dephosphorylates Y674 and/or Y675 [23]. However, we chose to study Y490 because phosphorylation of this residue results in a cascade of molecular events and survival effects in vitro that resembles the ones found in PTN-stimulated cells [10c]. We chose to study ALK (Tyr1278) because treatment of SH-SY5Y cells with midkine (MK), the only other member of the PTN family of cytokines that also binds PTPRZ1 and inhibits its phosphatase activity [25], causes a significant increase in the phosphorylation of ALK (Tyr1278) in SH-SY5Y cells [26]. We chose these concentrations of the inhibitors as relevant for the subsequent functional studies described below. Western blots probed with anti-phospho-TrkA antibodies demonstrated that steady state levels of tyrosine phosphorylation of TrkA increased 2–3-fold after treatment with both 10a and 12b (Figure 1). Western blots probed with anti-phospho-ALK antibodies demonstrated that both compounds caused a 2-fold increase in the phosphorylation of one isoform of ALK (the 140 kDa protein [27]) (Figure 1).
The data clearly demonstrate that two of the inhibitors, 10a and 12b, significantly increase the phosphorylation of key tyrosine residues of the PTPRZ1 substrates TrkA and ALK, in agreement with their IC50 values (Table 2). It is interesting to note that TrkA is the high affinity nerve growth factor (NGF) receptor. NGF activates the kinase activity of TrkA by increasing the phosphorylation of Y490 in TrkA, which is critical for NGF-induced survival and neuroprotective effects [28]. Increased phosphorylation of Y1278 in ALK is also involved in neuronal survival and differentiation [9]. The data suggest that the ability of PTPRZ1 inhibitors to increase the phosphorylation of the same residues in TrkA and ALK, by inhibiting the phosphatase activity of PTPRZ1 on its substrates, will induce similar neuroprotective effects. Accordingly, the endogenous inhibitor of the phosphatase activity of PTPRZ1, PTN, has been shown to prevent amphetamine-induced neuronal injury in vitro and in vivo [10b, 10c]. To test the possibility that PTPRZ1 inhibitors could protect cell cultures from amphetamine-induced toxicity, we used catecholaminergic PC12 cells, which express readable levels of PTPRZ1 [29]. PC12 cells were incubated for 24 hours with amphetamine (1 mM) and/or 10a or 12b (1.0 μM). Interestingly, 10a significantly prevented amphetamine-induced loss of PC12 cell viability (Figure 2a) and the same trend was observed with 12b (p = 0.06). These data demonstrate that PTPRZ1 inhibitors mimic the protective effects of PTN against amphetamine-induced toxicity in PC12 cells [10c].
Figure 2.
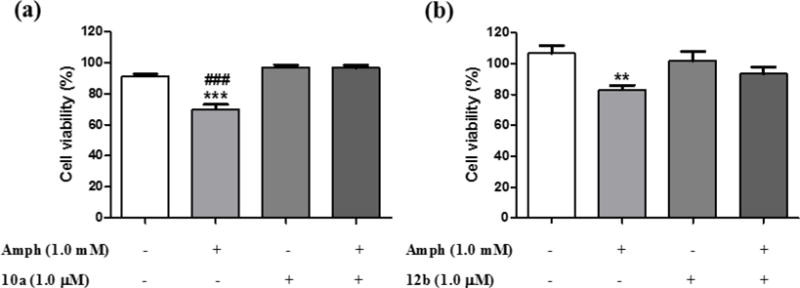
Effects of 10a and 12b on amphetamine-induced toxicity in PC12 cells. PC12 cells cultured with media supplemented with amphetamine (Amph; 1 mM) and/or 10a (1.0 μM) and/or 12b (1.0 μM) for 24 hours. Cellular viability was assessed by the MTT test. Results are expressed as mean ± S.E.M. **p < 0.01, ***p < 0.001 vs. vehicle-treated (control) cells. ###p < 0.001 vs. Amph+10a-treated cells.
In Vivo Permeability Study
To test the ability of the designed compounds to cross the BBB, compound 10a was selected to carry out an in vivo experiment. Samples were obtained from mice sacrificed 1h post-gavage with 60 mg/kg of 10a and analyzed by GC-MS. Extracted ion chromatograms (EIC) showed the presence of a peak at the same retention time as a standard 10a solution, in both plasma and brain samples, while this peak was not present in samples from untreated mice. A preliminary quantification of the permeability has been carried out, showing that the estimated concentration of 10a in brain samples is of 500–1000 ng/mL, while the concentration in plasma is 100–300 ng/mL. Moreover, we have recently shown that treatment with 10a completely blocked alcohol-induced reward in mice [30] and modulation of the rewarding effects of drugs of abuse such alcohol, requires direct actions in the brain.
Computational studies
Despite its biological interest, PTPRZ1 has not been thoroughly studied to date as a druggable target. The first crystal structure obtained for PTPRZ1 was deposited as 5AWX.19 However, inhibitors failed to crystalize along with the protein. To consider several conformations of the target macromolecule, together with the available apo form (5AWX), a computational study was carried out. Since no crystal structure was available for holo conformations of PTPRZ1, homology models were built using Receptor-type Tyrosine-Protein Phosphatase Gamma (PTPRG)19 as a template, based on the 73.61% of sequence homology between both phosphatases [31]. PTPRZ1 belongs to the receptor-associated Class I PTP subfamily, which contains a highly conserved phosphatase domain (PD1) and a cysteine in the active site [32]. The general structure of the PD1 is constituted by an active site motif C(X)5R [33] and several surrounding loops implicated in the catalytic cycle and substrate recognition process. The WPD-loop plays an acid-base role, carried out by the Trp-Pro-Asp motif and therefore is implicated in the recycling of the active site. The pTyr-loop [34] is implicated in the recognition of the phosphotyrosine substrate; and the Q-loop, has a relevant role in the activation of a key water molecule during the catalytic cycle. The templates used for the homology modeling can be classified according to the different conformations of the WPD-loop as follows: i) closed conformation: when the phosphatase domain is bound to a substrate-like ligand [35] or a competitive inhibitor (PDB code 3QCC) [35,36]; ii) a “superopen” conformation: when the phosphatase is bound to an inhibitor that alters the overall conformation of the WPD-loop (PDB code 3QCH) [35] by acting as a wedge between the WPD-loop and the rest of the protein; and iii) open conformation: when the protein is in the apo form (PDB code 5AWX) [33,37]. Models for PTPRZ1 were generated with the SWISS-MODEL web server [38]as described elsewhere [31]. They were subsequently used for docking experiments carried out as described in the experimental section, using compounds 4c, 4d, 5b, 10a, 12b and 12d, as ligands. The highest docking scores for all compounds inside the different conformations of PTPRZ1 were obtained for the “superopen” conformation. The general binding mode predicted for these compounds implies the enclosing of the trifluoromethylthiobenzyl moiety within the hydrophobic pocket that is accessible by the displacement of the WPD-loop. Among the amino acids that line the hydrophobic pocket, the main van der Waals interactions are established between the trifluoromethylthiobenzyl moiety and the aromatic side chains of Trp1989 and Phe1984, and the aliphatic and cycloaliphatic side chains of Arg1939 and Pro1905, respectively. The rest of the molecule, in all cases, interacts with the surroundings of the WPD-loop and the active site, with no direct interaction with the catalytic site (Table S1 and Figures S2, see Supplementary material).
To assess the stability of the proposed binding modes, 10 ns molecular dynamics (MD) simulations were carried out for all complexes. Binding energies were analyzed making use of the fast and versatile MM-ISMSA program [39]. Results confirm the complexes stability in the “superopen” conformation for all compounds bound to PTPRZ1 as can be seen by the stable RMSD values (Table S2 and Figure S3, see Supplementary material).
Given their high selectivity and drug-like properties, the binding modes for 4c, 10a and 12b were intensively inspected, and the most populated conformations explored during 97% (4c), 78% (10a) and 77% (12b) of the simulation time, respectively, were selected for the per-residue energy decompositions (Figure 3). Results show that the previously described van der Waals interactions established with the hydrophobic pocket in the initial docking binding models are maintained during most of the simulation time for the three complexes and that those having a higher energetic contribution are the π–π stacking and T-shaped interactions established between the trifluoromethylthiobenzyl moiety of the ligands and the aromatic side chains of Phe1984 and Trp1889. Furthermore, compound 4c interacts strongly with the side chains of Pro1900, Val1904 and the aliphatic part of the side chain of Glu1980 of the WPD-loop. In addition, the amide moiety of 4c stablishes alternating hydrogen bonding interactions to either the side chain NH of Gln1981 or the backbone carbonyls of Pro1905 and Glu1980. In the complex of 10a with PTPR1Z, the hydrophobic interactions persist, whereas electrostatic interactions are weaker, since the previously described hydrogen bonds cannot be established. This binding mode is shared by all the other studied compounds in their most populated conformers except for 12b-PTPRZ1 complex, because of its protonated state. Compound 12b can stablish strong interactions between the positively charged amine moiety and the side chains of Glu1980 and Asp1987 and with the backbone carbonyl of Glu1906, all three located at the entrance of the hydrophobic pocket. Furthermore, compound 12b, due to its high flexibility, folds to establish an intramolecular π–π interaction between the two trifluoromethylthiobenzyl moieties. In this folded conformation, compound 12b establishes van der Waals interactions between the trifluoromethylthiobenzyl moieties and the hydrophobic residues that line the hydrophobic pocket, some of which are located in the WPD-loop (Figure S4, see Supplementary material).
Figure 3.
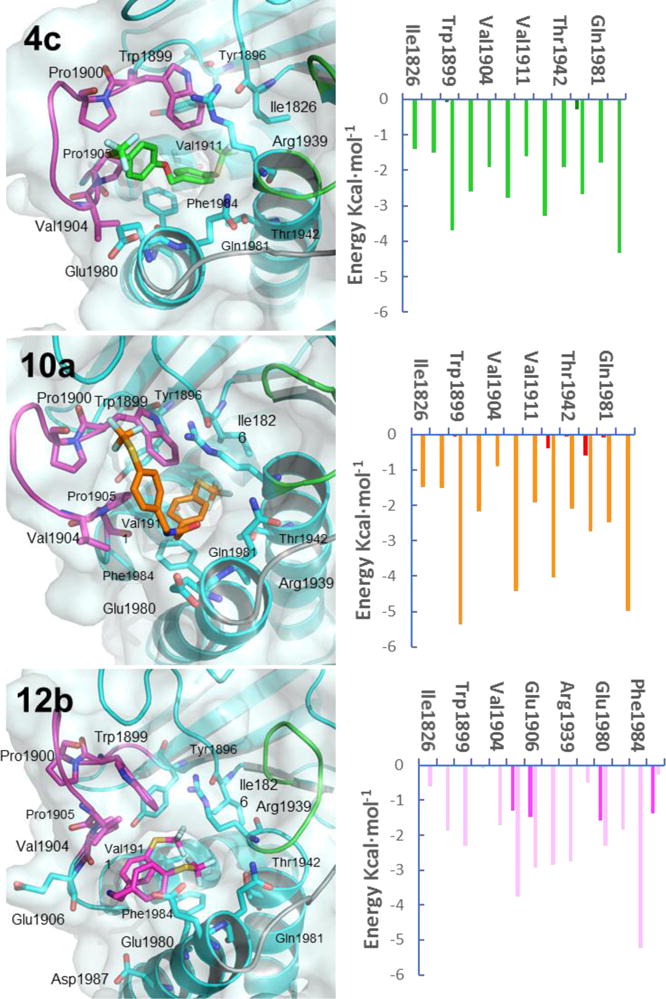
Proposed binding modes of 4c, 10a and 12b inside PTPRZ1. (Left) PyMOL stick and cartoon representation of the proposed binding modes for 4c, 10a and 12b inside PTPRZ1. Structures shown correspond to the most populated conformations along the MD simulations in the PTPRZ1 superopen conformation. 4c, 10a and 12b are shown in green, orange and light pink sticks, respectively. Residues involved in interactions are shown as sticks. The protein is colored cyan and the active site, WPD-loop and Q-loop are highlighted in green, magenta and grey, respectively. (Right) Per-residue energy decomposition of each complex with PTPRZ1. Orange, green and pale magenta bars represent the van der Waals contributions and deep green, red and magenta bars represent electrostatic binding energies for 4c, 10a and 12b.
With the aim of rationalizing the high selectivity displayed by 4c, 10a and 12b, the same computational approach was carried out for PTP1B. The PTP1B structures were obtained from the PDB in the closed and open conformation (PDB codes 1PTY and 3A5J, respectively) and a homology model for the “superopen” conformation was built using as template PDB structure 3QCH. Docking calculations were carried out as described previously and, in this case, the highest docking scores were obtained for all compounds bound to the closed conformation of PTP1B (Table S1, see Supplementary material) where they share an overall similar binding mode (Figure S5, see Supplementary material). Corresponding 10 ns MD simulations were carried out and the binding modes were further analyzed. Lower affinity towards PTP1B were found, as demonstrated by the unstable binding modes calculated through MM-ISMSA and the shifting RMSD values, that are comparatively higher than the ones obtained for the PTPRZ1 complexes (Table S2 and Figure S3, see supplementary material). It is noteworthy that the only stable binding modes in PTP1B, which have the most similar RMSD fluctuations if compared to those of PTPRZ1 complexes, are shown by compounds 4d and 5b along with 4c. The other compounds fluctuate during the MD simulation and slowly unbind from the original binding mode, resulting in completely different conformations that can be extracted from the MD simulations (Figure S6, see Supplementary material). The per-residue energy decompositions of the complexes between PTP1B and 4c, 10a and 12b were also carried out. The 4c-PTP1B complex showed worse energy interactions compared to those found for PTPRZ1. The main interactions established by compound 4c are with Tyr46, Asp48 and Val49 from the described P-loop, Phe182 from the WPD-loop, Ser216 and Ala217 from the active site, and Gln262 from the Q-loop (Figure S7, see Supplementary material). Meanwhile, compound 10a in complex with PTP1B, presented no stable binding mode during the simulations, which resulted in an unbounding from the active site, clearly observed by the rising RMSD values after 1.4 ns of the MD simulation and the reduced per-residue interactions of the binding. The complex of 12b and PTP1B rendered no stable binding within the active site during the simulation. Nevertheless, after ~ 2.5 ns 12b moves away from the active site and establishes a strong hydrogen bonding interaction between the protonated amine and the side chain of the Gln262 present in the Q-loop, which is maintained throughout the rest of the simulation (Figures S4 and S7, Supplementary material).
These results clearly account for the lower affinity towards PTP1B displayed by compounds 4c, 10a and 12b, and most importantly, they are in complete agreement with the remarkable PTPRZ1/PTP1B selectivity profile described above.
CONCLUSION
PTN has a protective role in the CNS through its interaction with PTPRZ1. The interaction of PTN with PTPRZ1 inactivates its intrinsic tyrosine phosphatase activity, increasing the phosphorylation level of substrates which are crucial to prevent neurodegenerative and drug addiction disorders.
In this work, we have designed and synthesized a series of small molecules capable of mimicking the activity of PTN, by interacting with the active site of the intracellular domain PD1 of PTPRZ1.
The most active compounds 10a and 12b (IC50 = 0,1 μM) are selective against PTP1B, and significantly increase the phosphorylation of key tyrosine residues of TrkA and ALK, two PTPRZ1 substrates involved in neuronal survival and differentiation. Moreover, both compounds mimic the PTN protective effects against amphetamine-induced toxicity in PC12 cells.
Docking and MD experiments have allowed to propose the binding mode of all these compounds inside PTPRZ1 and assess the stability of the corresponding complexes along the simulation time. More importantly, these methods have allowed to explain the remarkable PTPRZ1/PTP1B selectivity displayed by 4c and 10a.
We have demonstrated that 10a can cross the BBB and, therefore, we propose this compound as a promising candidate for the development of new drugs for the treatment of CNS disorders, such as addictive and neurodegenerative diseases.
EXPERIMENTAL SECTION
Chemistry
Materials
All reagents and solvents were obtained from different commercial sources (Sigma-Aldrich, Fluka, Acros Organics, Alfa Aesar or Scharlab) and used without further purification. Tetrahydrofuran and dichloromethane were dried by the solvent purification system Technical Bulletin AL-258.
General Methods
Reaction progress was monitored using analytical thin-layer chromatography (TLC) on Merck silica gel 60 F-254 plate. Visualization was achieved by UV light (254 nm). Flash chromatography was performed with Scharlau silica gel 60 (0.04–0.06 mm) packing. 1H and 13C spectra were recorded on a Bruker Advance III 400 MHz instrument. Signals are quoted as s (singlet), d (doublet), t (triplet), q (quartet) or m (multiplet). Chemical shifts (δ) are expressed in parts per million relative to solvent resonance as the internal standard. Coupling constants (J) are in hertz (Hz). Elemental analyses were performed by the Microanalytical Service Laboratory of the Universidad Complutense (Madrid) on a LECO CHNS-932 apparatus. Analyses indicated by the symbols of the elements or functions were within ± 0.4 % of the theoretical values. Melting points were determined on a Stuart Scientific (BIBBY) melting point apparatus. Mass spectrometry was performed by the CEMBIO Analytical Service Laboratory of the Universidad CEU San Pablo on a MS/IT Esquire 3000 Bruker Daltonics apparatus. All final products were analyzed for purity by reverse-phase high-performance liquid chromatography (HPLC) on Agilent Technologies 1260 Infinity II apparatus. HPLC methods used for each compound shown in Supplementary material.
1-((Trifluoromethyl)sulfonyl)-4-((4-((trifluoromethyl)sulfonyl)benzyl)oxy)benzene (1)
To a solution of 10a (153 mg, 0.39 mmol) in dry DCM (10 mL) was added MCPBA (688 mg, 3.98 mmol). The reaction mixture was stirred at 80 °C for 48 h in a sealed tube. The crude was washed with NaOH 1M, brine and dried (MgSO4). The drying agent was filtered off and the solvent was evaporated. The crude was purified by column chromatography on silica gel using hexane-EtOAc as eluent (8 : 2 v/v) to afford 120 mg (67%) of 1 as a white solid. Mp. 139–141 °C. 1H NMR (400 MHz, CDCl3): δ 8.11 (d, J = 8.2 Hz, 2H, ArH), 8.02 (d, J = 8.9 Hz, 2H, ArH), 7.76 (d, J = 8.2 Hz, 2H, ArH), 7.21 (d, J = 8.9 Hz, 2H, ArH), 5.33 (s, 2H, -CH2). 13C NMR (100 MHz, CDCl3): δ 164.6, 144.9, 133.5, 131.4, 131.4 (“q”, J = 2.0 Hz), 128.3, 123.4 (q, J = 2.0 Hz), 112.0 (q, JCF = 325.8 Hz), 112.0 (q, JCF = 325.8 Hz), 116.1, 69.3. HPLC Purity >98%, tR= 4.436, Method 1. Anal. C15H10F6O5S2 (C, H, F, O, S).
4-((Trifluoromethyl)thio)-N-(4-((trifluoromethyl)thio)phenyl)benzamide (4a)
To a solution of K2CO3 (174 mg, 1.26 mmol) in 4 mL of dry THF at 0 °C were subsequently added 11a (100 mg, 0.42 mmol) and aniline 3a (128 mg, 0.66 mmol). The resulting mixture was stirred at RT under argon till completion was seen by TLC (20 h). The solvent of the reaction was evaporated and the residue was suspended in H2O. The aqueous layer was extracted with EtOAc, and the combined organic layers were washed with 1M HCl and brine, dried (MgSO4), filtered and evaporated to give 4a as a white solid (31 mg, 19%). Mp. 162–164 °C (hexane/EtOAc). 1H NMR (400 MHz, CDCl3): δ 7.91 (d, J = 8.4 Hz, 2H, ArH), 7.87 (s, 1H, -NH), 7.80 (d, J = 8.4 Hz, 2H, ArH), 7.73 (d, J = 8.8 Hz, 2H, ArH), 7.68 (d, J = 8.8 Hz, 2H, ArH). 13C NMR (100 MHz, CDCl3): δ 164.8, 140.2, 137.8, 136.7, 136.4, 129.6(q, JCF = 308.3 Hz), 129.4 (q, JCF = 308.3 Hz), 129.3 (q, J = 2.0 Hz), 128.2, 120.8, 119.9(q, J = 2.0 Hz). HPLC Purity >98%, tR= 4.734, Method 5. LC/MS (ESI) m/z 398.0 [M+H]+. Anal. C15H9F6NOS2 (C, H, F, N, O, S).
N-2-Bis(4-((trifluoromethyl)thio)phenyl)acetamide (4b)
To a solution of acid 2b (200 mg, 0.64 mmol) in 2.5 mL of DMF were added HOBt (104 mg, 0.77 mmol), EDCI (139 mg, 0.90 mmol) and the corresponding aniline 3a (247 mg, 1.28 mmol). The resulting mixture was stirred at RT till completion was seen by TLC (72 h). The mixture was then diluted with EtOAc and washed with NaHCO3 saturated solution, NH4Cl and brine. The organic layer was dried (MgSO4), filtered and evaporated to give 4b as a white solid (208 mg, 78%). Mp. 163–165 °C (hexane/EtOAc). 1H NMR (400 MHz, CDCl3): δ 7.69 (d, J = 7.9 Hz, 2H, ArH), 7.59 (d, J = 8.5 Hz, 2H, ArH), 7.54 (d, J = 8.5 Hz, 2H, ArH), 7.41 (d, J = 7.9 Hz, 2H, ArH), 7.18 (s, 1H, -NH), 3.78 (s, 2H, -CH2). 13C NMR (100 MHz, CDCl3): δ 168.3, 140.1, 137.6, 137.1, 130.7, 129.6 (q, JCF = 308 Hz), 129.6 (q, JCF = 308 Hz), 124.1 (q, J = 2.0 Hz), 120.4, 119.5 (q, J = 2.0 Hz), 44.5. HPLC Purity >98%, tR= 3.690, Method 5. LC/MS (ESI) m/z 412.00 [M+H]+. Anal. C16H11F6NOS2 (C, H, F, N, O, S).
N-(4-((Trifluoromethyl)thio)benzyl)-2-(4-((trifluoromethyl)thio)phenyl)acetamide (4c)
To a solution of 2b (200 mg, 0.64 mmol) in 2.5 mL of DMF were added HOBt (104 mg, 0.77 mmol), EDCI (139 mg, 0.90 mmol) and amine 3b (199 mg, 0.96 mmol). The resulting mixture was stirred at RT till completion was seen by TLC (72 h). The mixture was then diluted with EtOAc and washed with NaHCO3 saturated solution, NH4Cl and brine. The organic layer was dried (MgSO4), filtered and evaporated to give 4c as a white solid (105 mg, 39%). Mp. 124–126 °C (hexane/EtOAc). 1H NMR (400 MHz, CDCl3): δ 7.65 (d, J = 8.1 Hz, 2H, ArH), 7.59 (d, J = 8.1 Hz, 2H, ArH), 7.35 (d, J = 8.2 Hz, 2H, ArH), 7.25 (d, J = 8.1 Hz, 2H, ArH), 5.75 (s, 1H, -NH), 4.47 (d, J = 6.0 Hz, 2H, -CH2-NH-), 3.65 (s, 2H, -CH2CO). 13C NMR (100 MHz, CDCl3) δ 170.1, 141.3, 137.9, 137.0, 136.8, 130.6, 129.6 (q, JCF = 308.2 Hz), 129.6 (q, JCF = 308.2 Hz), 128.6, 123.7 (q, J = 2.1 Hz), 123.6 (d, J = 2.1 Hz), 43.2, 43.1. HPLC Purity >97%, tR= 3.073, Method 6. LC/MS (ESI) m/z 425.9 [M+H]+. Anal. C17H13F6NOS2 (C, H, F, N, O, S).
4-((Trifluoromethyl)thio)-N-(4-((trifluoromethyl)thio)benzyl)benzamide (4d)
To a solution of K2CO3 (87 mg, 0.63 mmol) in 2 mL of dry THF at 0 °C were subsequently added chloride 2a (50 mg, 0.21 mmol) and amine 3b (68 mg, 0.33 mmol). The resulting mixture was stirred at RT under argon till completion was seen by TLC (20 h). The solvent of the reaction was evaporated and the residue was suspended in H2O. The aqueous layer was extracted with EtOAc, and the combined organic layers were washed with 1M HCl and brine, dried (MgSO4), filtered and evaporated. To give 4d as a white solid (119.5 mg, 34%). Mp. 117–119 °C (hexane/EtOAc). 1H NMR (400 MHz, CDCl3): δ 7.84 (d, J = 8.3 Hz, 2H, ArH), 7.74 (d, J = 8.3 Hz, 2H, ArH), 7.65 (d, J = 8.1 Hz, 2H, ArH), 7.41 (d, J = 8.1 Hz, 2H, ArH), 6.48 (s, 1H, -NH), 4.70 (d, J = 5.9 Hz, 2H, -CH2). 13C NMR (100 MHz, CDCl3) δ 166.5, 141.2, 136.9, 136.3, 129.7 (q, JCF = 308.3 Hz), 129.4 (q, JCF = 308.3 Hz), 128.9, 128.7 (q, J = 2.0 Hz), 128.1, 123.8 (q, J= 2.0 Hz), 43.7. HPLC Purity >98%, tR= 4.534, Method 5. LC/MS (ESI) m/z 412.00 [M+H]+. Anal. C16H11F6NOS2 (C, H, F, N, O, S).
4-((Trifluoromethyl)sulfonyl)-N-(4-((trifluoromethyl)sulfonyl)phenyl)benzamide (5a)
To a solution of 4a (65 mg, 0.16 mmol) in dry DCM (10 mL) was added MCPBA (276 mg, 1.6 mmol). The resulting mixture was stirred at 100 °C in a sealed tube until the reaction was completed (TLC) (72 h). The reaction mixture was washed with NaOH 2M, NaCl sat. and H2O. The organic layer was then dried (MgSO4), filtered and evaporated to give 5a as a white solid (40.6 mg, 55%). Mp. 194–196 °C (hexane/EtOAc). 1H NMR (400 MHz, CDCl3): δ 8.23 (d, J = 8.4 Hz, 2H, ArH), 8.17 (d, J = 8.4 Hz, 2H, ArH), 8.08 (d, J = 8.8 Hz, 2H, ArH), 8.00 (d, J = 8.8 Hz, 2H, ArH). 13C NMR (100 MHz, DMSO) δ 165.0, 147.0, 142.3, 132.4, 132.2, 131.2, 130.2, 123.0, 120.9, 119.5 (“q”, J = 324.11), 119.4 (“q”, J = 326.40). HPLC Purity >97%, tR= 4.277, Method 5. LC/MS (ESI) m/z 461.9 [M+H]+. Anal. C15H9F6NO5S2 (C, H, F, N, O, S).
N,2-bis(4-((trifluoromethyl)sulfonyl)phenyl)acetamide (5b)
To a solution of 4b (102 mg, 0.248 mmol) in dry DCM (10 mL) was added MCPBA (428 mg, 2.48 mmol). The resulting mixture was stirred at 100 °C in a sealed tube until the reaction was completed (TLC) (24 h). The reaction mixture was washed with NaOH 2M, NaCl sat. and H2O. The organic layer was then dried (MgSO4), filtered and evaporated to give 5b as a white solid (80 mg, 68%). Mp. 171–173 °C (hexane/EtOAc). 1H NMR (400 MHz, CDCl3): δ 8.07 (d, J = 8.3 Hz, 2H, ArH), 8.00 (d, J = 8.9 Hz, 2H, ArH), 7.83 (d, J = 8.9 Hz, 2H, ArH), 7.67 (d, J = 8.3 Hz, 2H, ArH), 7.57 (bs, 1H, -NH), 3.97 (s, 2H, -CH2).; 13C NMR (100 MHz, CDCl3) δ 167.6, 144.9, 143.0, 132.6, 131.5, 131.1, 130.8, 125.9,119.9, 119.9 (q, JCF = 325.7 Hz), 119.8 (q, JCF = 325.7 Hz), 44.4. HPLC Purity >97%, tR= 5.674, Method 3. LC/MS (ESI) m/z 475.9 [M+H]+. Anal. C16H11F6NO5S2 (C, H, F, N, O, S).
N-(4-((trifluoromethyl)sulfonyl)benzyl)-2-(4-((trifluoromethyl)sulfonyl)phenyl)acetamide (5c)
To a solution of 4c (42 mg, 0.1 mmol) in dry DCM (10 mL) was added MCPBA (260 mg, 1.5 mmol). The resulting mixture was stirred at 100 °C in a sealed tube until the reaction was completed (TLC) (48 h). The reaction mixture was washed with NaOH 2M, NaCl sat. and H2O. The organic layer was then dried (MgSO4), filtered and evaporated to give 5c as a white solid (33 mg, 67%). Mp. 164–170 °C (hexane/EtOAc). 1H NMR (400 MHz, CDCl3): δ 8.03 (d, J = 8.0 Hz, 2H, ArH), 7.99 (d, J = 8.0 Hz, 2H, ArH), 7.63 (d, J = 7.9 Hz, 2H, ArH), 7.54 (d, J = 7.9 Hz, 2H, ArH), 6.03 (bs, 1H, -NH), 4.60 (d, J = 6.1 Hz, 2H, -CH2NH), 3.77 (s, 2H, -CH2CO). 13C NMR (100 MHz, CDCl3) δ 169.1, 147.7, 140.03, 131.4, 131.0, 130.5, 130.47, 128.95, 119.9 (q, JCF = 323.7 Hz), 43.5, 43.3. HPLC Purity 95%, tR= 4.549, Method 3. LC/MS (ESI) m/z 489.9 [M+H]+. Anal. C17H13F6NO5S2 (C, H, F, N, O, S).
1-Fluoro-4-((trifluoromethyl)sulfonyl)benzene (7)
To a solution of 6 (100 mg, 0.51 mmol) in dry DCM (10 mL) MCPBA (440 mg, 2.55 mmol) was added and the mixture was heated at 80 °C in a sealed tube for 6 h. The reaction crude was diluted with DCM (10 mL) and the organic layer was washed with NaOH 1 M, brine and dried (MgSO4). The drying agent was filtered off and the solvent was removed at vacuum to give 7 as a light-orange oil. The compound was used in the next reaction without further purification.
(Trifluoromethyl)(4-((4-((trifluoromethyl)thio)benzyl)oxy)phenyl)sulfane (10a)
To a solution of 8 (303 mg, 1.46 mmol) in 5 mL of dry DMF, was added NaH (60%) (42 mg, 1.75 mmol) at 0 °C under an argon atmosphere. After stirring for 15 min, compound 6 (0.22 mL, 1.53 mmol) was added and the mixture was stirred at RT. The reaction was monitored with TLC until completion (24 h). The solvent was evaporated, the residue was solved in EtOAc, washed with brine and H2O, dried (MgSO4), filtered and evaporated. The crude was purified by column chromatography on silica gel using hexane-EtOAc as eluent (9.75 : 0.25 v/v) to afford 10a as a white solid (350 mg, 62%). Mp. 114–116 °C. 1H NMR (400 MHz, CDCl3): δ 7.69 (d, J = 8.2 Hz, 2H, ArH), 7.59 (d, J = 8.8 Hz, 2H, ArH), 7.48 (d, J = 8.2 Hz, 2H, ArH), 6.99 (d, J = 8.8 Hz, 2H, ArH), 5.12 (s, 2H, -CH2). 13C-NMR (100 MHz, CDCl3): δ 160.8, 139.5, 138.5, 136.8, 129.8 (q, JCF = 308 Hz), 129.7 (q, JCF = 308 Hz), 128.3, 124.4 (q, J = 2.1 Hz), 115.9, 115.8 (q, J = 2.1 Hz), 69.4. HPLC Purity >98%, tR = 5.215, Method 1. Anal. C15H10F6OS2 (C, H, F, O, S).
(Trifluoromethyl)(4-((4-((trifluoromethyl)sulfonyl)phenoxy)methyl)phenyl)sulfane (10b)
To a solution of 8 (106 mg, 0.51 mmol) in 5 mL of dry DMF, was added NaH (60%) (15 mg, 0.61 mmol) at 0 °C under an argon atmosphere. After stirring for 15 min, fluorine derivative 7 (116 mg, 0.51 mmol) was added, the mixture was stirred at RT and the reaction was monitored with TLC until completion (2 h). The reaction mixture was poured into a mixture of HCl (1M) / H2O (1:1) and then extracted with DCM. The organic layer was dried (MgSO4), filtered and evaporated. The crude was purified by column chromatography on silica gel using hexane-DCM as eluent (9:1 v/v) to afford 10b as a white solid (121 mg, 31%). Mp. 110–112 °C. 1H NMR (400 MHz, CDCl3): δ 7.98 (d, J = 8.9 Hz, 2H, ArH), 7.72 (d, J = 8.1 Hz, 2H, ArH), 7.49 (d, J = 8.1 Hz, 2H, ArH), 7.18 (d, J = 8.9 Hz, 2H, ArH), 5.22 (s, 2H, -CH2). 13C NMR (100 MHz, CDCl3): δ 164.8, 138.2, 136.7, 133.4, 129.5 (q, JCF = 308 Hz), 128.3, 124.7 (q, J = 2.0 Hz), 122.7 (q, J = 2.0 Hz), 119.9 (q, JCF = 308 Hz), 115.9, 69.8. HPLC Purity >97%, tR = 7.296, Method 2. Anal. C15H10F6O3S2 (C, H, F, O, S).
4-((4-((Trifluoromethyl)thio)phenoxy)methyl)pyridine (10c)
To a solution of 9 (111 mg, 1.02 mmol) in 5 mL of dry DMF, was added NaH (60%) (29 mg, 1.23 mmol) at 0 °C under an argon atmosphere. After stirring for 15 min, derivative 6 (0.15 mL, 1.02 mmol) was added, the mixture was stirred at RT until the reaction was completed (TLC) (72 h). The reaction mixture was poured into a mixture of H2O/Ice and the solution was taken to pH=7 with NaHCO3 saturated solution. The precipitate was then filtered and washed with H2O. The crude was purified by column chromatography on silica gel using hexane-EtOAc as eluent (2: 8 v/v) to afford 10c as a white solid (66 mg, 23%). Mp. 97–99 °C. 1H NMR (400 MHz, CDCl3): δ 8.63 (d, J = 4.6 Hz, 2H, ArH), 7.59 (d, J = 8.7 Hz, 2H, ArH), 7.34 (d, J = 5.2 Hz, 2H, ArH), 6.98 (d, J = 8.7 Hz, 2H, ArH), 5.11 (s, 2H,-CH2). 13C NMR (100 MHz, CDCl3): δ 160.4, 150.2, 145.3, 138.4, 129.6 (q, JCF = 308.2 Hz), 121.4, 115.9 (q, J = 2.0 Hz), 115.7, 68.3. HPLC Purity >98%, tR = 4.674, Method 3. LC/MS (ESI) m/z 286.0 [M+H]+. Anal. C13H10F3NOS (C, H, F, N, O, S).
4-((Trifluoromethyl)thio)-N-(4-((trifluoromethyl)thio)benzyl)aniline (12a)
To a solution of aldehyde 11a (200 mg, 1.04 mmol) and aniline 3a (214 mg, 1.04 mmol) in dry DCM (10 mL) were added, under argon, NaBH(OAc)3 (438 mg, 2.07 mmol) and CH3COOH (59 μL). The resulting solution was stirred at RT until the reaction was completed (TLC) (6 h). The reaction mixture was neutralized with NaOH 1M aqueous solution, and the organic phase was washed with brine and H2O, dried (MgSO4), filter and evaporated. Column chromatography on silica gel using as eluent hexane-EtOAc (9.5 : 0.5 v/v) gave 12a as a white solid (207 mg, 52%). Mp. 87–89 °C. 1H NMR (400 MHz, CDCl3): δ 7.65 (d, J = 8.1 Hz, 2H, ArH), 7.43 (d, J = 8.8 Hz, 2H, ArH), 7.41 (d, J = 8.1 Hz, 2H, ArH), 6.61 (d, J = 8.8 Hz, 2H, ArH), 4.42 (s, 2H, -CH2). 13C NMR (100 MHz, CDCl3): δ 149.9, 141.9, 138.5, 137.0, 130.0 (q, JCF = 308.1 Hz), 129.8 (q, JCF = 308.1 Hz), 128.5, 123.7 (q, J = 1.8 Hz), 113.6, 111.0 (q, J = 1.8 Hz), 47.6. HPLC Purity >98%, tR= 5.010, Method 4. Anal. C15H11F6NS2 (C, H, F, N, S).
Bis(4-((trifluoromethyl)thio)benzyl)amine hydrochloride (12b) & tris(4-((trifluoromethyl)thio)benzyl)amine (13)
To a solution of aldehyde 11a (237 mg, 1.15 mmol) and amine 3b (0.18 mL, 1.15 mmol) in dry DCM (10 mL) were added, under argon, NaBH(OAc)3 (1459 mg, 6.88 mmol) and CH3COOH (66 μL). The resulting solution was stirred at RT until the reaction was completed (TLC) (72 h). The reaction mixture was neutralized with NaOH 1M aqueous solution, and the organic phase was washed with brine and H2O, dried (MgSO4), filter and evaporated. Column chromatography on silica gel using hexane-EtOAc (gradient from 9.25:0.75 to pure EtOAc) as eluent, gave amines 13 (40 mg, 6%) and 12b (80 mg, 17%).
For 13: 1H NMR (400 MHz, CDCl3) δ 7.62 (d, J = 7.9 Hz, 6H, ArH), 7.45 (d, J = 7.9 Hz, 6H, ArH), 3.61 (s, 6H, 3x -CH2). Bubbling HCl(g) through an ethereal solution of the compound and isolation by filtration gave the corresponding hydrochloride (84%). Mp. 115–117 °C. HPLC Purity >94%, tR= 6.346, Method 5. LC/MS (ESI) m/z 588 [M+H]+. Anal. C24H19ClF9NS3 (C, H, Cl, F, N, S).
For 12b: 1H NMR (400 MHz, CDCl3): δ 7.59 (d, J = 7.9 Hz, 4H, ArH), 7.42 (d, J = 7.9 Hz, 4H, ArH); 3.60 (s, 4H, 2 × -CH2). 13C NMR (100 MHz, CDCl3): δ 142.1, 135.5, 128.6 (q, JCF= 309.0 Hz), 128.1, 121.8 (q, JCF= 2.0 Hz), 51.5.
Bubbling HCl(g) through an ethereal solution of the compound and isolation by filtration gave the corresponding hydrochloride (87%). Mp. 197–199 °C. HPLC Purity >98%, tR= 5.108, Method 5. LC/MS (ESI) m/z 398.00 [M+H]+. Anal. C16H14ClF6NS2 (C, H, Cl, F, N, S).
N-benzyl-1-(4-((trifluoromethyl)thio)phenyl)methanamine (12c)
To a solution of aldehyde 11c (0.048 mL, 0.483 mmol) and amine 3b (0.08 mL, 0.48 mmol) in dry DCM (5 mL) were added, under argon, NaBH(OAc)3 (204 mg, 0.97 mmol) and CF3COOH (96 μL). The resulting solution was stirred at RT until the reaction was completed (TLC) (48 h). The reaction mixture was neutralized with NaOH 1M aqueous solution, and the organic phase was washed with brine and H2O, dried (MgSO4), filtered and evaporated. Column chromatography on silica gel using as eluent hexane-DCM (gradient from 4:6 v/v to 1:1 v/v) gave 12c (50 mg, 34%). 1H NMR (400 MHz, CDCl3): δ 7.62 (d, J = 8.1 Hz, 2H), 7.42 (d, J = 8.1 Hz, 2H), 7.35–7.27 (m, 5H), 3.85 (s, 2H) 3.82 (s, 2H). 13C NMR (100 MHz, CDCl3): δ 143.5, 139.9, 136.6, 129.8 (q, JCF = 307.0 Hz), 129.3, 128.6, 128.3, 127.3, 122.8 (q, J = 2.0 Hz), 53.3, 52.5. Bubbling HCl(g) through an ethereal solution of the compound and isolation by filtration gave the corresponding hydrochloride (53%). Mp. 244–246 °C. HPLC Purity >97%, tR= 3.020, Method 4. LC/MS (ESI) m/z 298.00 [M+H]+. Anal. C15H15ClF3NS (C, H, Cl, F, N, S).
N-benzyl-4-((trifluoromethyl)thio)aniline (12d)
To a solution of aldehyde 11c (329 mg, 3.11 mmol) and aniline 3a (0.15 mL, 1.04 mmol) in dry DCM (10 mL) were added, under argon, NaBH(OAc)3 (1974 mg, 9.32 mmol) and CH3COOH (59 μL). The resulting solution was stirred at RT until the reaction was completed (TLC) (72 h). The reaction mixture was neutralized with NaOH 1M aqueous solution, and the organic phase was washed with brine and H2O, dried (MgSO4), filtered and evaporated. Column chromatography on silica gel using as eluent hexane-EtOAc (9.7:0.4 v/v) gave the 12d (83 mg, 73%). 1H NMR (400 MHz, CDCl3): δ 7.45 (d, J = 8.6 Hz, 2H, ArH), 7.39–7.33 (m, 5H, ArH), 6.63 (d, J = 8.6 Hz, 2H, ArH), 4.37 (s, 2H, -CH2). 13C NMR (100 MHz, CDCl3): δ 150.3, 138.5, 138.3, 129.9, (q, JCF = 308.0 Hz), 128.9, 127.7, 127.6, 113.3, 110.1 (q, J = 2.0 Hz), 48.0. Bubbling HCl(g) through an ethereal solution of the compound and isolation by filtration gave the corresponding hydrochloride (27%). Mp. 146–148 °C. HPLC Purity >98%, tR= 4.601, Method 5. LC/MS (ESI) m/z 284.00 [M+H]+. Anal. C14H13ClF3NS requires (C, H, Cl, F, N, S).
N-(4-chlorobenzyl)-4-((trifluoromethyl)thio)aniline (12e)
To a solution of aldehyde 11b (437 mg, 3.11 mmol) and aniline 3a (0.15 mL, 1.04 mmol) in dry DCM (10 mL) were added, under argon, NaBH(OAc)3 (1091 mg, 5.18 mmol) and then CH3COOH (1.05 mL). The resulting solution was stirred at RT until the reaction was completed (TLC) (48 h). The reaction mixture was neutralized with NaOH 1M aqueous solution, and the organic phase was washed with brine and H2O, dried (MgSO4), filtered and evaporated. Column chromatography on silica gel using as eluent hexane-DCM (9.8:0.2 v/v) gave 12e (160 mg, 49%). 1H NMR (400 MHz, CDCl3): δ 7.44 (d, J = 8.6 Hz, 2H, ArH), 7.34 (d, J= 8.5 Hz, 2H, ArH), 7.29 (d, J = 8.5 Hz, 2H, ArH), 6.60 (d, J = 8.7 Hz, 2H, ArH) 4.39 (“t”, J= 5.4 1H, -NH), 4.39 (d, J= 5.42 Hz, 2H, -CH2). 13C NMR (100 MHz, CDCl3): δ 150.0, 138.4, 137.1, 133.4, 129.8 (q, JCF = 308 Hz), 128.7, 128.4, 110.4 (q, J = 2 Hz), 47.3. Bubbling HCl(g) through an ethereal solution of the compound and isolation by filtration gave the corresponding hydrochloride (68%). Mp. 170–172 °C. HPLC Purity >98%, tR= 4.787, Method 5. LC/MS (ESI) m/z 318.00 [M+H]+. Anal. C14H12Cl2F3NS (C, H, Cl, F, N, S).
Molecular Modelling
The crystal structure of PTPRZ1 in the open conformation was obtained from the Protein Data Bank deposited under the accession code 5AWX. Homology models for the closed and “superopen” conformations of PTPRZ1 were built using the SWISS-MODEL web server [40] and using the phosphatase domain (PD1) of PTPRG in the PDB structures 3QCC and 3QCH, respectively, as templates. The crystal structures of the open (PDB code 3A5J) and closed (PDB code 1PTY) conformations of PTP1B were used, and the homology model of the “superopen” conformation of PTP1B was also built using the PDB structure 3QCH as template.
Using the Protein Preparation Wizard module of the Schrödinger Suite (http://www.Schrodinger.com), missing chains and residues were added to the crystal structures and the receptor geometries were optimized. The protonation states of charged amino acids were calculated with the PROPKA module of the Schrödinger Suite, and the catalytic cysteine is protonated in all PTPRZ1 and PTP1B structures. The described ligands were built with the LigPrep module of Maestro, generating all possible states at pH 7 +/- 2. Receptor grid was calculated using the catalytic cysteine as the center of the 13Å-size box that enclosed the catalytic site and the induced pocket from the superopen conformation, ensuring all possible ligand poses. Docking calculations were performed using extra precision (XP) mode of the GLIDE module and a van der Waals radii scale factor of 1.0/0.8. The best ligand poses were considered for further analysis of the ligand-receptor interactions using molecular dynamics (MD) simulations.
For the selected compounds, geometry optimization and charge distributions were calculated quantum mechanically (RHF/3–21G*//RHF/6–31G**) with Gaussian 03 [41]. MD simulations for each complex in the lowest energy docking pose for PTPRZ1 and PTP1B were carried out with the general AMBER14 (http://ambermd.org/) force field and the GAFF force field for the parametrization of the small molecules [42]. Systems were introduced in a truncated octahedron box of approximately 10 000 TIP3P water molecules with 13 Å cut-off distance and adding from 2 or 15 chlorine ions depending on the system. Smooth particle mesh Ewald (PME) [43] method with a spacing grid of 1Å was used for electrostatic interactions and SHAKE algorithm applied to all hydrogen bonds with 2.0 fs integration step [44]. An initial energy minimization of the water molecules and counter-ions was carried out on each system. The systems were further heated from 100K to 300K in 25 ps, and solvent molecules progressively allowed to move freely. To explore the complex between ligand and macromolecule 10 ns MD simulations where carried out without any restraints, generating snapshots each 20 ps for further analysis. The trajectories were collected and the cpptraj module [45] of AMBER14 was used to calculate the root mean square deviation (RMSD) of the atomic positions of the ligands and the most populated conformers. The MM-ISMSA program was used to calculate the total binding energies for each complex [39] giving the complex per-residue energy decomposition.
Biological Assays
ELISA
HeLa cells were cultured in RPMI-1640 medium (Sigma, Spain) supplemented with 10% fetal bovine serum (FBS; Sigma-Aldrich, Madrid, Spain), 0.005% Penicillin-Streptomycin and 0.005% Glutamine. Trypsin/EDTA (Sigma-Aldrich, Madrid, Spain) was used to release cells for subculturing. When cells reached 90% confluence they were seeded in 96 well-plates at 2 × 104 cells per well in triplicates. After 24 h of serum starvation, HeLa cells were treated for 10 min with 50 ng/mL epidermal growth factor (EGF; Sigma-Aldrich, Madrid, Spain) as a positive control to induce tyrosine phosphorylation. Cells were treated for 10 min with three different concentrations of each compound (10.0, 1.0 and 0.1 μM) and control 0.25% DMSO. A tyrosine phosphorylation ELISA Kit (Raybiotech, Norcross, GA, USA) was used following the manufacturer’s instructions.
In vitro dephosphorylation assays
Human recombinant PTPRZ1 and PTP1B proteins were purchased from Sigma (Madrid, Spain). The inhibitory activities of the studied compounds were determined using the phosphate sensor reagent (Thermofisher, Waltham, MA, USA) [46], using p-NPP (Sigma-Aldrich, Madrid, Spain) as substrate. PTPRZ1 and PTP1B were used at a concentration of 0.1 μM. Increasing concentrations of the different compounds were used (0.001–100 μM) to calculate the IC50. In each experiment, the hydrolysis of the p-NPP residue was determined as an increase in fluorescence at 450 nm (excitation at 430 nm) using a Hitachi F4500 Fluorescence Spectrometer.
Western-blots
The human neuroblastoma cell line SH-SY5Y was purchased from American Type Culture Collection (ATCC, Manassas, VA, USA) and was incubated at 37 °C in 5% CO2. SH-SY5Y cells were cultured in a 1:1 mixture of Eagle’s minimum essential medium (EMEM) and F12 medium containing 10% FBS. Just before treatments, medium was changed to 1:1 mixture of EMEM and F12 medium without serum. Cells were treated with 10a and 12b (1.0, 5.0 and 10.0 μM) and DMSO vehicle for 20 min. Cells were lysed in 20 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% Triton X-100, 2.5 mM sodium pyrophosphate, 1 mM β-glycerophosphate, leupeptin, 1 μg/mL aprotinin, and EDTA-free Complete Protease Inhibitor Cocktail tablets (Roche Diagnostics, Indianapolis, IN, USA). Lysate protein concentration was determined using the BCA Protein Assay Kit (Pierce, Rockford, IL, USA) and equal amounts of protein (20 μg) were subjected to sodium dodecyl sulfate–polyacrylamide gel electrophoresis and transferred to polyvinylidene difluoride membranes. Membranes incubated with anti-phospho-TrkA (Y490) and anti-phospho-ALK (Y1278) antibodies and reprobed with anti-TrkA (Upstate, Charlottesville, VA) and anti-ALK (Life Technologies, Carlsbad, CA) antibodies to confirm the identities of the proteins. Secondary antibodies were horseradish peroxidase-conjugated goat anti-mouse or anti-rabbit IgG. The membranes were developed with enhanced chemiluminescence detection reagents (Pierce, Rockford, IL, USA). Films were scanned using a Chemidoc System (Bio-Rad, Madrid, Spain) and densitometry was performed with ImageJ software.
Cell viability
Rat pheochromocytoma PC12 cells (ATCC, Manassas, VA, USA) were cultured with RPMI-1640 Medium supplemented with 10% FBS in 96-well plates (104 cells/well). To test the effects of 10a and 12b on amphetamine-induced toxicity in cell cultures, PC12 cells were incubated for 20 h with amphetamine (1.0 mM) and 10a or 12b (1.0 μM). Cellular viability was studied with the MTT assay (Sigma-Aldrich, Madrid, Spain). Briefly, cells were washed with PBS and incubated in fresh medium including 20 μL of 5 mg/mL MTT. MTT solution was removed after 4 h of incubation, DMSO added into each well and the plate was shaken for 10 min at RT. Absorbance was determined at 570 nm in a Versamax microplate reader (Bionova, Madrid, Spain).
Statistics
Data from Western blots and cell viability assays are presented as mean ± standard error of the mean (S.E.M.). Data were analyzed using one-way ANOVA followed by post-hoc comparisons with Tukey’s post-hoc tests. P < 0.05 was considered as statistically significant. All statistical analyses were performed using Graph-Pad Prism program (San Diego, CA, USA).
In vivo compound detection
Compound detection in plasma and brain tissue was performed using GC-MS analysis (Agilent 7890A gas chromatograph coupled to an Agilent 7200 accurate mass high resolution GC/Q-ToF). Separation was performed using an Agilent DB-5ms + DG capillary column (30 m × 0.25 mm i.d., 0.25 μm film thickness + 10 m Duraguard) using Helium as carrier gas. Mass analysis was operated on EI conditions, recording data in full-scan mode at 70 eV in a mass range of m/z 50 to 600. Ion source, quadrupole and transfer line temperatures were 250 °C, 150 °C and 290 °C respectively. The oven temperature program was: from 60 °C (1 min) to 325 °C (hold 10 min) at 20 °C/min.
Reliability of the method was based on the detection of a standard solution of 10a at a concentration of 2 mg/mL in ethanol. Then, a working standard solution was prepared by dilution of this stock solution at 5 μg/mL in heptane. Subsequent working solutions of the analyte were prepared by serial half dilution with heptane.
Biological samples were processed depending on their nature: i) frozen mouse brain tissues (50–100 mg) were placed in tubes containing CK14 ceramic beads from Precellys. ethyl acetate was added to each sample as extraction solvent and homogenization performed in a Precellys 24 Dual system equipped with a Cryolys cooler. Supernatants were evaporated to dryness in a speedvac concentrator and reconstituted in heptane before injection; ii) 100 μL of mouse plasma samples were subjected to standard protein precipitation. For this, three volumes of cold methanol were added and then samples maintained at -2 °C for 20 min. After centrifugation, supernatants were dried in a speedvac concentrator and the residue was reconstituted in heptane for further analysis.
Detection of 10a was performed on extracted ion chromatograms (EIC) at m/z 191.0137, corresponding to the fragment C8H6F3S. The 10a peak presented a limit of quantification (LOQ) in standards of 70 ng/mL, approximately.
Supplementary Material
Acknowledgments
This work was supported by Spanish MSSSI (PNSD2015I001), MINECO (CTQ2014-52604-R and SAF2014-53977-R) and the United States National Institute on Alcohol Abuse and Alcoholism (NIAAA, INIA consortium grant AA020912 to A.W.L.). B. di G, R. F-C and M. V-R thank Fundación Universitaria San Pablo CEU for FPI fellowships. B. di G. also thanks the Spanish MINECO for a FPU fellowship.
Abbreviations
- EDCI
1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide
- HOBt
hydroxybenzotriazole
- MCPBA
meta-chloroperbenzoic acid
- MK
Midkine
- PAMPA-BBB
membrane permeability assay for the blood-brain barrier
- PTN
Pleiotrophin
- PTPRZ1
Protein Tyrosine-Phosphatase Receptor Z1
- SDDB
strategic drug delivery to brain
- TPSA
topological polar surface area
Footnotes
SUPPLEMENTARY MATERIAL
HPLC data; 13C NMR and 1H NMR of spectra of compounds 1, 4a-d, 5a-c, 10a-c and 12a-e; Tyrosine phosphorylation levels in HeLa cells; Glide docking scores; MM-ISMSA binding energies; Pymol representations of complexes between PTPRZ1 or PTP1B and compounds 4c, 4d, 5b, 10a, 12b, and 12d; RMSD evolution for MD simulations.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
References
- 1.Kadomatsu K, Muramatsu T. Midkine and pleiotrophin in neural development and cancer. Cancer Lett. 2004;204:127–143. doi: 10.1016/S0304-3835(03)00450-6. [DOI] [PubMed] [Google Scholar]
- 2.Kadomatsu K, Kishida S, Tsubota S. The heparin-binding growth factor midkine: the biological activities and candidate receptors. J Biochem. 2013;153:511–521. doi: 10.1093/jb/mvt035. [DOI] [PubMed] [Google Scholar]
- 3.Deuel TF, Zhang N, Yeh HJ, Silos-Santiago I, Wang ZY. Pleiotrophin: a cytokine with diverse functions and a novel signaling pathway. Arch Biochem Biophys. 2002;397:162–171. doi: 10.1006/abbi.2001.2705. [DOI] [PubMed] [Google Scholar]
- 4.(a) Li YS, Milner PG, Chauhan AK, Watson MA, Hoffman RM, Kodner CM, Milbrandt J, Deuel TF. Cloning and expression of a developmentally regulated protein that induces mitogenic and neurite outgrowth activity. Science. 1990;250:1690–1694. doi: 10.1126/science.2270483. [DOI] [PubMed] [Google Scholar]; (b) Yeh HJ, He YY, Xu J, Hsu CY, Deuel TF. Upregulation of pleiotrophin gene expression in developing microvasculature, macrophages, and astrocytes after acute ischemic brain injury. J Neurosci. 1998;18:3699–3707. doi: 10.1523/JNEUROSCI.18-10-03699.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Kadomatsu K, Tomomura M, Muramatsu T. cDNA cloning and sequencing of a new gene intensely expressed in early differentiation stages of embryonal carcinoma cells and in mid-gestation period of mouse embryogenesis. Biochem Biophys Res Commun. 1988;151:1312–1318. doi: 10.1016/s0006-291x(88)80505-9. [DOI] [PubMed] [Google Scholar]
- 5.(a) Alguacil LF, Herradon G. Midkine and Pleiotrophin in the Treatment of Neurodegenerative Diseases and Drug Addiction. Recent Pat CNS Drug Discov. 2015;10:28–33. doi: 10.2174/1574889810666150326103916. [DOI] [PubMed] [Google Scholar]; (b) Martin YB, Herradon G, Ezquerra L. Uncovering new pharmacological targets to treat neuropathic pain by understanding how the organism reacts to nerve injury. Curr Pharm Res. 2011;17:434–448. doi: 10.2174/138161211795164130. [DOI] [PubMed] [Google Scholar]; (c) Muramatsu T. Midkine: a promising molecule for drug development to treat diseases of the central nervous system. Curr Pharm Res. 2011;17:410–423. doi: 10.2174/138161211795164167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.(a) Vicente-Rodriguez M, Rojo Gonzalez L, Gramage E, Fernandez-Calle R, Chen Y, Perez-Garcia C, Ferrer-Alcon M, Uribarri M, Bailey A, Herradon G. Pleiotrophin overexpression regulates amphetamine-induced reward and striatal dopaminergic denervation without changing the expression of dopamine D1 and D2 receptors: Implications for neuroinflammation. Eur Neuropsychopharm. 2016;29:1794–1805. doi: 10.1016/j.euroneuro.2016.09.002. [DOI] [PubMed] [Google Scholar]; (b) Vicente-Rodriguez M, Fernandez-Calle R, Gramage E, Perez-Garcia C, Ramos MP, Herradon G. Midkine is a novel regulator of amphetamine-induced striatal gliosis and cognitive impairment: evidence for a stimulus-dependent regulation of neuroinflammation by midkine. Mediators Inflamm. 2016;2016:9894504. doi: 10.1155/2016/9894504. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Fernandez-Calle R, Vicente-Rodriguez M, Gramage E, Pita J, Perez-Garcia C, Ferrer-Alcon M, Uribarri M, Ramos MP, Herradon G. Pleiotrophin regulates microglia-mediated neuroinflammation. J Neuroinflammation. 2017;14:46. doi: 10.1186/s12974-017-0823-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.(a) Wisniewski T, Lalowski M, Baumann M, Rauvala H, Raulo E, Nolo R, Frangione B. HB-GAM is a cytokine present in Alzheimer’s and Down’s syndrome lesions. NeuroReport. 1996;7:667–671. doi: 10.1097/00001756-199601310-00068. [DOI] [PubMed] [Google Scholar]; (b) Yasuhara O, Muramatsu H, Kim SU, Muramatsu T, Maruta H, McGeer PL. Midkine, a novel neurotrophic factor, is present in senile plaques of Alzheimer disease. Biochem Biophys Res Commun. 1993;192:246–251. doi: 10.1006/bbrc.1993.1406. [DOI] [PubMed] [Google Scholar]
- 8.Marchionini DM, Lehrmann E, Chu Y, He B, Sortwell CE, Becker KG, Freed WJ, Kordower JH, Collier TJ. Role of heparin binding growth factors in nigrostriatal dopamine system development and Parkinson’s disease. Brain Res. 2007;1147:77–88. doi: 10.1016/j.brainres.2007.02.028. [DOI] [PubMed] [Google Scholar]
- 9.Herradon G, Perez-Garcia C. Targeting midkine and pleiotrophin signalling pathways in addiction and neurodegenerative disorders: recent progress and perspectives. Br J Pharmacol. 2014;171:837–848. doi: 10.1111/bph.12312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.(a) Soto-Montenegro ML, Vicente-Rodriguez M, Perez-Garcia C, Gramage E, Desco M, Herradon G. Functional neuroimaging of amphetamine-induced striatal neurotoxicity in the pleiotrophin knockout mouse model. Neurosci Lett. 2015;591:132–137. doi: 10.1016/j.neulet.2015.02.041. [DOI] [PubMed] [Google Scholar]; (b) Gramage E, Rossi L, Granado N, Moratalla R, Herradon G. Genetic inactivation of pleiotrophin triggers amphetamine-induced cell loss in the substantia nigra and enhances amphetamine neurotoxicity in the striatum. Neuroscience. 2010;170:308–316. doi: 10.1016/j.neuroscience.2010.06.078. [DOI] [PubMed] [Google Scholar]; (c) Gramage E, Putelli A, Polanco MJ, Gonzalez-Martin C, Ezquerra L, Alguacil LF, Perez-Pinera P, Deuel TF, Herradon G. The neurotrophic factor pleiotrophin modulates amphetamine-seeking behaviour and amphetamine-induced neurotoxic effects: evidence from pleiotrophin knockout mice. Addict Biol. 2010;15:403–412. doi: 10.1111/j.1369-1600.2009.00202.x. [DOI] [PubMed] [Google Scholar]
- 11.(a) Gombash SE, Manfredsson FP, Mandel RJ, Collier TJ, Fischer DL, Kemp CJ, Kuhn NM, Wohlgenant SL, Fleming SM, Sortwell CE. Neuroprotective potential of pleiotrophin overexpression in the striatonigral pathway compared with overexpression in both the striatonigral and nigrostriatal pathways. Gene Ther. 2014;21(7):682–693. doi: 10.1038/gt.2014.42. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Gombash SE, Lipton JW, Collier TJ, Madhavan L, Steece-Collier K, Cole-Strauss A, Terpstra BT, Spieles-Engemann AL, Daley BF, Wohlgenant SL, Thompson VB, Manfredsson FP, Mandel RJ, Sortwell CE. Striatal pleiotrophin overexpression provides functional and morphological neuroprotection in the 6-hydroxydopamine model. Mol Ther. 2012;20:544–554. doi: 10.1038/mt.2011.216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gramage E, Perez-Garcia C, Vicente-Rodriguez M, Bollen S, Rojo L, Herradon G. Regulation of extinction of cocaine-induced place preference by midkine is related to a differential phosphorylation of peroxiredoxin 6 in dorsal striatum. Behav Brain Res. 2013;253:223–231. doi: 10.1016/j.bbr.2013.07.026. [DOI] [PubMed] [Google Scholar]
- 13.Gramage E, Vicente-Rodriguez M, Herradon G. Pleiotrophin modulates morphine withdrawal but has no effects on morphine-conditioned place preference. Neurosci Lett. 2015;604:75–79. doi: 10.1016/j.neulet.2015.07.022. [DOI] [PubMed] [Google Scholar]
- 14.(a) Vicente-Rodriguez M, Perez-Garcia C, Haro M, Ramos MP, Herradon G. Genetic inactivation of midkine modulates behavioural responses to ethanol possibly by enhancing GABA(A) receptor sensitivity to GABA(A) acting drugs. Behav Brain Res. 2014;274:258–263. doi: 10.1016/j.bbr.2014.08.023. [DOI] [PubMed] [Google Scholar]; (b) Vicente-Rodriguez M, Perez-Garcia C, Ferrer-Alcon M, Uribarri M, Sanchez-Alonso MG, Ramos MP, Herradon G. Pleiotrophin differentially regulates the rewarding and sedative effects of ethanol. J Neurochem. 2014;131:688–695. doi: 10.1111/jnc.12841. [DOI] [PubMed] [Google Scholar]; (c) Chen H, He D, Lasek AW. Midkine in the mouse ventral tegmental area limits ethanol intake and Ccl2 gene expression. Genes Brain Behav. 2017 Apr 11; doi: 10.1111/gbb.12384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Banks WA, Gertler A, Solomon G, Niv-Spector L, Shpilman M, Yi X, Batrakova E, Vinogradov S, Kabanov AV. Principles of strategic drug delivery to the brain (SDDB): development of anorectic and orexigenic analogs of leptin. Physiol Behav. 2011;105:145–149. doi: 10.1016/j.physbeh.2011.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.(a) Maeda N, Nishiwaki T, Shintani T, Hamanaka H, Noda M. 6B4 proteoglycan/phosphacan, an extracellular variant of receptor-like protein-tyrosine phosphatase zeta/RPTPbeta, binds pleiotrophin/heparin-binding growth-associated molecule (HB-GAM) J Biol Chem. 1996;271:21446–21452. doi: 10.1074/jbc.271.35.21446. [DOI] [PubMed] [Google Scholar]; (b) Meng K, Rodriguez-Pena A, Dimitrov T, Chen W, Yamin M, Noda M, Deuel TF. Pleiotrophin signals increased tyrosine phosphorylation of beta beta-catenin through inactivation of the intrinsic catalytic activity of the receptor-type protein tyrosine phosphatase beta/zeta. Proc Natl Acad Sci U S A. 2000;97:2603–2608. doi: 10.1073/pnas.020487997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pariser H, Ezquerra L, Herradon G, Perez-Pinera P, Deuel TF. Fyn is a downstream target of the pleiotrophin/receptor protein tyrosine phosphatase beta/zeta-signaling pathway: regulation of tyrosine phosphorylation of Fyn by pleiotrophin. Biochem Biophys Res Commun. 2005;332:664–669. doi: 10.1016/j.bbrc.2005.05.007. [DOI] [PubMed] [Google Scholar]
- 18.Perez-Pinera P, Zhang W, Chang Y, Vega JA, Deuel TF. Anaplastic lymphoma kinase is activated through the pleiotrophin/receptor protein-tyrosine phosphatase beta/zeta signaling pathway: an alternative mechanism of receptor tyrosine kinase activation. J Biol Chem. 2007;282:28683–28690. doi: 10.1074/jbc.M704505200. [DOI] [PubMed] [Google Scholar]
- 19.Fujikawa A, Nagahira A, Sugawara H, Ishii K, Imajo S, Matsumoto M, Kuboyama K, Suzuki R, Tanga N, Noda M, Uchiyama S, Tomoo T, Ogata A, Masumura M. Small-molecule inhibition of PTPRZ reduces tumor growth in a rat model of glioblastoma. Sci Rep. 2016;6:20473. doi: 10.1038/srep20473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huang P, Ramphal J, Wei J, Liang C, Jallal B, McMahon G, Tang C. Structure-based design and discovery of novel inhibitors of protein tyrosine phosphatases. Bioorg Med Chem. 2003;11:1835–1849. doi: 10.1016/s0968-0896(03)00039-7. [DOI] [PubMed] [Google Scholar]
- 21.Pajouhesh H, Lenz GR. Medicinal chemical properties of successful central nervous system drugs. NeuroRx. 2005;2:541–553. doi: 10.1602/neurorx.2.4.541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Verma M, Gupta SJ, Chaudhary A, Garg VK. Protein tyrosine phosphatase 1B inhibitors as antidiabetic agents - A brief review. Bioorg Chem. 2017;70:267–283. doi: 10.1016/j.bioorg.2016.12.004. [DOI] [PubMed] [Google Scholar]
- 23.Shintani T, Noda M. Protein tyrosine phosphatase receptor type Z dephosphorylates TrkA receptors and attenuates NGF-dependent neurite outgrowth of PC12 cells. J Biochem. 2008;144:259–266. doi: 10.1093/jb/mvn064. [DOI] [PubMed] [Google Scholar]
- 24.Abbott KL, Matthews RT, Pierce M. Receptor tyrosine phosphatase beta (RPTPbeta) activity and signaling are attenuated by glycosylation and subsequent cell surface galectin-1 binding. J Biol Chem. 2008;283:33026–33035. doi: 10.1074/jbc.M803646200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Maeda N, Ichihara-Tanaka K, Kimura T, Kadomatsu K, Muramatsu T, Noda M. A receptor-like protein-tyrosine phosphatase PTPzeta/RPTPbeta binds a heparin-binding growth factor midkine. Involvement of arginine 78 of midkine in the high affinity binding to PTPzeta. J Biol Chem. 1999;274:12474–12479. doi: 10.1074/jbc.274.18.12474. [DOI] [PubMed] [Google Scholar]
- 26.He D, Chen H, Muramatsu H, Lasek AW. Ethanol activates midkine and anaplastic lymphoma kinase signaling in neuroblastoma cells and in the brain. J Neurochem. 2015;135:508–521. doi: 10.1111/jnc.13252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Moog-Lutz C, Degoutin J, Gouzi JY, Frobert Y, Brunet-de Carvalho N, Bureau J, Creminon C, Vigny M. Activation and inhibition of anaplastic lymphoma kinase receptor tyrosine kinase by monoclonal antibodies and absence of agonist activity of pleiotrophin. J Biol Chem. 2005;280:26039–26048. doi: 10.1074/jbc.M501972200. [DOI] [PubMed] [Google Scholar]
- 28.Mok SA, Campenot RB. A nerve growth factor-induced retrograde survival signal mediated by mechanisms downstream of TrkA. Neuropharmacology. 2007;52:270–278. doi: 10.1016/j.neuropharm.2006.07.032. [DOI] [PubMed] [Google Scholar]
- 29.Gramage E, Alguacil LF, Herradon G. Pleiotrophin prevents cocaine-induced toxicity in vitro. Eur J Pharmacol. 2008;595:35–38. doi: 10.1016/j.ejphar.2008.07.067. [DOI] [PubMed] [Google Scholar]
- 30.Vicente-Rodriguez M, Fernandez-Calle R, Gramage E, Perez-Garcia C, Zapico JM, Coderch C, Pastor M, Di Geronimo B, De Pascual-Teresa B, Ramos A, Lasek AW, Herradon G. Preclinical development and evaluation of inhibitors of Receptor Protein Tyrosine Phosphatase beta/zeta for the treatment of alcohol use disorder. Alcohol Clin Exp Res. 2017;41(Suppl 1):110A. [Google Scholar]
- 31.Di Geronimo B, Coderch C, Herradón G, Ramos A, de Pascual-Teresa B. Exploring the Protein Tyrosine Phosphatase Receptor Z1 (PTPRZ1) in the search for new selective inhibitors for the prevention of alcohol abuse. Presented at 6th EUCHEMS Chemistry Congress; Seville, Spain. September 11-15, 2016; Abstract 999. [Google Scholar]
- 32.Tautz L, Critton DA, Grotegut S. Protein tyrosine phosphatases: structure, function, and implication in human disease. Methods Mol Biol. 2013;1053:179–221. doi: 10.1007/978-1-62703-562-0_13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Barr AJ, Ugochukwu E, Lee WH, King ON, Filippakopoulos P, Alfano I, Savitsky P, Burgess-Brown NA, Muller S, Knapp S. Large-scale structural analysis of the classical human protein tyrosine phosphatome. Cell. 2009;136:352–363. doi: 10.1016/j.cell.2008.11.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Brandao TA, Hengge AC, Johnson SJ. Insights into the reaction of protein-tyrosine phosphatase 1B: crystal structures for transition state analogs of both catalytic steps. J Biol Chem. 2010;285:15874–15883. doi: 10.1074/jbc.M109.066951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sheriff S, Beno BR, Zhai W, Kostich WA, McDonnell PA, Kish K, Goldfarb V, Gao M, Kiefer SE, Yanchunas J, Huang Y, Shi S, Zhu S, Dzierba C, Bronson J, Macor JE, Appiah KK, Westphal RS, O’Connell J, Gerritz SW. Small molecule receptor protein tyrosine phosphatase gamma (RPTPgamma) ligands that inhibit phosphatase activity via perturbation of the tryptophan-proline-aspartate (WPD) loop. J Med Chem. 2011;54:6548–6562. doi: 10.1021/jm2003766. [DOI] [PubMed] [Google Scholar]
- 36.Almo SC, Bonanno JB, Sauder JM, Emtage S, Dilorenzo TP, Malashkevich V, Wasserman SR, Swaminathan S, Eswaramoorthy S, Agarwal R, Kumaran D, Madegowda M, Ragumani S, Patskovsky Y, Alvarado J, Ramagopal UA, Faber-Barata J, Chance MR, Sali A, Fiser A, Zhang ZY, Lawrence DS, Burley SK. Structural genomics of protein phosphatases. J Struct Funct Genomics. 2007;8:121–140. doi: 10.1007/s10969-007-9036-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wiesmann C, Barr KJ, Kung J, Zhu J, Erlanson DA, Shen W, Fahr BJ, Zhong M, Taylor L, Randal M, McDowell RS, Hansen SK. Allosteric inhibition of protein tyrosine phosphatase 1B. Nat Struct Mol Biol. 2004;11:730–737. doi: 10.1038/nsmb803. [DOI] [PubMed] [Google Scholar]
- 38.Bordoli L, Kiefer F, Arnold K, Benkert P, Battey J, Schwede T. Protein structure homology modeling using SWISS-MODEL workspace. Nat Protoc. 2009;4:1–13. doi: 10.1038/nprot.2008.197. [DOI] [PubMed] [Google Scholar]
- 39.Klett J, Nunez-Salgado A, Dos Santos HG, Cortes-Cabrera A, Perona A, Gil-Redondo R, Abia D, Gago F, Morreale A. MM-ISMSA: an ultrafast and accurate scoring function for protein-protein docking. J Chem Theory Comput. 2012;8:3395–3408. doi: 10.1021/ct300497z. [DOI] [PubMed] [Google Scholar]
- 40.Guex N, Peitsch MC, Schwede T. Automated comparative protein structure modeling with SWISS-MODEL and Swiss-PdbViewer: a historical perspective. Electrophoresis. 2009;30(Suppl 1):S162–73. doi: 10.1002/elps.200900140. [DOI] [PubMed] [Google Scholar]
- 41.Frisch MJTGW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Montgomery JA, Jr, Vreven T, Kudin KN, Burant JC, Millam JM, Iyengar SS, Tomasi J, Barone V, Mennucci B, Cossi M, Scalmani G, Rega N, Petersson GA, Nakatsuji H, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Klene M, Li X, Knox JE, Hratchian HP, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Ayala PY, Morokuma K, Voth GA, Salvador P, Dannenberg JJ, Zakrzewski VG, Dapprich S, Daniels AD, Strain MC, Farkas O, Malick DK, Rabuck AD, Raghavachari K, Foresman JB, Ortiz JV, Cui Q, Baboul AG, Clifford S, Cioslowski J, Stefanov BB, Liu G, Liashenko A, Piskorz P, Komaromi I, Martin RL, Fox DJ, Keith T, Al-Laham MA, Peng CY, Nanayakkara A, Challacombe M, Gill PMW, Johnson B, Chen W, Wong MW, Gonzalez C, Pople JA. Gaussian 03, Revision C.02. Gaussian, Inc.; Wallingford CT: 2004. [Google Scholar]
- 42.Wang J, Wolf RM, Caldwell JW, Kollman PA, Case DA. Development and testing of a general amber force field. J Comput Chem. 2004;25:1157–1174. doi: 10.1002/jcc.20035. [DOI] [PubMed] [Google Scholar]
- 43.Darden T, York D, Pedersen L. Particle mesh Ewald: An N-log(N) method for Ewald sums in large systems. J Chem Phys. 1993;98:10089. [Google Scholar]
- 44.Lill MA. Efficient incorporation of protein flexibility and dynamics into molecular docking simulations. Biochemistry. 2011;50:6157–6169. doi: 10.1021/bi2004558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Roe DR, Cheatham TE., 3rd PTRAJ and CPPTRAJ: software for processing and analysis of molecular dynamics trajectory data. J Chem Theory Comput. 2013;9:3084–3095. doi: 10.1021/ct400341p. [DOI] [PubMed] [Google Scholar]
- 46.Balavenkatraman KK, Aceto N, Britschgi A, Mueller U, Bence KK, Neel BG, Bentires-Alj M. Epithelial protein-tyrosine phosphatase 1B contributes to the induction of mammary tumors by HER2/Neu but is not essential for tumor maintenance. Mol Cancer Res. 2011;9:1377–1384. doi: 10.1158/1541-7786.MCR-11-0198. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.